
The Molecular Basis and Clinical Consequences of Chronic Inflammation in Prostatic Diseases: Prostatitis, Benign Prostatic Hyperplasia, and Prostate Cancer

 ,
,  , , and
, , and 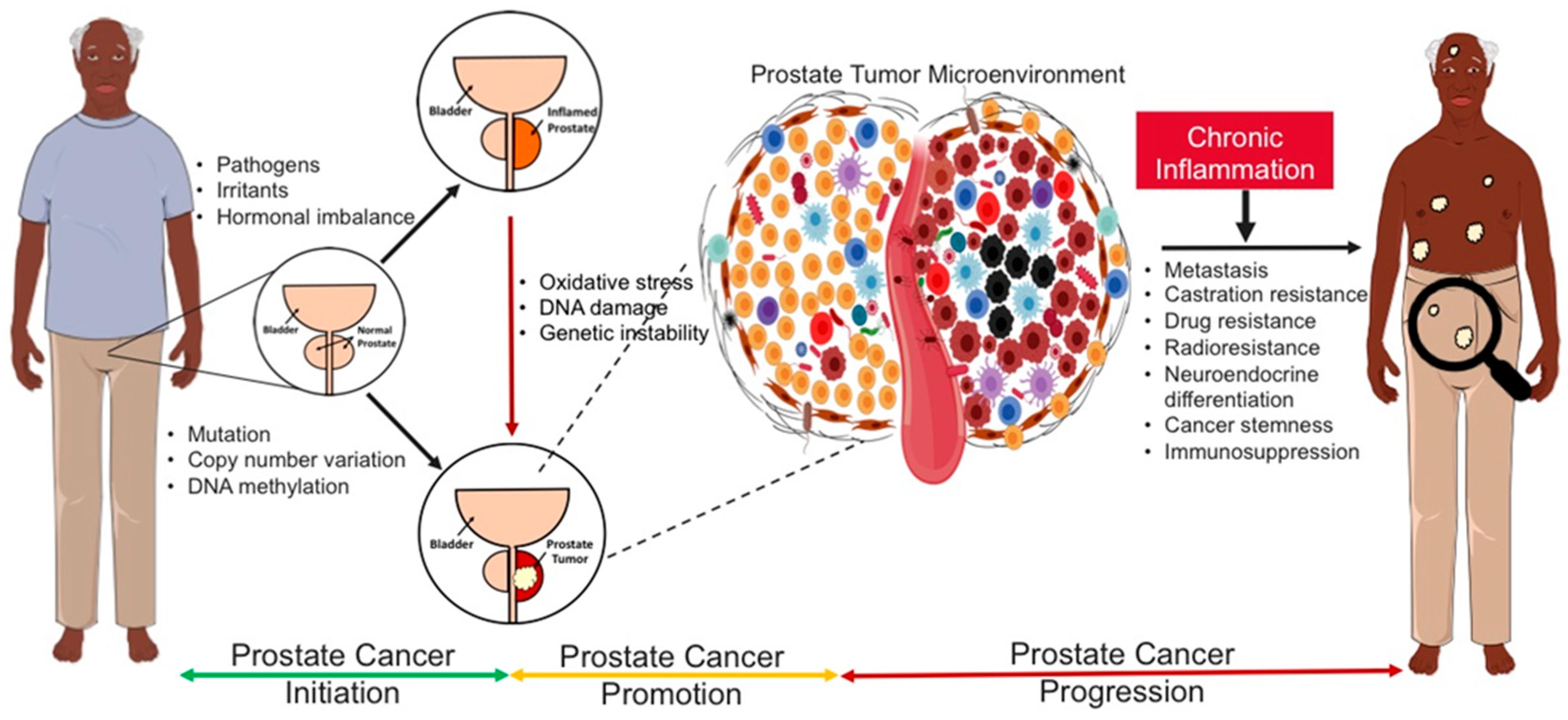{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Prostate Pathologies and Co-Morbidities
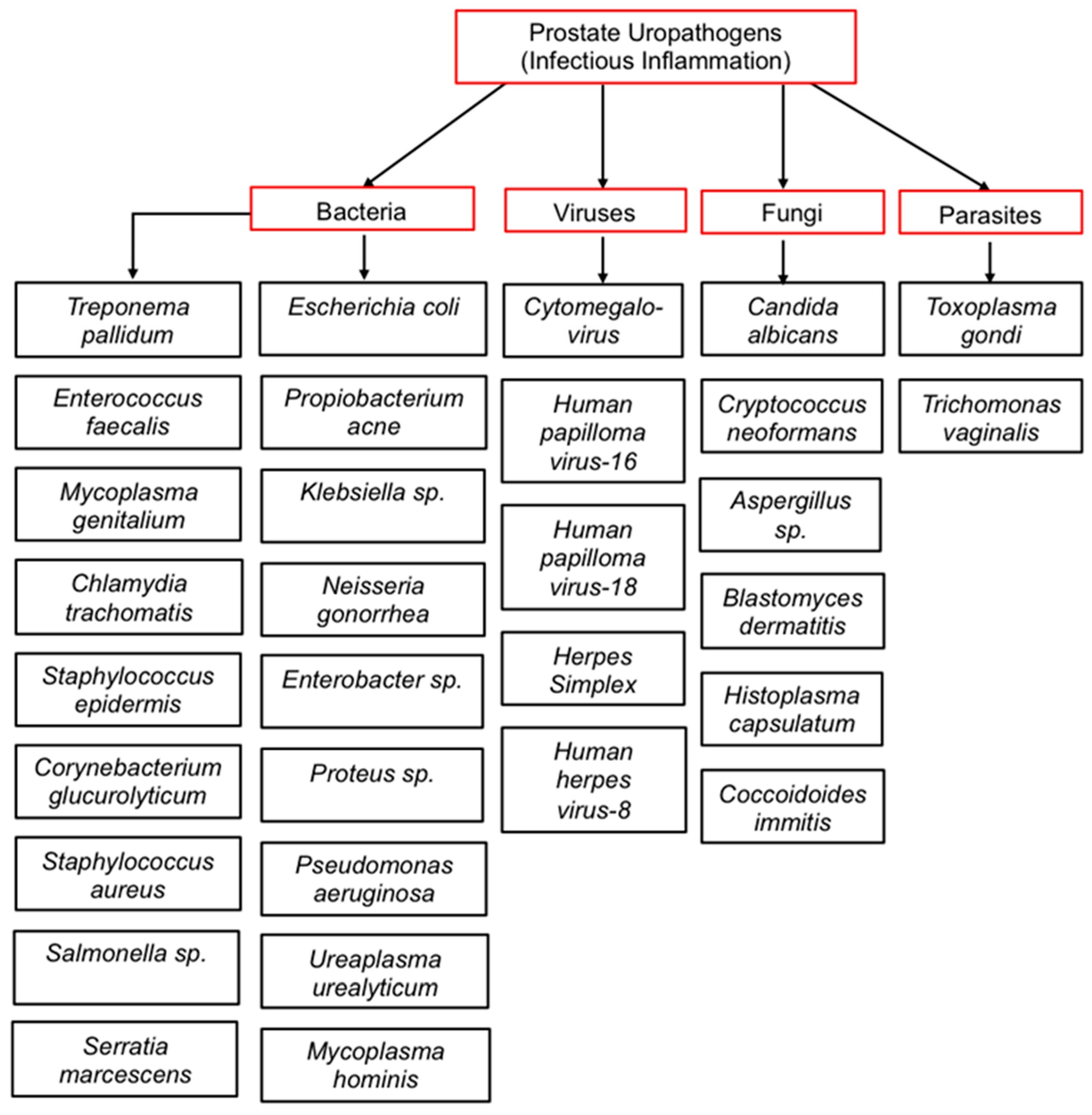
3. Causal Inflammatory Drivers of Prostate Inflammation (Prostatitis)
4. Causal Inflammatory Drivers of Benign Prostatic Hyperplasia/Enlargement (BPH/E)
5. Causal Inflammatory Drivers of Prostate Carcinogenesis
6. Role of The Human Microbiome in Tumor Development and Progression
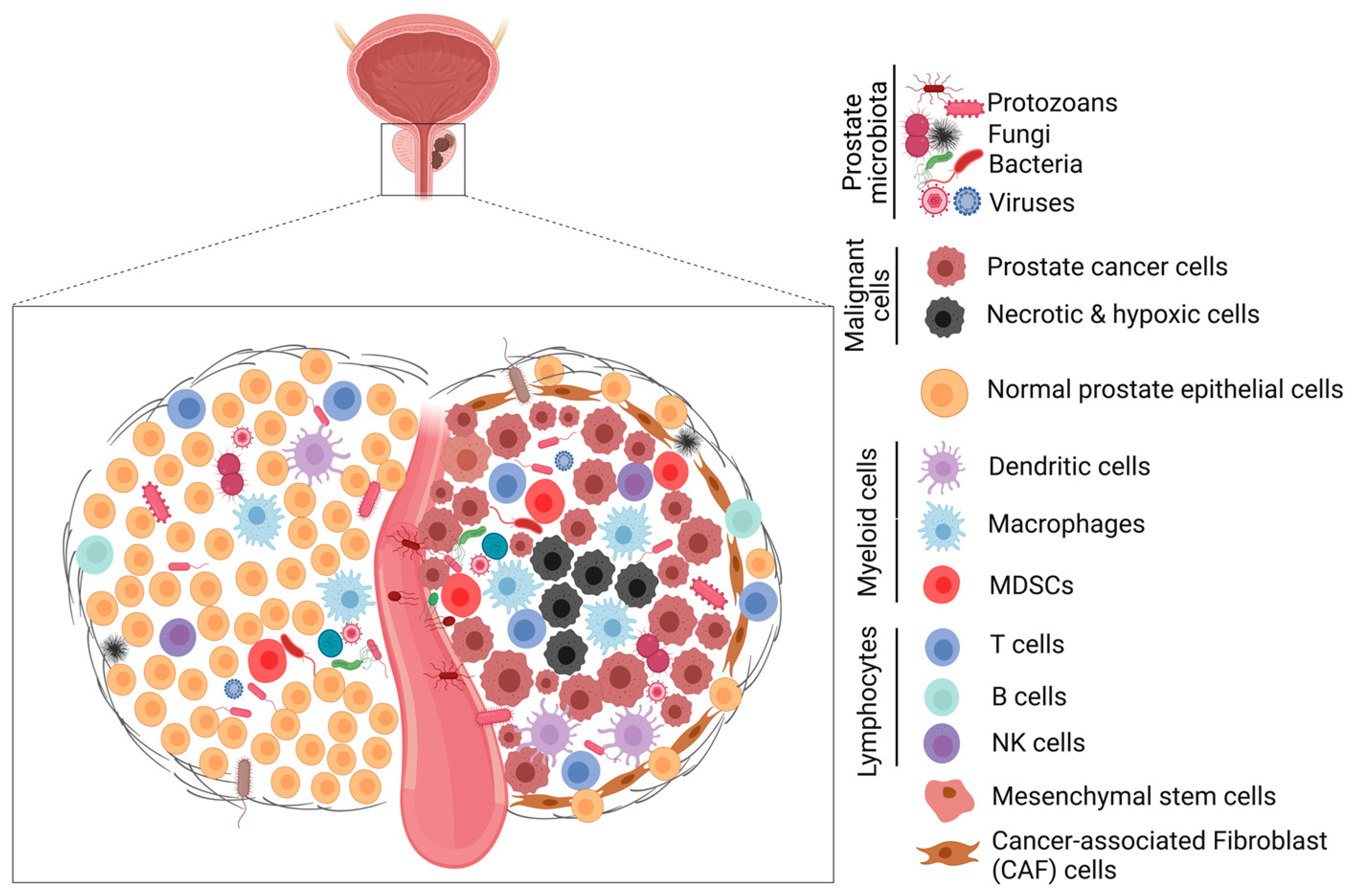
6.1. Role of The Prostate Microbiota in Prostate Tumorigenesis
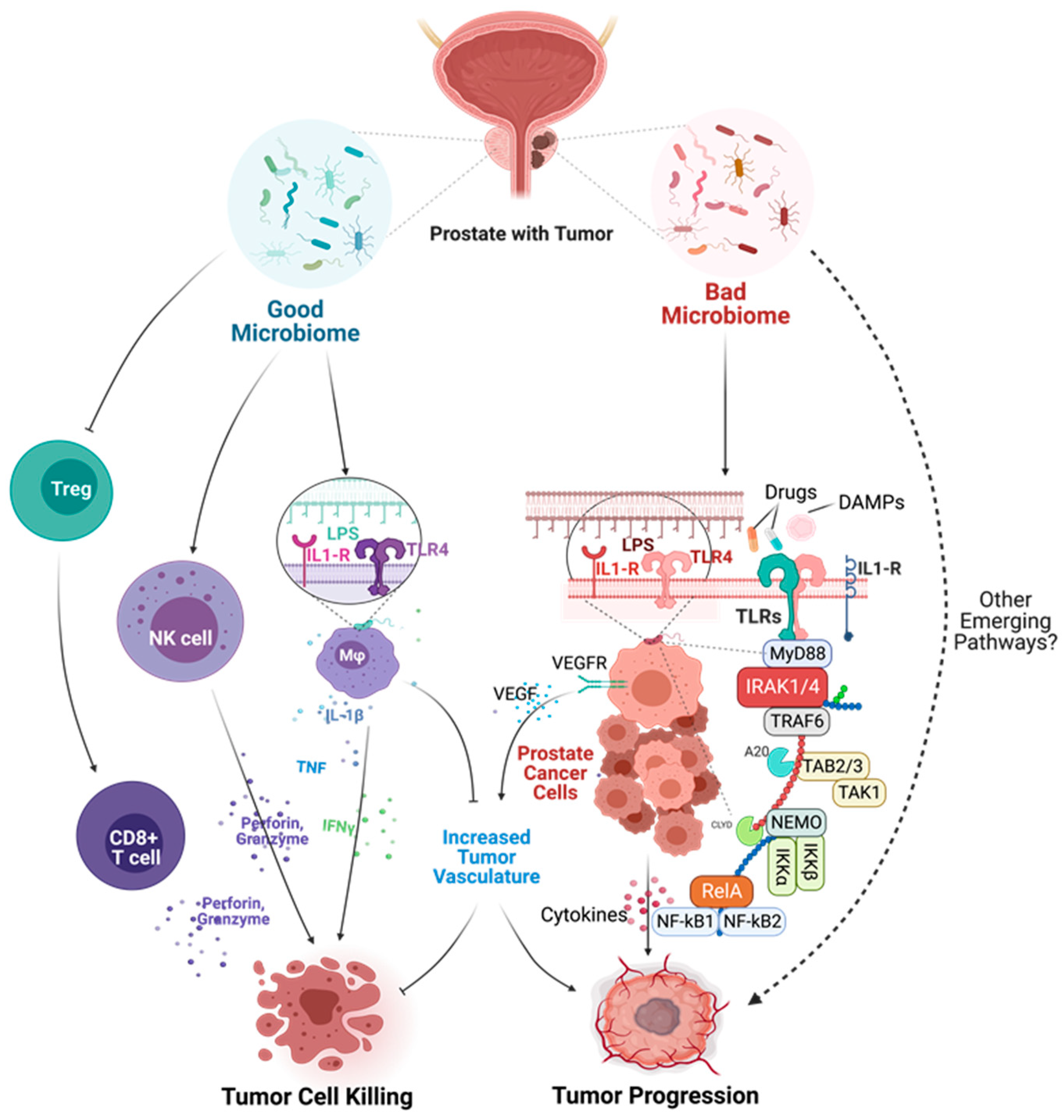
6.2. Mechanisms of Microbial Dysbiosis in Prostate Diseases
7. Role of Sterile Inflammation in Prostate Carcinogenesis
8. Impact of Racial Disparities in Chronic Inflammation-Driven Prostate Diseases
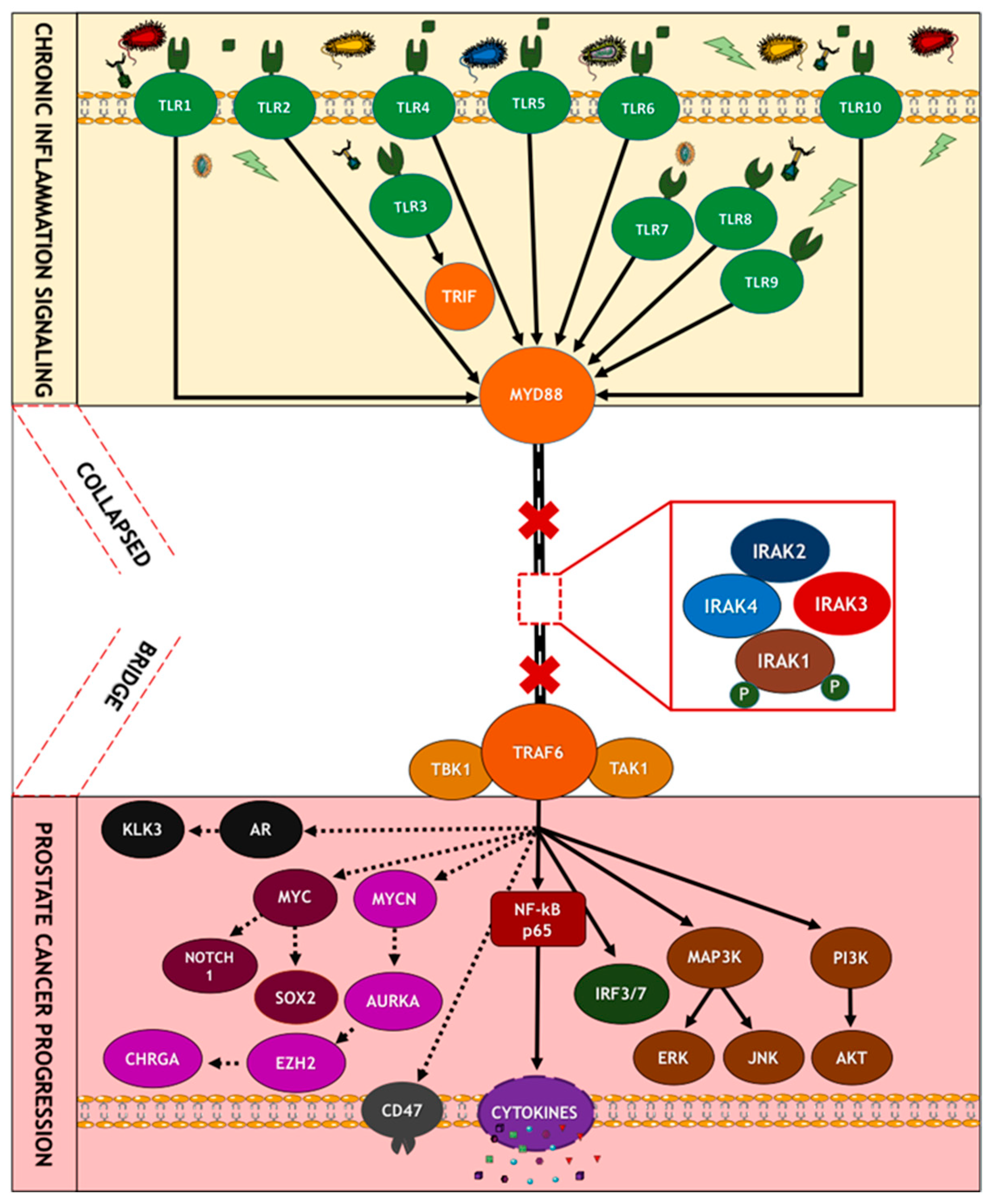
9. Mechanisms of Oncogenic Inflammatory Signal Transduction
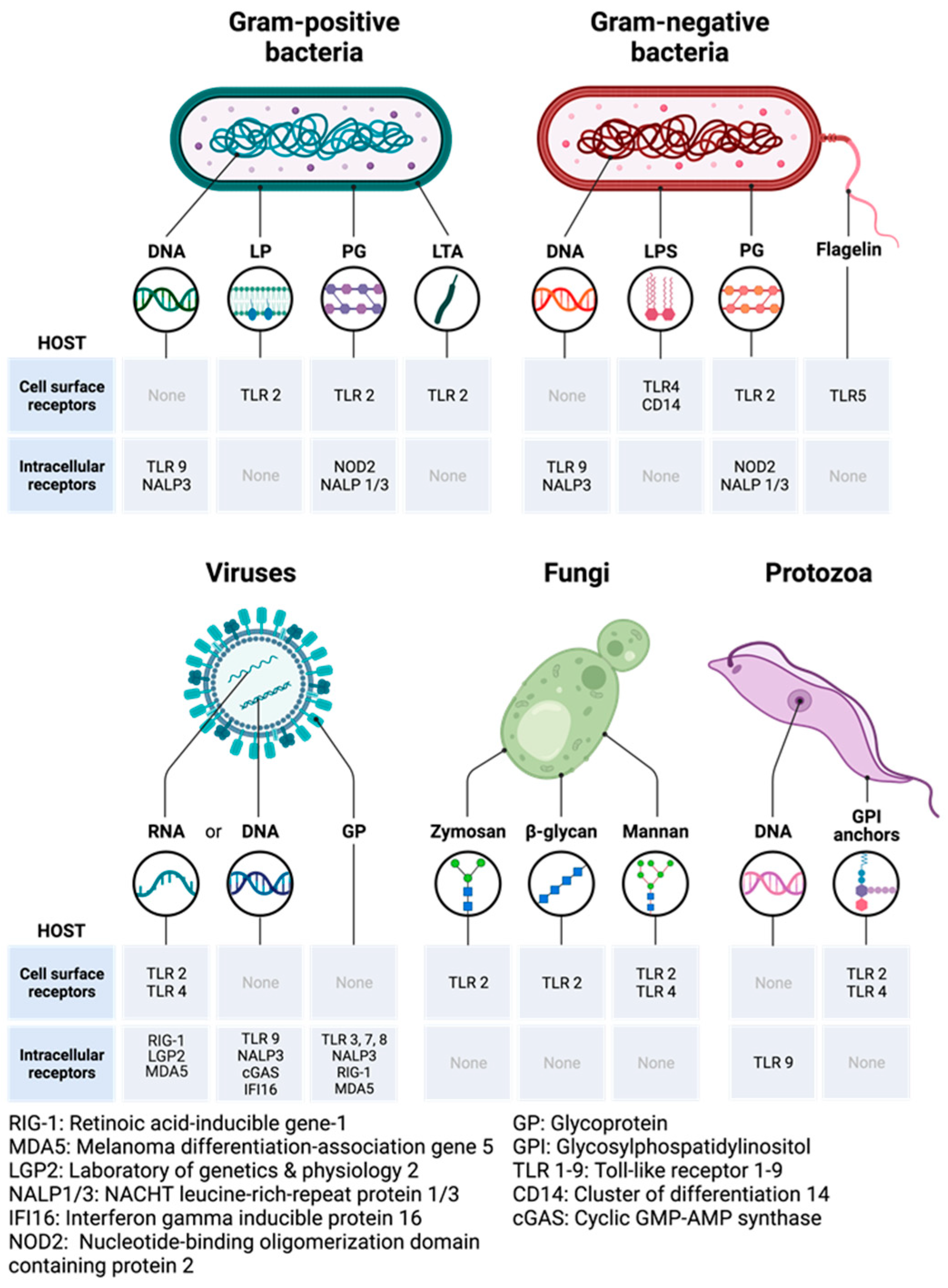
9.1. Pathogen-Associated Molecular and Damage-Associated Molecular Patterns
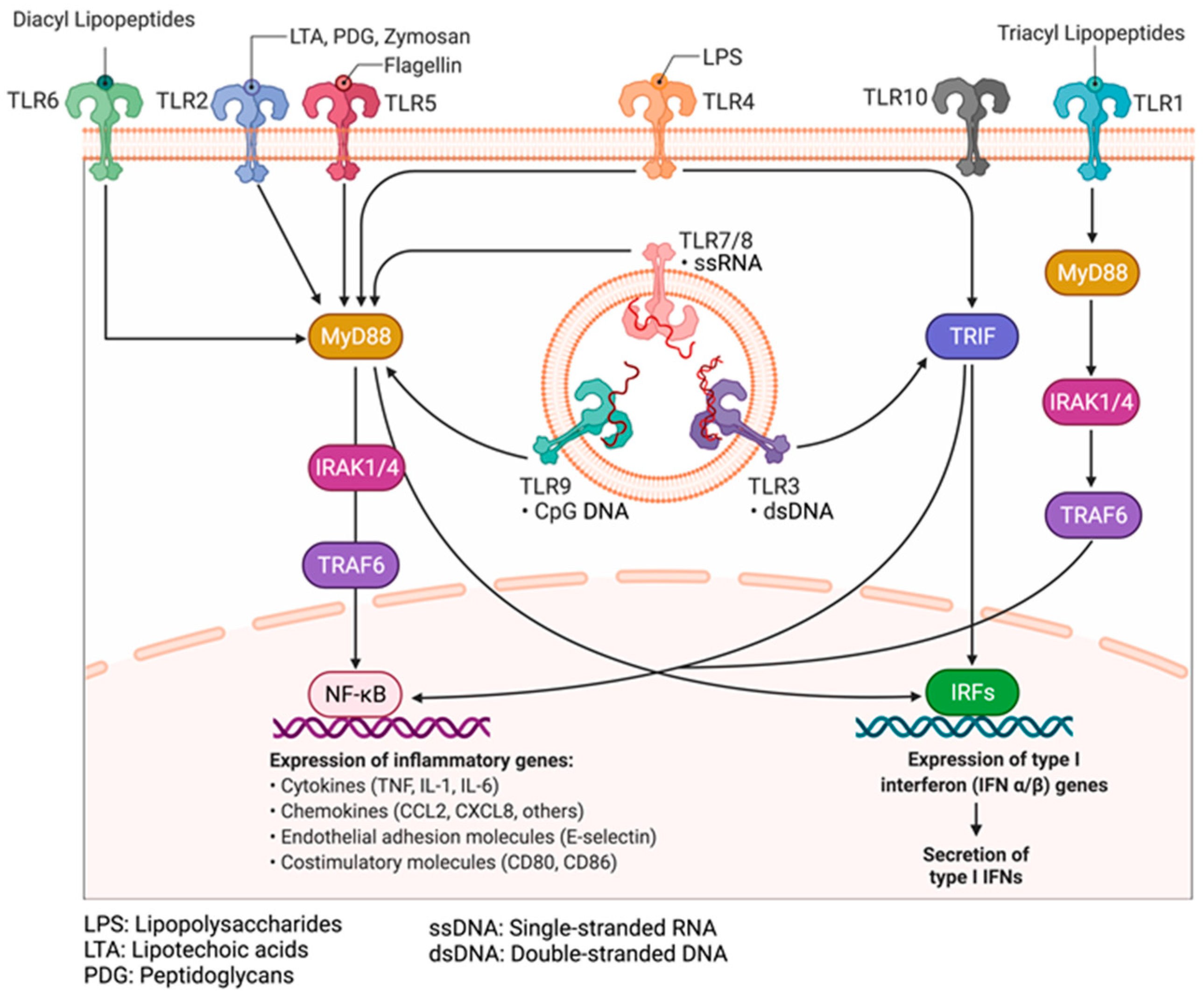
9.2. Pattern Recognition Receptors: Toll-like Receptors and Their Adaptors
9.3. Mechanisms of Activation of PRRs by PAMPs
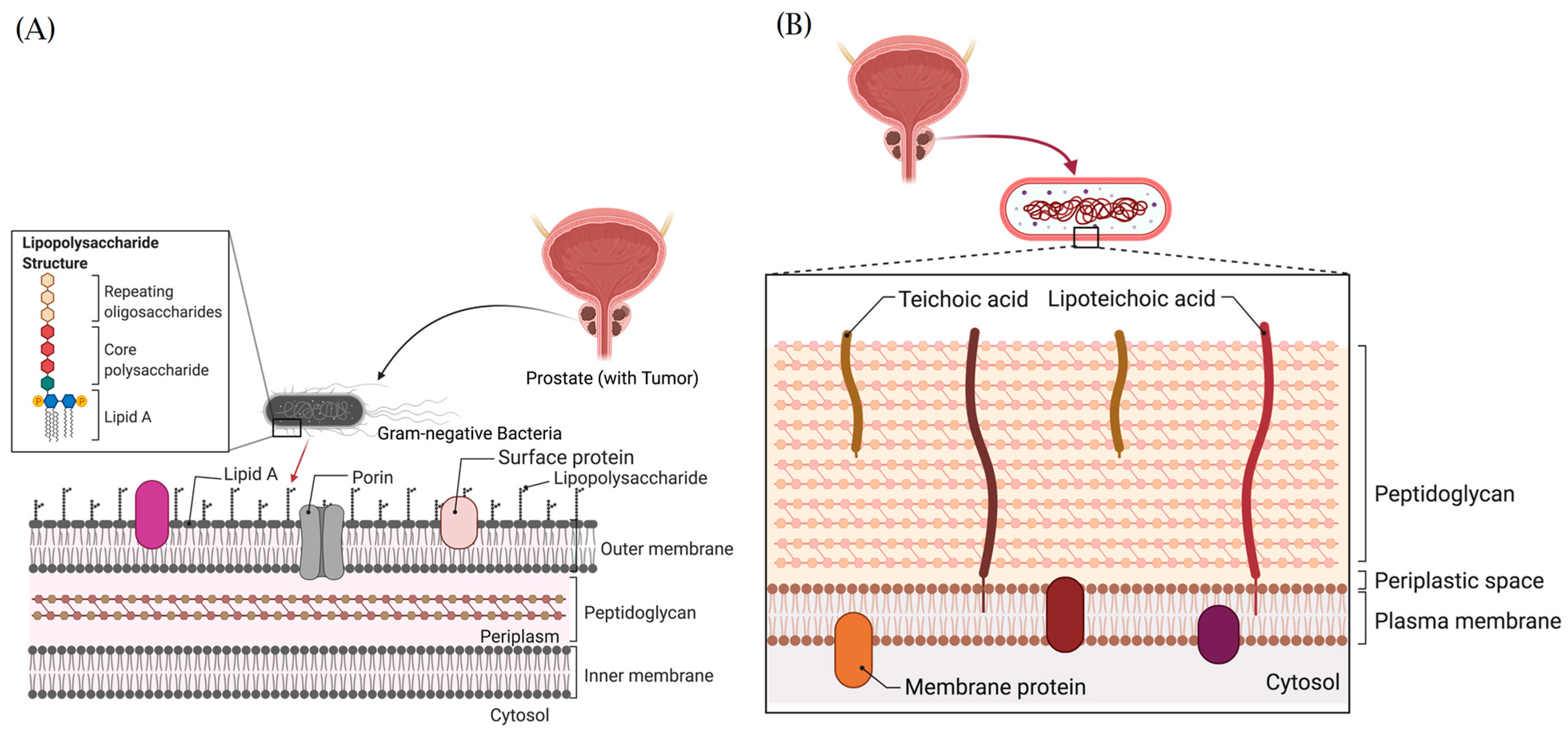
9.4. Structural Architecture and Oncogenic Functions of Bacteria Lipopolysaccharides and Lipoteichoic Acids
9.5. Mechanisms of Activation of PRRs by DAMPs
9.6. Oncogenic Role of TLR Signaling in Prostate Carcinogenesis
9.7. Oncogenic Significance of Inflammasomes and NOD-like Receptor (NLR) Signaling
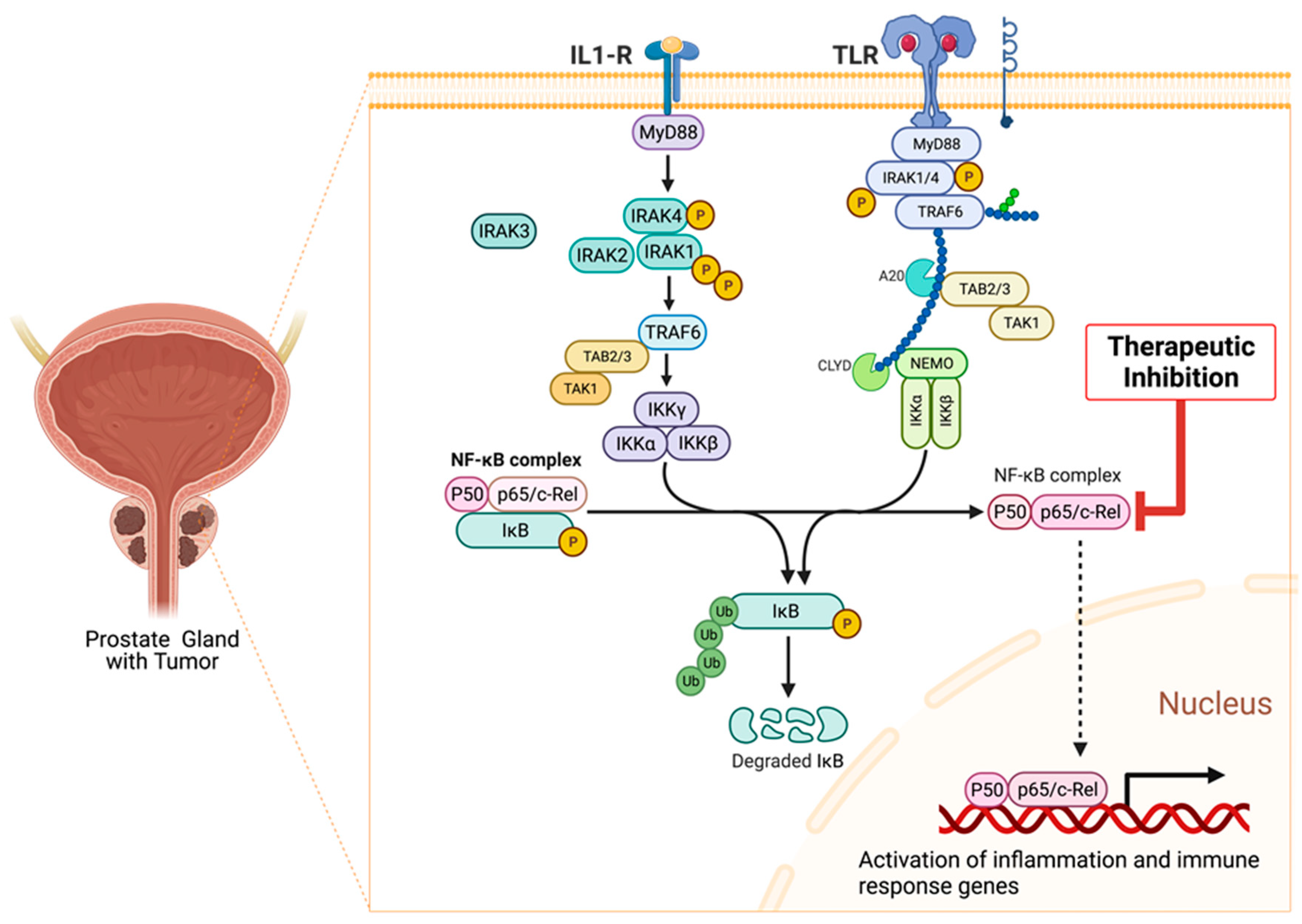
