Simple Summary
The interactions between circulating tumor cells (CTCs) and plasma proteins are critical for hematogenous metastasis, and understanding the molecular mechanisms behind these interactions can improve liquid-biopsy-based diagnostics and cancer therapies. This review summarizes recent literature on the surface molecules of CTCs that interact with coagulation proteins and their biological and clinical relevance. It also discusses future research directions to expand our knowledge of the CTC interactome, which can lead to the discovery of new molecular markers for diagnostics and additional targets for cancer therapies.
Abstract
Cancer metastasis is a complex process. After their intravasation into the circulation, the cancer cells are exposed to a harsh environment of physical and biochemical hazards. Whether circulating tumor cells (CTCs) survive and escape from blood flow defines their ability to metastasize. CTCs sense their environment with surface-exposed receptors. The recognition of corresponding ligands, e.g., fibrinogen, by integrins can induce intracellular signaling processes driving CTCs’ survival. Other receptors, such as tissue factor (TF), enable CTCs to induce coagulation. Cancer-associated thrombosis (CAT) is adversely connected to patients’ outcome. However, cancer cells have also the ability to inhibit coagulation, e.g., through expressing thrombomodulin (TM) or heparan sulfate (HS), an activator of antithrombin (AT). To that extent, individual CTCs can interact with plasma proteins, and whether these interactions are connected to metastasis or clinical symptoms such as CAT is largely unknown. In the present review, we discuss the biological and clinical relevance of cancer-cell-expressed surface molecules and their interaction with plasma proteins. We aim to encourage future research to expand our knowledge of the CTC interactome, as this may not only yield new molecular markers improving liquid-biopsy-based diagnostics but also additional targets for better cancer therapies.
1. Introduction
Cancer cells which shed from a primary tumor and circulate in the bloodstream are referred to as circulating tumor cells (CTCs). The presence of CTCs in blood has been largely associated with cancer progression [1,2,3], and it serves as an indicator of the systemic dissemination of cancer, identifying patients at a higher risk of developing metastasis [4,5]. However, the effective number of CTCs is low, as less than 0.01% of the tumor cells exposed to blood can survive [6] and their half-life is estimated to be only around 20 min [7]. CTCs face numerous challenges in the blood, including immune surveillance, the coagulation system and mechanical stress [8]. Only CTCs that can survive the hostile blood environment can form metastases. Critical to this process is also the CTC’s ability to escape from the circulation system through attaching to the blood vessel wall and transmigrating into the perivascular tissues [9]. In addition to their survival and escape abilities, CTCs may also possess the capacity to promote tumor growth through inducing angiogenesis and other tumor supportive pathways once they extravasated into the surrounding tissue. Overall, the complex interplay between CTCs and the blood microenvironment is a crucial determinant of the metastatic potential of cancer cells.
Accumulating studies suggest that the success of the metastatic process is conditioned via the molecular crosstalk between tumor cells and the hostile blood microenvironment [10]. During the metastatic process, CTCs have the opportunity to interact tightly with myeloid-derived suppressor cells (MDSCs) [11], neutrophils [12], NK cells [13], macrophages, T cells [14,15] and platelets [16]. Depending on the cancer cell repertoire, these cell–cell interactions may result in CTC elimination but could also empower them to escape immune recognition. MDSCs interact with CTCs via jagged canonical Notch ligand 1 (JAG1) and neurogenic locus notch homolog protein 1 (Notch1) supporting the adhesion of the cancer cell to the endothelium and extravasation [17]. Neutrophils were shown to bind to vascular adhesion molecule 1 (VCAM1) on CTCs, which promoted CTC proliferation and metastasis [18]. The extravasation process is fueled by inflammatory cytokines released from neutrophils, which induce the expression of adhesion molecules at the endothelium [19]. Interestingly, the binding of neutrophils to CTCs is believed to prevent the attack of NK cells [12]. In case the function of NK cells is not quenched, they were able to clear CTCs. The physical contact between NK and CTC, which is required for the elimination of the tumor cell by granzyme B, death receptor signaling or perforin, is facilitated via the NK cell exposed lectin-like receptor K1 (KLRK1) sensing the major histocompatibility complex class 1 (MHC1) on CTCs [13]. Similar to NK cells, cytotoxic T cells are also able to eliminate CTCs. Immunological synapse formation between the MHC-exposed antigens on CTCs and T cell receptors initiates the killing of the cancer cells via death receptor ligand binding [20]. Additionally, CTCs have been reported to bind platelets [16], which are considered to be tumor-supportive. It has been suggested that CTCs form a shield of platelets around themselves to protect against the attack of NK cells [21,22]. The detailed crosstalk between CTCs and blood cells is beyond the scope of the present review. We refer to excellent other reviews [6,23]. In the present article we aim to focus on CTC receptors that enable interaction with plasma proteins.
Besides blood cells, the plasma contains thousands of different proteins [24,25]. Among all plasma proteins, 93% are albumin and globulins. Most of the remaining 7% consists of fibrinogen and other coagulation factors. Although the interaction between CTCs and plasma proteins is obvious, and potentially connected to the fate of the CTC, only a few studies addressing this topic are currently available. For example, the interaction between CTCs and the components of the coagulation system is of particular interest, as it may contribute to cancer-associated thrombosis (CAT) [26,27]. CAT is reported to be the second most common cause of death in patients with cancer [28,29] and was first identified by Trousseau in 1865. As a systemic hypercoagulable state found in many cancer patients, CAT can present as venous thromboembolism (VTE) [30], pulmonary embolism (PE) [31], arterial thromboembolism (ATE) [32] or microthrombosis. VTE is the most common form of CAT and refers to the formation of blood clots in the veins, usually in the legs, which can break loose and travel to the lungs as a PE. ATE refers to the formation of blood clots in the arteries, which can cause a heart attack or stroke. Microthrombosis is the formation of small clots in the microvasculature, leading to tissue ischemia and organ dysfunction.
Many studies have focused on the impact of the tumor microenvironment on systemic effects such as thrombocytosis [33] or neutrophil build immunothrombosis through releasing neutrophil extracellular traps (NETs) [34,35]. However, research investigating CAT from the perspective of CTCs is scarce, and although it is known that CTCs can express procoagulant proteins [30,36] and that they appear as active part of thrombi [26,27], it remains largely unknown how CTCs respond to the binding of plasma proteins and whether the corresponding cellular response promotes metastasis or further coagulation.
The interactions between CTCs and plasma proteins are potentially mediated by various cell surface receptors, such as tissue factor (TF), integrins, thrombomodulin (TM) and heparan sulfate (HS). These receptors can recognize and bind to different plasma proteins, influencing the fate of the CTCs. TF, for example, is a key factor in coagulation and recognizes FVIIa, while integrins can bind to fibrinogen and fibronectin in the context of platelet activation. TM, on the other hand, can counteract TF-promoted coagulation through directly interacting with thrombin. HS has the potential to promote or suppress coagulation via binding to various plasma proteins such as antithrombin (AT), fibrinogen or von Willebrand factor (vWF).
The interaction between CTCs and different plasma proteins is evidently important, but our current understanding of its biological and clinical significance is limited. In this review, we aim to highlight the relevance of this interaction and encourage further research on this topic. Delineation of these interactions is crucial to better understand tumor progression and to identify potential therapeutic targets. In the following sections, these interactions are described in detail.
2. Tissue Factor
TF, also known as coagulation Factor III, CD142 or thromboplastin, is a 47 kDa cell-surface, transmembrane protein of the class II cytokine receptor family. Its expression correlates with the frequency of CAT in cancer patients [28,29]. Under normal homeostatic conditions, TF is only exposed to the blood upon vascular injury, implicating endothelial damage and, thus, contact of the blood to the subendothelial TF-expressing smooth muscle cells. In response to this, TF interacts with calcium and other coagulation factors in the blood to trigger hemostasis.
In contrast to this hemostatic situation, various studies document that CTCs express TF and that they shed TF-positive microvesicles into circulation [37]. Interestingly, TF expression is not a specialty limited to a few cancer cells but has shown to be significantly expressed on the cellular surface of tumor cells of different tumor entities [38,39,40,41]. Functional TF expressed by CTCs can initiate the extrinsic coagulation cascade [42,43] through binding to FVII, which is then rapidly converted to FVIIa. The active TF-FVIIa complex catalyzes the activation of FX, which then forms the prothrombinase complex that converts prothrombin to thrombin. Thrombin cleaves fibrinogen into fibrin and activates platelets via protease-activated receptors (PARs), promoting the formation of hemostatic clots. Additionally, thrombin activates FVIII, FXIII and FV, amplifying the coagulation response. Overall, the expression of functional TF by CTCs highlights its significance in the pathophysiology of cancer and its potential role in promoting thrombosis.
