
Acute Lymphoblastic Leukemia Immunotherapy Treatment: Now, Next, and Beyond

 ,
,  ,
,  , and
, and 
Simple Summary
Abstract
1. Introduction
2. Different Biological Characteristics in Pediatric and Adults ALL Patients
3. Evolution of ALL Treatment Applications
4. Immunotherapy for ALL
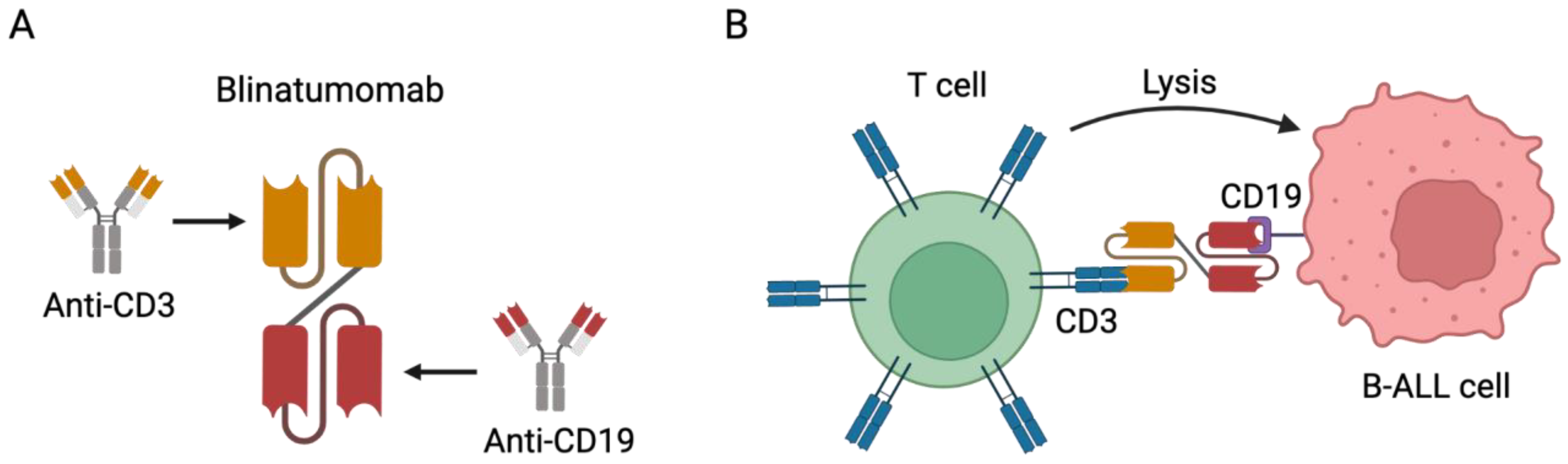
4.1. Bispecific Antibodies (BsAbs)
4.1.1. Blinatumomab in Adult Patients
4.1.2. Blinatumomab in Pediatric Patients
4.2. Antibody–Drug Conjugates (ADCs)
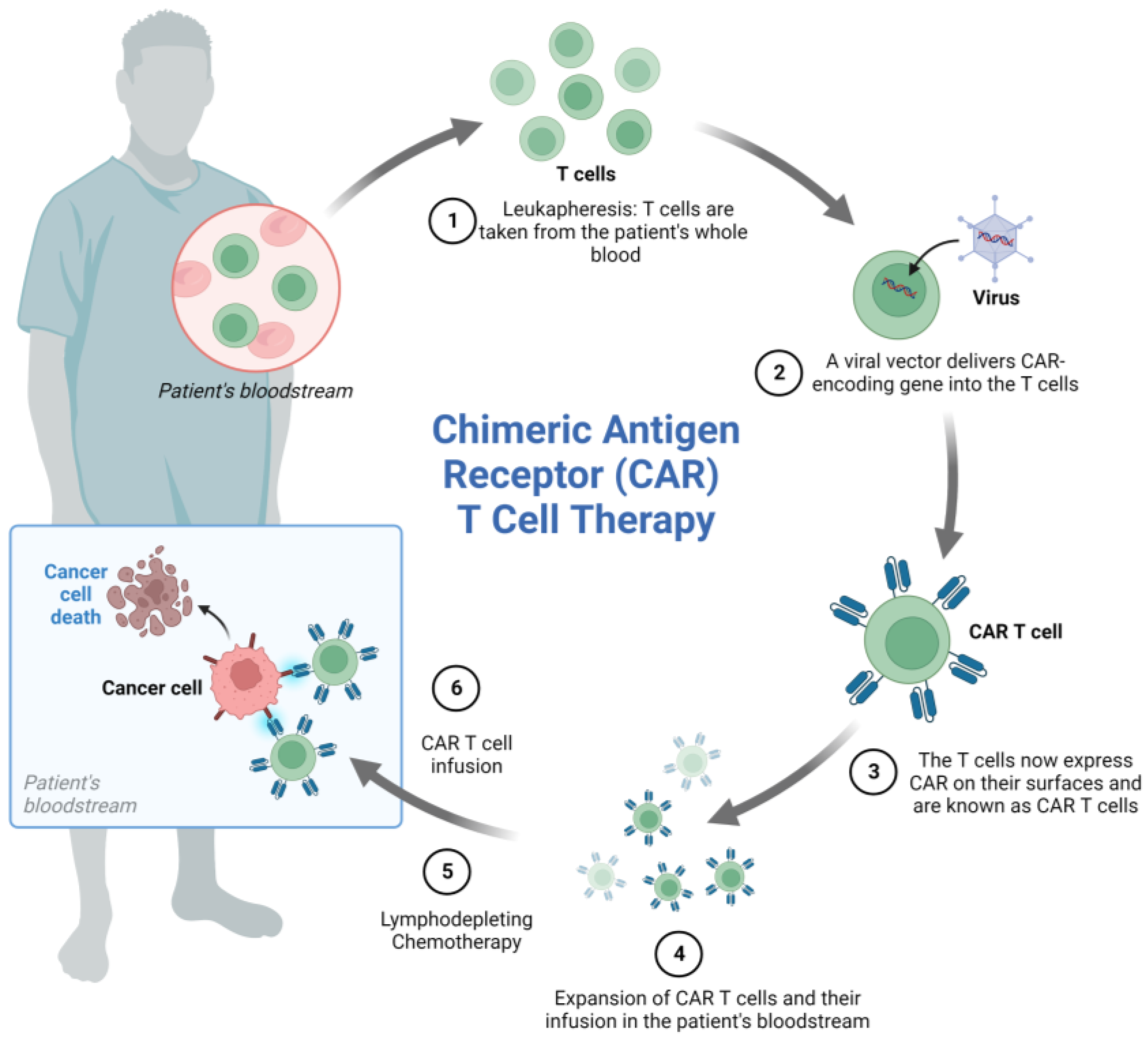
4.3. Chimeric Antigen Receptor (CAR)-Engineered Immune Cell Therapy in ALL
4.3.1. CART Cells
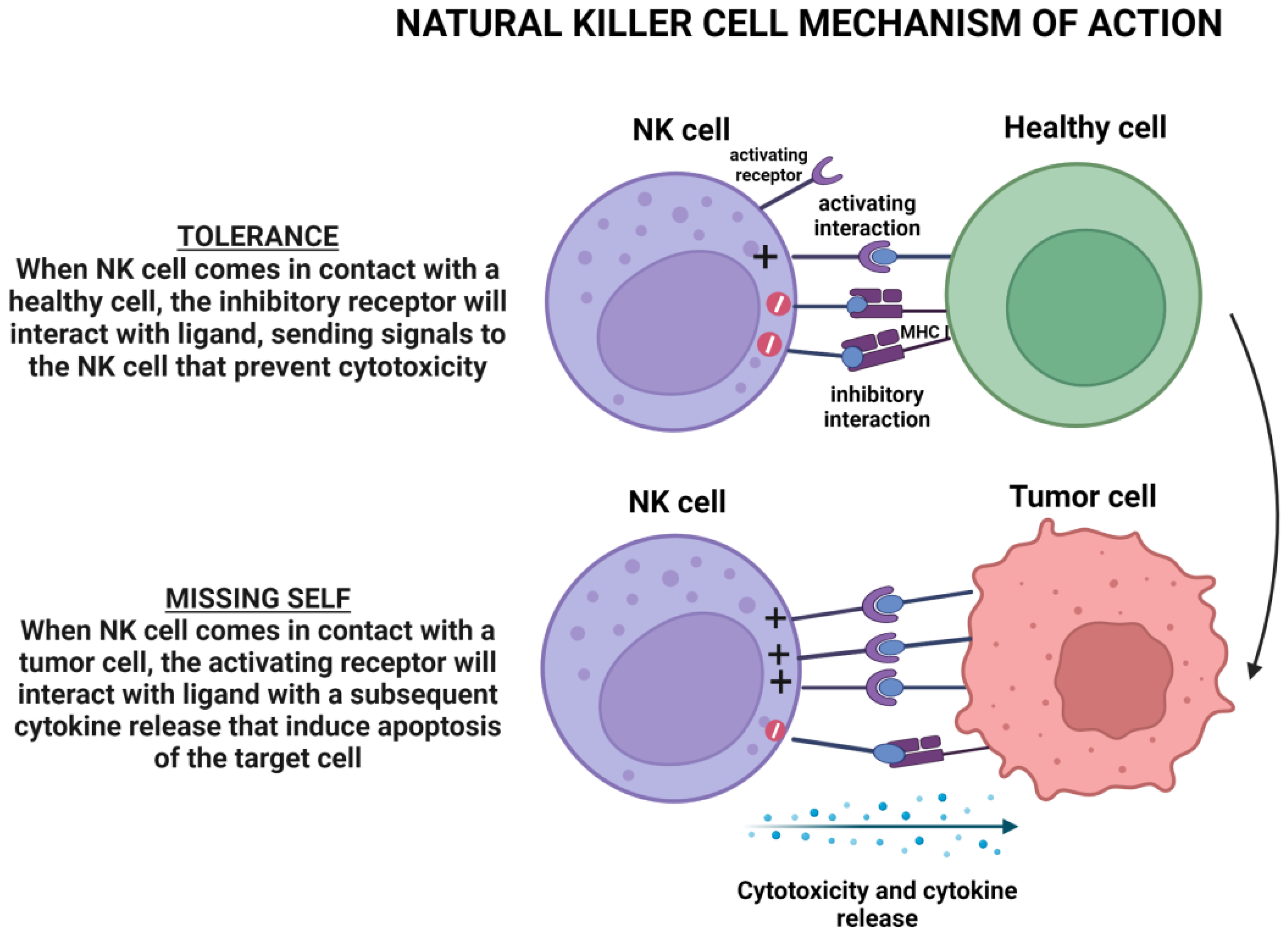
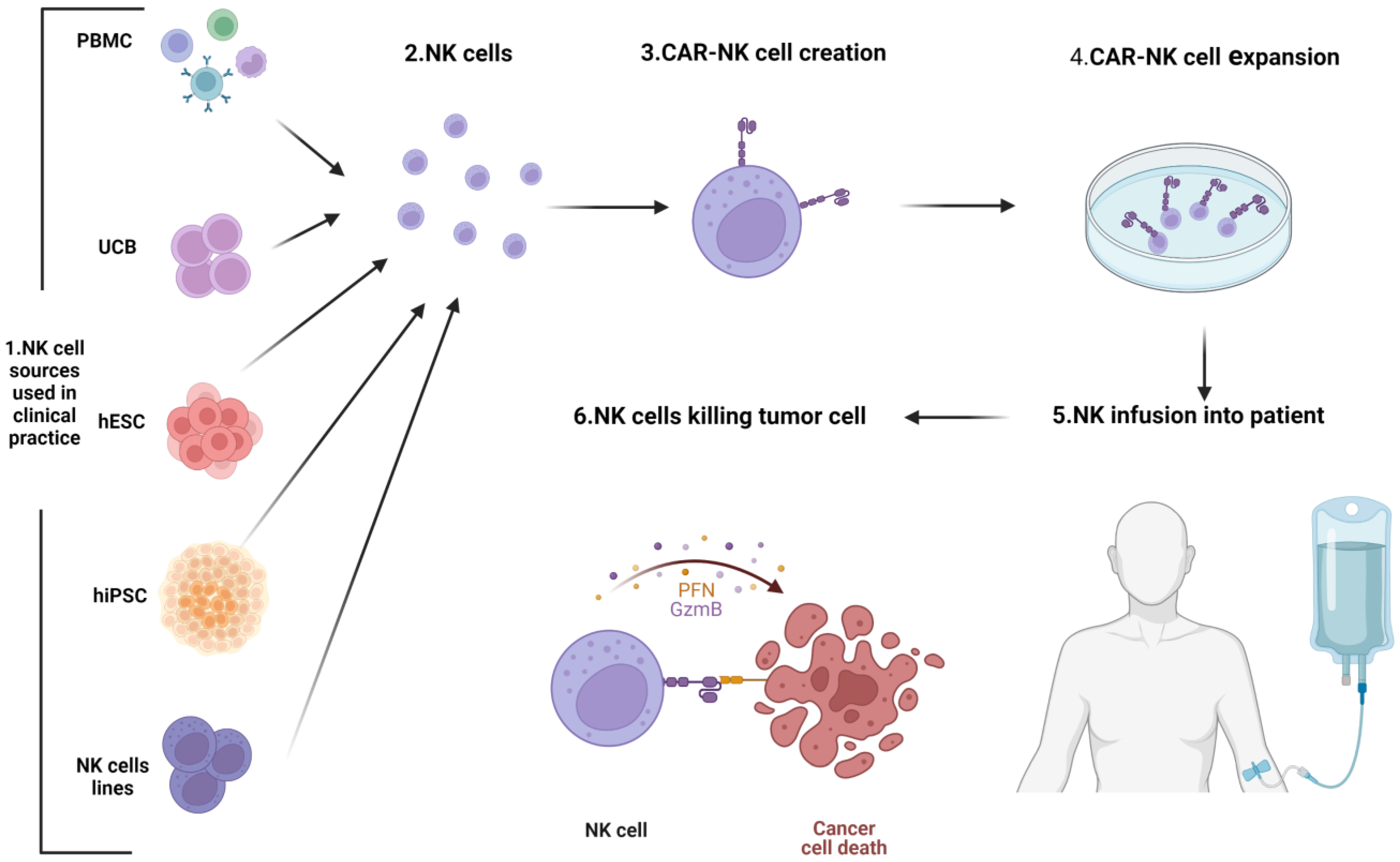
4.3.2. CARNK Cell Therapy
5. New Treatments under Investigation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Malard, F.; Mohty, M. Acute Lymphoblastic Leukaemia. Lancet 2020, 395, 1146–1162. [Google Scholar] [CrossRef]
- Terwilliger, T.; Abdul-Hay, M. Acute Lymphoblastic Leukemia: A Comprehensive Review and 2017 Update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef]
- Berg, S.L.; Steuber, P.; Poplack, D.G. Clinical Manifestations of Acute Lymphoblastic Leukemia. In Hematology, Basic Principles and Practice; Churchill Livingstone: Philadelphia, PA, USA, 2000. [Google Scholar]
- Del Principe, M.I.; Buzzatti, E.; Piciocchi, A.; Forghieri, F.; Bonifacio, M.; Lessi, F.; Imbergamo, S.; Orciuolo, E.; Rossi, G.; Fracchiolla, N.; et al. Clinical Significance of Occult Central Nervous System Disease in Adult Acute Lymphoblastic Leukemia. A Multicenter Report from the Campus ALL Network. Haematologica 2021, 106, 39–45. [Google Scholar] [CrossRef]
- Inaba, H.; Greaves, M.; Mullighan, C.G. Acute Lymphoblastic Leukaemia. Lancet 2013, 381, 1943–1955. [Google Scholar] [CrossRef]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.d.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th Edition of the World Health Organization Classification of Haemato lymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating Morphologic, Clinical, and Genomic Data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef]
- Spector, L.G.; Ross, J.A.; Robison, L.L.; Bhatia, S. Epidemiology and Etiology. In Childhood Leukemias, 2nd ed.; Cambridge University Press: Cambridge, UK, 2006; pp. 48–66. [Google Scholar] [CrossRef]
- Sehgal, S.; Mujtaba, S.; Gupta, D.; Aggarwal, R.; Marwaha, R.K. High Incidence of Epstein Barr Virus Infection in Childhood Acute Lymphocytic Lukemia: A Preliminary Study. Indian J. Pathol. Microbiol. 2010, 53, 63. [Google Scholar] [CrossRef]
- Geriniere, L.; Bastion, Y.; Dumontet, C.; Salles, G.; Espinouse, D.; Coiffier, B. Heterogeneity of Acute Lymphoblastic Leukemia in HFV-Seropositive Patients. Ann. Oncol. 1994, 5, 437–440. [Google Scholar] [CrossRef]
- Chessells, J.M.; Harrison, G.; Richards, S.M.; Bailey, C.C.; Hill, F.G.H.; Gibson, B.E.; Hann, I.M.; Bailey, C.C.; Barton, C.; Broadbent, V.; et al. Down’s Syndrome and Acute Lymphoblastic Leukaemia: Clinical Features and Response to Treatment. Arch. Dis. Child. 2001, 85, 321–325. [Google Scholar] [CrossRef]
- Dördelmann, M.; Schrappe, M.; Reiter, A.; Zimmermann, M.; Graf, N.; Schott, G.; Lampert, F.; Harbott, J.; Niemeyer, C.; Ritter, J.; et al. Down’s Syndrome in Childhood Acute Lymphoblastic Leukemia: Clinical Characteristics and Treatment Outcome in Four Consecutive BFM Trials. Leukemia 1998, 12, 645–651. [Google Scholar] [CrossRef]
- Bielorai, B.; Fisher, T.; Waldman, D.; Lerenthal, Y.; Nissenkorn, A.; Tohami, T.; Marek, D.; Amariglio, N.; Toren, A. Acute Lymphoblastic Leukemia in Early Childhood as the Presenting Sign of Ataxia-Telangiectasia Variant. Pediatr. Hematol. Oncol. 2013, 30, 574–582. [Google Scholar] [CrossRef]
- Toledano, S.R.; Lange, B.J. Ataxia-Telangiectasia and Acute Lymphoblastic Leukemia. Cancer 1980, 45, 1675–1678. [Google Scholar] [CrossRef]
- Lim, J.Y.S.; Bhatia, S.; Robison, L.L.; Yang, J.J. Genomics of Racial and Ethnic Disparities in Childhood Acute Lymphoblastic Leukemia. Cancer 2014, 120, 955–962. [Google Scholar] [CrossRef]
- Dores, G.M.; Devesa, S.S.; Curtis, R.E.; Linet, M.S.; Morton, L.M. Acute Leukemia Incidence and Patient Survival among Children and Adults in the United States, 2001–-2007. Blood 2012, 119, 34–43. [Google Scholar] [CrossRef]
- Feng, Q.; De Smith, A.J.; Vergara-Lluri, M.; Muskens, I.S.; McKean-Cowdin, R.; Kogan, S.; Brynes, R.; Wiemels, J.L. Trends in Acute Lymphoblastic Leukemia Incidence in the United States by Race/Ethnicity From 2000 to 2016. Am. J. Epidemiol. 2021, 190, 519–527. [Google Scholar] [CrossRef]
- Hallböök, H.; Gustafsson, G.; Smedmyr, B.; Söderhäll, S.; Heyman, M. Treatment Outcome in Young Adults and Children >10 Years of Age with Acute Lymphoblastic Leukemia in Sweden. Cancer 2006, 107, 1551–1561. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Redaelli, A.; Laskin, B.L.; Stephens, J.M.; Botteman, M.F.; Pashos, C.L. A Systematic Literature Review of the Clinical and Epidemiological Burden of Acute Lymphoblastic Leukaemia (ALL). Eur. J. Cancer Care 2005, 14, 53–62. [Google Scholar] [CrossRef]
- Allemani, C.; Weir, H.K.; Carreira, H.; Harewood, R.; Spika, D.; Wang, X.S.; Bannon, F.; Ahn, J.V.; Johnson, C.J.; Bonaventure, A.; et al. Global Surveillance of Cancer Survival 1995–2009: Analysis of Individual Data for 25 676 887 Patients from 279 Population-Based Registries in 67 Countries (CONCORD-2). Lancet 2015, 385, 977–1010. [Google Scholar] [CrossRef]
- Dong, Y.; Shi, O.; Zeng, Q.; Lu, X.; Wang, W.; Li, Y.; Wang, Q.; Wang, Q.; Wang, Q. Leukemia Incidence Trends at the Global, Regional, and National Level between 1990 and 2017. Exp. Hematol. Oncol. 2020, 9, 14. [Google Scholar] [CrossRef]
- Bassan, R.; Hoelzer, D. Modern Therapy of Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2011, 29, 532–543. [Google Scholar] [CrossRef]
- Pulte, D.; Gondos, A.; Brenner, H. Improvement in Survival in Younger Patients with Acute Lymphoblastic Leukemia from the 1980s to the Early 21st Century. Blood 2009, 113, 1408–1411. [Google Scholar] [CrossRef]
- Pui, C.H.; Pei, D.; Campana, D.; Bowman, W.P.; Sandlund, J.T.; Kaste, S.C.; Ribeiro, R.C.; Rubnitz, J.E.; Coustan-Smith, E.; Jeha, S.; et al. Improved Prognosis for Older Adolescents with Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2011, 29, 386–391. [Google Scholar] [CrossRef]
- Jabbour, E.; O’Brien, S.; Konopleva, M.; Kantarjian, H. New Insights into the Pathophysiology and Therapy of Adult Acute Lymphoblastic Leukemia. Cancer 2015, 121, 2517–2528. [Google Scholar] [CrossRef]
- Bonaventure, A.; Harewood, R.; Stiller, C.A.; Gatta, G.; Clavel, J.; Stefan, D.C.; Carreira, H.; Spika, D.; Marcos-Gragera, R.; Peris-Bonet, R.; et al. Worldwide Comparison of Survival from Childhood Leukaemia for 1995–2009, by Subtype, Age, and Sex (CONCORD-2): A Population-Based Study of Individual Data for 89 828 Children from 198 Registries in 53 Countries. Lancet Haematol. 2017, 4, e202–e217. [Google Scholar] [CrossRef]
- Ghosn, E.; Yoshimoto, M.; Nakauchi, H.; Weissman, I.L.; Herzenberg, L.A. Hematopoietic Stem Cell-Independent Hematopoiesis and the Origins of Innate-like B Lymphocytes. Development 2019, 146, dev170571. [Google Scholar] [CrossRef]
- Greaves, M.F.; Maia, A.T.; Wiemels, J.L.; Ford, A.M. Leukemia in Twins: Lessons in Natural History. Blood 2003, 102, 2321–2333. [Google Scholar] [CrossRef]
- Shin, S.Y.; Lee, H.; Lee, S.T.; Choi, J.R.; Jung, C.W.; Koo, H.H.; Kim, S.H. Recurrent Somatic Mutations and Low Germline Predisposition Mutations in Korean ALL Patients. Sci. Rep. 2021, 11, 8893. [Google Scholar] [CrossRef]