9.8. Association of Interleukin-1 Receptor (IL-1R) Family Signaling with Chronic Inflammation and Carcinogenesis
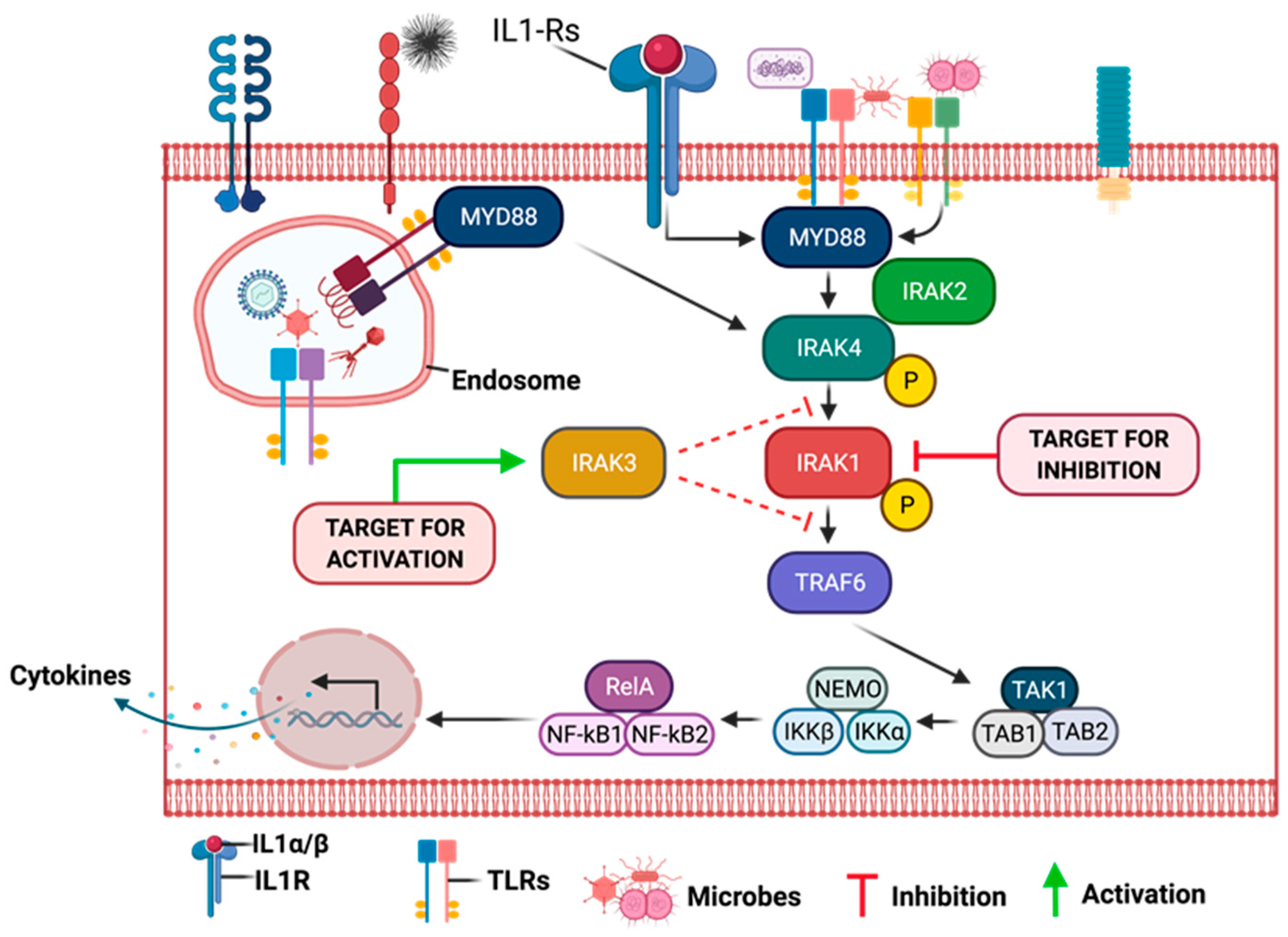
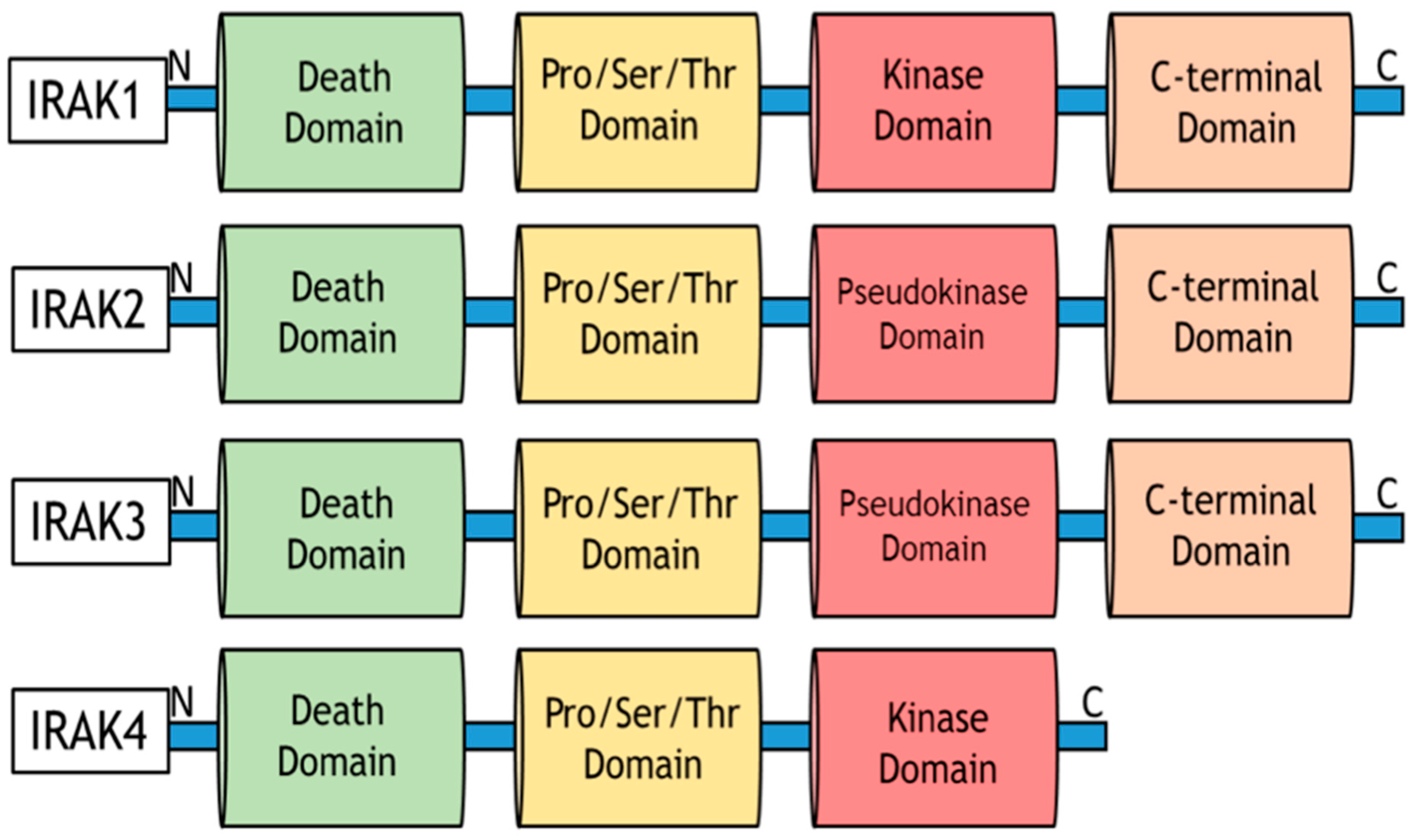
9.9. Role of IRAKs in Inflammation Signaling and Carcinogenesis
9.10. Molecular Mechanisms of IRAK1 Signaling Transduction
9.11. Clinical Significance of Aberrant IRAK1 Signaling in Cancer Progression
9.12. Targeting Aberrant IRAK1 Signaling in Cancers
9.13. Molecular Mechanisms of NF-κB Signaling in Cancer
9.14. Role of NF-κB p65 (RELA) Signaling in Prostate Carcinogenesis
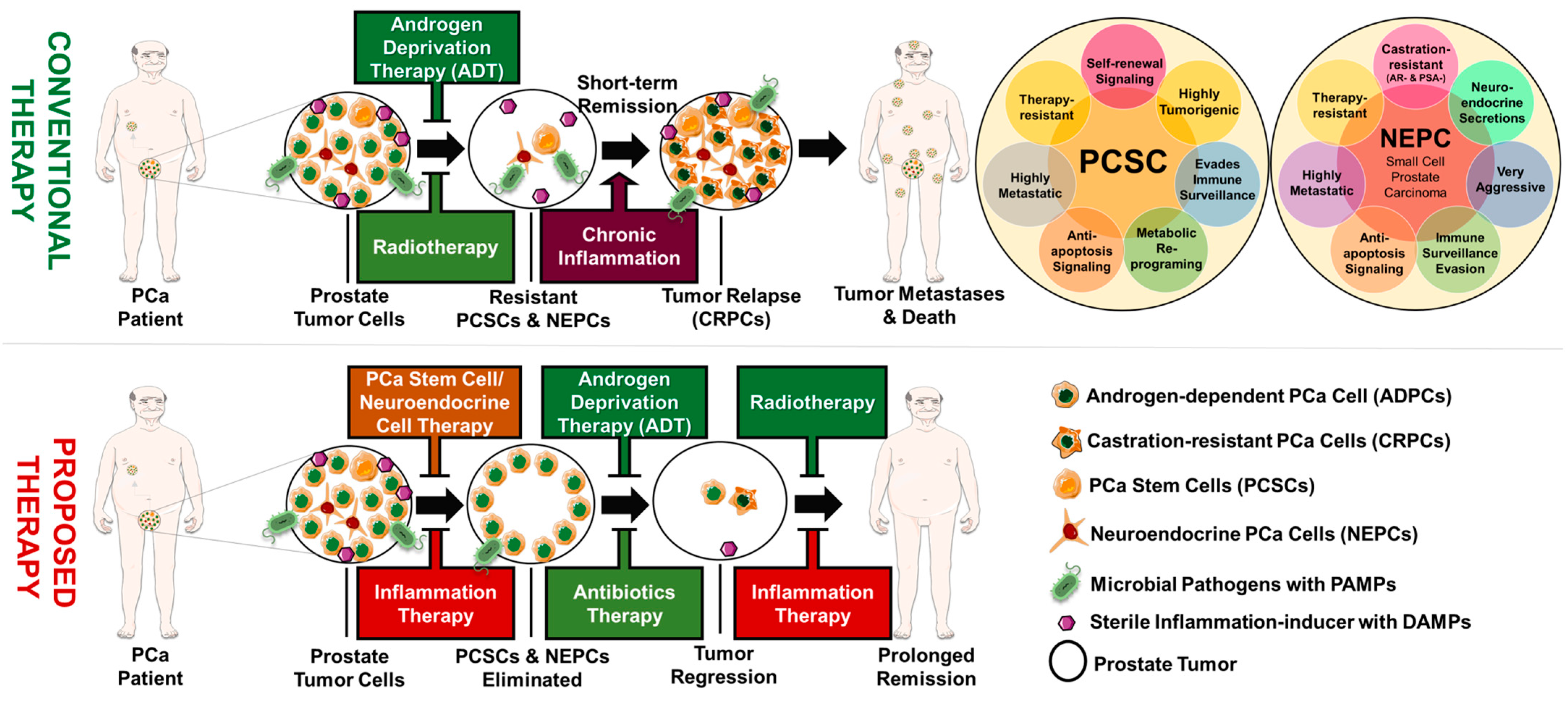
10. Prostate Tumor Heterogeneity and Progression
10.1. Role of Cancer Stemness in Prostate Cancer Progression
10.2. Linking Chronic Inflammation to Prostate Cancer Stemness
10.3. Role of Neuroendocrine Differentiation in Prostate Cancer Progression
10.4. Linking Chronic Inflammation to Neuroendocrine Differentiation
10.5. Linking Chronic Inflammation to Castration Resistance and Prostate Cancer Progression
11. Conclusions
- I.
- How do dysregulation and genetic alteration of each inflammatory molecule, including IL-1Rs, TLRs, TLR adapters (such as MyD88, Mal/TIRAP, CD14, TRAM, and TRIF), and midstream to downstream molecules (such as IRAKs, TRAF6, TAK1/2, TAB1/2/3, JNKs, ERK1/2, MAPK1-14, PI3K isoforms, AKTs, mTOR, NF-κB transcription factors), promote the development and progression of prostate diseases?
- II.
- What roles do inflammatory suppressors/repressors such as IRAK3, TOLLIP, SOCS1, RNF216, IL1RL1, IL36RN, and TNFAIP3 play in the pathogenesis of prostate diseases?
- III.
- Do IRAKs and other midstream inflammatory molecules have any clinical significance in the pathogenesis of prostate diseases?
- IV.