However, TF plays a vital role in tumor progression not only through initiating thrombosis. The TF-FVIIa complex at the CTC surface contributes to tumor cell proliferation, angiogenesis and metastasis [38,44] through activating PARs and mitogen-activated protein kinase (MAPK) pathway promoting, e.g., epithelial–mesenchymal transition (EMT) (Figure 1). EMT is a key process that enhances the ability of CTCs to disseminate as it drives vascular extravasation. Therefore, molecular markers of EMT and related signaling pathways may predict the ability of CTCs to metastasize [41,45,46,47,48]. In this context, it has been shown that the formation of the TF–FVIIa complex at the surface of breast cancer and lung carcinoma cells mediates PAR-2 signaling [49,50,51]. This results in MAPK pathway activation and further downstream effects such as the upregulation of cytokines and proangiogenic factors [52]. For the activation of the p44/42 MAPK pathway, as well as JAK2, p70/p85S6K and p90RSK, TF cytoplasmic domain is not necessary [53,54,55], but the extracellular domain which can bind FVIIa is required. Additionally, the TF–FVIIa-FXa complex-mediated activation of PAR-2 induced IL-8 expression and triggered cell migration [56]. Next to PAR-2, TF-mediated formation of thrombin was shown to activate PAR-1 in different tumor cells promoting, e.g., metastasis [57,58]. Interestingly, the prometastatic effect of PAR-1 requires co-expression with TF at the surface of the tumor cells, while the exclusive overexpression of PAR-1 was not sufficient [59].
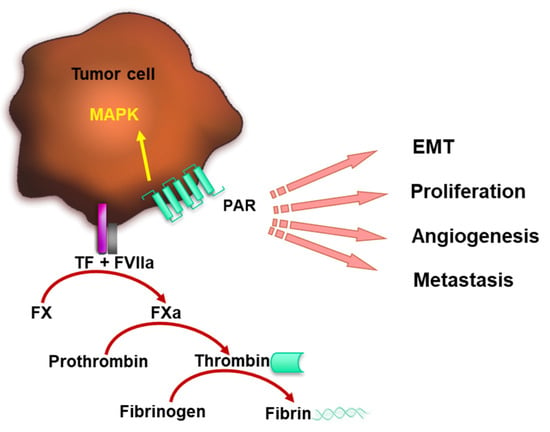
Figure 1.
TF–FVIIa complex promotes coagulation and tumor progression. TF forms a complex with FVIIa catalyzing FX activation, which leads to thrombin formation. Thrombin cleaves fibrinogen to fibrin and contributes to blood clotting. FVIIa bound to the tumor cell surface by TF mediates PAR signaling, which induces MAPK pathway activation, tumor cell EMT, proliferation, angiogenesis and metastasis.
3. Integrins
The binding of FVII to tumor cell surfaces via TF induces thrombin formation, which cleaves fibrinogen to fibrin and leads to blood clotting [60]. Additionally, another mechanism linking TF to metastasis is through the fibrinogen-dependent and platelet-dependent restriction in natural killer cell-mediated clearance of micrometastases [61]. Fibrinogen, which is one of the most abundant coagulation proteins, can directly bind to the surface of CTCs via integrins [62,63]. Integrins are transmembrane receptors ubiquitously expressed on cell surfaces and they play a critical role in regulating interactions between cells and their environment. These receptors are responsible for mediating cell–cell, cell–extracellular matrix and cell–protein interactions via responding to both intracellular and extracellular signals [64,65,66,67,68]. Integrins are obligate heterodimeric receptors consisting of an α and a β subunit. There are 18 α and 8 β subunits and various combinations comprise a family of 24 heterodimeric integrins members [62,69].
Most integrins can interact with multiple ligands [62]. Vice versa, one integrin ligand can interact with different integrins. Based on their structural similarity and ligand recognition patterns, integrins can be grouped into four classes: RGD-binding, collagen-binding, laminin-binding and leukocyte-specific integrin receptors [68]. The abbreviation RGD stands for the tripeptide sequence: Arg-Gly-Asp. Since the RGD peptide is the common feature of various plasmatic proteins [62,70,71], comprising, e.g., fibrinogen [72], fibronectin [73] and vWF [74,74,75,76], this review focuses exclusively on RGD-binding integrins. Most of the RGD-binding integrins are expressed at elevated levels in various cancer types, suggesting their crucial contribution to pathophysiological functions in cancer [77].
Plasma fibrinogen and its related fragments are ascribed to play an important role in the pathophysiology of tumor angiogenesis, invasion and metastasis [60,61]. It has been shown that a high pre-therapeutic plasma fibrinogen level independently predicts adverse events and the clinical outcome of patients with various cancer types [78,79,80,81]. Clinical studies have confirmed that elevated plasma fibrinogen levels are associated with poor outcome in patients with non-metastatic and metastatic renal cell carcinoma [82,83]. Though the β3 integrin family includes αIIbβ3 and αvβ3, often referred to as the fibrinogen receptor, fibrinogen can also bind to other integrins including α5β1 [84]. In lung cancer, the prevention of fibrinogen–integrin interaction via fibrinogen knockout promoted tumor growth and metastasis through serine/threonine kinase (also known as PKB) AKT signaling [85]. Fibrinogen binding to integrins α5β1 and αvβ3 promoted the activation of NF-κB and increased the expression of NF-κB-mediated inflammatory chemokines [86].
Fibronectin is a large, disulfide-linked dimeric glycoprotein that shares the same integrin binding receptor for αvβ3 and α5β1 with fibrinogen [62]. Fibronectin is present in a soluble form in plasma or as insoluble filaments deposited in the extracellular matrix [87]. During normal wound healing, the plasma fibronectin or insoluble fibrinogen filaments are released by damaged blood vessels and repair tissue through forming scaffolds with platelets [88]. Malignant tumors often develop at sites of chronic injury and permanent wounds, where the leaky vessels form fibrin clots around the tumoral lesion [89]. Hence, the formation of thrombotic clots via recruiting fibronectin to tumor cell surface integrins was thought to support CTC extravasation [90,91].
Under dynamic flow conditions, melanoma surface integrin αvβ3 has the unique ability to interact with immobilized fibrinogen and fibronectin and support cell arrest [92]. Previous studies also showed that integrin αIIbβ3 can catch blood-flowing fibrinogen in the context of platelet aggregation. This contributes to initial tumor cell arrest at the vascular endothelium during the early stage of tumor cell dissemination [93]. Lung cancer cell α5β1 integrin interacts with fibronectin, promoting cancer cell proliferation and metastasis [94]. However, direct interaction between fibrinogen and α5β1 integrins was proven only under static conditions. Under flow conditions, α5β1 integrins did not contribute to cancer cell arrest on immobilized fibrinogen [92]. The interaction of integrins with plasmatic proteins and its impact on tumor progression is schematically summarized in Figure 2.
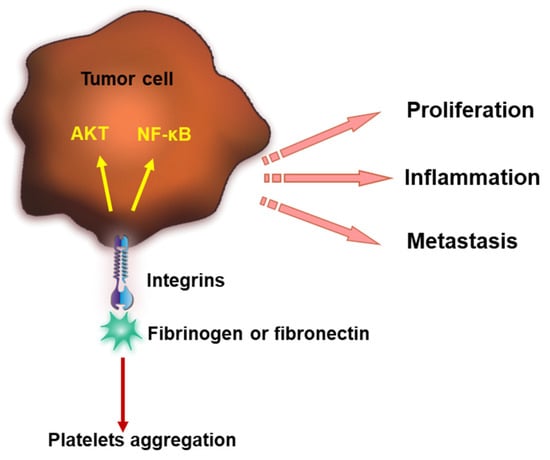
Figure 2.
Integrins interact with fibrinogen or fibronectin, inducing platelet aggregation and coagulation and further contributing to tumor progression. Integrins bind fibrinogen or fibronectin and lead to AKT signaling and NF-κB activation. This promotes tumor proliferation, inflammation and metastasis.
4. Thrombomodulin
Although the formation of thrombin can promote the generation of fibrin and, thus, blood clotting, thrombin can also directly bind to tumor cell surface-exposed TM. Once bound to the surface, the protein C (PC) anti-coagulant pathway is switched on, representing an important negative feedback regulator of the coagulation cascade.
Mature human TM is a 557-amino-acid residue, type 1 transmembrane glycoprotein. It was discovered in the 1980s [95]. TM interacts with thrombin, building the TM-thrombin complex, which catalyzes the activation of the zymogen PC. The activated protein C (APC) together with protein S (PS) form the APC–PS complex, preventing further activation of FV and FXIII [96]. The PC anti-coagulant pathway switches thrombin from a pro-coagulant to an anti-coagulant enzyme.
PS is not only a cofactor of activated PC, but also a binding partner of Tissue Factor Pathway Inhibitor (TFPI) [97]. TFPI is the physiological inhibitor of the TF pathway of blood coagulation. The binding of PS to TFPI inhibits FXa. The TFPI/FXa complex binds to the complex of TF and FVIIa to form an inactive quaternary TFPI/FXa/TF/FVIIa complex. The protein-S-promoted activity of PC is further enhanced in the presence of calcium and phospholipids [98]. The anticoagulative activity of APC is inhibited by the Protein C Inhibitor (PCI), also known as Plasminogen Activator Inhibitor 3 (PAI3).