- Mancini, M.; Scappaticci, D.; Cimino, G.; Nanni, M.; Derme, V.; Elia, L.; Tafuri, A.; Vignetti, M.; Vitale, A.; Cuneo, A.; et al. A Comprehensive Genetic Classification of Adult Acute Lymphoblastic Leukemia (ALL): Analysis of the GIMEMA 0496 Protocol. Blood 2005, 105, 3434–3441. [Google Scholar] [CrossRef]
- Groupe Francais de Cytogenetique Hematologique. Cytogenetic Abnormalities in Adult Acute Lymphoblastic Leukemia: Correlations with Hematologic Findings and Outcome. A Collaborative Study of the Groupe Français de Cytogέnέtique Hέmatologique: By the Groupe FranGais de Cytogέnέtique Hέmatologique (Partic). Blood 1996, 87, 3135–3142. [Google Scholar] [CrossRef]
- Advani, A.S.; Hunger, S.P.; Burnett, A.K. Acute Leukemia in Adolescents and Young Adults. Semin. Oncol. 2009, 36, 213–226. [Google Scholar] [CrossRef]
- Litzow, M.R. Antigen-Based Immunotherapy for the Treatment of Acute Lymphoblastic Leukemia: The Emerging Role of Blinatumomab. Immunotargets Ther. 2014, 3, 79–89. [Google Scholar] [CrossRef][Green Version]
- Stary, J.; Zimmermann, M.; Campbell, M.; Castillo, L.; Dibar, E.; Donska, S.; Gonzalez, A.; Izraeli, S.; Janic, D.; Jazbec, J.; et al. Intensive Chemotherapy for Childhood Acute Lymphoblastic Leukemia: Results of the Randomized Intercontinental Trial ALL IC-BFM 2002. J. Clin. Oncol. 2014, 32, 174–184. [Google Scholar] [CrossRef]
- Beldjord, K.; Chevret, S.; Asnafi, V.; Huguet, F.; Boulland, M.L.; Leguay, T.; Thomas, X.; Cayuela, J.M.; Grardel, N.; Chalandon, Y.; et al. Oncogenetics and Minimal Residual Disease Are Independent Outcome Predictors in Adult Patients with Acute Lymphoblastic Leukemia. Blood 2014, 123, 3739–3749. [Google Scholar] [CrossRef]
- Pinkel, D. Five-Year Follow-Up of Total Therapy of Childhood Lymphocytic Leukemia. JAMA 1971, 216, 648–652. [Google Scholar] [CrossRef]
- Aur, R.J.; Simone, J.; Hustu, H.O.; Walters, T.; Borella, L.; Pratt, C.; Pinkel, D. Central Nervous System Therapy and Combination Chemotherapy of Childhood Lymphocytic Leukemia. Blood 1971, 37, 272–281. [Google Scholar] [CrossRef]
- Bleyer, W.A.; Poplack, D.G. Prophylaxis and Treatment of Leukemia in the Central Nervous System and Other Sanctuaries. Semin. Oncol. 1985, 12, 131–148. [Google Scholar]
- Anderson, F.S.; Kunin-Batson, A.S. Neurocognitive Late Effects of Chemotherapy in Children: The Past 10 Years of Research on Brain Structure and Function. Pediatr. Blood Cancer 2009, 52, 159–164. [Google Scholar] [CrossRef]
- Kadan-Lottick, N.S.; Zeltzer, L.K.; Liu, Q.; Yasui, Y.; Ellenberg, L.; Gioia, G.; Robison, L.L.; Krull, K.R. Neurocognitive Functioning in Adult Survivors of Childhood Non-Central Nervous System Cancers. J. Natl. Cancer Inst. 2010, 102, 881–893. [Google Scholar] [CrossRef]
- Bleyer, W.A. Neurologic Sequelae of Methotrexate and Ionizing Radiation: A New Classification. Cancer Treat. Rep. 1981, 65 (Suppl. 1), 89–98. [Google Scholar] [PubMed]
- Oeffinger, K.C.; Mertens, A.C.; Sklar, C.A.; Kawashima, T.; Hudson, M.M.; Meadows, A.T.; Friedman, D.L.; Marina, N.; Hobbie, W.; Kadan-Lottick, N.S.; et al. Chronic Health Conditions in Adult Survivors of Childhood Cancer. N. Engl. J. Med. 2006, 355, 1572–1582. [Google Scholar] [CrossRef]
- Pui, C.-H.; Cheng, C.; Leung, W.; Rai, S.N.; Rivera, G.K.; Sandlund, J.T.; Ribeiro, R.C.; Relling, M.V.; Kun, L.E.; Evans, W.E.; et al. Extended Follow-up of Long-Term Survivors of Childhood Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2003, 349, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Hijiya, N.; Hudson, M.M.; Lensing, S.; Zacher, M.; Onciu, M.; Behm, F.G.; Razzouk, B.I.; Ribeiro, R.C.; Rubnitz, J.E.; Sandlund, J.T.; et al. Cumulative Incidence of Secondary Neoplasms as a First Event After Childhood Acute Lymphoblastic Leukemia. JAMA 2007, 297, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Geenen, M.M.; Cardous-Ubbink, M.C.; Kremer, L.C.M.; Van Den Bos, C.; Van Der Pal, H.J.H.; Heinen, R.C.; Jaspers, M.W.M.; Koning, C.C.E.; Oldenburger, F.; Langeveld, N.E.; et al. Medical Assessment of Adverse Health Outcomes in Long-Term Survivors of Childhood Cancer. JAMA 2007, 297, 2705–2715. [Google Scholar] [CrossRef]
- Waber, D.P.; Turek, J.; Catania, L.; Stevenson, K.; Robaey, P.; Romero, I.; Adams, H.; Alyman, C.; Jandet-Brunet, C.; Neuberg, D.S.; et al. Neuropsychological Outcomes from a Randomized Trial of Triple Intrathecal Chemotherapy Compared with 18 Gy Cranial Radiation as CNS Treatment in Acute Lymphoblastic Leukemia: Findings from Dana-Farber Cancer Institute ALL Consortium Protocol 95-01. J. Clin. Oncol. 2007, 25, 4914–4921. [Google Scholar] [CrossRef]
- Schrappe, M.; Reiter, A.; Ludwig, W.-D.; Harbott, J.; Zimmermann, M.; Hiddemann, W.; Niemeyer, C.; Henze, G.; Feldges, A.; Zintl, F.; et al. Improved Outcome in Childhood Acute Lymphoblastic Leukemia despite Reduced Use of Anthracyclines and Cranial Radiotherapy: Results of Trial ALL-BFM 90. Blood 2000, 95, 3310–3322. [Google Scholar] [CrossRef]
- Silverman, L.B.; Gelber, R.D.; Dalton, V.K.; Asselin, B.L.; Barr, R.D.; Clavell, L.A.; Hurwitz, C.A.; Moghrabi, A.; Samson, Y.; Schorin, M.A.; et al. Improved Outcome for Children with Acute Lymphoblastic Leukemia: Results of Dana-Farber Consortium Protocol 91-01. Blood 2001, 97, 1211–1218. [Google Scholar] [CrossRef]
- Lukenbill, J.; Advani, A.S. The Treatment of Adolescents and Young Adults with Acute Lymphoblastic Leukemia. Curr. Hematol. Malig. Rep. 2013, 8, 91–97. [Google Scholar] [CrossRef]
- Rowe, J.M.; Buck, G.; Burnett, A.K.; Chopra, R.; Wiernik, P.H.; Richards, S.M.; Lazarus, H.M.; Franklin, I.M.; Litzow, M.R.; Ciobanu, N.; et al. Induction Therapy for Adults with Acute Lymphoblastic Leukemia: Results of More than 1500 Patients from the International ALL Trial: MRC UKALL XII/ECOG E2993. Blood 2005, 106, 3760–3767. [Google Scholar] [CrossRef]
- Williams, M.T.S.; Yousafzai, Y.M.; Elder, A.; Rehe, K.; Bomken, S.; Frishman-Levy, L.; Tavor, S.; Sinclair, P.; Dormon, K.; Masic, D.; et al. The Ability to Cross the Blood–Cerebrospinal Fluid Barrier Is a Generic Property of Acute Lymphoblastic Leukemia Blasts. Blood 2016, 127, 1998–2006. [Google Scholar] [CrossRef]
- Krishnan, S.; Wade, R.; Moorman, A.V.; Mitchell, C.; Kinsey, S.E.; Eden, T.O.B.; Parker, C.; Vora, A.; Richards, S.; Saha, V. Temporal Changes in the Incidence and Pattern of Central Nervous System Relapses in Children with Acute Lymphoblastic Leukaemia Treated on Four Consecutive Medical Research Council Trials, 1985–2001. Leukemia 2009, 24, 450–459. [Google Scholar] [CrossRef]
- Akers, S.M.; Rellick, S.L.; Fortney, J.E.; Gibson, L.F. Cellular Elements of the Subarachnoid Space Promote ALL Survival during Chemotherapy. Leuk. Res. 2011, 35, 705–711. [Google Scholar] [CrossRef]
- Gaynes, J.S.; Jonart, L.M.; Zamora, E.A.; Naumann, J.A.; Gossai, N.P.; Gordon, P.M. The Central Nervous System Microenvironment Influences the Leukemia Transcriptome and Enhances Leukemia Chemo-Resistance. Haematologica 2017, 102, e136–e139. [Google Scholar] [CrossRef]
- Jonart, L.M.; Ebadi, M.; Basile, P.; Johnson, K.; Makori, J.; Gordon, P.M. Disrupting the Leukemia Niche in the Central Nervous System Attenuates Leukemia Chemoresistance. Haematologica 2020, 105, 2130–2140. [Google Scholar] [CrossRef]
- Fernández-Sevilla, L.M.; Valencia, J.; Flores-Villalobos, M.A.; Gonzalez-Murillo, Á.; Sacedón, R.; Jiménez, E.; Ramírez, M.; Varas, A.; Vicente, Á. The Choroid Plexus Stroma Constitutes a Sanctuary for Paediatric B-Cell Precursor Acute Lymphoblastic Leukaemia in the Central Nervous System. J. Pathol. 2020, 252, 189–200. [Google Scholar] [CrossRef]
- Locatelli, F.; Schrappe, M.; Bernardo, M.E.; Rutella, S. How I Treat Relapsed Childhood Acute Lymphoblastic Leukemia. Blood 2012, 120, 2807–2816. [Google Scholar] [CrossRef]