- Can inflammatory molecules be effectively used as diagnostic and prognostic biomarkers for predicting PCa progression, chronic prostatitis, and recurrent BPH?
- V.
- What is the oncogenic role of each IRAK family member in PCa chemoresistance, angiogenesis, NED, stemness, castration resistance, cancer proliferation, and metastasis, among others?
- VI.
- Which of the inflammatory genes play the biggest role in recurring prostatic disease conditions?
- VII.
- What are the best ways to therapeutically target aberrant and chronic inflammatory signaling in prostatic diseases?
- VIII.
- What is the role of each component of the Inflammasomes or NLR signaling in promoting the pathogenesis of prostate disease conditions?
- IX.
- What are the roles of the PI3K/AKT/mTOR pathway signaling in chronic inflammation-driven BPH and PCa?
- X.
- Does dysregulation or persistent activation of interferon signals affect prostate disease development, progression, or recurrence?
- XI.
- Does aberrant RIG-1-like receptor (RLR) signaling play any significant role in the persistence of prostatic disease conditions?
- XII.
- What roles do cGAS-STING signaling play in chronic prostatitis, BPH, and PCa?
- XIII.
- Are there microbial signatures to distinguish between prostatitis, BPH, and PCa?
- XIV.
- Does microbial dysbiosis contribute to racial disparities in prostate disease conditions?
- XV.
- Can microbial dysbiosis signatures be used as diagnostic and prognostic biomarkers for prostatic diseases?
- XVI.
- What are the molecular mechanisms linking prostatic diseases with other chronic inflammatory conditions, including inflammatory bowel disease (IBD) and diabetes mellitus (DM)?
- XVII.
- Are there novel strategies that can be explored to develop prophylactic or therapeutic vaccines against chronic prostatitis, BPH, and PCa?