In the context of tumor progression, TM is supposed to have tumor-suppressive properties. Numerous preclinical and clinical studies indicate that the dampening of cell proliferation, invasion and metastasis is mediated by TM (Figure 3) [99,100,101]. A mouse model expressing a mutant form of TM with reduced thrombin affinity exhibited a strongly prometastatic phenotype, suggesting TM is a powerful determinant of hematogenous metastasis [101]. The antitumor effects of TM depend also on the activation of PC. In a mouse model of pancreatic cancer, recombinant TM interfered with tumor growth through inhibiting NF-κB and thrombin-induced PAR-1 activation [102]. There is also evidence that TM expression at the surface of tumor cells correlates with better prognosis in patients suffering from bladder, breast, colon, prostate, lung or oral epithelium cancer [103,104].
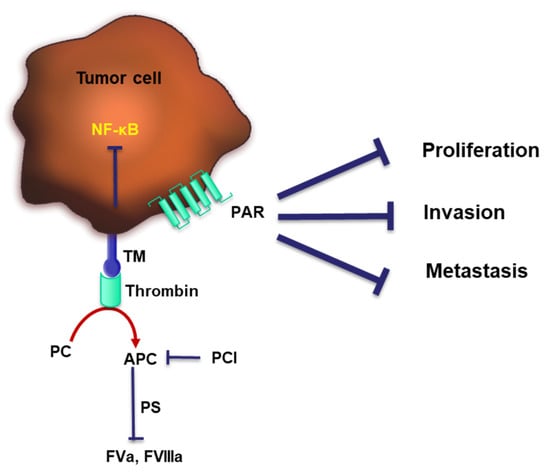
Figure 3.
TM switches thrombin from a pro-coagulant to an anti-coagulant and prevents tumor progression. TM interacts with thrombin to subsequently activate PC. Activated PC (APC) in concert with PS switch on the PC anti-coagulant pathway. Thrombin can directly activate PAR-1, whereas the complex of thrombin and TM can active NF-κB, both pathways were shown to exert tumor-suppressing properties through dampening cell proliferation, invasion and metastasis.
Anticoagulation is an effective process that involves a large panel of molecular players. Tumor cells may serve as prothrombotic triggers through aberrant regulation of receptor expression. Therefore, anticoagulation of cancer patients via direct oral anticoagulants or low-molecular-weight heparins appears to be a valid strategy to counteract hypercoagulation [105,106]. Heparins are close relatives of HS, which is an abundant glycan expressed by every mammalian cell including cancer cells. To which extent the HS of cancer cells has the potential to interfere with the coagulation system is discussed in the following section.
5. Heparan Sulfate
HS is a glycan covalently connected to a protein backbone forming so-called heparan sulfate proteoglycans (HSPG). HSPGs are ubiquitously present in mammals and exist as soluble or membrane-bound proteins. Membrane-bound proteins which are, thus, exposed at cell surfaces are glypicans and syndecans. While glypicans are connected to the plasma membrane via glycophosphatidylinositol anchors, syndecans are transmembrane proteins. The synthesis of the glycan chains of proteoglycan takes place within the Golgi apparatus and involves the action of various enzymes [107]. After the elongation of the HS chain by exostosins, several sulfotransferases introduce sulfate groups at different molecular positions. These modifications are not obligate but depend on the expression of the acting sulfotransferases and the availability of the required substrates [108,109]. Accordingly, the HS chain exhibits vast structural diversity, which is also reflected by the long list of biological properties and molecular interaction partners [110,111]. The ability of HS to bind human plasma proteins has previously been explored via pull-down assays coupled to a mass-spectrometry-based analysis [112]. In total, 99 plasma proteins with HS-binding properties have been identified. However, only a few studies investigated the (patho)physiological relevance of this interaction.
HS is a strongly negatively charged carbohydrate, and the binding of proteins depends at least in part on electrostatic interaction. Previously, Cardin and Weintraub described the amino acid consensus sequences that are involved in electrostatic interactions [113]. This so-called “Cardin Weintraub” motif contains the basic amino acids arginine or lysine in close proximity. Cardin and Weintraub postulated the HS binding site as one connected linear stretch of amino acids. However, insights that are more recent revealed that the spatial proximity of the required amino acids due to a specific three-dimensional configuration of the protein creates an HS binding cleft with high affinity [114]. Accordingly, binding affinities and specificities depend on the conformation of the HS binding site of the protein [115]. However, the structure of HS also defines this interaction. For instance, the binding of AT to HS is favored by a concise pentasaccharide sequence, which also inspired the synthesis of fondaparinux, a clinically relevant anticoagulant [116]. Recent studies showed that enhanced 3-O-sulfation of HS promoted the binding of AT to cancer cells [117].
AT, also known as AT III, is the main inhibitor of coagulation next to APC. As a plasmatic protein, AT has the ability to bind to the HS of CTCs, preventing the activation of thrombin and FXa. Once bound to HS, AT undergoes a conformational change. This enhances its reactivity towards thrombin, thereby promoting the formation of an AT/thrombin complex. Upon binding to HS, AT showed a strongly increased activity in neutralizing the enzymatic activity of thrombin [117,118,119]. Moreover, HS can interact with TFPI, which is a potent inhibitor of the blood coagulation factors FXa and FVIIa, counteracting coagulation [120,121].
The role of HS in coagulation is complex, since besides being anticoagulant, HS may also exhibit pro-coagulant properties. The interaction of HS with fibrinogen has been documented to reduce anticoagulant activity through forming an HS–fibrinogen–thrombin ternary complex, reducing the capacity of HS to bind to AT [122]. Moreover, and as previously mentioned, fibrinogen is recruited to the cancer cell surface by integrins. Interestingly, the activity of integrins is at least partially regulated by cell-surface-exposed HS, indicating complex and interwoven communication between the HS exposed by CTCs and the coagulation system [123].
In addition to its well-established anticoagulant function, AT has also been reported to exert anti-tumor properties. The binding of AT to cell-surface HS competes with the binding of growth factors blocking their proangiogenic effects [124]. AT can also attenuate inflammatory events through inhibiting NF-κB. Interestingly, the inhibitory potency of AT depends on the interaction of AT with cell-surface HS [125]. Similarly, it was reported that AT could also act as a modulator of tumor cell migration and invasion in different tumor cells. The inhibitory process required the activation of AT by HS [126]. Figure 4 illustrates the dual functions of HS in coagulation and its anti-tumor effects through AT binding.
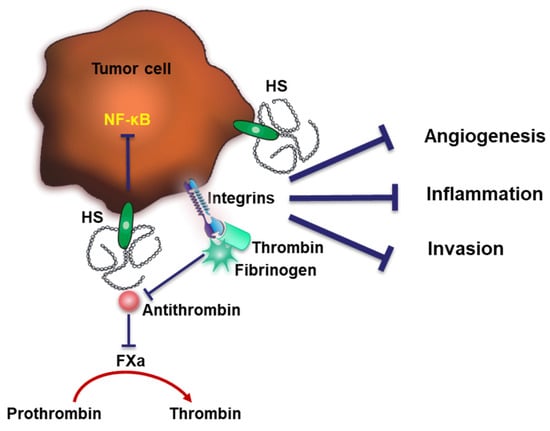
Figure 4.
Dual functions of HS in coagulation. Cell-surface HS interacts with AT and neutralizes thrombin to inhibit coagulation. HS may also exhibit pro-coagulating capacities through forming a complex with fibrinogen and thrombin. The binding of AT to HS was shown to exert anti-tumor effects through inhibiting tumor angiogenesis, inflammation and invasion.
The crosstalk between AT and HS is a prominent example reflecting the relevance of HS in regulating coagulation. However, similar effects can be expected for other plasma proteins. We have recently shown that HS exposed by CTCs together with αvβ3 integrins triggers the binding of vWF to the cell surface [108] (Figure 5). VWF is a complex multimeric plasma glycoprotein critical for normal hemostatic function. VWF has two main roles in hemostasis: first, to recruit and tether platelets at sites of vascular injury, facilitating platelet aggregation. Second, vWF acts as a protective carrier molecule for procoagulant FVIII. The hemostatic function of these vWF multimers is tightly regulated by the vWF-specific protease ADAMTS13 (a disintegrin and metalloproteinase with thrombospondin type 1 motif, 13). Interestingly, thrombin inhibits ADAMTS13, which further amplifies platelet recruitment and coagulation. Endothelial cells release vWF constitutively into the plasma at low levels. Increased secretion of vWF is followed upon endothelial stimulation with thrombin or tumor-cell derived vascular endothelial growth factor A [127,128]. This is in line with clinical studies which showed elevated vWF levels in the plasma of cancer patients. Together with a reduced plasma concentration of ADAMTS13, more plasmatic vWF was associated with a higher risk of developing venous thromboembolism [129].