- Goldstone, A.H.; Richards, S.M.; Lazarus, H.M.; Tallman, M.S.; Buck, G.; Fielding, A.K.; Burnett, A.K.; Chopra, R.; Wiernik, P.H.; Foroni, L.; et al. In Adults with Standard-Risk Acute Lymphoblastic Leukemia, the Greatest Benefit Is Achieved from a Matched Sibling Allogeneic Transplantation in First Complete Remission, and an Autologous Transplantation Is Less Effective than Conventional Consolidation. Blood 2008, 111, 1827–1833. [Google Scholar] [CrossRef]
- Cornelissen, J.J.; van der Holt, B.; Verhoef, G.E.; van’t Veer, M.B.; van Oers, M.H.; Schouten, H.C.; Ossenkoppele, G.; Sonneveld, P.; Maertens, J.; van Marwijk Kooy, M.; et al. Myeloablative Allogeneic versus Autologous Stem Cell Transplantation in Adult Patients with Acute Lymphoblastic Leukemia in First Remission: A Prospective Sibling Donor versus No-Donor Comparison. Blood 2009, 113, 1375–1382. [Google Scholar] [CrossRef]
- Jamieson, C.H.M.; Amylon, M.D.; Wong, R.M.; Blume, K.G. Allogeneic Hematopoietic Cell Transplantation for Patients with High-Risk Acute Lymphoblastic Leukemia in First or Second Complete Remission Using Fractionated Total-Body Irradiation and High-Dose Etoposide: A 15-Year Experience. Exp. Hematol. 2003, 31, 981–986. [Google Scholar] [CrossRef]
- Gruen, M.; Bommert, K.; Bargou, R.C. T-Cell-Mediated Lysis of B Cells Induced by a CD19xCD3 Bispecific Single-Chain Antibody Is Perforin Dependent and Death Receptor Independent. Cancer Immunol. Immunother. 2004, 53, 625–632. [Google Scholar] [CrossRef]
- Thiery, J.; Keefe, D.; Boulant, S.; Boucrot, E.; Walch, M.; Martinvalet, D.; Goping, I.S.; Bleackley, R.C.; Kirchhausen, T.; Lieberman, J. Perforin Pores in the Endosomal Membrane Trigger the Release of Endocytosed Granzyme B into the Cytosol of Target Cells. Nat. Immunol. 2011, 12, 770–777. [Google Scholar] [CrossRef]
- Trapani, J.A. Target Cell Apoptosis Induced by Cytotoxic T Cells and Natural Killer Cells Involves Synergy between the Pore-Forming Protein, Perforin, and the Serine Protease, Granzyme B. Aust. N. Z. J. Med. 1995, 25, 793–799. [Google Scholar] [CrossRef]
- Brischwein, K.; Schlereth, B.; Guller, B.; Steiger, C.; Wolf, A.; Lutterbuese, R.; Offner, S.; Locher, M.; Urbig, T.; Raum, T.; et al. MT110: A Novel Bispecific Single-Chain Antibody Construct with High Efficacy in Eradicating Established Tumors. Mol. Immunol. 2006, 43, 1129–1143. [Google Scholar] [CrossRef]
- Nagorsen, D.; Baeuerle, P.A. Immunomodulatory Therapy of Cancer with T Cell-Engaging BiTE Antibody Blinatumomab. Exp. Cell Res. 2011, 317, 1255–1260. [Google Scholar] [CrossRef]
- Hoffmann, P.; Hofmeister, R.; Brischwein, K.; Brandl, C.; Crommer, S.; Bargou, R.; Itin, C.; Prang, N.; Baeuerle, P.A. Serial Killing of Tumor Cells by Cytotoxic T Cells Redirected with a CD19-/CD3-Bispecific Single-Chain Antibody Construct. Int. J. Cancer 2005, 115, 98–104. [Google Scholar] [CrossRef]
- Lee, K.J.; Chow, V.; Weissman, A.; Tulpule, S.; Aldoss, I.; Akhtari, M. Clinical Use of Blinatumomab for B-Cell Acute Lymphoblastic Leukemia in Adults. Ther. Clin. Risk Manag. 2016, 12, 1301–1310. [Google Scholar] [CrossRef]
- Topp, M.S.; Gökbuget, N.; Stein, A.S.; Zugmaier, G.; O’Brien, S.; Bargou, R.C.; Dombret, H.; Fielding, A.K.; Heffner, L.; Larson, R.A.; et al. Safety and Activity of Blinatumomab for Adult Patients with Relapsed or Refractory B-Precursor Acute Lymphoblastic Leukaemia: A Multicentre, Single-Arm, Phase 2 Study. Lancet Oncol. 2015, 16, 57–66. [Google Scholar] [CrossRef]
- Shukla, N.; Sulis, M.L. Blinatumomab for Treatment of Children with High-Risk Relapsed B-Cell Acute Lymphoblastic Leukemia. JAMA 2021, 325, 830–832. [Google Scholar] [CrossRef]
- Gökbuget, N.; Dombret, H.; Bonifacio, M.; Reichle, A.; Graux, C.; Faul, C.; Diedrich, H.; Topp, M.S.; Brüggemann, M.; Horst, H.A.; et al. Blinatumomab for Minimal Residual Disease in Adults with B-Cell Precursor Acute Lymphoblastic Leukemia. Blood 2018, 131, 1522–1531. [Google Scholar] [CrossRef]
- Queudeville, M.; Ebinger, M. Blinatumomab in Pediatric Acute Lymphoblastic Leukemia—From Salvage to First Line Therapy (A Systematic Review). J. Clin. Med. 2021, 10, 2544. [Google Scholar] [CrossRef]
- van der Sluis, I.M.; de Lorenzo, P.; Kotecha, R.S.; Attarbaschi, A.; Escherich, G.; Nysom, K.; Stary, J.; Ferster, A.; Brethon, B.; Locatelli, F.; et al. Blinatumomab Added to Chemotherapy in Infant Lymphoblastic Leukemia. N. Engl. J. Med. 2023, 388, 1572–1581. [Google Scholar] [CrossRef]
- Stein, A.; Franklin, J.L.; Chia, V.M.; Arrindell, D.; Kormany, W.; Wright, J.; Parson, M.; Amouzadeh, H.R.; Choudhry, J.; Joseph, G. Benefit-Risk Assessment of Blinatumomab in the Treatment of Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Drug Saf. 2019, 42, 587–601. [Google Scholar] [CrossRef]
- Gaballa, M.R.; Banerjee, P.; Milton, D.R.; Jiang, X.; Ganesh, C.; Khazal, S.; Nandivada, V.; Islam, S.; Kaplan, M.; Daher, M.; et al. Blinatumomab Maintenance after Allogeneic Hematopoietic Cell Transplantation for B-Lineage Acute Lymphoblastic Leukemia. Blood 2022, 139, 1908–1919. [Google Scholar] [CrossRef]
- Sakaguchi, H.; Umeda, K.; Kato, I.; Sakaguchi, K.; Hiramatsu, H.; Ishida, H.; Yabe, H.; Goto, H.; Kawahara, Y.; Yamashita, Y.I.; et al. Safety and Efficacy of Post-Haematopoietic Cell Transplantation Maintenance Therapy with Blinatumomab for Relapsed/Refractory CD19-Positive B-Cell Acute Lymphoblastic Leukaemia: Protocol for a Phase I-II, Multicentre, Non-Blinded, Non-Controlled Trial (JPLSG SCT-ALL-BLIN21). BMJ Open 2023, 13, e070051. [Google Scholar] [CrossRef]
- Kantarjian, H.; Stein, A.; Gökbuget, N.; Fielding, A.K.; Schuh, A.C.; Ribera, J.-M.; Wei, A.; Dombret, H.; Foà, R.; Bassan, R.; et al. Blinatumomab versus Chemotherapy for Advanced Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2017, 376, 836–847. [Google Scholar] [CrossRef]
- Martinelli, G.; Boissel, N.; Chevallier, P.; Ottmann, O.; Gökbuget, N.; Topp, M.S.; Fielding, A.K.; Rambaldi, A.; Ritchie, E.K.; Papayannidis, C.; et al. Complete Hematologic and Molecular Response in Adult Patients with Relapsed/Refractory Philadelphia Chromosome-Positive B-Precursor Acute Lymphoblastic Leukemia Following Treatment with Blinatumomab: Results from a Phase II, Single-Arm, Multicenter Study. J. Clin. Oncol. 2017, 35, 1795–1802. [Google Scholar] [CrossRef]
- Foà, R.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Puzzolo, M.-C.; Canichella, M.; Viero, P.; Ferrara, F.; Lunghi, M.; et al. Dasatinib–Blinatumomab for Ph-Positive Acute Lymphoblastic Leukemia in Adults. N. Engl. J. Med. 2020, 383, 1613–1623. [Google Scholar] [CrossRef]
- Chiaretti, S.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Ferrara, F.; Lunghi, M.; Fabbiano, F.; Bonifacio, M.; Fracchiolla, N.; et al. S1617 A Dasatinib-Blinatumomab combination for the front-line treatment of adult Ph+ all patients. preliminary results of the gimema lal2116 d-alba trial; on behalf of gimema acute leukemia working party. Hemasphere 2019, 3, 746. [Google Scholar] [CrossRef]
- Handgretinger, R.; Zugmaier, G.; Henze, G.; Kreyenberg, H.; Lang, P.; Von Stackelberg, A. Complete Remission after Blinatumomab-Induced Donor T-Cell Activation in Three Pediatric Patients with Post-Transplant Relapsed Acute Lymphoblastic Leukemia. Leukemia 2010, 25, 181–184. [Google Scholar] [CrossRef]
- Schlegel, P.; Lang, P.; Zugmaier, G.; Ebinger, M.; Kreyenberg, H.; Witte, K.E.; Feucht, J.; Pfeiffer, M.; Teltschik, H.M.; Kyzirakos, C.; et al. Pediatric Posttransplant Relapsed/Refractory B-Precursor Acute Lymphoblastic Leukemia Shows Durable Remission by Therapy with the T-Cell Engaging Bispecific Antibody Blinatumomab. Haematologica 2014, 99, 1212–1219. [Google Scholar] [CrossRef]