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, C.H.; Akin-Olugbade, O.; Kirschenbaum, A. Overview of Prostate Anatomy, Histology, and Pathology. Endocrinol. Metab. Clin. N. Am. 2011, 40, 565–575. [Google Scholar] [CrossRef]
- Pelucchi, C.; Talamini, R.; Negri, E.; Franceschi, S.; La Vecchia, C. Genital and Urinary Tract Diseases and Prostate Cancer Risk. Eur. J. Cancer Prev. 2006, 15, 254–257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Yang, P.; Guo, P.; Miele, L.; Sarkar, F.H.; Wang, Z.; Zhou, Q. Unraveling the Mystery of Cancer Metabolism in the Genesis of Tumor-Initiating Cells and Development of Cancer. Biochim. Biophys. Acta 2013, 1836, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and Cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Brüggemann, H.; Al-Zeer, M.A. Bacterial Signatures and Their Inflammatory Potentials Associated with Prostate Cancer. APMIS 2020, 128, 80–91. [Google Scholar] [CrossRef]
- Crew, K.D.; Neugut, A.I. Epidemiology of Gastric Cancer. World J. Gastroenterol. 2006, 12, 354–362. [Google Scholar] [CrossRef]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory Mast Cells Up-Regulate Angiogenesis during Squamous Epithelial Carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef] [PubMed]
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef]
- Stark, T.; Livas, L.; Kyprianou, N. Inflammation in Prostate Cancer Progression and Therapeutic Targeting. Transl. Androl. Urol. 2015, 4, 455–463. [Google Scholar]
- Sfanos, K.S.; De Marzo, A.M. Prostate Cancer and Inflammation: The Evidence. Histopathology 2012, 60, 199–215. [Google Scholar] [CrossRef]
- Poutahidis, T.; Cappelle, K.; Levkovich, T.; Lee, C.-W.; Doulberis, M.; Ge, Z.; Fox, J.G.; Horwitz, B.H.; Erdman, S.E. Pathogenic Intestinal Bacteria Enhance Prostate Cancer Development via Systemic Activation of Immune Cells in Mice. PLoS ONE 2013, 8, e73933. [Google Scholar] [CrossRef] [PubMed]
- Karan, D.; Dubey, S. From Inflammation to Prostate Cancer: The Role of Inflammasomes. Adv. Urol. 2016, 2016, 3140372. [Google Scholar] [CrossRef] [PubMed]
- Ittmann, M. Anatomy and Histology of the Human and Murine Prostate. Cold Spring Harb. Perspect. Med. 2018, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Moodie, L.; Ilie, G.; Rutledge, R.; Andreou, P.; Kirkland, S. Assessment of Current Mental Health Status in a Population-Based Sample of Canadian Men with and without a History of Prostate Cancer Diagnosis: An Analysis of the Canadian Longitudinal Study on Aging (CLSA). Front. Psychiatry 2020, 11, 586260. [Google Scholar] [CrossRef]
- Begley, L.A.; Kasina, S.; MacDonald, J.; Macoska, J.A. The Inflammatory Microenvironment of the Aging Prostate Facilitates Cellular Proliferation and Hypertrophy. Cytokine 2008, 43, 194–199. [Google Scholar] [CrossRef]
- Liu, P.H.; Shah, R.B.; Li, Y.; Arora, A.; Ung, P.M.U.; Raman, R.; Gorbatenko, A.; Kozono, S.; Zhou, X.Z.; Brechin, V.; et al. An IRAK1–PIN1 Signalling Axis Drives Intrinsic Tumour Resistance to Radiation Therapy. Nat. Cell Biol. 2019, 21, 203–213. [Google Scholar] [CrossRef]
- Nickel, J.C.; Nyberg, L.M.; Hennenfent, M. Research Guidelines for Chronic Prostatitis: Consensus Report from the First National Institutes of Health International Prostatitis Collaborative Network. Urology 1999, 54, 229–233. [Google Scholar] [CrossRef]
- Du, C.; Bhatia, M.; Tang, S.C.W.; Zhang, M.; Steiner, T. Mediators of Inflammation: Inflammation in Cancer, Chronic Diseases, and Wound Healing. Mediat. Inflamm. 2015, 2015, 570653. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Anuja, K.; Roy, S.; Ghosh, C.; Gupta, P.; Bhattacharjee, S.; Banerjee, B. Prolonged Inflammatory Microenvironment Is Crucial for Pro-Neoplastic Growth and Genome Instability: A Detailed Review. Inflamm. Res. 2017, 66, 119–128. [Google Scholar] [CrossRef]
- Cai, T.; Santi, R.; Tamanini, I.; Galli, I.C.; Perletti, G.; Bjerklund Johansen, T.E.; Nesi, G. Current Knowledge of the Potential Links between Inflammation and Prostate Cancer. Int. J. Mol. Sci. 2019, 20, 3833. [Google Scholar] [CrossRef] [PubMed]
- Nickel, J.C. Prostatitis. Can. Urol. Assoc. J. 2011, 55, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Pirola, G.M.; Verdacchi, T.; Rosadi, S.; Annino, F.; De Angelis, M. Chronic Prostatitis: Current Treatment Options. Res. Rep. Urol. 2019, 11, 165–174. [Google Scholar] [CrossRef]
- Hua, V.N.; Williams, D.H.; Schaeffer, A.J. Role of Bacteria in Chronic Prostatitis/Chronic Pelvic Pain Syndrome. Curr. Prostate Rep. 2005, 3, 87–93. [Google Scholar] [CrossRef]
- Porcaro, A.B.; Novella, G.; Balzarro, M.; Martignoni, G.; Brunelli, M.; Cacciamani, G.; Cerruto, M.A.; Artibani, W. Prostate Chronic Inflammation Type IV and Prostate Cancer Risk in Patients Undergoing First Biopsy Set: Results of a Large Cohort Study. Asian J. Urol. 2015, 2, 224–232. [Google Scholar] [CrossRef]
- Weir, K. Microbiology: Inflammatory Evidence. Nature 2015, 528, S130–S131. [Google Scholar] [CrossRef] [PubMed]
- Bergh Drott, J. The Role of Microorganisms in Prostate Cancer Development. Ph.D. Thesis, Umeå Universitet, Umeå, Sweden, 2012. [Google Scholar]
- Sarma, A.V.; McLaughlin, J.C.; Wallner, L.P.; Dunn, R.L.; Cooney, K.A.; Schottenfeld, D.; Montie, J.E.; Wei, J.T. Sexual Behavior, Sexually Transmitted Diseases and Prostatitis: The Risk of Prostate Cancer in Black Men. J. Urol. 2006, 176, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Bien, J.; Sokolova, O.; Bozko, P. Role of Uropathogenic Escherichia coli Virulence Factors in Development of Urinary Tract Infection and Kidney Damage. Int. J. Nephrol. 2012, 2012, 681473. [Google Scholar] [CrossRef]
- Ho, D.R. Prostate Inflammation: A Brief Review. Urol. Sci. 2017, 28, 113–118. [Google Scholar] [CrossRef]
- Untergasser, G.; Madersbacher, S.; Berger, P. Benign Prostatic Hyperplasia: Age-Related Tissue-Remodeling. Exp. Gerontol. 2005, 40, 121–128. [Google Scholar] [CrossRef]
- Oesterling, J.E. The Origin and Development of Benign Prostatic Hyperplasia an Age-Dependent Process. J. Androl. 1991, 12, 348–355. [Google Scholar]
- Roehrborn, C.G.; Nuckolls, J.G.; Wei, J.T.; Steers, W. The Benign Prostatic Hyperplasia Registry and Patient Survey: Study Design, Methods and Patient Baseline Characteristics. BJU Int. 2007, 100, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Di Silverio, F.; Gentile, V.; De Matteis, A.; Mariotti, G.; Giuseppe, V.; Luigi, P.A.; Sciarra, A. Distribution of Inflammation, Pre-Malignant Lesions, Incidental Carcinoma in Histologically Confirmed Benign Prostatic Hyperplasia: A Retrospective Analysis. Eur. Urol. 2003, 43, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Fibbi, B.; Penna, G.; Morelli, A.; Adorini, L.; Maggi, M. Chronic Inflammation in the Pathogenesis of Benign Prostatic Hyperplasia. Int. J. Androl. 2010, 33, 475–488. [Google Scholar] [CrossRef]
- De Nunzio, C.; Albisinni, S.; Gacci, M.; Tubaro, A. The Role of Inflammation in the Progression of Benign Prostatic Hyperplasia. Curr. Bladder Dysfunct. Rep. 2013, 8, 142–149. [Google Scholar] [CrossRef]
- De Nunzio, C.; Kramer, G.; Marberger, M.; Montironi, R.; Nelson, W.; Schröder, F.; Sciarra, A.; Tubaro, A. The Controversial Relationship between Benign Prostatic Hyperplasia and Prostate Cancer: The Role of Inflammation. Eur. Urol. 2011, 60, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Roehrborn, C.G. Combination Medical Therapy for Lower Urinary Tract Symptoms and Benign Prostatic Hyperplasia. Rev. Urol. 2005, 7 (Suppl. S8), S43–S51. [Google Scholar]
- McVary, K.T. BPH: Epidemiology and Comorbidities. Am. J. Manag. Care 2006, 12 (Suppl. S5), S122–S128. [Google Scholar]
- Liu, D.; Shoag, J.E.; Poliak, D.; Goueli, R.S.; Ravikumar, V.; Redmond, D.; Vosoughi, A.; Fontugne, J.; Pan, H.; Lee, D.; et al. Integrative Multiplatform Molecular Profiling of Benign Prostatic Hyperplasia Identifies Distinct Subtypes. Nat. Commun. 2020, 11, 1987. [Google Scholar] [CrossRef]
- Shi, Y.F.; Yu, D.J.; Jiang, C.Y.; Wang, X.J.; Zhu, Y.P.; Zhao, R.Z.; Lv, Z.; Sun, X.W. TRAF6 Regulates Proliferation of Stromal Cells in the Transition and Peripheral Zones of Benign Prostatic Hyperplasia via Akt/MTOR Signaling. Prostate 2018, 78, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Chughtai, B.; Lee, R.; Te, A.; Kaplan, S. Role of Inflammation in Benign Prostatic Hyperplasia. Rev. Urol. 2011, 13, 147–150. [Google Scholar] [PubMed]
- Norström, M.M.; Rådestad, E.; Sundberg, B.; Mattsson, J.; Henningsohn, L.; Levitsky, V.; Uhlin, M. Progression of Benign Prostatic Hyperplasia Is Associated with Pro-Inflammatory Mediators and Chronic Activation of Prostate-Infiltrating Lymphocytes. Oncotarget 2016, 7, 23581–23593. [Google Scholar] [CrossRef] [PubMed]
- Penna, G.; Fibbi, B.; Amuchastegui, S.; Corsiero, E.; Laverny, G.; Silvestrini, E.; Chavalmane, A.; Morelli, A.; Sarchielli, E.; Vannelli, G.B.; et al. The Vitamin D Receptor Agonist Elocalcitol Inhibits IL-8-Dependent Benign Prostatic Hyperplasia Stromal Cell Proliferation and Inflammatory Response by Targeting the RhoA/Rho Kinase and NF-KappaB Pathways. Prostate 2009, 69, 480–493. [Google Scholar] [CrossRef]
- Steiner, G.E.; Newman, M.E.; Paikl, D.; Stix, U.; Memaran-Dagda, N.; Lee, C.; Marberger, M.J. Expression and Function of Pro-Inflammatory Interleukin IL-17 and IL-17 Receptor in Normal, Benign Hyperplastic, and Malignant Prostate. Prostate 2003, 56, 171–182. [Google Scholar] [CrossRef]
- Smith, D.K.; Hasanali, S.L.; Wang, J.; Kallifatidis, G.; Morera, D.S.; Jordan, A.R.; Terris, M.K.; Klaassen, Z.; Bollag, R.; Lokeshwar, V.B.; et al. Promotion of Epithelial Hyperplasia by Interleukin-8-CXCR Axis in Human Prostate. Prostate 2020, 80, 938–949. [Google Scholar] [CrossRef]
- Shah, U.S.; Arlotti, J.; Dhir, R.; Lu, S.; Pirozzi, G.; Prakash, K.; Getzenberg, R.H. Androgen Regulation of JM-27 Is Associated with the Diseased Prostate. J. Androl. 2004, 25, 618–624. [Google Scholar] [CrossRef]
- Kramer, G.; Mitteregger, D.; Marberger, M. Is Benign Prostatic Hyperplasia (BPH) an Immune Inflammatory Disease? Eur. Urol. 2006, 51, 1202–1216. [Google Scholar] [CrossRef]
- Taoka, R.; Kakehi, Y. The Influence of Asymptomatic Inflammatory Prostatitis on the Onset and Progression of Lower Urinary Tract Symptoms in Men with Histologic Benign Prostatic Hyperplasia. Asian J. Urol. 2017, 4, 158–163. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Sandeep, N.; Ahmedin, J. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Pernar, C.H.; Ebot, E.M.; Wilson, K.M.; Mucci, L.A. The Epidemiology of Prostate Cancer. Cold Spring Harb. Perspect. Med. 2018, 8, a030361. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Meyers, D.; Freije, D.; Isaacs, S.; Wiley, K.; Nusskern, D.; Ewing, C.; Wilkens, E.; Bujnovszky, P.; Steven Bova, G.; et al. Evidence for a Prostate Cancer Susceptibility Locus on the X Chromosome. Nat. Genet. 1998, 20, 175–179. [Google Scholar] [CrossRef]
- Gambert, S.R. Screening for Prostate Cancer. Int. Urol. Nephrol. 2001, 33, 249–257. [Google Scholar] [CrossRef]
- Bristow, R.G.; Berlin, A.; Dal Pra, A. An Arranged Marriage for Precision Medicine: Hypoxia and Genomic Assays in Localized Prostate Cancer Radiotherapy. Br. J. Radiol. 2014, 87, 20130753. [Google Scholar] [CrossRef] [PubMed]
- Hynes, P.G.; Kelly, K. Prostate Cancer Stem Cells: The Case for Model Systems. J. Carcinog. 2012, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Rycaj, K.; Cho, E.J.; Liu, X.; Chao, H.-P.; Liu, B.; Li, Q.; Devkota, A.K.; Zhang, D.; Chen, X.; Moore, J.; et al. Longitudinal Tracking of Subpopulation Dynamics and Molecular Changes during LNCaP Cell Castration and Identification of Inhibitors That Could Target the PSA Castration-Resistant Cells. Oncotarget 2016, 7, 14220. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Godoy, A.; Azzouni, F.; Wilton, J.H.; Ip, C.; Mohler, J.L. Prostate Cancer Cells Differ in Testosterone Accumulation, Dihydrotestosterone Conversion, and Androgen Receptor Signaling Response to Steroid 5α-Reductase Inhibitors. Prostate 2013, 73, 1470–1482. [Google Scholar] [CrossRef]
- Wu, C.-T.; Chen, M.-F.; Chen, W.-C.; Hsieh, C.-C. The Role of IL-6 in the Radiation Response of Prostate Cancer. Radiat. Oncol. 2013, 8, 159. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Alekseyenko, A.V.; Wu, J.; Peters, B.A.; Jacobs, E.J.; Gapstur, S.M.; Purdue, M.P.; Abnet, C.C.; Stolzenberg-Solomon, R.; Miller, G.; et al. Human Oral Microbiome and Prospective Risk for Pancreatic Cancer: A Population-Based Nested Case-Control Study. Gut 2018, 67, 120–127. [Google Scholar] [CrossRef]
- Shrestha, E.; White, J.R.; Yu, S.H.; Kulac, I.; Ertunc, O.; De Marzo, A.M.; Yegnasubramanian, S.; Mangold, L.A.; Partin, A.W.; Sfanos, K.S. Profiling the Urinary Microbiome in Men with Positive versus Negative Biopsies for Prostate Cancer. J. Urol. 2018, 199, 161–171. [Google Scholar] [CrossRef]
- Shannon, B.A.; Garrett, K.L.; Cohen, R.J. Links between Propionibacterium Acnes and Prostate Cancer. Future Oncol. 2006, 2, 225–232. [Google Scholar] [CrossRef]
- Li, Q.; Deng, Q.; Chao, H.P.; Liu, X.; Lu, Y.; Lin, K.; Liu, B.; Tang, G.W.; Zhang, D.; Tracz, A.; et al. Linking Prostate Cancer Cell AR Heterogeneity to Distinct Castration and Enzalutamide Responses. Nat. Commun. 2018, 9, 3600. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; McCubrey, J.A.; Jefferson, H.S.; Paine, M.S.; Chappell, W.H.; Terrian, D.M. A Dominant Role for P53-Dependent Cellular Senescence in Radiosensitization of Human Prostate Cancer Cells. Cell Cycle 2007, 6, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef]
- Feng, Y.; Ramnarine, V.R.; Bell, R.; Volik, S.; Davicioni, E.; Hayes, V.M.; Ren, S.; Collins, C.C. Metagenomic and Metatranscriptomic Analysis of Human Prostate Microbiota from Patients with Prostate Cancer. BMC Genom. 2019, 20, 146. [Google Scholar] [CrossRef]
