
Targeting the Interplay of Independent Cellular Pathways and Immunity: A Challenge in Cancer Immunotherapy

 , ,
, ,  ,
,  and
and
Abstract
Simple Summary
Abstract
1. Introduction
2. Oxidative Stress in Cancer Immunotherapy
2.1. Oxidative Stress in the Tumor Microenvironment
2.2. Dynamic ROS Levels in Tumor Immune Microenvironment
2.3. ROS in Cancer Immunotherapy
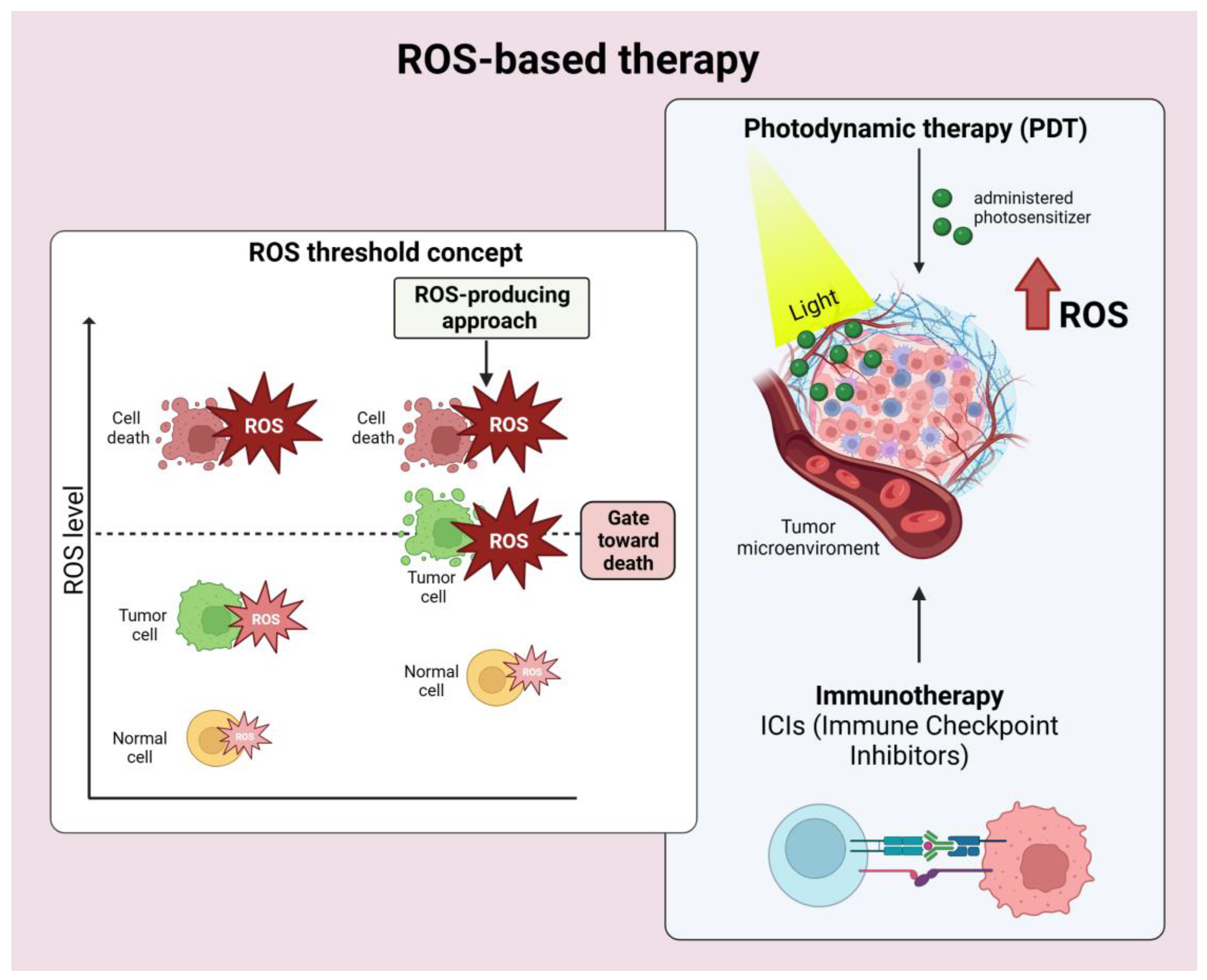