- Von Stackelberg, A.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; Trippett, T.M.; Rizzari, C.; Bader, P.; O’brien, M.M.; Brethon, B.; Bhojwani, D.; et al. Phase I/Phase II Study of Blinatumomab in Pediatric Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2016, 34, 4381–4389. [Google Scholar] [CrossRef]
- Mejstríková, E.; Hrusak, O.; Borowitz, M.J.; Whitlock, J.A.; Brethon, B.; Trippett, T.M.; Zugmaier, G.; Gore, L.; Von Stackelberg, A.; Locatelli, F. CD19-Negative Relapse of Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia Following Blinatumomab Treatment. Blood Cancer J. 2017, 7, 659. [Google Scholar] [CrossRef]
- Zoghbi, A.; zur Stadt, U.; Winkler, B.; Müller, I.; Escherich, G. Lineage Switch under Blinatumomab Treatment of Relapsed Common Acute Lymphoblastic Leukemia without MLL Rearrangement. Pediatr. Blood Cancer 2017, 64, e26594. [Google Scholar] [CrossRef]
- Wadhwa, A.; Kutny, M.A.; Xavier, A.C. Blinatumomab Activity in a Patient with Down Syndrome B-Precursor Acute Lymphoblastic Leukemia. Pediatr. Blood Cancer 2018, 65, e26824. [Google Scholar] [CrossRef]
- Gore, L.; Locatelli, F.; Zugmaier, G.; Handgretinger, R.; O’Brien, M.M.; Bader, P.; Bhojwani, D.; Schlegel, P.G.; Tuglus, C.A.; von Stackelberg, A. Survival after Blinatumomab Treatment in Pediatric Patients with Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Blood Cancer J. 2018, 8, 80. [Google Scholar] [CrossRef]
- Wölfl, M.; Rasche, M.; Eyrich, M.; Schmid, R.; Reinhardt, D.; Schlegel, P.G. Spontaneous Reversion of a Lineage Switch Following an Initial Blinatumomab-Induced ALL-to-AML Switch in MLL-Rearranged Infant ALL. Blood Adv. 2018, 2, 1382–1385. [Google Scholar] [CrossRef]
- Elitzur, S.; Arad-Cohen, N.; Barzilai-Birenboim, S.; Ben-Harush, M.; Bielorai, B.; Elhasid, R.; Feuerstein, T.; Gilad, G.; Gural, A.; Kharit, M.; et al. Blinatumomab as a Bridge to Further Therapy in Cases of Overwhelming Toxicity in Pediatric B-Cell Precursor Acute Lymphoblastic Leukemia: Report from the Israeli Study Group of Childhood Leukemia. Pediatr. Blood Cancer 2019, 66, e27898. [Google Scholar] [CrossRef]
- Keating, A.K.; Gossai, N.; Phillips, C.L.; Maloney, K.; Campbell, K.; Doan, A.; Bhojwani, D.; Burke, M.J.; Verneris, M.R. Reducing Minimal Residual Disease with Blinatumomab Prior to HCT for Pediatric Patients with Acute Lymphoblastic Leukemia. Blood Adv. 2019, 3, 1926–1929. [Google Scholar] [CrossRef]
- Mouttet, B.; Vinti, L.; Ancliff, P.; Bodmer, N.; Brethon, B.; Cario, G.; Chen-Santel, C.; Elitzur, S.; Hazar, V.; Kunz, J.; et al. Durable Remissions in TCF3-HLF Positive Acute Lymphoblastic Leukemia with Blinatumomab and Stem Cell Transplantation. Haematologica 2019, 104, e244–e247. [Google Scholar] [CrossRef]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Gore, L.; et al. A Randomized Phase 3 Trial of Blinatumomab Vs. Chemotherapy As Post-Reinduction Therapy in High and Intermediate Risk (HR/IR) First Relapse of B-Acute Lymphoblastic Leukemia (B-ALL) in Children and Adolescents/Young Adults (AYAs) Demonstrates Superior Eff. Blood 2019, 134, LBA-1. [Google Scholar] [CrossRef]
- Locatelli, F.; Whitlock, J.A.; Peters, C.; Chen-Santel, C.; Chia, V.; Dennis, R.M.; Heym, K.M.; Katz, A.J.; Kelsh, M.A.; Sposto, R.; et al. Blinatumomab versus Historical Standard Therapy in Pediatric Patients with Relapsed/Refractory Ph-Negative B-Cell Precursor Acute Lymphoblastic Leukemia. Leukemia 2020, 34, 2473–2478. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Mergen, N.; Bader, P.; Jeha, S.; Schlegel, P.G.; Bourquin, J.P.; Handgretinger, R.; Brethon, B.; Rossig, C.; et al. Blinatumomab in Pediatric Patients with Relapsed/Refractory Acute Lymphoblastic Leukemia: Results of the RIALTO Trial, an Expanded Access Study. Blood Cancer J. 2020, 10, 77. [Google Scholar] [CrossRef]
- Clesham, K.; Rao, V.; Bartram, J.; Ancliff, P.; Ghorashian, S.; O’Connor, D.; Pavasovic, V.; Rao, A.; Samarasinghe, S.; Cummins, M.; et al. Blinatumomab for Infant Acute Lymphoblastic Leukemia. Blood 2020, 135, 1501–1504. [Google Scholar] [CrossRef]
- Ampatzidou, M.; Kattamis, A.; Baka, M.; Paterakis, G.; Anastasiou, T.; Tzanoudaki, M.; Kaisari, A.; Avgerinou, G.; Doganis, D.; Papadakis, V.; et al. Insights from the Greek Experience of the Use of Blinatumomab in Pediatric Relapsed and Refractory Acute Lymphoblastic Leukemia Patients. Neoplasma 2020, 67, 1424–1430. [Google Scholar] [CrossRef]
- Horibe, K.; Morris, J.D.; Tuglus, C.A.; Dos Santos, C.; Kalabus, J.; Anderson, A.; Goto, H.; Ogawa, C. A Phase 1b Study of Blinatumomab in Japanese Children with Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Int. J. Hematol. 2020, 112, 223–233. [Google Scholar] [CrossRef]
- Mikhailova, E.; Illarionova, O.; Shelikhova, L.; Zerkalenkova, E.; Molostova, O.; Olshanskaya, Y.; Novichkova, G.; Maschan, A.; Maschan, M.; Popov, A. Immunophenotypic Changes in Leukemic Blasts in Children with Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia after Treatment with CD19-Directed Chimeric Antigen Receptor (CAR)-Expressing T Cells. Haematologica 2022, 107, 970–974. [Google Scholar] [CrossRef]
- Contreras, C.F.; Higham, C.S.; Behnert, A.; Kim, K.; Stieglitz, E.; Tasian, S.K. Clinical Utilization of Blinatumomab and Inotuzumab Immunotherapy in Children with Relapsed or Refractory B-Acute Lymphoblastic Leukemia. Pediatr. Blood Cancer 2021, 68, e28718. [Google Scholar] [CrossRef]
- Brethon, B.; Lainey, E.; Caye-Eude, A.; Grain, A.; Fenneteau, O.; Yakouben, K.; Roupret-Serzec, J.; Le Mouel, L.; Cavé, H.; Baruchel, A. Case Report: Targeting 2 Antigens as a Promising Strategy in Mixed Phenotype Acute Leukemia: Combination of Blinatumomab with Gemtuzumab Ozogamicin in an Infant With a KMT2A-Rearranged Leukemia. Front. Oncol. 2021, 11, 255. [Google Scholar] [CrossRef]
- Queudeville, M.; Schlegel, P.; Heinz, A.T.; Lenz, T.; Döring, M.; Holzer, U.; Hartmann, U.; Kreyenberg, H.; von Stackelberg, A.; Schrappe, M.; et al. Blinatumomab in Pediatric Patients with Relapsed/Refractory B-Cell Precursor Acute Lymphoblastic Leukemia. Eur. J. Haematol. 2021, 106, 473–483. [Google Scholar] [CrossRef]
- Sutton, R.; Pozza, L.D.; Khaw, S.L.; Fraser, C.; Revesz, T.; Chamberlain, J.; Mitchell, R.; Trahair, T.N.; Bateman, C.M.; Venn, N.C.; et al. Outcomes for Australian Children with Relapsed/Refractory Acute Lymphoblastic Leukaemia Treated with Blinatumomab. Pediatr. Blood Cancer 2021, 68, e28922. [Google Scholar] [CrossRef]
- Brown, P.A.; Ji, L.; Xu, X.; Devidas, M.; Hogan, L.E.; Borowitz, M.J.; Raetz, E.A.; Zugmaier, G.; Sharon, E.; Bernhardt, M.B.; et al. Effect of Postreinduction Therapy Consolidation with Blinatumomab vs Chemotherapy on Disease-Free Survival in Children, Adolescents, and Young Adults with First Relapse of B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 833–842. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Rizzari, C.; Morris, J.D.; Gruhn, B.; Klingebiel, T.; Parasole, R.; Linderkamp, C.; Flotho, C.; Petit, A.; et al. Effect of Blinatumomab vs Chemotherapy on Event-Free Survival Among Children with High-Risk First-Relapse B-Cell Acute Lymphoblastic Leukemia: A Randomized Clinical Trial. JAMA 2021, 325, 843–854. [Google Scholar] [CrossRef]
- Locatelli, F.; Zugmaier, G.; Mergen, N.; Bader, P.; Jeha, S.; Schlegel, P.G.; Bourquin, J.P.; Handgretinger, R.; Brethon, B.; Rössig, C.; et al. Blinatumomab in Pediatric Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia: RIALTO Expanded Access Study Final Analysis. Blood Adv. 2022, 6, 1004–1014. [Google Scholar] [CrossRef]
- Locatelli, F.; Maschan, A.; Boissel, N.; Strocchio, L.; Alam, N.; Pezzani, I.; Brescianini, A.; Kreuzbauer, G.; Baruchel, A. Pediatric Patients with Acute Lymphoblastic Leukemia Treated with Blinatumomab in a Real-World Setting: Results from the NEUF Study. Pediatr. Blood Cancer 2022, 69, e29562. [Google Scholar] [CrossRef]