- Garcia-Martinez, I.; Shaker, M.E.; Mehal, W.Z. Therapeutic Opportunities in Damage-Associated Molecular Pattern-Driven Metabolic Diseases. Antioxid. Redox Signal. 2015, 23, 1305–1315. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, J.; Han, J.H.; Lee, J.Y.; Chang, I.H.; Kim, T.H.; Do Kim, K.; Myung, S.C. Variants in Interleukin-1 Receptor-Associated Kinase 4 (IRAK4) Gene Are Associated with Susceptibility to Prostate Cancer. Korean J. Urol. Oncol. 2012, 10, 27–33. [Google Scholar]
- Kimman, T.G.; Barius, S.; Reijmerink, N.; Reimerink, J.; Stelma, F.F.; Koppelman, G.H.; Thijs, C.; Postman, D.S.; Kerkhof, M. Association of Interacting Genes in the Toll-like Receptor Signaling Pathway and the Antibody Response to Pertussis Vaccination. PLoS ONE 2008, 3, e3665. [Google Scholar] [CrossRef]
- Rogers, E.N.; Jones, D.Z.; Kidd, N.C.; Yeyeodu, S.; Brock, G.; Ragin, C.; Jackson, M.; McFarlane-Anderson, N.; Tulloch-Reid, M.; Sean Kimbro, K.; et al. Toll-like Receptor-Associated Sequence Variants and Prostate Cancer Risk among Men of African Descent. Genes Immun. 2013, 14, 347–355. [Google Scholar] [CrossRef]
- Kwon, O.J.; Zhang, B.; Zhang, L.; Xin, L. High Fat Diet Promotes Prostatic Basal-to-Luminal Differentiation and Accelerates Initiation of Prostate Epithelial Hyperplasia Originated from Basal Cells. Stem Cell Res. 2016, 16, 682–691. [Google Scholar] [CrossRef]
- Barton, M.K. Daily aspirin may reduce mortality from prostate cancer with risk of high recurrence. CA A Cancer J. Clin. 2015, 65, 83–84. [Google Scholar] [CrossRef]
- Cui, L.; Morris, A.; Ghedin, E. The Human Mycobiome in Health and Disease. Genome Med. 2013, 5, 63. [Google Scholar] [CrossRef]
- Apidianakis, Y.; Pitsouli, C.; Perrimon, N.; Rahme, L. Synergy between Bacterial Infection and Genetic Predisposition in Intestinal Dysplasia. Proc. Natl. Acad. Sci. USA 2009, 106, 20883–20888. [Google Scholar] [CrossRef]
- Kwon, O.-J.; Zhang, L.; Ittmann, M.M.; Xin, L. Prostatic Inflammation Enhances Basal-to-Luminal Differentiation and Accelerates Initiation of Prostate Cancer with a Basal Cell Origin. Proc. Natl. Acad. Sci. USA 2014, 111, E592–E600. [Google Scholar] [CrossRef]
- Santoni, M.; Conti, A.; Burattini, L.; Berardi, R.; Scarpelli, M.; Cheng, L.; Lopez-Beltran, A.; Cascinu, S.; Montironi, R. Neuroendocrine Differentiation in Prostate Cancer: Novel Morphological Insights and Future Therapeutic Perspectives. Biochim. Biophys. Acta BBA Rev. Cancer 2014, 1846, 630–637. [Google Scholar] [CrossRef]
- Hernandez, C.; Huebener, P.; Schwabe, R.F. Damage-Associated Molecular Patterns in Cancer: A Double-Edged Sword. Oncogene 2016, 35, 5931–5941. [Google Scholar] [CrossRef]
- Eloe-Fadrosh, E.A.; Rasko, D.A. The Human Microbiome: From Symbiosis to Pathogenesis. Annu. Rev. Med. 2013, 64, 145–163. [Google Scholar] [CrossRef]
- Harmon, K. Bugs Inside: What Happens When the Microbes That Keep Us Healthy Disappear? Scientific American, 16 December 2009. [Google Scholar]
- Human Microbiome Project Consortium. A Framework for Human Microbiome Research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef]
- Kidane, D.; Chae, W.J.; Czochor, J.; Eckert, K.A.; Glazer, P.M.; Bothwell, A.L.M.; Sweasy, J.B. Interplay between DNA Repair and Inflammation, and the Link to Cancer. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 116–139. [Google Scholar] [CrossRef] [PubMed]
- Massari, F.; Mollica, V.; Di Nunno, V.; Gatto, L.; Santoni, M.; Scarpelli, M.; Cimadamore, A.; Lopez-Beltran, A.; Cheng, L.; Battelli, N.; et al. The Human Microbiota and Prostate Cancer: Friend or Foe? Cancers 2019, 11, 459. [Google Scholar] [CrossRef]
- Francescone, R.; Hou, V.; Grivennikov, S.I. Microbiome, Inflammation, and Cancer. Cancer J. 2014, 20, 181. [Google Scholar] [CrossRef]
- Feng, X.; Han, L.; Ma, S.; Zhao, L.; Wang, L.; Zhang, K.; Yin, P.; Guo, L.; Jing, W.; Li, Q. Microbes in Tumoral In Situ Tissues and in Tumorigenesis. Front. Cell. Infect. Microbiol. 2020, 10, 572570. [Google Scholar] [CrossRef]
- Markowski, M.C.; Boorjian, S.A.; Burton, J.P.; Hahn, N.M.; Ingersoll, M.A.; Vareki, S.M.; Pal, S.K.; Sfanos, K.S. The Microbiome and Genitourinary Cancer: A Collaborative Review. Eur. Urol. 2019, 75, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Zackular, J.P.; Baxter, N.T.; Iverson, K.D.; Sadler, W.D.; Petrosino, J.F.; Chen, G.Y.; Schloss, P.D. The Gut Microbiome Modulates Colon Tumorigenesis. mBio 2013, 4, e00692-13. [Google Scholar] [CrossRef] [PubMed]
- Katongole, P.; Sande, O.J.; Joloba, M.; Reynolds, S.J.; Niyonzima, N. The Human Microbiome and Its Link in Prostate Cancer Risk and Pathogenesis. Infect. Agent Cancer 2020, 15, 53. [Google Scholar] [CrossRef]
- Cavarretta, I.; Mancini, N.; Salonia, A. Analysis of the Enteric Microbiome: First Tentative Steps Towards a Comprehensive Work-up of Prostate Cancer? Eur. Urol. 2018, 74, 583–584. [Google Scholar] [CrossRef]
- Cavarretta, I.; Ferrarese, R.; Cazzaniga, W.; Saita, D.; Lucianò, R.; Ceresola, E.R.; Locatelli, I.; Visconti, L.; Lavorgna, G.; Briganti, A.; et al. The Microbiome of the Prostate Tumor Microenvironment. Eur. Urol. 2017, 72, 625–631. [Google Scholar] [CrossRef]
- Miyake, M.; Ohnishi, K.; Hori, S.; Nakano, A.; Nakano, R.; Yano, H.; Ohnishi, S.; Owari, T.; Morizawa, Y.; Itami, Y.; et al. Mycoplasma Genitalium Infection and Chronic Inflammation in Human Prostate Cancer: Detection Using Prostatectomy and Needle Biopsy Specimens. Cells 2019, 8, 212. [Google Scholar] [CrossRef]
- Liss, M.A.; White, J.R.; Goros, M.; Gelfond, J.; Leach, R.; Johnson-Pais, T.; Lai, Z.; Rourke, E.; Basler, J.; Ankerst, D.; et al. Metabolic Biosynthesis Pathways Identified from Fecal Microbiome Associated with Prostate Cancer. Eur. Urol. 2018, 74, 575–582. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Yegnasubramanian, S.; Nelson, W.G.; De Marzo, A.M. The Inflammatory Microenvironment and Microbiome in Prostate Cancer Development. Nat. Rev. Urol. 2018, 15, 11–24. [Google Scholar] [CrossRef]
- Porter, C.M.; Shrestha, E.; Peiffer, L.B.; Sfanos, K.S. The Microbiome in Prostate Inflammation and Prostate Cancer. Prostate Cancer Prostatic Dis. 2018, 21, 345–354. [Google Scholar] [CrossRef]
- Sha, S.; Ni, L.; Stefil, M.; Dixon, M.; Mouraviev, V. The Human Gastrointestinal Microbiota and Prostate Cancer Development and Treatment. Investig. Clin. Urol. 2020, 61, S43–S50. [Google Scholar] [CrossRef]
- Harada, N.; Hanaoka, R.; Horiuchi, H.; Kitakaze, T.; Mitani, T.; Inui, H.; Yamaji, R. Castration Influences Intestinal Microflora and Induces Abdominal Obesity in High-Fat Diet-Fed Mice. Sci. Rep. 2016, 6, 23001. [Google Scholar] [CrossRef]
- Clarke, G.; Stilling, R.M.; Kennedy, P.J.; Stanton, C.; Cryan, J.F.; Dinan, T.G. Minireview: Gut Microbiota: The Neglected Endocrine Organ. Mol. Endocrinol. 2014, 28, 1221–1238. [Google Scholar] [CrossRef] [PubMed]
- Xuan, C.; Shamonki, J.M.; Chung, A.; DiNome, M.L.; Chung, M.; Sieling, P.A.; Lee, D.J. Microbial Dysbiosis Is Associated with Human Breast Cancer. PLoS ONE 2014, 9, e83744. [Google Scholar] [CrossRef] [PubMed]
- Elkahwaji, J.E.; Hauke, R.J.; Brawner, C.M. Chronic Bacterial Inflammation Induces Prostatic Intraepithelial Neoplasia in Mouse Prostate. Br. J. Cancer 2009, 101, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Duncan, S.H.; Louis, P.; Flint, H.J. Cultivable Bacterial Diversity from the Human Colon. Lett. Appl. Microbiol. 2007, 44, 343–350. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium Persistence and Antibiotic Response in Colorectal Cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef]
- Sfanos, K.S.; Isaacs, W.B.; De Marzo, A.M. Infections and Inflammation in Prostate Cancer. Am. J. Clin. Exp. Urol. 2013, 1, 3–11. [Google Scholar] [PubMed]
- Plottel, C.S.; Blaser, M.J. Microbiome and Malignancy. Cell Host Microbe 2011, 10, 324–335. [Google Scholar] [CrossRef]
- De Marzo, A.M.; Platz, E.A.; Sutcliffe, S.; Xu, J.; Grönberg, H.; Drake, C.G.; Nakai, Y.; Isaacs, W.B.; Nelson, W.G. Inflammation in Prostate Carcinogenesis. Nat. Rev. Cancer 2007, 7, 256–269. [Google Scholar] [CrossRef] [PubMed]
- Pettaway, C.A.; Lamerato, L.E.; Eaddy, M.T.; Edwards, J.K.; Hogue, S.L.; Crane, M.M. Benign Prostatic Hyperplasia: Racial Differences in Treatment Patterns and Prostate Cancer Prevalence. BJU Int. 2011, 108, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Kristal, A.R.; Arnold, K.B.; Schenk, J.M.; Neuhouser, M.L.; Weiss, N.; Goodman, P.; Antvelink, C.M.; Penson, D.F.; Thompson, I.M. Race/Ethnicity, Obesity, Health Related Behaviors and the Risk of Symptomatic Benign Prostatic Hyperplasia: Results from the Prostate Cancer Prevention Trial. J. Urol. 2007, 177, 1395–1400. [Google Scholar] [CrossRef] [PubMed]
- Gillard, M.; Javier, R.; Ji, Y.; Lilly Zheng, S.; Xu, J.; Brendler, C.B.; Crawford, S.E.; Pierce, B.L.; Vander Griend, D.J.; Franco, O.E. Elevation of Stromal-Derived Mediators of Inflammation Promote Prostate Cancer Progression in African-American Men. Cancer Res. 2018, 78, 6134–6145. [Google Scholar] [CrossRef] [PubMed]
- Hoke, G.P.; McWilliams, G.W. Epidemiology of Benign Prostatic Hyperplasia and Comorbidities in Racial and Ethnic Minority Populations. Am. J. Med. 2008, 121, S3–S10. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef]
- Del Prete, A.; Allavena, P.; Santoro, G.; Fumarulo, R.; Corsi, M.M.; Mantovani, A. Molecular Pathways in Cancer-Related Inflammation. Biochem. Med. 2011, 21, 264–275. [Google Scholar] [CrossRef]
- Su, J.; Xie, Q.; Wilson, I.; Li, L. Differential Regulation and Role of Interleukin-1 Receptor Associated Kinase-M in Innate Immunity Signaling. Cell. Signal. 2007, 19, 1596–1601. [Google Scholar] [CrossRef]
- Afsharimoghaddam, A.; Soleimani, M.; Lashay, A.; Dehghani, M.; Sepehri, Z. Controversial Roles Played by Toll like Receptor 4 in Urinary Bladder Cancer; A Systematic Review. Life Sci. 2016, 158, 31–36. [Google Scholar] [CrossRef]
- Oseni, S.O.; Pavlovic, M.; Hartmann, J.; Kumi-Diaka, J. Abstract 995: Integrative Genomic Characterization and CRISPR-Mediated Gene Editing Studies Identify IRAKs as Novel Therapeutic Targets for Inflammation-Driven Prostate Tumorigenesis. Cancer Res. 2020, 80, 995. [Google Scholar] [CrossRef]
- Oseni, S.O.; Branly, R.; Pavlovic, M.; Kumi-Diaka, J. Synergistic Effects of Metabolic Inhibitors on Radiochemosensitized Spheroid Prostate Cancer Cells. Cancer Res. 2017, 77, 5422. [Google Scholar] [CrossRef]
- Oseni, S.O.; Naar, C.; Philemy, M.; Laguer, G.A.; Ambrin, F.; Metayer, S.; Perez, A.; Betty, J.; Praveen, P.; Hartmann, J.; et al. Abstract 1753: Deregulation and Therapeutic Potential of Targeting IRAK3 as Chronic Inflammation Suppressor in Prostate Cancer. Cancer Res. 2021, 81, 1753. [Google Scholar] [CrossRef]
- Choi, H.W.; Klessig, D.F. DAMPs, MAMPs, and NAMPs in Plant Innate Immunity. BMC Plant Biol. 2016, 16, 232. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s That Spur Autophagy and Immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Pandey, S.; Singh, S.; Anang, V.; Bhatt, A.N.; Natarajan, K.; Dwarakanath, B.S. Pattern Recognition Receptors in Cancer Progression and Metastasis. Cancer Growth Metastasis 2015, 8, CGM-S24314. [Google Scholar] [CrossRef]
- Ibrahim, Z.A.; Armour, C.L.; Phipps, S.; Sukkar, M.B. RAGE and TLRs: Relatives, Friends or Neighbours? Mol. Immunol. 2013, 56, 739–744. [Google Scholar] [CrossRef]
- Salaun, B.; Romero, P.; Lebecque, S. Toll-like Receptors’ Two-Edged Sword: When Immunity Meets Apoptosis. Eur. J. Immunol. 2007, 37, 3311–3318. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kawai, T. Toll-like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Du, J.; Nicolaes, G.A.F.; Kruijswijk, D.; Versloot, M.; Van Der Poll, T.; Van’t Veer, C. The Structure Function of the Death Domain of Human IRAK-M. Cell Commun. Signal. 2014, 12, 77. [Google Scholar] [CrossRef]
- Chaturvedi, A.; Pierce, S.K. How Location Governs Toll-Like Receptor Signaling. Traffic 2009, 10, 621–628. [Google Scholar] [CrossRef]
- Gorina, R.; Font-Nieves, M.; Márquez-Kisinousky, L.; Santalucia, T.; Planas, A.M. Astrocyte TLR4 Activation Induces a Proinflammatory Environment through the Interplay between MyD88-Dependent NFκB Signaling, MAPK, and Jak1/Stat1 Pathways. Glia 2011, 59, 242–255. [Google Scholar] [CrossRef]
- Liu, P.H.; Sidi, S. Targeting the Innate Immune Kinase IRAK1 in Radioresistant Cancer: Double-Edged Sword or One-Two Punch? Front. Oncol. 2019, 9, 1174. [Google Scholar] [CrossRef]
- Singh, A.R.; Peirce, S.K.; Joshi, S.; Durden, D.L. PTEN and PI-3 Kinase Inhibitors Control LPS Signaling and the Lymphoproliferative Response in the CD19+ B Cell Compartment. Exp. Cell Res. 2014, 327, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Gambara, G.; De Cesaris, P.; De Nunzio, C.; Ziparo, E.; Tubaro, A.; Filippini, A.; Riccioli, A. Toll-like Receptors in Prostate Infection and Cancer between Bench and Bedside. J. Cell. Mol. Med. 2013, 17, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Block, M.S.; Vierkant, R.A.; Rambau, P.F.; Winham, S.J.; Wagner, P.; Traficante, N.; Tołoczko, A.; Tiezzi, D.G.; Taran, F.A.; Sinn, P.; et al. MyD88 and TLR4 Expression in Epithelial Ovarian Cancer. Mayo Clin. Proc. 2018, 93, 307–320. [Google Scholar] [CrossRef]
- Cui, J.; Chen, Y.; Wang, H.Y.; Wang, R.-F. Mechanisms and Pathways of Innate Immune Activation and Regulation in Health and Cancer. Hum. Vaccines Immunother. 2014, 10, 3270–3285. [Google Scholar] [CrossRef]