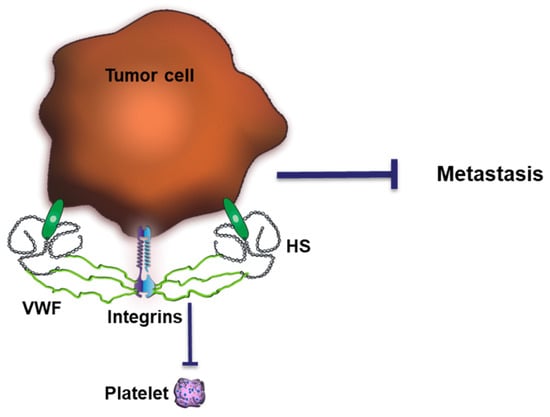
Figure 5.
Impact of HS-mediated binding of vWF on tumor progression. VWF forms a complex with tumor cell surface HS and integrins, preventing platelet binding and hematogenous metastasis.
We found that the abundance and length of the cell-surface-exposed HS chain coordinate the recruitment of plasmatic vWF to the cancer cell surface, which blocked, e.g., the binding of platelets. Murine tumor models and analysis of patients’ samples indicated that loss of this anti-coagulative HS at the cancer cell surface increased the ability of the CTC to metastasize, which was in turn connected to reduced patients’ survival [108].
6. Conclusions
The communication of CTCs with their environment defines their ability to survive within the blood stream and to form metastasis. In the present review, we shed some light on the interaction of CTCs with plasma proteins. This interaction is mediated by various cell-surface-exposed receptors and triggers various intracellular signaling pathways as well as extracellular reactions such as blood coagulation. Which plasma proteins can be recognized by CTCs and to which extent this interaction affects the fate of the CTC are largely unknown and comprehensive research is still missing. In this review article, we aimed to emphasize that improved knowledge of the CTC interactome is relevant to understanding the strategies of CTCs to survive and metastasize. However, the crosstalk between CTCs and plasma proteins is a double-edged sword. While the coagulation system is often considered to be exploited by the cancer cells, they are also a relevant part of the innate immune system and, as such, a powerful weapon to eliminate CTCs. This complex situation is also reflected in clinical data. Although the positive correlation between CAT and tumor progression suggests the coagulation system to be tumor supportive, therapy of cancer patients with anticoagulants limits successful thrombosis but has most often no direct tumor suppressive effects. Therefore, we assume that some interactions with plasma proteins support CTCs, while others have the potential to hinder further cancer progression. The exact biological impact of each molecular interaction depends most likely on the cancer entity, the stage of the disease and on the genetic repertoire of individual CTCs. The detection and evaluation of CTCs have become valuable tools in diagnostics to monitor therapy and patients’ outcomes. In recent years, the development of single-cell analysis techniques such as single cell RNA sequencing enable the in-depth characterization and advanced classification of CTCs. Further insights into the biological meaning of gene or protein expression patterns might be possible through investigating the interaction of single CTCs with blood components such as plasma proteins. Currently, there is a lack of studies that specifically describe methodologies to analyze the interactions between CTCs and plasma proteins in a clinical setting. CTC detection and isolation from patients’ blood is based on different technologies such as CTC enrichment through surface markers (i.e., epithelial cell adhesion molecule, EpCAM) or biophysical properties (i.e., size, density) [130,131,132,133]. The identification of CTCs in the blood of patients may also offer the opportunity to detect CTC-bound plasma proteins. Applicable methods may range from immunofluorescence microscopy and flow cytometry to single-cell mass spectrometry. These could not only improve our understanding of cancer metastasis but may allow better detection of cancer cells in liquid biopsy and differentiation between malignant and benign CTCs. Research has shown that TF-targeting therapeutics can effectively eradicate cancer stem cells isolated from breast, lung and ovarian cancer without the development of drug resistance. Given the high expression of TF in CTCs, this approach may also hold promise for the treatment of CTCs and metastatic disease. Overall, a better understanding of the CTC interactome and its impact on cancer progression is essential for the development of more effective diagnostic and therapeutic strategies for cancer patients [134].
Author Contributions
Writing—original draft preparation, Y.W.; review and editing, S.W.S. and C.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bayarri-Lara, C.; Ortega, F.G.; De Guevara, A.C.L.; Puche, J.L.; Zafra, J.R.; De Miguel-Pérez, D.; Ramos, A.S.-P.; Giraldo-Ospina, C.F.; Gómez, J.A.N.; Delgado-Rodríguez, M.; et al. Circulating Tumor Cells Identify Early Recurrence in Patients with Non-Small Cell Lung Cancer Undergoing Radical Resection. PLoS ONE 2016, 11, e0148659. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Ureña, M.; Ortega, F.G.; de Miguel-Pérez, D.; Rodriguez-Martínez, A.; García-Puche, J.L.; Ilyine, H.; Lorente, J.A.; Exposito-Hernandez, J.; Garrido-Navas, M.C.; Delgado-Ramirez, M.; et al. Circulating tumor cells criteria (CyCAR) versus standard RECIST criteria for treatment response assessment in metastatic colorectal cancer patients. J. Transl. Med. 2018, 16, 251. [Google Scholar] [CrossRef] [PubMed]
- Mamdouhi, T.; Twomey, J.D.; McSweeney, K.M.; Zhang, B. Fugitives on the run: Circulating tumor cells (CTCs) in metastatic diseases. Cancer Metastasis Rev. 2019, 38, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Tsai, W.-S.; Chen, J.-S.; Shao, H.-J.; Wu, J.-C.; Lai, J.-M.; Lu, S.-H.; Hung, T.-F.; Chiu, Y.-C.; You, J.-F.; Hsieh, P.-S.; et al. Circulating Tumor Cell Count Correlates with Colorectal Neoplasm Progression and Is a Prognostic Marker for Distant Metastasis in Non-Metastatic Patients. Sci. Rep. 2016, 6, 24517. [Google Scholar] [CrossRef] [PubMed]
- Al-Mehdi, A.B.; Tozawa, K.; Fisher, A.B.; Shientag, L.; Lee, A.; Muschel, R.J. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: A new model for metastasis. Nat. Med. 2000, 6, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Rejniak, K.A. Circulating Tumor Cells: When a Solid Tumor Meets a Fluid Microenvironment. Adv. Exp. Med. Biol. 2016, 936, 93–106. [Google Scholar] [PubMed]
- Tao, J.; Zhu, L.; Yakoub, M.; Reißfelder, C.; Loges, S.; Schölch, S. Cell–Cell Interactions Drive Metastasis of Circulating Tumor Microemboli. Cancer Res. 2022, 82, 2661–2671. [Google Scholar] [CrossRef]
- Chaffer, C.L.; Weinberg, R.A. A Perspective on Cancer Cell Metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef]
- Lin, D.; Shen, L.; Luo, M.; Zhang, K.; Li, J.; Yang, Q.; Zhu, F.; Zhou, D.; Zheng, S.; Chen, Y.; et al. Circulating tumor cells: Biology and clinical significance. Signal Transduct. Target. Ther. 2021, 6, 404. [Google Scholar] [CrossRef]
- Ward, M.P.; EKane, L.; ANorris, L.; Mohamed, B.M.; Kelly, T.; Bates, M.; JO’Leary, J. Platelets, immune cells and the coagulation cascade; friend or foe of the circulating tumour cell? Mol. Cancer 2021, 20, 59. [Google Scholar] [CrossRef]
- Liu, Q.; Liao, Q.; Zhao, Y. Myeloid-derived suppressor cells (MDSC) facilitate distant metastasis of malignancies by shielding circulating tumor cells (CTC) from immune surveillance. Med. Hypotheses 2016, 87, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, A.; Brooks, M.W.; Houshyar, S.; Reinhardt, F.; Ardolino, M.; Fessler, E.; Weinberg, R.A. Suppress Intraluminal NK Cell-Mediated Tumor Cell Clearance and Enhance Extravasation of Disseminated Carcinoma Cells. Cancer Discov. 2016, 6, 630–649. [Google Scholar] [CrossRef] [PubMed]