- Beneduce, G.; De Matteo, A.; Stellato, P.; Testi, A.M.; Bertorello, N.; Colombini, A.; Putti, M.C.; Rizzari, C.; Cesaro, S.; Cellini, M.; et al. Blinatumomab in Children and Adolescents with Relapsed/Refractory B Cell Precursor Acute Lymphoblastic Leukemia: A Real-Life Multicenter Retrospective Study in Seven AIEOP (Associazione Italiana Di Ematologia e Oncologia Pediatrica) Centers. Cancers 2022, 14, 426. [Google Scholar] [CrossRef]
- Su, Z.; Xiao, D.; Xie, F.; Liu, L.; Wang, Y.; Fan, S.; Zhou, X.; Li, S. Antibody–Drug Conjugates: Recent Advances in Linker Chemistry. Acta Pharm. Sin. B 2021, 11, 3889–3907. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody-Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Kalim, M.; Chen, J.; Wang, S.; Lin, C.; Ullah, S.; Liang, K.; Ding, Q.; Chen, S.; Zhan, J.B. Intracellular Trafficking of New Anticancer Therapeutics: Antibody–Drug Conjugates. Drug Des. Devel. Ther. 2017, 11, 2265–2276. [Google Scholar] [CrossRef]
- Staudacher, A.H.; Brown, M.P. Antibody Drug Conjugates and Bystander Killing: Is Antigen-Dependent Internalisation Required? Br. J. Cancer 2017, 117, 1736–1742. [Google Scholar] [CrossRef]
- Kantarjian, H.M.; DeAngelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef]
- DeAngelo, D.J.; Advani, A.S.; Marks, D.I.; Stelljes, M.; Liedtke, M.; Stock, W.; Gökbuget, N.; Jabbour, E.; Merchant, A.; Wang, T.; et al. Inotuzumab Ozogamicin for Relapsed/Refractory Acute Lymphoblastic Leukemia: Outcomes by Disease Burden. Blood Cancer J. 2020, 10, 81. [Google Scholar] [CrossRef]
- Özcan, M.; Cassaday, R.D.; Singh, P.; Zarzycka, E.; Zhang, X.; Nègre, E.; Vandendries, E.; Altuntas, F. The Efficacy and Safety of Low-Dose Inotuzumab Ozogamicin in Patients with Relapsed or Refractory Acute Lymphoblastic Leukemia: Interim Results of a Phase 4 Study. Blood 2021, 138, 1208. [Google Scholar] [CrossRef]
- Kebriaei, P.; Cutler, C.; De Lima, M.; Giralt, S.; Lee, S.J.; Marks, D.; Merchant, A.; Stock, W.; Van Besien, K.; Stelljes, M. Management of Important Adverse Events Associated with Inotuzumab Ozogamicin: Expert Panel Review. Bone Marrow Transplant. 2018, 53, 449–456. [Google Scholar] [CrossRef]
- O’Brien, M.M.; Ji, L.; Shah, N.N.; Rheingold, S.R.; Bhojwani, D.; Yuan, C.M.; Xu, X.; Yi, J.S.; Harris, A.C.; Brown, P.A.; et al. Phase II Trial of Inotuzumab Ozogamicin in Children and Adolescents with Relapsed or Refractory B-Cell Acute Lymphoblastic Leukemia: Children’s Oncology Group Protocol AALL1621. J. Clin. Oncol. 2022, 40, 956–967. [Google Scholar] [CrossRef]
- Brivio, E.; Locatelli, F.; Lopez-Yurda, M.; Malone, A.; Díaz-de-Heredia, C.; Bielorai, B.; Rossig, C.; van der Velden, V.H.J.; Ammerlaan, A.C.J.; Thano, A.; et al. A Phase 1 Study of Inotuzumab Ozogamicin in Pediatric Relapsed/Refractory Acute Lymphoblastic Leukemia (ITCC-059 Study). Blood 2021, 137, 1582–1590. [Google Scholar] [CrossRef]
- Jabbour, E.; Sasaki, K.; Ravandi, F.; Huang, X.; Short, N.J.; Khouri, M.; Kebriaei, P.; Burger, J.; Khoury, J.; Jorgensen, J.; et al. Chemoimmunotherapy with Inotuzumab Ozogamicin Combined with Mini-Hyper-CVD, with or without Blinatumomab, Is Highly Effective in Patients with Philadelphia Chromosome–Negative Acute Lymphoblastic Leukemia in First Salvage. Cancer 2018, 124, 4044–4055. [Google Scholar] [CrossRef]
- Jabbour, E.J.; Sasaki, K.; Ravandi, F.; Short, N.J.; Garcia-Manero, G.; Daver, N.; Kadia, T.; Konopleva, M.; Jain, N.; Cortes, J.; et al. Inotuzumab Ozogamicin in Combination with Low-Intensity Chemotherapy (Mini-HCVD) with or without Blinatumomab versus Standard Intensive Chemotherapy (HCVAD) as Frontline Therapy for Older Patients with Philadelphia Chromosome-Negative Acute Lymphoblastic. Cancer 2019, 125, 2579–2586. [Google Scholar] [CrossRef]
- Sasaki, K.; Kantarjian, H.M.; Ravandi, F.; Short, N.J.; Kebriaei, P.; Huang, X.; Rytting, M.E.; Jain, N.; Konopleva, M.Y.; Garcia-Manero, G.; et al. Sequential Combination of Inotuzumab Ozogamicin (InO) with Low-Intensity Chemotherapy (Mini-Hyper-CVD) with or without Blinatumomab Is Highly Effective in Patients (Pts) with Philadelphia Chromosome-Negative Acute Lymphoblastic Leukemia (ALL) in First Rel. Blood 2019, 134, 3806. [Google Scholar] [CrossRef]
- Kantarjian, H.; Ravandi, F.; Short, N.J.; Huang, X.; Jain, N.; Sasaki, K.; Daver, N.; Pemmaraju, N.; Khoury, J.D.; Jorgensen, J.; et al. Inotuzumab Ozogamicin in Combination with Low-Intensity Chemotherapy for Older Patients with Philadelphia Chromosome-Negative Acute Lymphoblastic Leukaemia: A Single-Arm, Phase 2 Study. Lancet Oncol. 2018, 19, 240–248. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; Kantarjian, H.M.; Short, N.J.; Ravandi, F.; Ferrajoli, A.; Schroeder, H.M.; Garcia-Manero, G.; Montalban Bravo, G.; Cortes, J.E.; Kwari, M.; et al. Updated Results from the Phase II Study of Hyper-CVAD in Sequential Combination with Blinatumomab in Newly Diagnosed Adults with B-Cell Acute Lymphoblastic Leukemia (B-ALL). Blood 2019, 134, 3807. [Google Scholar] [CrossRef]
- Henze, A.T.; Mazzone, M. The Impact of Hypoxia on Tumor-Associated Macrophages. J. Clin. Investig. 2016, 126, 3672–3679. [Google Scholar] [CrossRef]
- Hughes, R.; Qian, B.Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef]
- Gravante, G.; Ong, S.L.; Metcalfe, M.S.; Sorge, R.; Sconocchia, G.; Orlando, G.; Lloyd, D.M.; Dennison, A.R. Cytokine Response to Ischemia/Reperfusion Injury in an Ex Vivo Perfused Porcine Liver Model. Transplant. Proc. 2009, 41, 1107–1112. [Google Scholar] [CrossRef]
- Ohno, S.; Ohno, Y.; Suzuki, N.; Kamei, T.; Koike, K.; Inagawa, H.; Kohchi, C.; Soma, G.I.; Inoue, M. Correlation of Histological Localization of Tumor-Associated Macrophages with Clinicopathological Features in Endometrial Cancer. Anticancer Res. 2004, 24, 3335–3342. [Google Scholar]
- Vito, A.; El-Sayes, N.; Mossman, K. Hypoxia-Driven Immune Escape in the Tumor Microenvironment. Cells 2020, 9, 992. [Google Scholar] [CrossRef]
- Khalaf, K.; Hana, D.; Chou, J.T.T.; Singh, C.; Mackiewicz, A.; Kaczmarek, M. Aspects of the Tumor Microenvironment Involved in Immune Resistance and Drug Resistance. Front. Immunol. 2021, 12, 1764. [Google Scholar] [CrossRef]
- Duell, J.; Lurati, S.; Dittrich, M.; Bedke, T.; Pule, M.; Einsele, H.; Rossig, C.; Topp, M.S. First Generation Chimeric Antigen Receptor Display Functional Defects in Key Signal Pathways Upon Antigen Stimulation. Blood 2010, 116, 2088. [Google Scholar] [CrossRef]
- Van Der Stegen, S.J.C.; Hamieh, M.; Sadelain, M. The Pharmacology of Second-Generation Chimeric Antigen Receptors. Nat. Rev. Drug Discov. 2015, 14, 499–509. [Google Scholar] [CrossRef]
- Zhong, X.S.; Matsushita, M.; Plotkin, J.; Riviere, I.; Sadelain, M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell–Mediated Tumor Eradication. Mol. Ther. 2010, 18, 413–420. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The Fourth Generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an Old Dog New Tricks: Next-Generation CAR T Cells. Br. J. Cancer 2018, 120, 26–37. [Google Scholar] [CrossRef]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-Cells: Utilizing Cytokines and pro-Inflammatory Ligands to Enhance CAR T-Cell Anti-Tumour Efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef]
- Miller, B.C.; Maus, M.V. CD19-Targeted CAR T Cells: A New Tool in the Fight against B Cell Malignancies. Oncol. Res. Treat. 2015, 38, 683–690. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor–Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Brentjens, R.J.; Rivière, I.; Park, J.H.; Davila, M.L.; Wang, X.; Stefanski, J.; Taylor, C.; Yeh, R.; Bartido, S.; Borquez-Ojeda, O.; et al. Safety and Persistence of Adoptively Transferred Autologous CD19-Targeted T Cells in Patients with Relapsed or Chemotherapy Refractory B-Cell Leukemias. Blood 2011, 118, 4817–4828. [Google Scholar] [CrossRef]