- Rubartelli, A.; Lotze, M.T.; Latz, E.; Manfredi, A.A. Mechanisms of Sterile Inflammation. Front. Immunol. 2013, 4, 398. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen Recognition by the Innate Immune System. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile Inflammation: Sensing and Reacting to Damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Franz, K.M.; Kagan, J.C. Innate Immune Receptors as Competitive Determinants of Cell Fate. Mol. Cell 2017, 66, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Standiford, T.J.; Keshamouni, V.G. Breaking the Tolerance for Tumor. Oncoimmunology 2012, 1, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, A.E.; Lentschat, A.; Wahl, L.M.; Golenbock, D.T.; Vogel, S.N. Dysregulation of LPS-Induced Toll-like Receptor 4-MyD88 Complex Formation and IL-1 Receptor-Associated Kinase 1 Activation in Endotoxin-Tolerant Cells. J. Immunol. 2002, 169, 5209–5216. [Google Scholar] [CrossRef] [PubMed]
- Gentile, L.F.; Moldawer, L.L. DAMPs, PAMPs, and the Origins of SIRS in Bacterial Sepsis. Shock 2013, 39, 113–114. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B. Inferences, Questions and Possibilities in Toll-like Receptor Signalling. Nature 2004, 430, 257–263. [Google Scholar] [CrossRef]
- Drage, M.G.; Pecora, N.D.; Hise, A.G.; Febbraio, M.; Silverstein, R.L.; Golenbock, D.T.; Boom, W.H.; Harding, C.V. TLR2 and Its Co-Receptors Determine Responses of Macrophages and Dendritic Cells to Lipoproteins of Mycobacterium Tuberculosis. Cell. Immunol. 2009, 258, 29–37. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Microbial Recognition by Toll-like Receptors. J. Dermatol. Sci. 2004, 34, 73–82. [Google Scholar] [CrossRef]
- Cimadamore, A.; Santoni, M.; Massari, F.; Gasparrini, S.; Cheng, L.; Lopez-Beltran, A.; Montironi, R.; Scarpelli, M. Microbiome and Cancers, with Focus on Genitourinary Tumors. Front. Oncol. 2019, 9, 178. [Google Scholar] [CrossRef]
- Sabbatucci, M.; Purificato, C.; Fantuzzi, L.; Gessani, S. Toll-like Receptor Cross-Talk in Human Monocytes Regulates CC-Chemokine Production, Antigen Uptake and Immune Cell Recruitment. Immunobiology 2011, 216, 1135–1142. [Google Scholar] [CrossRef]
- Altmann, S.; Korytář, T.; Kaczmarzyk, D.; Nipkow, M.; Kühn, C.; Goldammer, T.; Rebl, A. Toll-like Receptors in Maraena Whitefish: Evolutionary Relationship among Salmonid Fishes and Patterns of Response to Aeromonas Salmonicida. Fish Shellfish Immunol. 2016, 54, 391–401. [Google Scholar] [CrossRef]
- Wang, D.; Liu, K.; Wake, H.; Teshigawara, K.; Mori, S.; Nishibori, M. Anti-High Mobility Group Box-1 (HMGB1) Antibody Inhibits Hemorrhage-Induced Brain Injury and Improved Neurological Deficits in Rats. Sci. Rep. 2017, 7, 46243. [Google Scholar] [CrossRef]
- Jain, S.; Dash, P.; Minz, A.P.; Satpathi, S.; Samal, A.G.; Behera, P.K.; Satpathi, P.S.; Senapati, S. Lipopolysaccharide (LPS) Enhances Prostate Cancer Metastasis Potentially through NF-ΚB Activation and Recurrent Dexamethasone Administration Fails to Suppress It in Vivo. Prostate 2019, 79, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Suklabaidya, S.; Das, B.; Raghav, S.K.; Batra, S.K.; Senapati, S. TLR4 Activation by Lipopolysaccharide Confers Survival Advantage to Growth Factor Deprived Prostate Cancer Cells. Prostate 2015, 75, 1020–1033. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Huang, Q.; Zheng, W.; Huang, Y.; Hua, J.; Yang, S.; Zhuang, J.; Wang, J.; Chang, J.; Xu, J.; et al. LPS Upregulated VEGFR-3 Expression Promote Migration and Invasion in Colorectal Cancer via a Mechanism of Increased NF-ΚB Binding to the Promoter of VEGFR-3. Cell. Physiol. Biochem. 2016, 39, 1665–1678. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Qian, S.; Zhang, J.; Feng, J.; Luo, K.; Sun, L.; Zhao, L.; Ran, Y.; Sun, L.; Wang, J.; et al. Lipopolysaccharide Promotes Metastasis via Acceleration of Glycolysis by the Nuclear Factor-ΚB/Snail/Hexokinase3 Signaling Axis in Colorectal Cancer. Cancer Metab. 2021, 9, 23. [Google Scholar] [CrossRef]
- Chen, X.; Li, C.; He, T.; Mao, J.; Li, C.; Lyu, J.; Meng, Q.H. Metformin Inhibits Prostate Cancer Cell Proliferation, Migration, and Tumor Growth through Upregulation of PEDF Expression. Cancer Biol. Ther. 2016, 17, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Liu, Z.; Liu, R.; Lu, C.; Wang, L.; Mao, W.; Zhu, Q.; Shou, H.; Zhang, K.; Li, Y.; et al. Epigenetic Induction of Tumor Stemness via the Lipopolysaccharide-TET3-HOXB2 Signaling Axis in Esophageal Squamous Cell Carcinoma. Cell Commun. Signal. 2020, 18, 17. [Google Scholar] [CrossRef]
- Kim, H.J.; Park, J.W.; Cho, Y.S.; Cho, C.H.; Kim, J.S.; Shin, H.W.; Chung, D.H.; Kim, S.J.; Chun, Y.S. Pathogenic Role of HIF-1α in Prostate Hyperplasia in the Presence of Chronic Inflammation. Biochim. Biophys. Acta 2013, 1832, 183–194. [Google Scholar] [CrossRef]
- Xia, G.; Kohler, T.; Peschel, A. The Wall Teichoic Acid and Lipoteichoic Acid Polymers of Staphylococcus Aureus. Int. J. Med. Microbiol. 2010, 300, 148–154. [Google Scholar] [CrossRef]
- Navarre, W.W.; Schneewind, O. Surface Proteins of Gram-Positive Bacteria and Mechanisms of Their Targeting to the Cell Wall Envelope. Microbiol. Mol. Biol. Rev. 1999, 63, 174–229. [Google Scholar] [CrossRef]
- Schneewind, O.; Missiakas, D. Lipoteichoic Acids, Phosphate-Containing Polymers in the Envelope of Gram-Positive Bacteria. J. Bacteriol. 2014, 196, 1133–1142. [Google Scholar] [CrossRef]
- Irvine, K.L.; Hopkins, L.J.; Gangloff, M.; Bryant, C.E. The Molecular Basis for Recognition of Bacterial Ligands at Equine TLR2, TLR1 and TLR6. Vet. Res. 2013, 44, 50. [Google Scholar] [CrossRef] [PubMed]
- Rezania, S.; Amirmozaffari, N.; Rashidi, N.; Mirzadegan, E.; Zarei, S.; Ghasemi, J.; Zarei, O.; Katouzian, L.; Zarnani, A.H. The Same and Not the Same: Heterogeneous Functional Activation of Prostate Tumor Cells by TLR Ligation. Cancer Cell Int. 2014, 14, 54. [Google Scholar] [CrossRef] [PubMed]
- Hattar, K.; Reinert, C.P.; Sibelius, U.; Gökyildirim, M.Y.; Subtil, F.S.B.; Wilhelm, J.; Eul, B.; Dahlem, G.; Grimminger, F.; Seeger, W.; et al. Lipoteichoic Acids from Staphylococcus aureus Stimulate Proliferation of Human Non-Small-Cell Lung Cancer Cells in Vitro. Cancer Immunol. Immunother. 2017, 66, 799–809. [Google Scholar] [CrossRef]
- Murphy, S.F.; Schaeffer, A.J.; Done, J.D.; Quick, M.L.; Acar, U.; Thumbikat, P. Commensal Bacterial Modulation of the Host Immune Response to Ameliorate Pain in a Murine Model of Chronic Prostatitis. Pain 2017, 158, 1517–1527. [Google Scholar] [CrossRef]
- Komai, K.; Shichita, T.; Ito, M.; Kanamori, M.; Chikuma, S.; Yoshimura, A. Role of Scavenger Receptors as Damage-Associated Molecular Pattern Receptors in Toll-like Receptor Activation. Int. Immunol. 2017, 29, 59–70. [Google Scholar] [CrossRef]
- Brown, J.; Wang, H.; Hajishengallis, G.N.; Martin, M. TLR-Signaling Networks. J. Dent. Res. 2011, 90, 417–427. [Google Scholar] [CrossRef]
- Kurowska-Stolarska, M.; Stolarski, B.; Kewin, P.; Murphy, G.; Corrigan, C.J.; Ying, S.; Pitman, N.; Mirchandani, A.; Rana, B.; van Rooijen, N.; et al. IL-33 Amplifies the Polarization of Alternatively Activated Macrophages That Contribute to Airway Inflammation. J. Immunol. 2009, 183, 6469–6477. [Google Scholar] [CrossRef]
- Krysko, O.; Aaes, T.L.; Bachert, C.; Vandenabeele, P.; Krysko, D.V. Many Faces of DAMPs in Cancer Therapy. Cell Death Dis. 2013, 4, e631. [Google Scholar] [CrossRef] [PubMed]
- Paone, A.; Galli, R.; Gabellini, C.; Lukashev, D.; Starace, D.; Gorlach, A.; De Cesaris, P.; Ziparo, E.; Del Bufalo, D.; Sitkovsky, M.V.; et al. Toll-like Receptor 3 Regulates Angiogenesis and Apoptosis in Prostate Cancer Cell Lines through Hypoxia-Inducible Factor 1α. Neoplasia 2010, 12, 539–549. [Google Scholar] [CrossRef]
- Chang, Z.L. Important Aspects of Toll-like Receptors, Ligands and Their Signaling Pathways. Inflamm. Res. 2010, 59, 791–808. [Google Scholar] [CrossRef] [PubMed]
- Kutikhin, A.G.; Yuzhalin, A.E. Are Toll-like Receptor Gene Polymorphisms Associated with Prostate Cancer? Cancer Manag. Res. 2012, 2012, 23–29. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Y.; Zhang, Q.; Wang, F.; Zhang, D. Toll-like Receptors and Prostate Cancer. Front. Immunol. 2014, 5, 352. [Google Scholar] [CrossRef]
- González-Reyes, S.; Fernández, J.M.; González, L.O.; Aguirre, A.; Suárez, A.; González, J.M.; Escaff, S.; Vizoso, F.J. Study of TLR3, TLR4, and TLR9 in Prostate Carcinomas and Their Association with Biochemical Recurrence. Cancer Immunol. Immunother. 2011, 60, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.; Young Lee, J. Intrinsic and Extrinsic Regulation of Innate Immune Receptors. Yonsei Med. J. 2011, 52, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Mäkinen, L.K.; Atula, T.; Häyry, V.; Jouhi, L.; Datta, N.; Lehtonen, S.; Ahmed, A.; Mäkitie, A.A.; Haglund, C.; Hagström, J. Predictive Role of Toll-like Receptors 2, 4, and 9 in Oral Tongue Squamous Cell Carcinoma. Oral. Oncol. 2015, 51, 96–102. [Google Scholar] [CrossRef]
- Guenther, M.K.; Graab, U.; Fulda, S. Synthetic Lethal Interaction between PI3K/Akt/MTOR and Ras/MEK/ERK Pathway Inhibition in Rhabdomyosarcoma. Cancer Lett. 2013, 337, 200–209. [Google Scholar] [CrossRef]
- Sang, M.; Hulsurkar, M.; Zhang, X.; Song, H.; Zheng, D.; Zhang, Y.; Li, M.; Xu, J.; Zhang, S.; Ittmann, M.; et al. GRK3 Is a Direct Target of CREB Activation and Regulates Neuroendocrine Differentiation of Prostate Cancer Cells. Oncotarget 2016, 7, 45171–45185. [Google Scholar] [CrossRef]
- Song, J.; Duncan, M.J.; Li, G.; Chan, C.; Grady, R.; Stapleton, A.; Abraham, S.N. A Novel TLR4-Mediated Signaling Pathway Leading to IL-6 Responses in Human Bladder Epithelial Cells. PLoS Pathog. 2007, 3, e60. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.; Payvandi, F.; Edelstein, L.C.; Amenta, P.S.; Zong, W.X.; Gélinas, C.; Rabson, A.B. Mechanisms of Constitutive NF-ΚB Activation in Human Prostate Cancer Cells. Prostate 2002, 52, 183–200. [Google Scholar] [CrossRef]
- Oseni, S.O.; Branly, R.; Pavlovic, M.; Kumi-Diaka, J. Abstract 5730: Co-Targeting Toll-like Receptor and PI3K Survival Signaling Pathways in Stem-like Castration Resistant Prostate Cancer Cells. Cancer Res. 2018, 78, 5730. [Google Scholar] [CrossRef]
- Zhai, C.; Cheng, J.; Mujahid, H.; Wang, H.; Kong, J.; Yin, Y.; Li, J.; Zhang, Y.; Ji, X.; Chen, W.; et al. Selective Inhibition of PI3K/Akt/MTOR Signaling Pathway Regulates Autophagy of Macrophage and Vulnerability of Atherosclerotic Plaque. PLoS ONE 2014, 9, e90563. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, D.A. Phosphoinositide 3-Kinase Signalling Pathways. J. Cell Sci. 2001, 114, 1439–1445. [Google Scholar] [CrossRef]
- Alvarado, A.G.; Thiagarajan, P.S.; Mulkearns-Hubert, E.E.; Silver, D.J.; Hale, J.S.; Alban, T.J.; Turaga, S.M.; Jarrar, A.; Reizes, O.; Longworth, M.S.; et al. Glioblastoma Cancer Stem Cells Evade Innate Immune Suppression of Self-Renewal through Reduced TLR4 Expression. Cell Stem Cell 2017, 20, 450–461.e4. [Google Scholar] [CrossRef] [PubMed]
- Zeuner, M.T.; Krüger, C.L.; Volk, K.; Bieback, K.; Cottrell, G.S.; Heilemann, M.; Widera, D. Biased Signalling Is an Essential Feature of TLR4 in Glioma Cells. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 3084–3095. [Google Scholar] [CrossRef]
- Chiarini, F.; Evangelisti, C.; Mccubrey, J.A.; Martelli, A.M. Current Treatment Strategies for Inhibiting MTOR in Cancer. Trends Pharmacol. Sci. 2015, 36, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Väisänen, M.R.; Väisänen, T.; Jukkola-Vuorinen, A.; Vuopala, K.S.; Desmond, R.; Selander, K.S.; Vaarala, M.H. Expression of Toll-like Receptor-9 Is Increased in Poorly Differentiated Prostate Tumors. Prostate 2010, 70, 817–824. [Google Scholar] [CrossRef] [PubMed]
- König, J.E.; Senge, T.; Allhoff, E.P.; König, W. Analysis of the Inflammatory Network in Benign Prostate Hyperplasia and Prostate Cancer. Prostate 2004, 58, 121–129. [Google Scholar] [CrossRef]
- Schulz, W.A.; Alexa, A.; Jung, V.; Hader, C.; Hoffmann, M.J.; Yamanaka, M.; Fritzsche, S.; Wlazlinski, A.; Müller, M.; Lengauer, T.; et al. Factor Interaction Analysis for Chromosome 8 and DNA Methylation Alterations Highlights Innate Immune Response Suppression and Cytoskeletal Changes in Prostate Cancer. Mol. Cancer 2007, 6, 14. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Feldman, N.; Rotter-Maskowitz, A.; Okun, E. DAMPs as Mediators of Sterile Inflammation in Aging-Related Pathologies. Ageing Res. Rev. 2015, 24, 29–39. [Google Scholar] [CrossRef]
- Lamkanfi, M. Emerging Inflammasome Effector Mechanisms. Nat. Rev. Immunol. 2011, 11, 213–220. [Google Scholar] [CrossRef]
- Kolb, R.; Liu, G.H.; Janowski, A.M.; Sutterwala, F.S.; Zhang, W. Inflammasomes in Cancer: A Double-Edged Sword. Protein Cell 2014, 5, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Kantono, M.; Guo, B. Inflammasomes and Cancer: The Dynamic Role of the Inflammasome in Tumor Development. Front. Immunol. 2017, 8, 1132. [Google Scholar] [CrossRef]
- Huyen Trang Ha Thi, R.; Hong, S. Inflammasome as a Therapeutic Target for Cancer Prevention and Treatment. J. Cancer Prev. 2017, 22, 62. [Google Scholar]
- Veeranki, S. Role of Inflammasomes and Their Regulators in Prostate Cancer Initiation, Progression and Metastasis. Cell. Mol. Biol. Lett. 2013, 18, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Karan, D.; Tawfik, O.; Dubey, S. Expression Analysis of Inflammasome Sensors and Implication of NLRP12 Inflammasome in Prostate Cancer. Sci. Rep. 2017, 7, 4378. [Google Scholar] [CrossRef] [PubMed]
- Rider, P.; Carmi, Y.; Guttman, O.; Braiman, A.; Cohen, I.; Voronov, E.; White, M.R.; Dinarello, C.A.; Apte, R.N. IL-1α and IL-1β Recruit Different Myeloid Cells and Promote Different Stages of Sterile Inflammation. J. Immunol. 2011, 187, 4835–4843. [Google Scholar] [CrossRef]
- Patel, M.N.; Carroll, R.G.; Galván-Peña, S.; Mills, E.L.; Olden, R.; Triantafilou, M.; Wolf, A.I.; Bryant, C.E.; Triantafilou, K.; Masters, S.L. Inflammasome Priming in Sterile Inflammatory Disease. Trends Mol. Med. 2017, 23, 165–180. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin-1 in the Pathogenesis and Treatment of Inflammatory Diseases. Blood 2011, 117, 3720–3732. [Google Scholar] [CrossRef]