- López-Soto, A.; Gonzalez, S.; Smyth, M.J.; Galluzzi, L. Control of Metastasis by NK Cells. Cancer Cell 2017, 32, 135–154. [Google Scholar] [CrossRef] [PubMed]
- Xue, D.; Xia, T.; Wang, J.; Chong, M.; Wang, S.; Zhang, C. Role of regulatory T cells and CD8+ T lymphocytes in the dissemination of circulating tumor cells in primary invasive breast cancer. Oncol. Lett. 2018, 16, 3045–3053. [Google Scholar] [CrossRef]
- Palicelli, A.; Bonacini, M.; Croci, S.; Bisagni, A.; Zanetti, E.; De Biase, D.; Sanguedolce, F.; Ragazzi, M.; Zanelli, M.; Chaux, A.; et al. What Do We Have to Know about PD-L1 Expression in Prostate Cancer? A Systematic Literature Review. Part 7: PD-L1 Expression in Liquid Biopsy. J. Pers. Med. 2021, 11, 1312. [Google Scholar] [CrossRef]
- Anvari, S.; Osei, E.; Maftoon, N. Interactions of platelets with circulating tumor cells contribute to cancer metastasis. Sci. Rep. 2021, 11, 15477. [Google Scholar] [CrossRef]
- Sprouse, M.L.; Welte, T.; Boral, D.; Liu, H.N.; Yin, W.; Vishnoi, M.; Goswami-Sewell, D.; Li, L.; Pei, G.; Jia, P.; et al. PMN-MDSCs Enhance CTC Metastatic Properties through Reciprocal Interactions via ROS/Notch/Nodal Signaling. Int. J. Mol. Sci. 2019, 20, 1916. [Google Scholar] [CrossRef]
- Szczerba, B.M.; Castro-Giner, F.; Vetter, M.; Krol, I.; Gkountela, S.; Landin, J.; Scheidmann, M.C.; Donato, C.; Scherrer, R.; Singer, J.; et al. Neutrophils escort circulating tumour cells to enable cell cycle progression. Nature 2019, 566, 553–557. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Bauer, A.T.; Kirschfink, M.; Ding, P.; Gebhardt, C.; Borsig, L.; Tuting, T.; Renne, T.; Haffner, K.; et al. Neutrophils activated by membrane attack complexes increase the permeability of melanoma blood vessels. Proc. Natl. Acad. Sci. USA 2022, 119, e2122716119. [Google Scholar] [CrossRef]
- Chalfin, H.J.; Pramparo, T.; Mortazavi, A.; Niglio, S.A.; Schonhoft, J.D.; Jendrisak, A.; Chu, Y.-L.; Richardson, R.; Krupa, R.; Anderson, A.K.; et al. Circulating Tumor Cell Subtypes and T-cell Populations as Prognostic Biomarkers to Combination Immunotherapy in Patients with Metastatic Genitourinary Cancer. Clin. Cancer Res. 2021, 27, 1391–1398. [Google Scholar] [CrossRef]
- Maurer, S.; Kropp, K.N.; Klein, G.; Steinle, A.; Haen, S.P.; Walz, J.S.; Hinterleitner, C.; Märklin, M.; Kopp, H.-G.; Salih, H.R. Platelet-mediated shedding of NKG2D ligands impairs NK cell immune-surveillance of tumor cells. OncoImmunology 2018, 7, e1364827. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Jirousková, M.; Degen, J.L. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell–mediated elimination of tumor cells. Blood 2005, 105, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Navas, C.; de Miguel-Pérez, D.; Exposito-Hernandez, J.; Bayarri, C.; Amezcua, V.; Ortigosa, A.; Valdivia, J.; Guerrero, R.; Puche, J.L.G.; Lorente, J.A.; et al. Cooperative and Escaping Mechanisms between Circulating Tumor Cells and Blood Constituents. Cells 2019, 8, 1382. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Lee, E.; Hestir, K.; Leo, C.; Huang, M.; Bosch, E.; Halenbeck, R.; Wu, G.; Zhou, A.; Behrens, D.; et al. Discovery of a Cytokine and Its Receptor by Functional Screening of the Extracellular Proteome. Science 2008, 320, 807–811. [Google Scholar] [CrossRef]
- Williams, S.A.; Kivimaki, M.; Langenberg, C.; Hingorani, A.D.; Casas, J.P.; Bouchard, C.; Jonasson, C.; Sarzynski, M.A.; Shipley, M.J.; Alexander, L.; et al. Plasma protein patterns as comprehensive indicators of health. Nat. Med. 2019, 25, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N.; Hisada, Y. Circulating Tumor Cells and Cancer-Associated Venous Thrombosis: A Missing Link. Arter. Thromb. Vasc. Biol. 2023, 43, 160–162. [Google Scholar] [CrossRef]
- Gi, T.; Kuwahara, A.; Yamashita, A.; Matsuda, S.; Maekawa, K.; Moriguchi-Goto, S.; Sato, Y.; Asada, Y. Histopathological Features of Cancer-Associated Venous Thromboembolism: Presence of Intrathrombus Cancer Cells and Prothrombotic Factors. Arter. Thromb. Vasc. Biol. 2023, 43, 146–159. [Google Scholar] [CrossRef]
- Lima, L.G.; Monteiro, R.Q. Activation of blood coagulation in cancer: Implications for tumour progression. Biosci. Rep. 2013, 33, e00064. [Google Scholar] [CrossRef]
- Donati, M.B. Cancer and thrombosis: From Phlegmasia alba dolens to transgenic mice. Thromb. Haemost. 1995, 74, 278–281. [Google Scholar] [CrossRef]
- Khorana, A.A.; Mackman, N.; Falanga, A.; Pabinger, I.; Noble, S.; Ageno, W.; Lee, A.Y. Cancer-associated venous thromboembolism. Nat. Rev. Dis. Prim. 2022, 8, 11. [Google Scholar] [CrossRef]
- Gimbel, I.A.; Mulder, F.I.; Bosch, F.T.; Freund, J.E.; Guman, N.; van Es, N.; Kamphuisen, P.W.; Büller, H.R.; Middeldorp, S. Pulmonary embolism at autopsy in cancer patients. J. Thromb. Haemost. 2021, 19, 1228–1235. [Google Scholar] [CrossRef]
- Grover, S.P.; Hisada, Y.M.; Kasthuri, R.S.; Reeves, B.N.; Mackman, N. Cancer Therapy-Associated Thrombosis. Arter. Thromb. Vasc. Biol. 2021, 41, 1291–1305. [Google Scholar] [CrossRef] [PubMed]
- Simanek, R.; Vormittag, R.; Ay, C.; Alguel, G.; Dunkler, D.; Schwarzinger, I.; Steger, G.; Jaeger, U.; Zielinski, C.; Pabinger, I. High platelet count associated with venous thromboembolism in cancer patients: Results from the Vienna Cancer and Thrombosis Study (CATS). J. Thromb. Haemost. 2009, 8, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Demers, M.; Wagner, D.D. NETosis: A New Factor in Tumor Progression and Cancer-Associated Thrombosis. Semin. Thromb. Hemost. 2014, 40, 277–283. [Google Scholar] [CrossRef]
- Burmeister, A.; Vidal, Y.S.S.; Liu, X.; Mess, C.; Wang, Y.; Konwar, S.; Tschongov, T.; Haffner, K.; Huck, V.; Schneider, S.W.; et al. Impact of neutrophil extracellular traps on fluid properties, blood flow and complement activation. Front. Immunol. 2022, 13, 1078891. [Google Scholar] [CrossRef] [PubMed]
- Hisada, Y.; Mackman, N. Cancer-associated pathways and biomarkers of venous thrombosis. Blood 2017, 130, 1499–1506. [Google Scholar] [CrossRef] [PubMed]
- Owens, A.P., 3rd; Mackman, N. Microparticles in hemostasis and thrombosis. Circ. Res. 2011, 108, 1284–1297. [Google Scholar] [CrossRef]
- Li, H.; Yu, Y.; Gao, L.; Zheng, P.; Liu, X.; Chen, H. Tissue factor: A neglected role in cancer biology. J. Thromb. Thrombolysis 2022, 54, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Nadir, Y.; Brenner, B. Novel strategies of coagulation inhibition for reducing tumor growth and angiogenesis. Thromb. Res. 2018, 164 (Suppl. S1), S153–S156. [Google Scholar] [CrossRef]
- Santagostino, E.; Negrier, C.; Klamroth, R.; Tiede, A.; Pabinger-Fasching, I.; Voigt, C.; Morfini, M. Safety and pharmacokinetics of a novel recombinant fusion protein linking coagulation factor IX with albumin (rIX-FP) in hemophilia B patients. Blood 2012, 120, 2405–2411. [Google Scholar] [CrossRef]
- Bourcy, M.; Suarez-Carmona, M.; Lambert, J.; Francart, M.E.; Schroeder, H.; Delierneux, C.; Gilles, C. Tissue Factor Induced by Epithelial-Mesenchymal Transition Triggers a Procoagulant State That Drives Metastasis of Circulating Tumor Cells. Cancer Res. 2016, 76, 4270–4282. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. The Role of Tissue Factor and Factor VIIa in Hemostasis. Anesth. Analg. 2009, 108, 1447–1452. [Google Scholar] [CrossRef]