- Park, J.H.; Rivière, I.; Gonen, M.; Wang, X.; Sénéchal, B.; Curran, K.J.; Sauter, C.; Wang, Y.; Santomasso, B.; Mead, E.; et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 449–459. [Google Scholar] [CrossRef]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T Cells Expressing CD19 Chimeric Antigen Receptors for Acute Lymphoblastic Leukaemia in Children and Young Adults: A Phase 1 Dose-Escalation Trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Turtle, C.J.; Hanafi, L.A.; Berger, C.; Gooley, T.A.; Cherian, S.; Hudecek, M.; Sommermeyer, D.; Melville, K.; Pender, B.; Budiarto, T.M.; et al. CD19 CAR–T Cells of Defined CD4+:CD8+ Composition in Adult B Cell ALL Patients. J. Clin. Investig. 2016, 126, 2123–2138. [Google Scholar] [CrossRef]
- Lee, D.W.; Stetler-Stevenson, M.; Yuan, C.M.; Shah, N.N.; Delbrook, C.; Yates, B.; Zhang, H.; Zhang, L.; Kochenderfer, J.N.; Rosenberg, S.A.; et al. Long-Term Outcomes Following CD19 CAR T Cell Therapy for B-ALL Are Superior in Patients Receiving a Fludarabine/Cyclophosphamide Preparative Regimen and Post-CAR Hematopoietic Stem Cell Transplantation. Blood 2016, 128, 218. [Google Scholar] [CrossRef]
- Shah, B.D.; Bishop, M.R.; Oluwole, O.O.; Logan, A.C.; Baer, M.R.; Donnellan, W.B.; O’Dwyer, K.M.; Holmes, H.; Arellano, M.L.; Ghobadi, A.; et al. KTE-X19 Anti-CD19 CAR T-Cell Therapy in Adult Relapsed/Refractory Acute Lymphoblastic Leukemia: ZUMA-3 Phase 1 Results. Blood 2021, 138, 11–22. [Google Scholar] [CrossRef]
- Shah, B.D.; Ghobadi, A.; Oluwole, O.O.; Logan, A.; Boissel, N.; Cassaday, R.D.; Forcade, E.; Bishop, M.R.; Topp, M.S.; Tzachanis, D.; et al. Phase 2 Results of the ZUMA-3 Study Evaluating KTE-X19, an Anti-CD19 Chimeric Antigen Receptor (CAR) T-Cell Therapy, in Adult Patients (Pts) with Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia (R/R B-ALL). J. Clin. Oncol. 2021, 39, 7002. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507. [Google Scholar] [CrossRef]
- Talekar, M.K.; Maude, S.L.; Hucks, G.E.; Motley, L.S.; Callahan, C.; White, C.M.; Baniewicz, D.; Barrett, D.M.; Rheingold, S.R.; Lacey, S.F.; et al. Effect of Chimeric Antigen Receptor-Modified T (CAR-T) Cells on Responses in Children with Non-CNS Extramedullary Relapse of CD19+ Acute Lymphoblastic Leukemia (ALL). J. Clin. Oncol. 2017, 35, 10507. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Y.; Ruan, M.; Li, J.; Zhong, M.; Li, Z.; Liu, F.; Wang, S.; Chen, Y.; Liu, L.; et al. Treatment of Testicular Relapse of B-Cell Acute Lymphoblastic Leukemia with CD19-Specific Chimeric Antigen Receptor T Cells. Clin. Lymphoma Myeloma Leuk. 2020, 20, 366–370. [Google Scholar] [CrossRef]
- Rheingold, S.R.; Chen, L.N.; Maude, S.L.; Aplenc, R.; Barker, C.; Barrett, D.M.; Callahan, C.; Cebry, K.; Kulikovskaya, I.; Lacey, S.F.; et al. Efficient Trafficking of Chimeric Antigen Receptor (CAR)-Modified T Cells to CSF and Induction of Durable CNS Remissions in Children with CNS/Combined Relapsed/Refractory ALL. Blood 2015, 126, 3769. [Google Scholar] [CrossRef]
- Gardner, R.A.; Finney, O.; Annesley, C.; Brakke, H.; Summers, C.; Leger, K.; Bleakley, M.; Brown, C.; Mgebroff, S.; Kelly-Spratt, K.S.; et al. Intent-to-Treat Leukemia Remission by CD19 CAR T Cells of Defined Formulation and Dose in Children and Young Adults. Blood 2017, 129, 3322–3331. [Google Scholar] [CrossRef]
- Maude, S.L.; Teachey, D.T.; Rheingold, S.R.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Barker, C.S.; Callahan, C.; Frey, N.V.; Nazimuddin, F.; et al. Sustained Remissions with CD19-Specific Chimeric Antigen Receptor (CAR)-Modified T Cells in Children with Relapsed/Refractory ALL. J. Clin. Oncol. 2016, 34, 3011. [Google Scholar] [CrossRef]
- Newman, H.; Leahy, A.E.B.; Li, Y.; Liu, H.; Myers, R.M.; DiNofia, A.M.; Dolan, J.G.; Callahan, C.; Devine, K.J.; Wray, L.; et al. CD19-Targeted Chimeric Antigen Receptor (CAR) T Cells in CNS Relapsed Acute Lymphoblastic Leukemia (ALL). J. Clin. Oncol. 2020, 38, 10511. [Google Scholar] [CrossRef]
- Zhang, X.; Lu, X.A.; Yang, J.; Zhang, G.; Li, J.; Song, L.; Su, Y.; Shi, Y.; Zhang, M.; He, J.; et al. Efficacy and Safety of Anti-CD19 CAR T-Cell Therapy in 110 Patients with B-Cell Acute Lymphoblastic Leukemia with High-Risk Features. Blood Adv. 2020, 4, 2325–2338. [Google Scholar] [CrossRef]
- Rubinstein, J.D.; Krupski, C.; Nelson, A.S.; O’Brien, M.M.; Davies, S.M.; Phillips, C.L. Chimeric Antigen Receptor T Cell Therapy in Patients with Multiply Relapsed or Refractory Extramedullary Leukemia. Biol. Blood Marrow Transplant. 2020, 26, e280–e285. [Google Scholar] [CrossRef]
- Lee, D.W.; Gardner, R.; Porter, D.L.; Louis, C.U.; Ahmed, N.; Jensen, M.; Grupp, S.A.; Mackall, C.L. Current Concepts in the Diagnosis and Management of Cytokine Release Syndrome. Blood 2014, 124, 188–195. [Google Scholar] [CrossRef]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef]
- Frey, N.V.; Porter, D.L. Cytokine Release Syndrome with Novel Therapeutics for Acute Lymphoblastic Leukemia. Hematology 2016, 2016, 567–572. [Google Scholar] [CrossRef]
- Frey, N.V.; Shaw, P.A.; Hexner, E.O.; Pequignot, E.; Gill, S.; Luger, S.M.; Mangan, J.K.; Loren, A.W.; Perl, A.E.; Maude, S.L.; et al. Optimizing Chimeric Antigen Receptor T-Cell Therapy for Adults with Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2020, 38, 415–422. [Google Scholar] [CrossRef]
- Gust, J.; Taraseviciute, A.; Turtle, C.J. Neurotoxicity Associated with CD19-Targeted CAR-T Cell Therapies. CNS Drugs 2018, 32, 1091–1101. [Google Scholar] [CrossRef]
- Neelapu, S.S. Managing the Toxicities of CAR T-Cell Therapy. Hematol. Oncol. 2019, 37, 48–52. [Google Scholar] [CrossRef]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142.e17. [Google Scholar] [CrossRef]
- Arnold, D.E.; Maude, S.L.; Callahan, C.A.; DiNofia, A.M.; Grupp, S.A.; Heimall, J.R. Subcutaneous Immunoglobulin Replacement Following CD19-Specific Chimeric Antigen Receptor T-Cell Therapy for B-Cell Acute Lymphoblastic Leukemia in Pediatric Patients. Pediatr. Blood Cancer 2020, 67, e28092. [Google Scholar] [CrossRef]
- Doan, A.; Pulsipher, M.A. Hypogammaglobulinemia Due to CAR T-Cell Therapy. Pediatr. Blood Cancer 2018, 65, e26914. [Google Scholar] [CrossRef]
- Porter, D.L.; Hwang, W.T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric Antigen Receptor T Cells Persist and Induce Sustained Remissions in Relapsed Refractory Chronic Lymphocytic Leukemia. Sci. Transl. Med. 2015, 7, 303ra139. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-Cell Depletion and Remissions of Malignancy along with Cytokine-Associated Toxicity in a Clinical Trial of Anti-CD19 Chimeric-Antigen-Receptor–Transduced T Cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef]
- Kochenderfer, J.N.; Wilson, W.H.; Janik, J.E.; Dudley, M.E.; Stetler-Stevenson, M.; Feldman, S.A.; Maric, I.; Raffeld, M.; Nathan, D.A.N.; Lanier, B.J.; et al. Eradication of B-Lineage Cells and Regression of Lymphoma in a Patient Treated with Autologous T Cells Genetically Engineered to Recognize CD19. Blood 2010, 116, 4099–4102. [Google Scholar] [CrossRef]
- Perez, E.E.; Orange, J.S.; Bonilla, F.; Chinen, J.; Chinn, I.K.; Dorsey, M.; El-Gamal, Y.; Harville, T.O.; Hossny, E.; Mazer, B.; et al. Update on the Use of Immunoglobulin in Human Disease: A Review of Evidence. J. Allergy Clin. Immunol. 2017, 139, S1–S46. [Google Scholar] [CrossRef]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with Car T-Cell Therapy in Patients with B-Cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef]