- Li, B.-H.; Yan, S.-Y.; Luo, L.-S.; Zeng, X.-T.; Wang, Y.-B.; Wang, X.-H. Ten Interleukins and Risk of Prostate Cancer. Front. Oncol. 2023, 13, 1108633. [Google Scholar] [CrossRef]
- Vollmer, S.; Strickson, S.; Zhang, T.; Gray, N.; Lee, K.L.; Rao, V.R.; Cohen, P. The Mechanism of Activation of IRAK1 and IRAK4 by Interleukin-1 and Toll-like Receptor Agonists. Biochem. J. 2017, 474, 2027–2038. [Google Scholar] [CrossRef]
- Voronov, E.; Shouval, D.S.; Krelin, Y.; Cagnano, E.; Benharroch, D.; Iwakura, Y.; Dinarello, C.A.; Apte, R.N. IL-1 Is Required for Tumor Invasiveness and Angiogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 2645–2650. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in Innate Immunity and Inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Dinarello, C.A.; Mantovani, A. The Interleukin-1 Family: Back to the Future. Immunity 2013, 39, 1003–1018. [Google Scholar] [CrossRef]
- Ricote, M.; García-Tuñón, I.; Bethencourt, F.R.; Fraile, B.; Paniagua, R.; Royuela, M. Interleukin-1 (IL-1alpha and IL-1beta) and Its Receptors (IL-1RI, IL-1RII, and IL-1Ra) in Prostate Carcinoma. Cancer 2004, 100, 1388–1396. [Google Scholar] [CrossRef] [PubMed]
- Freeman, L.C.; Ting, J.P.-Y. The Pathogenic Role of the Inflammasome in Neurodegenerative Diseases. J. Neurochem. 2016, 136, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.P.; Bray, T.M.; Ho, E. Induction of Proinflammatory Response in Prostate Cancer Epithelial Cells by Activated Macrophages. Cancer Lett. 2009, 276, 38–46. [Google Scholar] [CrossRef]
- Singh, S.; Xiao, Z.; Bavisi, K.; Roszik, J.; Melendez, B.D.; Wang, Z.; Cantwell, M.J.; Davis, R.E.; Lizee, G.; Hwu, P.; et al. IL-1α Mediates Innate and Acquired Resistance to Immunotherapy in Melanoma. J. Immunol. 2021, 206, 1966–1975. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qiao, Q.; Ferrao, R.; Shen, C.; Hatcher, J.M.; Buhrlage, S.J.; Gray, N.S.; Wu, H. Crystal Structure of Human IRAK1. Proc. Natl. Acad. Sci. USA 2017, 114, 13507–13512. [Google Scholar] [CrossRef]
- Hossen, M.J.; Yang, W.S.; Kim, D.; Aravinthan, A.; Kim, J.H.; Cho, J.Y. Thymoquinone: An IRAK1 Inhibitor with in Vivo and in Vitro Anti-Inflammatory Activities. Sci. Rep. 2017, 7, 42995. [Google Scholar] [CrossRef] [PubMed]
- Gosu, V.; Basith, S.; Durai, P.; Choi, S. Molecular Evolution and Structural Features of IRAK Family Members. PLoS ONE 2012, 7, e49771. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Wiklund, F.; Hsu, F.-C.; Bälter, K.; Zheng, S.L.; Johansson, J.-E.; Chang, B.; Liu, W.; Li, T.; Turner, A.R.; et al. Interactions of Sequence Variants in Interleukin-1 Receptor-Associated Kinase4 and the Toll-Like Receptor 6-1-10 Gene Cluster Increase Prostate Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2006, 15, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Xiao, H.; Affolter, J.; Kim, T.W.; Bulek, K.; Chaudhuri, S.; Carlson, D.; Hamilton, T.; Mazumder, B.; Stark, G.R.; et al. Interleukin-1 Receptor-Associated Kinase 2 Is Critical for Lipopolysaccharide-Mediated Post-Transcriptional Control. J. Biol. Chem. 2009, 284, 10367–10375. [Google Scholar] [CrossRef]
- Chou, C.K.; Chi, S.Y.; Huang, C.H.; Chou, F.F.; Huang, C.C.; Liu, R.T.; Kang, H.Y. IRAK1, a Target of MiR-146b, Reduces Cell Aggressiveness of Human Papillary Thyroid Carcinoma. J. Clin. Endocrinol. Metab. 2016, 101, 4357–4366. [Google Scholar] [CrossRef]
- Beverly, L.J.; Starczynowski, D.T. IRAK1: Oncotarget in MDS and AML. Oncotarget 2014, 5, 1699–1700. [Google Scholar] [CrossRef]
- Wee, Z.N.; Yatim, S.M.J.M.; Kohlbauer, V.K.; Feng, M.; Goh, J.Y.; Bao, Y.; Lee, P.L.; Zhang, S.; Wang, P.P.; Lim, E.; et al. IRAK1 Is a Therapeutic Target That Drives Breast Cancer Metastasis and Resistance to Paclitaxel. Nat. Commun. 2015, 6, 8746. [Google Scholar] [CrossRef]
- Ye, Z.H.; Gao, L.; Wen, D.Y.; He, Y.; Pang, Y.Y.; Chen, G. Diagnostic and Prognostic Roles of IRAK1 in Hepatocellular Carcinoma Tissues: An Analysis of Immunohistochemistry and RNA-Sequencing Data from the Cancer Genome Atlas. OncoTargets Ther. 2017, 10, 1711–1723. [Google Scholar] [CrossRef]
- Zhang, D.; Li, L.; Jiang, H.; Knolhoff, B.L.; Lockhart, A.C.; Wang-Gillam, A.; Denardo, D.G.; Ruzinova, M.B.; Lim, K.-H. Constitutive IRAK4 Activation Underlies Poor Prognosis and Chemoresistance in Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2017, 23, 1748–1759. [Google Scholar] [CrossRef]
- Cheng, B.Y.; Lau, E.Y.; Leung, H.-W.; Leung, C.O.-N.; Ho, N.P.; Gurung, S.; Cheng, L.K.; Lin, C.H.; Lo, R.C.-L.; Ma, S.; et al. IRAK1 Augments Cancer Stemness and Drug Resistance via the AP-1/AKR1B10 Signaling Cascade in Hepatocellular Carcinoma. Cancer Res. 2018, 78, 2332–2342. [Google Scholar] [CrossRef]
- Kim, Y.-I.; Park, J.-E.; Kwon, K.H.; Hong, C.Y.; Yi, A.-K. Interleukin-1 Receptor-Associated Kinase 2 and Protein Kinase D1-Dependent Regulation of IRAK-Monocyte Expression by CpG DNA. PLoS ONE 2012, 7, 43970. [Google Scholar] [CrossRef] [PubMed]
- Heiseke, A.F.; Jeuk, B.H.; Markota, A.; Straub, T.; Lehr, H.-A.; Reindl, W.; Krug, A.B. IRAK1 Drives Intestinal Inflammation by Promoting the Generation of Effector Th Cells with Optimal Gut-Homing Capacity. J. Immunol. 2015, 195, 5787–5794. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.; Peggie, M.; Campbell, D.G.; Vandermoere, F.; Carrick, E.; Cohen, P. Identification of the Phosphorylation Sites on the E3 Ubiquitin Ligase Pellino That Are Critical for Activation by IRAK1 and IRAK4. Proc. Natl. Acad. Sci. USA 2009, 106, 4584–4590. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Tsutsumi, N.; Tochio, H.; Ohnishi, H.; Kubota, K.; Kato, Z.; Shirakawa, M.; Kondo, N. Functional Assessment of the Mutational Effects of Human IRAK4 and MyD88 Genes. Mol. Immunol. 2014, 58, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Sode, J.; Vogel, U.; Bank, S.; Andersen, P.S.; Hetland, M.L.; Locht, H.; Heegaard, N.H.H.; Andersen, V. Confirmation of an IRAK3 Polymorphism as a Genetic Marker Predicting Response to Anti-TNF Treatment in Rheumatoid Arthritis. Pharm. J. 2018, 18, 81–86. [Google Scholar] [CrossRef]
- Goh, E.T.H.; Arthur, J.S.C.; Cheung, P.C.F.; Akira, S.; Toth, R.; Cohen, P. Identification of the Protein Kinases That Activate the E3 Ubiquitin Ligase Pellino 1 in the Innate Immune System. Biochem. J. 2012, 441, 339–346. [Google Scholar] [CrossRef]
- Uematsu, S.; Sato, S.; Yamamoto, M.; Hirotani, T.; Kato, H.; Takeshita, F.; Matsuda, M.; Coban, C.; Ishii, K.J.; Kawai, T.; et al. Interleukin-1 Receptor-Associated Kinase-1 Plays an Essential Role for Toll-like Receptor (TLR)7- and TLR9-Mediated Interferon-α Induction. J. Exp. Med. 2005, 201, 915–923. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, T.; Sane, D.C.; Li, L. IRAK1 Serves as a Novel Regulator Essential for Lipopolysaccharide-Induced Interleukin-10 Gene Expression. J. Biol. Chem. 2004, 279, 51697–51703. [Google Scholar] [CrossRef]
- Singer, J.W.; Fleischman, A.; Al-Fayoumi, S.; Mascarenhas, J.O.; Yu, Q.; Agarwal, A. Inhibition of Interleukin-1 Receptor-Associated Kinase 1 (IRAK1) as a Therapeutic Strategy. Oncotarget 2018, 9, 33416–33439. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, N.; Kohsaka, H.; Murakami, Y.; Mizoguchi, F.; Saito, T. Degradation in Macrophages Effect through Accelerated IRAK1 Exerts an Anti-Inflammatory INK4a P16. J. Immunol. 2012, 189, 5066–5072. [Google Scholar] [CrossRef]
- Rhyasen, G.W.; Starczynowski, D.T. IRAK Signalling in Cancer. Br. J. Cancer 2015, 112, 232–237. [Google Scholar] [CrossRef]
- Li, Z.; Younger, K.; Gartenhaus, R.; Joseph, A.M.; Hu, F.; Baer, M.R.; Brown, P.; Davila, E. Inhibition of IRAK1/4 Sensitizes T Cell Acute Lymphoblastic Leukemia to Chemotherapies. J. Clin. Investig. 2015, 125, 1081–1097. [Google Scholar] [CrossRef] [PubMed]
- Orjalo, A.V.; Bhaumik, D.; Gengler, B.K.; Scott, G.K.; Campisi, J. Cell Surface-Bound IL-1alpha Is an Upstream Regulator of the Senescence-Associated IL-6/IL-8 Cytokine Network. Proc. Natl. Acad. Sci. USA 2009, 106, 17031–17036. [Google Scholar] [CrossRef]
- Oseni, S.O.; Adebayo, O.; Adebayo, A.; Kwakye, A.; Pavlovic, M.; Asghar, W.; Hartmann, J.; Fields, G.B.; Kumi-Diaka, J. Integrative Genomic and Epigenomic Analyses Identified IRAK1 as a Novel Target for Chronic Inflammation-Driven Prostate Tumorigenesis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Adams, A.K.; Bolanos, L.C.; Dexheimer, P.J.; Karns, R.A.; Aronow, B.J.; Komurov, K.; Jegga, A.G.; Casper, K.A.; Patil, Y.J.; Wilson, K.M.; et al. IRAK1 Is a Novel DEK Transcriptional Target and Is Essential for Head and Neck Cancer Cell Survival. Oncotarget 2015, 6, 43395–43407. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Duan, X.; Wang, Y.; Zhang, Z. Interleukin-1 Receptor-Associated Kinase 1 Correlates with Metastasis and Invasion in Endometrial Carcinoma. J. Cell. Biochem. 2018, 119, 2545–2555. [Google Scholar] [CrossRef] [PubMed]
- Rhyasen, G.W.; Bolanos, L.; Fang, J.; Jerez, A.; Wunderlich, M.; Rigolino, C.; Mathews, L.; Ferrer, M.; Southall, N.; Guha, R.; et al. Targeting IRAK1 as a Therapeutic Approach for Myelodysplastic Syndrome. Cancer Cell 2013, 24, 90–104. [Google Scholar] [CrossRef]
- Ngo, V.N.; Young, R.M.; Scmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.-H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically Active MYD88 Mutations in Human Lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef]
- Wu, X.; Wang, L.; Qiu, Y.; Zhang, B.; Hu, Z.; Jin, R. Cooperation of IRAK1/4 Inhibitor and ABT-737 in Nanoparticles for Synergistic Therapy of T Cell Acute Lymphoblastic Leukemia. Int. J. Nanomed. 2017, 12, 8025–8034. [Google Scholar] [CrossRef]
- Srivastava, R.; Geng, D.; Liu, Y.; Zheng, L.; Li, Z.; Joseph, M.A.; McKenna, C.; Bansal, N.; Ochoa, A.; Davila, E. Augmentation of Therapeutic Responses in Melanoma by Inhibition of IRAK-1,-4. Cancer Res. 2012, 72, 6209–6216. [Google Scholar] [CrossRef]
- Jin, R.; Yi, Y.; Yull, F.E.; Blackwell, T.S.; Clark, P.E.; Koyama, T.; Smith, J.A.; Matusik, R.J.; Matusik, R.J. NF-ΚB Gene Signature Predicts Prostate Cancer Progression. Cancer Res. 2014, 74, 2763–2772. [Google Scholar] [CrossRef] [PubMed]
- Fusco, F.; Pescatore, A.; Bal, E.; Ghoul, A.; Paciolla, M.; Lioi, M.B.; D’Urso, M.; Rabia, S.H.; Bodemer, C.; Bonnefont, J.P.; et al. Alterations of the IKBKG Locus and Diseases: An Update and a Report of 13 Novel Mutations. Hum. Mutat. 2008, 29, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Lessard, L.; Bégin, L.R.; Gleave, M.E.; Mes-Masson, A.M.; Saad, F. Nuclear Localisation of Nuclear Factor-KappaB Transcription Factors in Prostate Cancer: An Immunohistochemical Study. Br. J. Cancer 2005, 93, 1019–1023. [Google Scholar] [CrossRef]
- Wang, J.; Yi, S.; Zhou, J.; Zhang, Y.; Guo, F. The NF-B Subunit RelB Regulates the Migration and Invasion Abilities and the Radio-Sensitivity of Prostate Cancer Cells. Int. J. Oncol. 2016, 49, 381–392. [Google Scholar] [CrossRef]
- Nuñez, C.; Cansino, J.R.; Bethencourt, F.; Pérez-Utrilla, M.; Fraile, B.; Martínez-Onsurbe, P.; Olmedilla, G.; Paniagua, R.; Royuela, M. TNF/IL-1/NIK/NF-ΚB Transduction Pathway: A Comparative Study in Normal and Pathological Human Prostate (Benign Hyperplasia and Carcinoma). Histopathology 2008, 53, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-ΚB, an Active Player in Human Cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef]
- Lopez-Granados, E.; Keenan, J.E.; Kinney, M.C.; Leo, H.; Jain, N.; Ma, C.A.; Quinones, R.; Gelfand, E.W.; Jain, A. A Novel Mutation in NFKBIA/IKBA Results in a Degradation-Resistant N-Truncated Protein and Is Associated with Ectodermal Dysplasia with Immunodeficiency. Hum. Mutat. 2008, 29, 861–868. [Google Scholar] [CrossRef]
- Darnay, B.G.; Ni, J.; Moore, P.A.; Aggarwal, B.B. Activation of NF-ΚB by Rank Requires Tumor Necrosis Factor Receptor- Associated Factor (TRAF) 6 and NF-ΚB-Inducing Kinase. Identification of a Novel TRAF6 Interaction Motif. J. Biol. Chem. 1999, 274, 7724–7731. [Google Scholar] [CrossRef]
- Palvimo, J.J.; Reinikainen, P.; Ikonen, T.; Kallio, P.J.; Moilanen, A.; Jänne, O.A. Mutual Transcriptional Interference between RelA and Androgen Receptor. J. Biol. Chem. 1996, 271, 24151–24156. [Google Scholar] [CrossRef]
- Pei, Z.; Li, H.; Guo, Y.; Jin, Y.; Lin, D. Sodium Selenite Inhibits the Expression of VEGF, TGFβ1and IL-6 Induced by LPS in Human PC3 Cells via TLR4-NF-KB Signaling Blockage. Int. Immunopharmacol. 2010, 10, 50–56. [Google Scholar] [CrossRef]
- Peng, H.; Wen, J.; Li, H.; Chang, J.; Zhou, X. Drug Inhibition Profile Prediction for NFkB Pathway in Multiple Myeloma. PLoS ONE 2011, 6, e14750. [Google Scholar] [CrossRef] [PubMed]
- Rajput, S.; Volk-Draper, L.D.; Ran, S. TLR4 Is a Novel Determinant of the Response to Paclitaxel in Breast Cancer. Mol. Cancer Ther. 2013, 12, 1676–1687. [Google Scholar] [CrossRef]
- Davoudi, Z.; Akbarzadeh, A.; Rahmatiyamchi, M.; Movassaghpour, A.A.; Alipour, M.; Nejati-Koshki, K.; Zarghami, N. Molecular Target Therapy of AKT and NF-KB Signaling Pathways and Multidrug Resistance by Specific Cell Penetrating Inhibitor Peptides in HL-60 Cells. Asian Pac. J. Cancer Prev. 2014, 15, 4353–4358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Yang, S.; Chen, X.; Stauffer, S.; Yu, F.; Lele, S.M.; Fu, K.; Datta, K.; Palermo, N.; Chen, Y.; et al. The Hippo Pathway Effector YAP Regulates Motility, Invasion, and Castration-Resistant Growth of Prostate Cancer Cells. Mol. Cell. Biol. 2015, 35, 1350–1362. [Google Scholar] [CrossRef] [PubMed]
- Al Nakouzi, N.; Le Moulec, S.; Albigès, L.; Wang, C.; Beuzeboc, P.; Gross-Goupil, M.; de La Motte Rouge, T.; Guillot, A.; Gajda, D.; Massard, C.; et al. Cabazitaxel Remains Active in Patients Progressing After Docetaxel Followed by Novel Androgen Receptor Pathway Targeted Therapies. Eur. Urol. 2015, 68, 228–235. [Google Scholar] [CrossRef] [PubMed]