- Morrissey, J.H. Tissue Factor: A Key Molecule in Hemostatic and Nonhemostatic Systems. Int. J. Hematol. 2004, 79, 103–108. [Google Scholar] [CrossRef]
- Hueng, D.-Y.; Lin, G.-J.; Huang, S.-H.; Liu, L.-W.; Ju, D.-T.; Chen, Y.-W.; Sytwu, H.-K.; Chang, C.; Huang, S.-M.; Yeh, Y.-S.; et al. Inhibition of Nodal suppresses angiogenesis and growth of human gliomas. J. Neuro-Oncology 2010, 104, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Li, J.F.; Su, L.; Zang, M.; Fan, Z.; Yu, B.; Liu, B. ALEX1, a novel tumor suppressor gene, inhibits gastric cancer metastasis via the PAR-1/Rho GTPase signaling pathway. J. Gastroenterol. 2018, 53, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Otsuki, T.; Fujimoto, D.; Hirono, Y.; Goi, T.; Yamaguchi, A. Thrombin conducts epithelial-mesenchymal transition via protease-activated receptor-1 in human gastric cancer. Int. J. Oncol. 2014, 45, 2287–2294. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.-C.; Zhang, T.; Di, W.; Li, W.-P. Thrombin promotes epithelial ovarian cancer cell invasion by inducing epithelial-mesenchymal transition. J. Gynecol. Oncol. 2013, 24, 265–272. [Google Scholar] [CrossRef]
- Nzeteu, G.A.N.; Geismann, C.; Arlt, A.; Hoogwater, F.J.H.; Nijkamp, M.W.; Meyer, N.H.; Bockhorn, M. Role of Epithelial-to-Mesenchymal Transition for the Generation of Circulating Tumors Cells and Cancer Cell Dissemination. Cancers 2022, 14, 5483. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Schaffner, F.; Kerver, M.; Petersen, H.H.; Ahamed, J.; Felding-Habermann, B.; Takada, Y.; Mueller, B.M.; Ruf, W. Inhibition of tissue factor signaling suppresses tumor growth. Blood 2008, 111, 190–199. [Google Scholar] [CrossRef]
- Bluff, J.E.; Brown, N.J.; Reed, M.W.; Staton, C.A. Tissue factor, angiogenesis and tumour progression. Breast Cancer Res. 2008, 10, 204. [Google Scholar] [CrossRef]
- Belting, M.; Dorrell, M.I.; Sandgren, S.; Aguilar, E.; Ahamed, J.; Dorfleutner, A.; Carmeliet, P.; Mueller, B.M.; Friedlander, M.; Ruf, W. Regulation of angiogenesis by tissue factor cytoplasmic domain signaling. Nat. Med. 2004, 10, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Gajsiewicz, J.M.; Morrissey, J.H. Structure–Function Relationship of the Interaction between Tissue Factor and Factor VIIa. Semin. Thromb. Hemost. 2015, 41, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, B.B.; Freskgård, P.O.; Nielsen, L.S.; Rao LV, M.; Ezban, M.; Petersen, L.C. Factor VIIa-induced p44/42 mitogen-activated protein kinase activation requires the proteolytic activity of factor VIIa and is independent of the tissue factor cytoplasmic domain. J. Biol. Chem. 1999, 274, 21349–21354. [Google Scholar] [CrossRef]
- Versteeg, H.H.; Spek, C.A.; Slofstra, S.H.; Diks, S.H.; Richel, D.J.; Peppelenbosch, M.P. FVIIa:TF induces cell survival via G12/G13-dependent Jak/STAT activation and BclXL production. Circ. Res. 2004, 94, 1032–1040. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.; Sørensen, B.B.; Slofstra, S.H.; Brande, J.H.M.V.D.; Stam, J.C.; Henegouwen, P.M.P.V.B.E.; Richel, D.J.; Petersen, L.C.; Peppelenbosch, M. VIIa/Tissue Factor Interaction Results in a Tissue Factor Cytoplasmic Domain-independent Activation of Protein Synthesis, p70, and p90 S6 Kinase Phosphorylation. J. Biol. Chem. 2002, 277, 27065–27072. [Google Scholar] [CrossRef]
- Hjortoe, G.M.; Petersen, L.C.; Albrektsen, T.; Sorensen, B.B.; Norby, P.L.; Mandal, S.K.; Pendurthi, U.R.; Rao, L.V.M. Tissue factor-factor VIIa–specific up-regulation of IL-8 expression in MDA-MB-231 cells is mediated by PAR-2 and results in increased cell migration. Blood 2004, 103, 3029–3037. [Google Scholar] [CrossRef]
- Yang, Y.; Stang, A.; Schweickert, P.G.; Lanman, N.A.; Paul, E.N.; Monia, B.P.; Revenko, A.S.; Palumbo, J.S.; Mullins, E.S.; Elzey, B.D.; et al. Thrombin Signaling Promotes Pancreatic Adenocarcinoma through PAR-1–Dependent Immune Evasion. Cancer Res 2019, 79, 3417–3430. [Google Scholar] [CrossRef]
- Nierodzik, M.L.; Chen, K.; Takeshita, K.; Li, J.J.; Huang, Y.Q.; Feng, X.S.; D′Andrea, M.R.; Andrade-Gordon, P.; Karpatkin, S. Protease-activated receptor 1 (PAR-1) is required and rate-limiting for thrombin-enhanced experimental pulmonary metastasis. Blood 1998, 92, 3694–3700. [Google Scholar] [CrossRef]
- Bromberg, M.E.; Bailly, M.A.; Konigsberg, W.H. Role of protease-activated receptor 1 in tumor metastasis promoted by tissue factor. Thromb. Haemost. 2001, 86, 1210–1214. [Google Scholar]
- Sahni, A.; Simpson-Haidaris, P.J.; Sahni, S.K.; Vaday, G.G.; Francis, C.W. Fibrinogen synthesized by cancer cells augments the proliferative effect of fibroblast growth factor-2 (FGF-2). J. Thromb. Haemost. 2007, 6, 176–183. [Google Scholar] [CrossRef]
- Palumbo, J.S.; Talmage, K.E.; Massari, J.V.; La Jeunesse, C.M.; Flick, M.J.; Kombrinck, K.W.; Hu, Z.; Barney, K.A.; Degen, J.L. Tumor cell–associated tissue factor and circulating hemostatic factors cooperate to increase metastatic potential through natural killer cell–dependent and–independent mechanisms. Blood 2007, 110, 133–141. [Google Scholar] [CrossRef]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Ivaska, J. Every step of the way: Integrins in cancer progression and metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed]
- Bouvard, D.; Pouwels, J.; De Franceschi, N.; Ivaska, J. Integrin inactivators: Balancing cellular functions in vitro and in vivo. Nat. Rev. Mol. Cell Biol. 2013, 14, 430–442. [Google Scholar] [CrossRef]
- LaFoya, B.; Munroe, J.A.; Miyamoto, A.; Detweiler, M.A.; Crow, J.J.; Gazdik, T.; Albig, A.R. Beyond the Matrix: The Many Non-ECM Ligands for Integrins. Int. J. Mol. Sci. 2018, 19, 449. [Google Scholar] [CrossRef] [PubMed]
- Moser, M.; Legate, K.R.; Zent, R.; Fässler, R. The Tail of Integrins, Talin, and Kindlins. Science 2009, 324, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Reddy, D.S. Integrins as receptor targets for neurological disorders. Pharmacol. Ther. 2012, 134, 68–81. [Google Scholar] [CrossRef]
- Zheng, Y.; Leftheris, K. Insights into Protein–Ligand Interactions in Integrin Complexes: Advances in Structure Determinations. J. Med. Chem. 2020, 63, 5675–5696. [Google Scholar] [CrossRef]
- Pan, L.; Zhao, Y.; Yuan, Z.; Qin, G. Research advances on structure and biological functions of integrins. Springerplus 2016, 5, 1094. [Google Scholar] [CrossRef]
- Suzuki, S.; Oldberg, A.; Hayman, E.G.; Pierschbacher, M.D.; Ruoslahti, E. Complete amino acid sequence of human vitronectin deduced from cDNA. Similarity of cell attachment sites in vitronectin and fibronectin. EMBO J. 1985, 4, 2519–2524. [Google Scholar] [CrossRef]
- Oldberg, A.; Franzén, A.; Heinegård, D. Cloning and sequence analysis of rat bone sialoprotein (osteopontin) cDNA reveals an Arg-Gly-Asp cell-binding sequence. Proc. Natl. Acad. Sci. USA 1986, 83, 8819–8823. [Google Scholar] [CrossRef] [PubMed]
- Hantgan, R.R.; Stahle, M.C.; Lord, S.T. Dynamic regulation of fibrinogen: Integrin αIIbβ3 binding. Biochemistry 2010, 49, 9217–9225. [Google Scholar] [CrossRef] [PubMed]
- Pierschbacher, M.D.; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar] [CrossRef]
- Plow, E.F.; Pierschbacher, M.D.; Ruoslahti, E.; Marguerie, G.A.; Ginsberg, M.H. The effect of Arg-Gly-Asp-containing peptides on fibrinogen and von Willebrand factor binding to platelets. Proc. Natl. Acad. Sci. USA 1985, 82, 8057–8061. [Google Scholar] [CrossRef] [PubMed]