- Hill, J.A.; Li, D.; Hay, K.A.; Green, M.L.; Cherian, S.; Chen, X.; Riddell, S.R.; Maloney, D.G.; Boeckh, M.; Turtle, C.J. Infectious Complications of CD19-Targeted Chimeric Antigen Receptor–Modified T-Cell Immunotherapy. Blood 2018, 131, 121–130. [Google Scholar] [CrossRef]
- Kansagra, A.J.; Frey, N.V.; Bar, M.; Laetsch, T.W.; Carpenter, P.A.; Savani, B.N.; Heslop, H.E.; Bollard, C.M.; Komanduri, K.V.; Gastineau, D.A.; et al. Clinical Utilization of Chimeric Antigen Receptor T Cells in B Cell Acute Lymphoblastic Leukemia: An Expert Opinion from the European Society for Blood and Marrow Transplantation and the American Society for Blood and Marrow Transplantation. Biol. Blood Marrow Transplant. 2019, 25, e76–e85. [Google Scholar] [CrossRef]
- Hensley, M.K.; Bain, W.G.; Jacobs, J.; Nambulli, S.; Parikh, U.; Cillo, A.; Staines, B.; Heaps, A.; Sobolewski, M.D.; Rennick, L.J.; et al. Intractable Coronavirus Disease 2019 (COVID-19) and Prolonged Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Replication in a Chimeric Antigen Receptor-Modified T-Cell Therapy Recipient: A Case Study. Clin. Infect. Dis. 2021, 73, e815–e821. [Google Scholar] [CrossRef]
- Biassoni, R. Natural Killer Cell Receptors. Adv. Exp. Med. Biol. 2008, 640, 35–52. [Google Scholar] [CrossRef]
- Mrózek, E.; Anderson, P.; Caligiuri, M. Role of Interleukin-15 in the Development of Human CD56+ Natural Killer Cells from CD34+ Hematopoietic Progenitor Cells. Blood 1996, 87, 2632–2640. [Google Scholar] [CrossRef]
- Sconocchia, G.; Fujiwara, H.; Rezvani, K.; Keyvanfar, K.; El Ouriaghli, F.; Grube, M.; Melenhorst, J.; Hensel, N.; Barrett, A.J. G-CSF-Mobilized CD34+ Cells Cultured in Interleukin-2 and Stem Cell Factor Generate a Phenotypically Novel Monocyte. J. Leukoc. Biol. 2004, 76, 1214–1219. [Google Scholar] [CrossRef][Green Version]
- Coppola, A.; Arriga, R.; Lauro, D.; del Principe, M.I.; Buccisano, F.; Maurillo, L.; Palomba, P.; Venditti, A.; Sconocchia, G. NK Cell Inflammation in the Clinical Outcome of Colorectal Carcinoma. Front. Med. 2015, 2, 33. [Google Scholar] [CrossRef]
- Sconocchia, G.; Arriga, R.; Tornillo, L.; Terracciano, L.; Ferrone, S.; Spagnoli, G.C. Melanoma Cells Inhibit NK Cell Functions. Cancer Res. 2012, 72, 5428–5429. [Google Scholar] [CrossRef]
- Sconocchia, G.; Zlobec, I.; Lugli, A.; Calabrese, D.; Iezzi, G.; Karamitopoulou, E.; Patsouris, E.S.; Peros, G.; Horcic, M.; Tornillo, L.; et al. Tumor Infiltration by FcγRIII (CD16)+ Myeloid Cells Is Associated with Improved Survival in Patients with Colorectal Carcinoma. Int. J. Cancer 2011, 128, 2663–2672. [Google Scholar] [CrossRef]
- Sconocchia, G.; Eppenberger, S.; Spagnoli, G.C.; Tornillo, L.; Droeser, R.; Caratelli, S.; Ferrelli, F.; Coppola, A.; Arriga, R.; Lauro, D.; et al. NK Cells and T Cells Cooperate during the Clinical Course of Colorectal Cancer. Oncoimmunology 2014, 3, e952197. [Google Scholar] [CrossRef]
- Ruggeri, L.; Capanni, M.; Casucci, M.; Volpi, I.; Tosti, A.; Perruccio, K.; Urbani, E.; Negrin, R.S.; Martelli, M.F.; Velardi, A. Role of Natural Killer Cell Alloreactivity in HLA-Mismatched Hematopoietic Stem Cell Transplantation. Blood 1999, 94, 333–339. [Google Scholar] [CrossRef]
- Sconocchia, G.; Lau, M.; Provenzano, M.; Rezvani, K.; Wongsena, W.; Fujiwara, H.; Hensel, N.; Melenhorst, J.; Li, J.; Ferrone, S.; et al. The Antileukemia Effect of HLA-Matched NK and NK-T Cells in Chronic Myelogenous Leukemia Involves NKG2D–Target-Cell Interactions. Blood 2005, 106, 3666–3672. [Google Scholar] [CrossRef]
- Schmidt, P.; Raftery, M.J.; Pecher, G. Engineering NK Cells for CAR Therapy—Recent Advances in Gene Transfer Methodology. Front. Immunol. 2021, 11, 3404. [Google Scholar] [CrossRef]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.J. Off-the-Shelf Cell Therapy with Induced Pluripotent Stem Cell-Derived Natural Killer Cells. Semin. Immunopathol. 2018, 41, 59–68. [Google Scholar] [CrossRef]
- Pan, K.; Farrukh, H.; Chittepu, V.C.S.R.; Xu, H.; Pan, C.X.; Zhu, Z. CAR Race to Cancer Immunotherapy: From CAR T, CAR NK to CAR Macrophage Therapy. J. Exp. Clin. Cancer Res. 2022, 41, 119. [Google Scholar] [CrossRef]
- Chang, Y.H.; Connolly, J.; Shimasaki, N.; Mimura, K.; Kono, K.; Campana, D. A Chimeric Receptor with NKG2D Specificity Enhances Natural Killer Cell Activation and Killing of Tumor Cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef]
- Klingemann, H. Are Natural Killer Cells Superior CAR Drivers? Oncoimmunology 2014, 3, e28147. [Google Scholar] [CrossRef]
- Rafei, H.; Daher, M.; Rezvani, K. Chimeric Antigen Receptor (CAR) Natural Killer (NK)-Cell Therapy: Leveraging the Power of Innate Immunity. Br. J. Haematol. 2021, 193, 216–230. [Google Scholar] [CrossRef]
- Williams, B.A.; Law, A.D.; Routy, B.; DenHollander, N.; Gupta, V.; Wang, X.-H.; Chaboureau, A.; Viswanathan, S.; Keating, A.; Williams, B.A.; et al. A Phase I Trial of NK-92 Cells for Refractory Hematological Malignancies Relapsing after Autologous Hematopoietic Cell Transplantation Shows Safety and Evidence of Efficacy. Oncotarget 2017, 8, 89256–89268. [Google Scholar] [CrossRef]
- Oelsner, S.; Waldmann, A.; Billmeier, A.; Röder, J.; Lindner, A.; Ullrich, E.; Marschalek, R.; Dotti, G.; Jung, G.; Große-Hovest, L.; et al. Genetically Engineered CAR NK Cells Display Selective Cytotoxicity against FLT3-Positive B-ALL and Inhibit in Vivo Leukemia Growth. Int. J. Cancer 2019, 145, 1935–1945. [Google Scholar] [CrossRef]
- Haddad, F.G.; Short, N.J. What Is the Optimal Tyrosine Kinase Inhibitor for Adults with Newly Diagnosed Philadelphia Chromosome–Positive Acute Lymphoblastic Leukemia? Hematology 2022, 2022, 213–217. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.; Konopleva, M.; Desikan, S.P.P.; Jain, N.; Ravandi, F.; Huang, X.; Wierda, W.G.; Borthakur, G.; Sasaki, K.; et al. Updated Results of a Phase II Study of Ponatinib and Blinatumomab for Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Blood 2021, 138, 2298. [Google Scholar] [CrossRef]
- Jabbour, E.; Short, N.J.; Ravandi, F.; Huang, X.; Daver, N.; DiNardo, C.D.; Konopleva, M.; Pemmaraju, N.; Wierda, W.; Garcia-Manero, G.; et al. Combination of Hyper-CVAD with Ponatinib as First-Line Therapy for Patients with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukaemia: Long-Term Follow-up of a Single-Centre, Phase 2 Study. Lancet Haematol. 2018, 5, e618–e627. [Google Scholar] [CrossRef]
- Ribera, J.-M.; Garcia, O.; Ribera, J.; Montesinos, P.; Cano, I.; Martínez, P.; Esteve, J.; Esteban, D.; García-Fortes, M.; Alonso, N.; et al. Ponatinib and Chemotherapy in Adults with De Novo Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Final Results of Ponalfil Clinical Trial. Blood 2021, 138, 1230. [Google Scholar] [CrossRef]
- Roddie, C.; O’Reilly, M.A.; Marzolini, M.A.V.; Wood, L.; Dias, J.; Cadinanos Garai, A.; Bosshard, L.; Abbasian, M.; Lowdell, M.W.; Wheeler, G.; et al. ALLCAR19: Updated Data Using AUTO1, a Novel Fast-Off Rate CD19 CAR in Relapsed/Refractory B-Cell Acute Lymphoblastic Leukaemia and Other B-Cell Malignancies. Blood 2020, 136, 3–4. [Google Scholar] [CrossRef]
- Roddie, C.; Dias, J.; O’Reilly, M.A.; Abbasian, M.; Cadinanos-Garai, A.; Vispute, K.; Bosshard-Carter, L.; Mitsikakou, M.; Mehra, V.; Roddy, H.; et al. Durable Responses and Low Toxicity after Fast Off-Rate Cd19 Chimeric Antigen Receptor-t Therapy in Adults with Relapsed or Refractory b-Cell Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2021, 39, 3352–3364. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T Cell Expansion and Prolonged Persistence in Pediatric Patients with ALL Treated with a Low-Affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef]