- Vlachostergios, P.J.; Puca, L.; Beltran, H. Emerging Variants of Castration-Resistant Prostate Cancer. Curr. Oncol. Rep. 2017, 19, 32. [Google Scholar] [CrossRef]
- Reis, L.; Billis, A.; Ferreira, U.; Denardi, F.; Domeniconi, R.; Scarano, W.; Seiva, F.; Pinheiro, P.; Fávaro, W. Differential Effects of Steroid Hormone Receptors (SHRS) on Toll-like Receptors (TLRS) in Nodular Hyperplasia (NH), High-Grade Prostatic Intraepithelial Neoplasia (HGPIN) and Cancer (CA): Novel Prostate Cancer Stem Cells (PCSC) Signaling Pathways. J. Urol. 2012, 187, e701. [Google Scholar] [CrossRef]
- Bitting, R.L.; Armstrong, A.J. Targeting the PI3K/Akt/MTOR Pathway in Castration-Resistant Prostate Cancer. Endocr. Relat. Cancer 2013, 20, R83–R99. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.P.; Li, J.; Yadav, S.S.; Tewari, A.K. Recent Insights into NF-ΚB Signalling Pathways and the Link between Inflammation and Prostate Cancer. BJU Int. 2014, 114, 168–176. [Google Scholar] [CrossRef]
- Rybak, A.P.; He, L.; Kapoor, A.; Cutz, J.C.; Tang, D. Characterization of Sphere-Propagating Cells with Stem-like Properties from DU145 Prostate Cancer Cells. Biochim. Biophys. Acta Mol. Cell Res. 2011, 1813, 683–694. [Google Scholar] [CrossRef]
- Finocchiaro, G. TLRgeting Evasion of Immune Pathways in Glioblastoma. Cell Stem Cell 2017, 20, 422–424. [Google Scholar] [CrossRef] [PubMed]
- Yeh, D.-W.; Huang, L.-R.; Chen, Y.-W.; Huang, C.-Y.F.; Chuang, T.-H. Interplay between Inflammation and Stemness in Cancer Cells: The Role of Toll-Like Receptor Signaling. J. Immunol. Res. 2016, 2016, 4368101. [Google Scholar] [CrossRef]
- Deng, Q.; Tang, D.G. Androgen Receptor and Prostate Cancer Stem Cells: Biological Mechanisms and Clinical Implications. Endocr. Relat. Cancer 2015, 22, T209–T220. [Google Scholar] [CrossRef] [PubMed]
- Yun, E.-J.; Lo, U.-G.; Hsieh, J.-T. The Evolving Landscape of Prostate Cancer Stem Cell (PCSC): Therapeutic Implications and Future Challenges. Asian J. Urol. 2016, 3, 203–210. [Google Scholar] [CrossRef]
- Menendez, J.A.; Alarcón, T. Metabostemness: A New Cancer Hallmark. Front. Oncol. 2014, 4, 262. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zhang, Y.; Zheng, J.; Pan, J. Reactive Oxygen Species in Cancer Stem Cells. Antioxid. Redox Signal. 2012, 16, 1215–1228. [Google Scholar] [CrossRef]
- Oseni, S.O.; Kwasi Kumi-Diaka, J. Emerging Role of Cancer Stemness in Prostate Cancer Recurrence and Management. Acta Sci. Cancer Biol. 2018, 2, 2–5. [Google Scholar]
- Chen, K.; Huang, Y.; Chen, J. Understanding and Targeting Cancer Stem Cells: Therapeutic Implications and Challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Rowehl, R.A.; Crawford, H.; Dufour, A.; Ju, J.; Botchkina, G.I. Genomic Analysis of Prostate Cancer Stem Cells Isolated from a Highly Metastatic Cell Line. Cancer Genom. Proteom. 2008, 5, 301–310. [Google Scholar]
- Rybak, A.P.; Bristow, R.G.; Kapoor, A. Prostate Cancer Stem Cells: Deciphering the Origins and Pathways Involved in Prostate Tumorigenesis and Aggression. Oncotarget 2015, 6, 1900–1919. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Alvero, A.B.; Chen, R.; Fu, H.H.; Montagna, M.; Schwartz, P.E.; Rutherford, T.; Silasi, D.A.; Steffensen, K.D.; Waldstrom, M.; Visintin, I.; et al. Molecular Phenotyping of Human Ovarian Cancer Stem Cells Unravels the Mechanisms for Repair and Chemoresistance. Cell Cycle 2009, 8, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.L.; Neering, S.J.; Upchurch, D.; Grimes, B.; Howard, D.S.; Rizzieri, D.A.; Luger, S.M.; Jordan, C.T. Nuclear Factor-KappaB Is Constitutively Activated in Primitive Human Acute Myelogenous Leukemia Cells. Blood 2001, 98, 2301–2307. [Google Scholar] [CrossRef] [PubMed]
- Garner, J.M.; Fan, M.; Yang, C.H.; Du, Z.; Sims, M.; Davidoff, A.M.; Pfeffer, L.M. Constitutive Activation of Signal Transducer and Activator of Transcription 3 (STAT3) and Nuclear Factor ΚB Signaling in Glioblastoma Cancer Stem Cells Regulates the Notch Pathway. J. Biol. Chem. 2013, 288, 26167–26176. [Google Scholar] [CrossRef]
- Murohashi, M.; Hinohara, K.; Kuroda, M.; Isagawa, T.; Tsuji, S.; Kobayashi, S.; Umezawa, K.; Tojo, A.; Aburatani, H.; Gotoh, N. Gene Set Enrichment Analysis Provides Insight into Novel Signalling Pathways in Breast Cancer Stem Cells. Br. J. Cancer 2010, 102, 206–212. [Google Scholar] [CrossRef]
- Moltzahn, F.; Thalmann, G.N. Cancer Stem Cells in Prostate Cancer. Transl. Androl. Urol. 2013, 2, 242–253. [Google Scholar] [CrossRef]
- Birnie, R.; Bryce, S.D.; Roome, C.; Dussupt, V.; Droop, A.; Lang, S.H.; Berry, P.A.; Hyde, C.F.; Lewis, J.L.; Stower, M.J.; et al. Gene Expression Profiling of Human Prostate Cancer Stem Cells Reveals a Pro-Inflammatory Phenotype and the Importance of Extracellular Matrix Interactions. Genome Biol. 2008, 9, R83. [Google Scholar] [CrossRef]
- Rajasekhar, V.K.; Studer, L.; Gerald, W.; Socci, N.D.; Scher, H.I. Tumour-Initiating Stem-like Cells in Human Prostate Cancer Exhibit Increased NF-ΚB Signalling. Nat. Commun. 2011, 2, 162. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Maitland, N.J.; Collins, A.T. Inflammation as the Primary Aetiological Agent of Human Prostate Cancer: A Stem Cell Connection? J. Cell. Biochem. 2008, 105, 931–939. [Google Scholar] [CrossRef]
- Dicken, H.; Hensley, P.J.; Kyprianou, N. Prostate Tumor Neuroendocrine Differentiation via EMT: The Road Less Traveled. Asian J. Urol. 2019, 6, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Sun, Y.; Hu, S.; Luo, J.; Li, L.; Li, X.; Yeh, S.; Jin, J.; Chang, C. Neuroendocrine Prostate Cancer (NEPCa) Increased the Neighboring PCa Chemoresistance via Altering the PTHrP/P38/Hsp27/Androgen Receptor (AR)/P21 Signals. Oncogene 2016, 35, 6065–6076. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-D.; Choo, R.; Huang, J. Neuroendocrine Differentiation in Prostate Cancer: A Mechanism of Radioresistance and Treatment Failure. Front. Oncol. 2015, 5, 90. [Google Scholar] [CrossRef] [PubMed]
- Monn, M.F.; Montironi, R.; Lopez-Beltran, A.; Cheng, L. Emerging Molecular Pathways and Targets in Neuroendocrine Prostate Cancer. Transl. Cancer Res. 2016, 5, S282–S285. [Google Scholar] [CrossRef]
- Da Silva, J.; Gioeli, D.; Weber, M.J.; Parsons, S.J. The Neuroendocrine-Derived Peptide Parathyroid Hormone-Related Protein Promotes Prostate Cancer Cell Growth by Stabilizing the Androgen Receptor. Cancer Res. 2009, 69, 7402–7411. [Google Scholar] [CrossRef]
- Chen, R.; Dong, X.; Gleave, M. Molecular Model for Neuroendocrine Prostate Cancer Progression. BJU Int. 2018, 122, 560–570. [Google Scholar] [CrossRef]
- Yadav, S.S.; Li, J.; Stockert, J.A.; Herzog, B.; O’Connor, J.; Garzon-Manco, L.; Parsons, R.; Tewari, A.K.; Yadav, K.K. Induction of Neuroendocrine Differentiation in Prostate Cancer Cells by Dovitinib (TKI-258) and Its Therapeutic Implications. Transl. Oncol. 2017, 10, 357–366. [Google Scholar] [CrossRef]
- Zafarghandi, R.M.; Kalantari, M.R.; Rezayat, A.A.; Asadpour, A.A. Large Cell Neuroendocrine Carcinoma of Prostate: A Rare Interesting Case and Literature Review. Nephro Urol. 2017, 9, e45086. [Google Scholar] [CrossRef]
- Chen, W.-Y.; Thuy Dung, P.V.; Yeh, H.-L.; Chen, W.-H.; Jiang, K.-C.; Li, H.-R.; Chen, Z.-Q.; Hsiao, M.; Huang, J.; Wen, Y.-C.; et al. Targeting PKLR/MYCN/ROMO1 Signaling Suppresses Neuroendocrine Differentiation of Castration-Resistant Prostate Cancer. Redox Biol. 2023, 62, 102686. [Google Scholar] [CrossRef]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis Molecular Pathways and Its Role in Cancer Progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, Y.; Buttyan, R.; Dong, X. Implications of PI3K/AKT Inhibition on REST Protein Stability and Neuroendocrine Phenotype Acquisition in Prostate Cancer Cells. Oncotarget 2017, 8, 84863–84876. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, D.; Zhou, T.; Song, H.; Hulsurkar, M.; Su, N.; Liu, Y.; Wang, Z.; Shao, L.; Ittmann, M.; et al. Androgen Deprivation Promotes Neuroendocrine Differentiation and Angiogenesis through CREB-EZH2-TSP1 Pathway in Prostate Cancers. Nat. Commun. 2018, 9, 4080. [Google Scholar] [CrossRef]
- Li, Y.; Donmez, N.; Sahinalp, C.; Xie, N.; Wang, Y.; Xue, H.; Mo, F.; Beltran, H.; Gleave, M.; Wang, Y.; et al. SRRM4 Drives Neuroendocrine Transdifferentiation of Prostate Adenocarcinoma Under Androgen Receptor Pathway Inhibition. Eur. Urol. 2017, 71, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Gururajan, M.; Cavassani, K.A.; Sievert, M.; Duan, P.; Lichterman, J.; Huang, J.M.; Smith, B.; You, S.; Nandana, S.; Chu, G.C.Y.; et al. SRC Family Kinase FYN Promotes the Neuroendocrine Phenotype and Visceral Metastasis in Advanced Prostate Cancer. Oncotarget 2015, 6, 44072–44083. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S. Defining and Targeting the Oncogenic Drivers of Neuroendocrine Prostate Cancer. Cancer Cell 2016, 29, 431–432. [Google Scholar] [CrossRef]
- Gupta, K.; Gupta, S. Neuroendocrine Differentiation in Prostate Cancer: Key Epigenetic Players. Transl. Cancer Res. 2017, 6, S104–S108. [Google Scholar] [CrossRef]
- Grigore, A.D.; Ben-Jacob, E.; Farach-Carson, M.C. Prostate Cancer and Neuroendocrine Differentiation: More Neuronal, Less Endocrine? Front. Oncol. 2015, 5, 37. [Google Scholar] [CrossRef]
- Lee, J.K.; Phillips, J.W.; Smith, B.A.; Park, J.W.; Stoyanova, T.; McCaffrey, E.F.; Baertsch, R.; Sokolov, A.; Meyerowitz, J.G.; Mathis, C.; et al. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef]
- Berger, A.; Brady, N.J.; Bareja, R.; Robinson, B.; Conteduca, V.; Augello, M.A.; Puca, L.; Ahmed, A.; Dardenne, E.; Lu, X.; et al. N-Myc-Mediated Epigenetic Reprogramming Drives Lineage Plasticity in Advanced Prostate Cancer. J. Clin. Investig. 2019, 129, 3924–3940. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, C.; Cui, Y.; Nadiminty, N.; Lou, W.; Gao, A.C. Interleukin-6 Induces Neuroendocrine Differentiation (NED) through Suppression of RE-1 Silencing Transcription Factor (REST). Prostate 2014, 74, 1086–1094. [Google Scholar] [CrossRef] [PubMed]
- Bishop, J.L.; Thaper, D.; Vahid, S.; Davies, A.; Ketola, K.; Kuruma, H.; Jama, R.; Nip, K.M.; Angeles, A.; Johnson, F.; et al. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor–Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef]
- Nguyen, D.P.; Li, J.; Tewari, A.K. Inflammation and Prostate Cancer: The Role of Interleukin 6 (IL-6). BJU Int. 2014, 113, 986–992. [Google Scholar] [CrossRef]
- Boehm, B.J.; Colopy, S.A.; Jerde, T.J.; Loftus, C.J.; Bushman, W. Acute Bacterial Inflammation of the Mouse Prostate. Prostate 2012, 72, 307–317. [Google Scholar] [CrossRef]
- Adisetiyo, H.; Liang, M.; Liao, C.-P.; Jeong, J.H.; Cohen, M.B.; Roy-Burman, P.; Frenkel, B. Dependence of Castration-Resistant Prostate Cancer (CRPC) Stem Cells on CRPC-Associated Fibroblasts. J. Cell. Physiol. 2014, 229, 1170–1176. [Google Scholar] [CrossRef]
- Patel, K.J.; Chandana, S.R.; Wiese, D.A.; Olsen, B.; Conley, B.A. Unusual Presentation of Large-Cell Poorly Differentiated Neuroendocrine Carcinoma of the Epiglottis. J. Clin. Oncol. 2010, 28, e461–e463. [Google Scholar] [CrossRef]
- Punnen, S.; Cooperberg, M.R. The Epidemiology of High-Risk Prostate Cancer. Curr. Opin. Urol. 2013, 23, 331–336. [Google Scholar] [CrossRef]
- Syson Chan, K.; Espinosa, I.; Chao, M.; Wong, D.; Ailles, L.; Diehn, M.; Gill, H.; Presti, J.; Chang, H.Y.; van de Rijn, M.; et al. Identification, Molecular Characterization, Clinical Prognosis, and Therapeutic Targeting of Human Bladder Tumor-Initiating Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 14016–14021. [Google Scholar] [CrossRef] [PubMed]
- Saini, S.; Majid, S.; Shahryari, V.; Arora, S.; Yamamura, S.; Chang, I.; Zaman, M.S.; Deng, G.; Tanaka, Y.; Dahiya, R. MiRNA-708 Control of CD44+ Prostate Cancer-Initiating Cells. Cancer Res. 2012, 72, 3618–3630. [Google Scholar] [CrossRef]
- Zhang, W.; Liao, C.Y.; Chtatou, H.; Incrocci, L.; Van Gent, D.C.; Van Weerden, W.M.; Nonnekens, J. Apalutamide Sensitizes Prostate Cancer to Ionizing Radiation via Inhibition of Non-Homologous End-Joining DNA Repair. Cancers 2019, 11, 1593. [Google Scholar] [CrossRef]
- Mandal, R.K.; George, G.P.; Mittal, R.D. Association of Toll-like Receptor (TLR) 2, 3 and 9 Genes Polymorphism with Prostate Cancer Risk in North Indian Population. Mol. Biol. Rep. 2012, 39, 7263–7269. [Google Scholar] [CrossRef]














Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oseni, S.O.; Naar, C.; Pavlović, M.; Asghar, W.; Hartmann, J.X.; Fields, G.B.; Esiobu, N.; Kumi-Diaka, J. The Molecular Basis and Clinical Consequences of Chronic Inflammation in Prostatic Diseases: Prostatitis, Benign Prostatic Hyperplasia, and Prostate Cancer. Cancers 2023, 15, 3110. https://doi.org/10.3390/cancers15123110
Oseni SO, Naar C, Pavlović M, Asghar W, Hartmann JX, Fields GB, Esiobu N, Kumi-Diaka J. The Molecular Basis and Clinical Consequences of Chronic Inflammation in Prostatic Diseases: Prostatitis, Benign Prostatic Hyperplasia, and Prostate Cancer. Cancers. 2023; 15(12):3110. https://doi.org/10.3390/cancers15123110
Chicago/Turabian StyleOseni, Saheed Oluwasina, Corey Naar, Mirjana Pavlović, Waseem Asghar, James X. Hartmann, Gregg B. Fields, Nwadiuto Esiobu, and James Kumi-Diaka. 2023. "The Molecular Basis and Clinical Consequences of Chronic Inflammation in Prostatic Diseases: Prostatitis, Benign Prostatic Hyperplasia, and Prostate Cancer" Cancers 15, no. 12: 3110. https://doi.org/10.3390/cancers15123110
APA StyleOseni, S. O., Naar, C., Pavlović, M., Asghar, W., Hartmann, J. X., Fields, G. B., Esiobu, N., & Kumi-Diaka, J. (2023). The Molecular Basis and Clinical Consequences of Chronic Inflammation in Prostatic Diseases: Prostatitis, Benign Prostatic Hyperplasia, and Prostate Cancer. Cancers, 15(12), 3110. https://doi.org/10.3390/cancers15123110