- Erpenbeck, L.; Schön, M.P. Deadly allies: The fatal interplay between platelets and metastasizing cancer cells. Blood 2010, 115, 3427–3436. [Google Scholar] [CrossRef]
- Terraube, V.; Pendu, R.; Baruch, D.; Gebbink, M.F.B.G.; Meyer, D.; Lenting, P.J.; Denis, C.V. Increased metastatic potential of tumor cells in von Willebrand factor-deficient mice. J. Thromb. Haemost. 2006, 4, 519–526. [Google Scholar] [CrossRef]
- Ludwig, B.S.; Kessler, H.; Kossatz, S.; Reuning, U. RGD-Binding Integrins Revisited: How Recently Discovered Functions and Novel Synthetic Ligands (Re-)Shape an Ever-Evolving Field. Cancers 2021, 13, 1711. [Google Scholar] [CrossRef]
- Polterauer, S.; Grimm, C.; Seebacher, V.; Concin, N.; Marth, C.; Tomovski, C.; Husslein, H.; Leipold, H.; Hefler-Frischmuth, K.; Tempfer, C.; et al. Plasma Fibrinogen Levels and Prognosis in Patients with Ovarian Cancer: A Multicenter Study. Oncol. 2009, 14, 979–985. [Google Scholar] [CrossRef]
- Seebacher, V.; Polterauer, S.; Grimm, C.; Husslein, H.; Leipold, H.; Hefler-Frischmuth, K.; Tempfer, C.; Reinthaller, A.; Hefler, L. The prognostic value of plasma fibrinogen levels in patients with endometrial cancer: A multi-centre trial. Br. J. Cancer 2010, 102, 952–956. [Google Scholar] [CrossRef]
- Tang, L.; Liu, K.; Wang, J.; Wang, C.; Zhao, P.; Liu, J. High preoperative plasma fibrinogen levels are associated with distant metastases and impaired prognosis after curative resection in patients with colorectal cancer. J. Surg. Oncol. 2010, 102, 428–432. [Google Scholar] [CrossRef]
- Tanaka, N.; Kikuchi, E.; Matsumoto, K.; Hayakawa, N.; Ide, H.; Miyajima, A.; Nakamura, S.; Oya, M. Prognostic value of plasma fibrinogen levels in patients with localized upper tract urothelial carcinoma. BJU Int. 2012, 111, 857–864. [Google Scholar] [CrossRef]
- Pichler, M.; Hutterer, G.C.; Stojakovic, T.; Mannweiler, S.; Pummer, K.; Zigeuner, R. High plasma fibrinogen level represents an independent negative prognostic factor regarding cancer-specific, metastasis-free, as well as overall survival in a European cohort of non-metastatic renal cell carcinoma patients. Br. J. Cancer 2013, 109, 1123–1129. [Google Scholar] [CrossRef]
- Casamassima, A.; Picciariello, M.; Quaranta, M.; Berardino, R.; Ranieri, C.; Paradiso, A.; Lorusso, V.; Guida, M. C-Reactive Protein: A Biomarker of Survival in Patients with Metastatic Renal Cell Carcinoma Treated with Subcutaneous Interleukin-2 Based Immunotherapy. J. Urol. 2005, 173, 52–55. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Yan, D.; Liu, Y.; Huang, P.; Cui, H. The Roles of Integrin α5β1 in Human Cancer. Onco. Targets Ther. 2020, 13, 13329–13344. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhang, G.; Zhang, Y.; Cui, X.; Wang, S.; Gao, S.; Wang, Y.; Liu, Y.; Bae, J.H.; Yang, W.-H.; et al. Fibrinogen Alpha Chain Knockout Promotes Tumor Growth and Metastasis through Integrin–AKT Signaling Pathway in Lung Cancer. Mol. Cancer Res. 2020, 18, 943–954. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Sahni, S.K.; Sahni, A.; Francis, C.W. Fibrinogen regulates the expression of inflammatory chemokines through NF-kappaB activation of endothelial cells. Thromb. Haemost. 2004, 92, 858–866. [Google Scholar] [CrossRef]
- Abe, Y.; Bui-Thanh, N.-A.; Ballantyne, C.M.; Burns, A.R. Extra domain A and type III connecting segment of fibronectin in assembly and cleavage. Biochem. Biophys. Res. Commun. 2005, 338, 1640–1647. [Google Scholar] [CrossRef]
- To, W.S.; Midwood, K.S. Plasma and cellular fibronectin: Distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair 2011, 4, 21. [Google Scholar] [CrossRef]
- Schäfer, M.; Werner, S. Cancer as an overhealing wound: An old hypothesis revisited. Nat. Rev. Mol. Cell Biol. 2008, 9, 628–638. [Google Scholar] [CrossRef]
- Knowles, L.M.; Gurski, L.A.; Engel, C.; Gnarra, J.R.; Maranchie, J.K.; Pilch, J. Integrin αvβ3 and Fibronectin Upregulate Slug in Cancer Cells to Promote Clot Invasion and Metastasis. Cancer Res 2013, 73, 6175–6184. [Google Scholar] [CrossRef]
- Malik, G.; Knowles, L.M.; Dhir, R.; Xu, S.; Yang, S.; Ruoslahti, E.; Pilch, J. Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer Res. 2010, 70, 4327–4334. [Google Scholar] [CrossRef] [PubMed]
- Pilch, J.; Habermann, R.; Felding-Habermann, B. Unique ability of integrin alpha(v)beta 3 to support tumor cell arrest under dynamic flow conditions. J. Biol. Chem. 2002, 277, 21930–21938. [Google Scholar] [CrossRef] [PubMed]
- Echtler, K.; Konrad, I.; Lorenz, M.; Schneider, S.; Hofmaier, S.; Plenagl, F.; Stark, K.; Czermak, T.; Tirniceriu, A.; Eichhorn, M.; et al. Platelet GPIIb supports initial pulmonary retention but inhibits subsequent proliferation of melanoma cells during hematogenic metastasis. PLoS ONE 2017, 12, e0172788. [Google Scholar] [CrossRef] [PubMed]
- Caccavari, F.; Valdembri, D.; Sandri, C.; Bussolino, F.; Serini, G. Integrin signaling and lung cancer. Cell Adhes. Migr. 2010, 4, 124–129. [Google Scholar] [CrossRef]
- Esmon, C.T.; Owen, W.G. The discovery of thrombomodulin. J. Thromb. Haemost. 2004, 2, 209–213. [Google Scholar] [CrossRef]
- Mehic, D.; Colling, M.; Pabinger, I.; Gebhart, J. Natural anticoagulants: A missing link in mild to moderate bleeding tendencies. Haemophilia 2021, 27, 701–709. [Google Scholar] [CrossRef]
- Amiral, J.; Seghatchian, J. Revisiting the activated protein C-protein S-thrombomodulin ternary pathway: Impact of new understanding on its laboratory investigation. Transfus. Apher. Sci. 2019, 58, 538–544. [Google Scholar] [CrossRef]
- Dahm, A.E.; Sandset, P.M.; Rosendaal, F.R. The association between protein S levels and anticoagulant activity of tissue factor pathway inhibitor type 1. J. Thromb. Haemost. 2008, 6, 393–395. [Google Scholar] [CrossRef]
- Hosaka, Y.; Higuchi, T.; Tsumagari, M.; Ishii, H. Inhibition of invasion and experimental metastasis of murine melanoma cells by human soluble thrombomodulin. Cancer Lett. 2000, 161, 231–240. [Google Scholar] [CrossRef]
- Chang, Y.-J.; Cheng, Y.-W.; Lin, R.-K.; Huang, C.-C.; Chen, W.T.-L.; Ke, T.-W.; Wei, P.-L. Thrombomodulin Influences the Survival of Patients with Non-Metastatic Colorectal Cancer through Epithelial-To-Mesenchymal Transition (EMT). PLoS ONE 2016, 11, e0160550. [Google Scholar] [CrossRef]
- Horowitz, N.A.; Blevins, E.A.; Miller, W.M.; Perry, A.R.; Talmage, K.E.; Mullins, E.S.; Flick, M.J.; Queiroz, K.C.S.; Shi, K.; Spek, C.A.; et al. Thrombomodulin is a determinant of metastasis through a mechanism linked to the thrombin binding domain but not the lectin-like domain. Blood 2011, 118, 2889–2895. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Uwagawa, T.; Shiba, H.; Shimada, Y.; Horiuchi, T.; Saito, N.; Yanaga, K. Recombinant thrombomodulin suppresses tumor growth of pancreatic cancer by blocking thrombin-induced PAR1 and NF-κB activation. Surgery 2017, 161, 1675–1682. [Google Scholar] [CrossRef] [PubMed]
- Hanly, A.; Hayanga, A.; Winter, D.; Bouchier-Hayes, D. Thrombomodulin: Tumour biology and prognostic implications. Eur. J. Surg. Oncol. (EJSO) 2005, 31, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Loghmani, H.; Conway, E.M. Exploring traditional and nontraditional roles for thrombomodulin. Blood 2018, 132, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Key, N.S.; Khorana, A.A.; Kuderer, N.M.; Bohlke, K.; Lee, A.Y.; Arcelus, J.I.; Wong, S.L.; Balaban, E.P.; Flowers, C.R.; Francis, C.W.; et al. Venous Thromboembolism Prophylaxis and Treatment in Patients with Cancer: ASCO Clinical Practice Guideline Update. J. Clin. Oncol. 2020, 38, 496–520. [Google Scholar] [CrossRef] [PubMed]