- Pan, J.; Niu, Q.; Deng, B.; Liu, S.; Wu, T.; Gao, Z.; Liu, Z.; Zhang, Y.; Qu, X.; Zhang, Y.; et al. CD22 CAR T-Cell Therapy in Refractory or Relapsed B Acute Lymphoblastic Leukemia. Leukemia 2019, 33, 2854–2866. [Google Scholar] [CrossRef]
- Dai, H.; Wu, Z.; Jia, H.; Tong, C.; Guo, Y.; Ti, D.; Han, X.; Liu, Y.; Zhang, W.; Wang, C.; et al. Bispecific CAR-T Cells Targeting Both CD19 and CD22 for Therapy of Adults with Relapsed or Refractory B Cell Acute Lymphoblastic Leukemia. J. Hematol. Oncol. 2020, 13, 30. [Google Scholar] [CrossRef]
- Gao, J.; Liu, W.J. Prognostic Value of the Response to Prednisone for Children with Acute Lymphoblastic Leukemia: A Meta-Analysis. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7858–7866. [Google Scholar] [CrossRef]
- Berg, S.L.; Blaney, S.M.; Devidas, M.; Lampkin, T.A.; Murgo, A.; Bernstein, M.; Billett, A.; Kurtzberg, J.; Reaman, G.; Gaynon, P.; et al. Phase II Study of Nelarabine (Compound 506U78) in Children and Young Adults with Refractory T-Cell Malignancies: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2005, 23, 3376–3382. [Google Scholar] [CrossRef]
- Dunsmore, K.P.; Winter, S.S.; Devidas, M.; Wood, B.L.; Esiashvili, N.; Chen, Z.; Eisenberg, N.; Briegel, N.; Hayashi, R.J.; Gastier-Foster, J.M.; et al. Children’s Oncology Group AALL0434: A Phase III Randomized Clinical Trial Testing Nelarabine in Newly Diagnosed t-Cell Acute Lymphoblastic Leukemia. J. Clin. Oncol. 2020, 38, 3282–3293. [Google Scholar] [CrossRef]
- Gökbuget, N.; Basara, N.; Baurmann, H.; Beck, J.; Brüggemann, M.; Diedrich, H.; Güldenzoph, B.; Hartung, G.; Horst, H.A.; Hüttmann, A.; et al. High Single-Drug Activity of Nelarabine in Relapsed T-Lymphoblastic Leukemia/Lymphoma Offers Curative Option with Subsequent Stem Cell Transplantation. Blood 2011, 118, 3504–3511. [Google Scholar] [CrossRef]
- Chun-fung, S.; Wan, T.M.; Mohan, A.A.M.; Qiu, Y.; Jiao, A. Bortezomib Is Effective in Treating T-ALL, Inducting G2/M Cell Cycle Arrest and WEE1 Downregulation. Blood 2021, 138, 4360. [Google Scholar] [CrossRef]
- Zheng, R.; Li, M.; Wang, S.; Liu, Y. Advances of Target Therapy on NOTCH1 Signaling Pathway in T-Cell Acute Lymphoblastic Leukemia. Exp. Hematol. Oncol. 2020, 9, 31. [Google Scholar] [CrossRef]
- Jaramillo, S.; Hennemann, H.; Horak, P.; Teleanu, V.; Heilig, C.E.; Hutter, B.; Stenzinger, A.; Glimm, H.; Goeppert, B.; Müller-Tidow, C.; et al. Ruxolitinib Is Effective in the Treatment of a Patient with Refractory T-ALL. EJHaem 2021, 2, 139–142. [Google Scholar] [CrossRef]
- Richard-Carpentier, G.; Jabbour, E.; Short, N.J.; Rausch, C.R.; Savoy, J.M.; Bose, P.; Yilmaz, M.; Jain, N.; Borthakur, G.; Ohanian, M.; et al. Clinical Experience with Venetoclax Combined With Chemotherapy for Relapsed or Refractory T-Cell Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2020, 20, 212–218. [Google Scholar] [CrossRef]
- Bride, K.L.; Vincent, T.L.; Im, S.Y.; Aplenc, R.; Barrett, D.M.; Carroll, W.L.; Carson, R.; Dai, Y.; Devidas, M.; Dunsmore, K.P.; et al. Preclinical Efficacy of Daratumumab in T-Cell Acute Lymphoblastic Leukemia. Blood 2018, 131, 995–999. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B-lymphoblasticleukemia/lymphoma |
| B-lymphoblasticleukemia/lymphoma, NOS |
| B-lymphoblastic leukemia/lymphoma with recurrent genetic abnormalities |
| B-lymphoblastic leukemia/lymphoma with t(9;22)(q34.1;q11.2);BCR-ABL1 |
| B-lymphoblastic leukemia/lymphoma with t(v;11q23.3);KMT2A rearranged |
| B-lymphoblastic leukemia/lymphoma with t(12;21)(p13.2;q22.1);ETV6-RUNX1 |
| B-lymphoblastic leukemia/lymphoma with hyperdiploidy |
| B-lymphoblastic leukemia/lymphoma with hypodiploidy |
| B-lymphoblastic leukemia/lymphoma with t(5;14)(q31.1;q32.3)IL3-IGH |
| B-lymphoblastic leukemia/lymphoma with t(1;19)(q23;p13.3);TCF3-PBX1 |
| Provisional entity:B-lymphoblastic leukemia/lymphoma with translocations involving tyrosine kinases or cytokine receptors (“BCR-ABL1–like”) |
| Provisional entity:B-lymphoblastic leukemia/lymphoma with intrachromosomal amplification of chromosome 21 (iAMP21) |
| T-lymphoblastic leukemia/lymphoma (can only be differentiated from B-ALL/LBL based on IHC and/or flow cytometry). |
| Provisional entity: Early T-cell precursor lymphoblastic leukemia |
| Provisional entity: NK cell lymphoblastic leukemia/lymphoma |
| Publication Year (Ref) | Participating Countries | Patient Selection |
|---|---|---|
| 2014 [82] | Germany | 9 R/R-BCP-ALL patients post-HSCT |
| 2016 [83] | 26 European and US Centers | 70 R/R-ALL patients (out of 93) who received the recommended dose of blinatumomab 5/15 µg/m2/day |
| 2017 [84] | Czech Republic, US, Canada, France, Germany, Italy | 18 BCP-ALL patients (4 with CD19-negative relapse) |
| 2017 [85] | Germany | 1 relapsed ALL patient without MLL rearrangement (case report) |
| 2018 [86] | US-Birmingham, Alabama | 1 BCP-ALL patient with Down syndrome (case report) |
| 2018 [87] | 26 European and US Centers | 70 R/R-ALL patients—follow-up study |
| 2018 [88] | Germany | 1 ALL patient without MLL rearrangement(case report) |
| 2019 [89] | Israeli | 11 BCP-ALL patients with overwhelming toxicity |
| 2019 [90] | US | 15 R/R-ALL patients with residue MRD |
| 2019 [91] | European experience from International BFM Study group | 9 B-ALL patients with t(17;19)(q22;p13)/TCF3-HLF |
| 2019 [92] | US, Austria, Canada, France, Germany, Italy, Netherlands | 59 R/R BCP-ALL patients (MT103-205 single-arm multicenter phase 2 study) |
| 2020 [93] | US, Austria, Canada, France, Germany, Italy, Netherlands | 70 R/R Ph-BCP-ALL patients (MT103-205 single-arm multicenter phase 2 study)—blinatumomab vs. standard therapy |
| 2020 [94] | US, Austria, France, Germany, Italy, Switzerland, UK | 110 R/R ALL patients—RIALTO expanded access study |
| 2020 [95] | UK, Ireland | 11 infants with persistent MRD |
| 2020 [96] | Greece | 9 R/R-ALL patients |
| 2020 [97] | Japan | 9 R/R-ALL patients |
| 2020 [98] | Russia | 90 R/R-BCP-ALL patients |
| 2021 [99] | Spain | 27 R/R B-ALL patients (children/AYA) |
| 2021 [100] | France | 1 infant with KMT2A rearranged ALL (case report) |
| 2021 [101] | Germany | 38 R/R BCP-ALL patients |
| 2021 [102] | Australia | 24 R/R BCP-ALL patients |
| 2021 [103] | US, Canada, Australia, New Zealand | 208 first relapsed B-ALL patients (aged from 1 to 30 years) |
| 2021 [104] | Europe, Australia, Israeli | 108 first relapsed B-ALL patients(aged 28 days to 18 years) |
| 2022 [105] | US, Austria, France, Germany, Italy, Switzerland, UK | 110 patients RIALTO expanded access study-FINAL ANALYSIS |
| 2022 [106] | France, Italy, Russia, Spain, United Kingdom | 72 with R/R Ph−BCP-ALL and 41 with MRD+, either Ph−or Ph+:retrospective observational study |
| 2022 [107] | Italy | 39 R/R ALL patients: real-life multicenter retrospective study in 7 AIEOP Centers |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aureli, A.; Marziani, B.; Venditti, A.; Sconocchia, T.; Sconocchia, G. Acute Lymphoblastic Leukemia Immunotherapy Treatment: Now, Next, and Beyond. Cancers 2023, 15, 3346. https://doi.org/10.3390/cancers15133346
Aureli A, Marziani B, Venditti A, Sconocchia T, Sconocchia G. Acute Lymphoblastic Leukemia Immunotherapy Treatment: Now, Next, and Beyond. Cancers. 2023; 15(13):3346. https://doi.org/10.3390/cancers15133346
Chicago/Turabian StyleAureli, Anna, Beatrice Marziani, Adriano Venditti, Tommaso Sconocchia, and Giuseppe Sconocchia. 2023. "Acute Lymphoblastic Leukemia Immunotherapy Treatment: Now, Next, and Beyond" Cancers 15, no. 13: 3346. https://doi.org/10.3390/cancers15133346
APA StyleAureli, A., Marziani, B., Venditti, A., Sconocchia, T., & Sconocchia, G. (2023). Acute Lymphoblastic Leukemia Immunotherapy Treatment: Now, Next, and Beyond. Cancers, 15(13), 3346. https://doi.org/10.3390/cancers15133346