- Haist, M.; Stege, H.; Pemler, S.; Heinz, J.; Fleischer, M.I.; Graf, C.; Ruf, W.; Loquai, C.; Grabbe, S. Anticoagulation with Factor Xa Inhibitors Is Associated with Improved Overall Response and Progression-Free Survival in Patients with Metastatic Malignant Melanoma Receiving Immune Checkpoint Inhibitors—A Retrospective, Real-World Cohort Study. Cancers 2021, 13, 5103. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Kusche-Gullberg, M. Heparan Sulfate: Biosynthesis, Structure, and Function. Int. Rev. Cell Mol. Biol. 2016, 325, 215–273. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Obser, T.; Bauer, A.T.; Heyes, M.; Starzonek, S.; Zulal, M.; Opitz, K.; Ott, L.; Riethdorf, S.; et al. Heparan sulfate dependent binding of plasmatic von Willebrand factor to blood circulating melanoma cells attenuates metastasis. Matrix Biol. 2022, 111, 76–94. [Google Scholar] [CrossRef]
- von Palubitzki, L.; Wang, Y.; Hoffmann, S.; Vidal-y-Sy, S.; Zobiak, B.; Failla, A.V.; Gorzelanny, C. Differences of the tumour cell glycocalyx affect binding of capsaicin-loaded chitosan nanocapsules. Sci. Rep. 2020, 10, 22443. [Google Scholar] [CrossRef]
- Vallet, S.D.; Berthollier, C.; Ricard-Blum, S. The glycosaminoglycan interactome 2.0. Am. J. Physiol. Physiol. 2022, 322, C1271–C1278. [Google Scholar] [CrossRef]
- Vallet, S.D.; Clerc, O.; Ricard-Blum, S. Glycosaminoglycan–Protein Interactions: The First Draft of the Glycosaminoglycan Interactome. J. Histochem. Cytochem. 2020, 69, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Gesslbauer, B.; Derler, R.; Handwerker, C.; Seles, E.; Kungl, A.J. Exploring the glycosaminoglycan-protein interaction network by glycan-mediated pull-down proteomics. Electrophoresis 2016, 37, 1437–1447. [Google Scholar] [CrossRef]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Rudd, T.R.; Preston, M.D.; Yates, E.A. The nature of the conserved basic amino acid sequences found among 437 heparin binding proteins determined by network analysis. Mol. Biosyst. 2017, 13, 852–865. [Google Scholar] [CrossRef] [PubMed]
- O′Donnell, J.S.; O′Sullivan, J.M.; Preston, R.J.S. Advances in understanding the molecular mechanisms that maintain normal haemostasis. Br. J. Haematol. 2019, 186, 24–36. [Google Scholar] [CrossRef]
- Petitou, M.; Casu, B.; Lindahl, U. 1976–1983, a critical period in the history of heparin: The discovery of the antithrombin binding site. Biochimie 2003, 85, 83–89. [Google Scholar] [CrossRef]
- Weiss, R.J.; Spahn, P.N.; Toledo, A.G.; Chiang, A.W.T.; Kellman, B.P.; Li, J.; Benner, C.; Glass, C.K.; Gordts, P.L.S.M.; Lewis, N.E.; et al. ZNF263 is a transcriptional regulator of heparin and heparan sulfate biosynthesis. Proc. Natl. Acad. Sci. USA 2020, 117, 9311–9317. [Google Scholar] [CrossRef]
- Nadir, Y.; Brenner, B.; Gingis-Velitski, S.; Levy-Adam, F.; Ilan, N.; Zcharia, E.; Nadir, E.; Vlodavsky, I. Heparanase induces tissue factor pathway inhibitor expression and extracellular accumulation in endothelial and tumor cells. Thromb. Haemost. 2008, 99, 133–141. [Google Scholar] [CrossRef]
- Nadir, Y. Effect of Heparanase and Heparan Sulfate Chains in Hemostasis. Semin. Thromb. Hemost. 2021, 47, 254–260. [Google Scholar] [CrossRef]
- Valentin, S.; Larnkjaer, A.; Østergaard, P.; Nielsen, J.I.; Nordfang, O. Characterization of the binding between tissue factor pathway inhibitor and glycosaminoglycans. Thromb. Res. 1994, 75, 173–183. [Google Scholar] [CrossRef]
- Wesselschmidt, R.; Likert, K.; Huang, Z.; MacPhail, L.; Broze, G.J. Structural requirements for tissue factor pathway inhibitor interactions with factor Xa and heparin. Blood Coagul. Fibrinolysis 1993, 4, 619–661. [Google Scholar] [CrossRef] [PubMed]
- Fredenburgh, J.C.; Leslie, B.A.; Stafford, A.R.; Lim, T.; Chan, H.H.; Weitz, J.I. Zn2+ Mediates High Affinity Binding of Heparin to the αC Domain of Fibrinogen. J. Biol. Chem. 2013, 288, 29394–29402. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, V.; Weber, H.; Wang, Y.; Schlesinger, M.; Gorzelanny, C.; Bendas, G. Exostosin 1 Knockdown Induces Chemoresistance in MV3 Melanoma Cells by Upregulating JNK and MEK/ERK Signaling. Int. J. Mol. Sci. 2023, 24, 5452. [Google Scholar] [CrossRef]
- Zhang, W.; Swanson, R.; Xiong, Y.; Richard, B.; Olson, S.T. Antiangiogenic antithrombin blocks the heparan sulfate-dependent binding of proangiogenic growth factors to their endothelial cell receptors: Evidence for differential binding of antiangiogenic and anticoagulant forms of antithrombin to proangiogenic heparan sulfate domains. J. Biol. Chem. 2006, 281, 37302–37310. [Google Scholar] [PubMed]
- Oelschläger, C.; Römisch, J.; Staubitz, A.; Stauss, H.; Leithäuser, B.; Tillmanns, H.; Hölschermann, H. Antithrombin III inhibits nuclear factor kappaB activation in human monocytes and vascular endothelial cells. Blood 2002, 99, 4015–4020. [Google Scholar] [CrossRef]
- Luengo-Gil, G.; Calvo, M.I.; Martín-Villar, E.; Águila, S.; Bohdan, N.; Antón, A.I.; Espín, S.; de la Peña, F.A.; Vicente, V.; Corral, J.; et al. Antithrombin controls tumor migration, invasion and angiogenesis by inhibition of enteropeptidase. Sci. Rep. 2016, 6, 27544. [Google Scholar] [CrossRef]
- Desch, A.; Strozyk, E.A.; Bauer, A.T.; Huck, V.; Niemeyer, V.; Wieland, T.; Schneider, S.W. Highly invasive melanoma cells activate the vascular endothelium via an MMP-2/integrin αvβ5-induced secretion of VEGF-A. Am. J. Pathol. 2012, 181, 693–705. [Google Scholar] [CrossRef]
- John, A.; Robador, J.R.; Vidal, Y.S.S.; Houdek, P.; Wladykowski, E.; Gunes, C.; Bolenz, C.; Schneider, S.W.; Bauer, A.T.; Gorzelanny, C. Urothelial Carcinoma of the Bladder Induces Endothelial Cell Activation and Hypercoagulation. Mol. Cancer Res. 2020, 18, 1099–1109. [Google Scholar] [CrossRef]
- Obermeier, H.L.; Riedl, J.; Ay, C.; Koder, S.; Quehenberger, P.; Bartsch, R.; Kaider, A.; Zielinski, C.C.; Pabinger, I. The role of ADAMTS-13 and von Willebrand factor in cancer patients: Results from the Vienna Cancer and Thrombosis Study. Res. Pr. Thromb. Haemost. 2019, 3, 503–514. [Google Scholar] [CrossRef]
- Bankó, P.; Lee, S.Y.; Nagygyörgy, V.; Zrínyi, M.; Chae, C.H.; Cho, D.H.; Telekes, A. Technologies for circulating tumor cell separation from whole blood. J. Hematol. Oncol. 2019, 12, 48. [Google Scholar] [CrossRef]
- Ferreira, M.M.; Ramani, V.C.; Jeffrey, S.S. Circulating tumor cell technologies. Mol. Oncol. 2016, 10, 374–394. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Fu, R.; Wang, S.; Zhang, T.; Tian, X.; Yang, D. Clinical Utility of Circulating Tumor Cells in Patients with Esophageal Cancer. Front. Oncol. 2022, 12, 1080. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, Z.; Wang, B. Size- and deformability-based isolation of circulating tumor cells with microfluidic chips and their applications in clinical studies. AIP Adv. 2018, 8, 120701. [Google Scholar] [CrossRef]
- Hu, Z.; Xu, J.; Cheng, J.; McMichael, E.; Yu, L.; Iii, W.E.C. Targeting tissue factor as a novel therapeutic oncotarget for eradication of cancer stem cells isolated from tumor cell lines, tumor xenografts and patients of breast, lung and ovarian cancer. Oncotarget 2017, 8, 1481–1494. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).