2.4. ROS-Based Cancer Immunotherapy
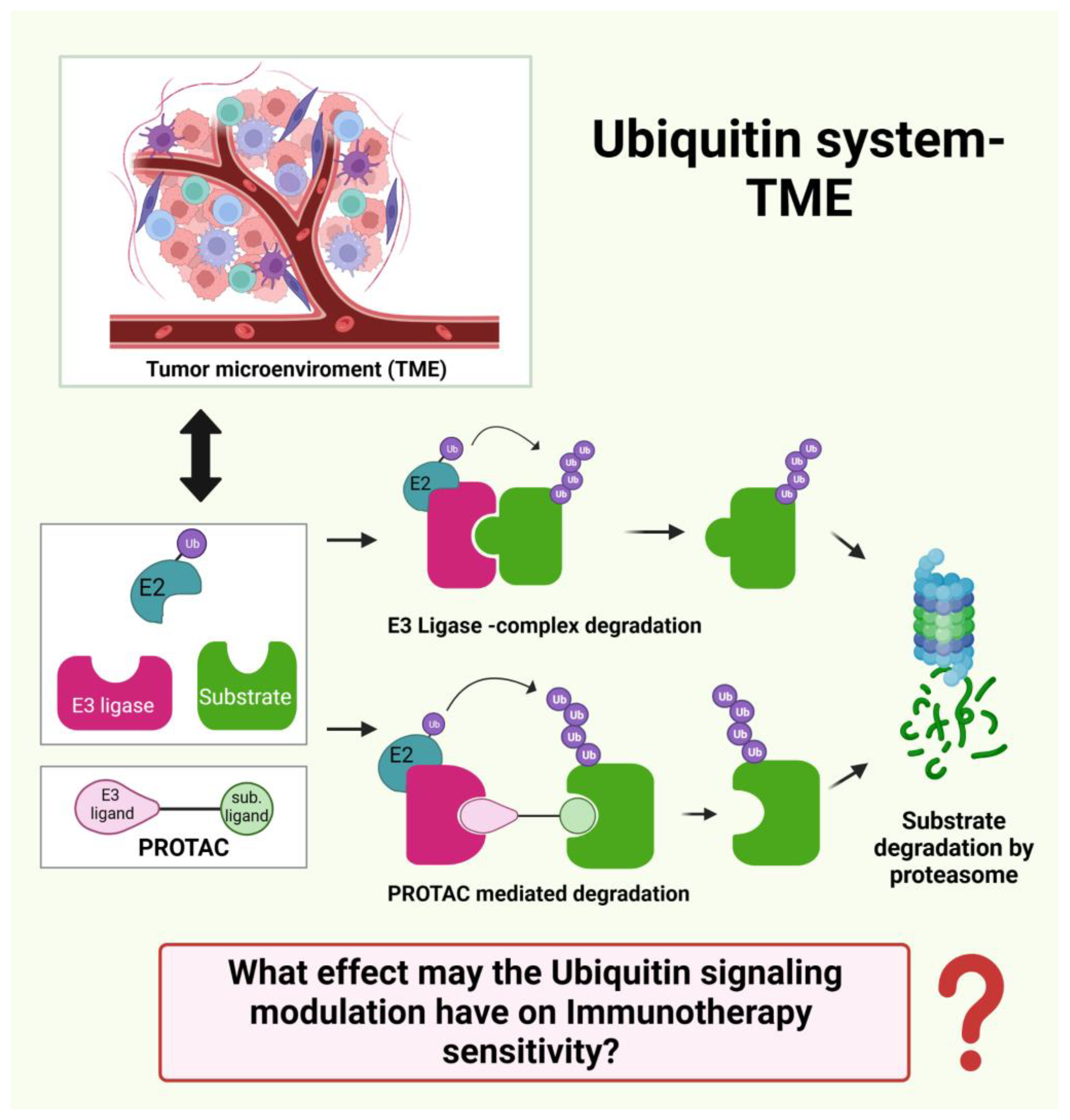
3. Ubiquitin Ligases in Cancer Immunotherapy
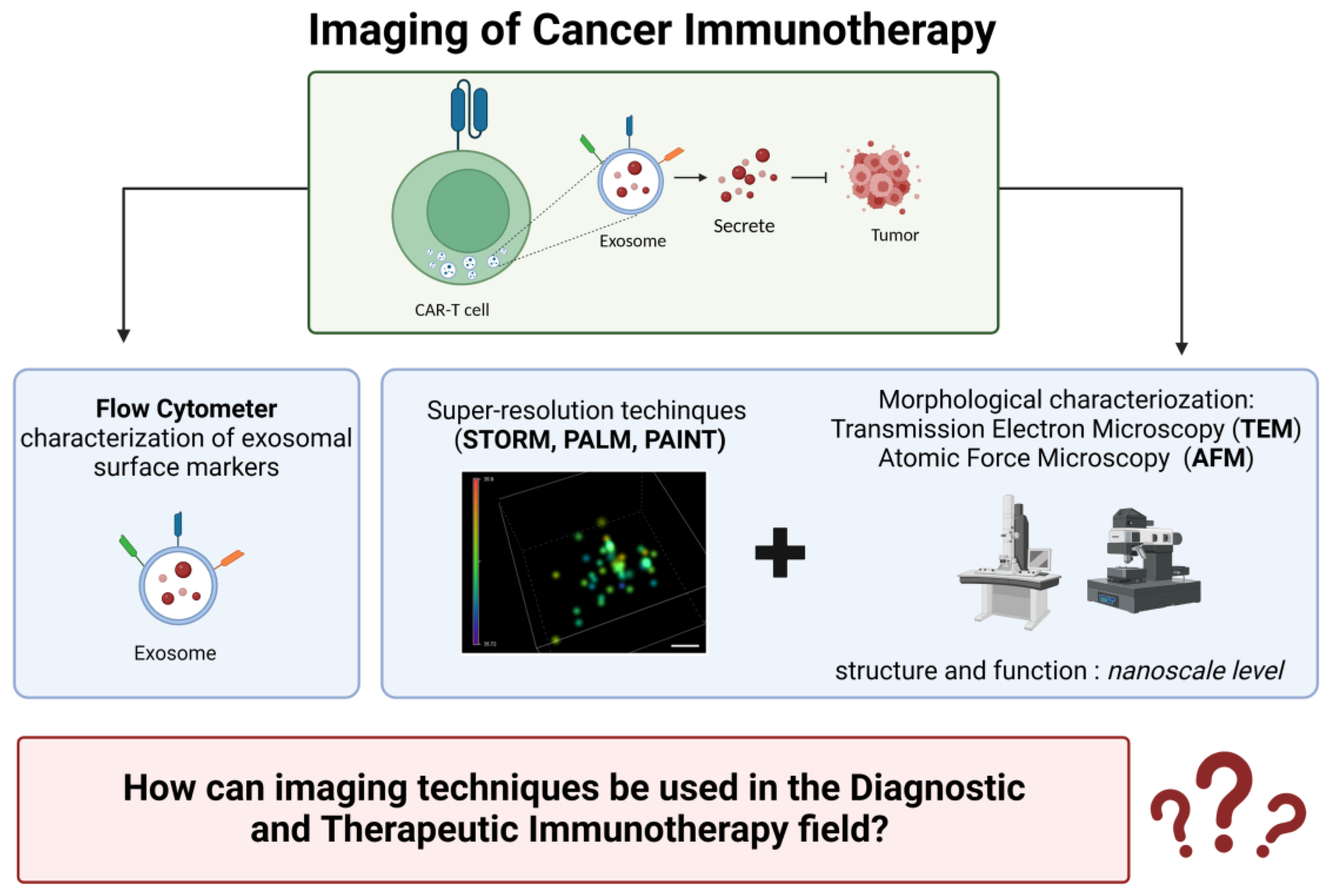
4. Imaging in Cancer Immunotherapy
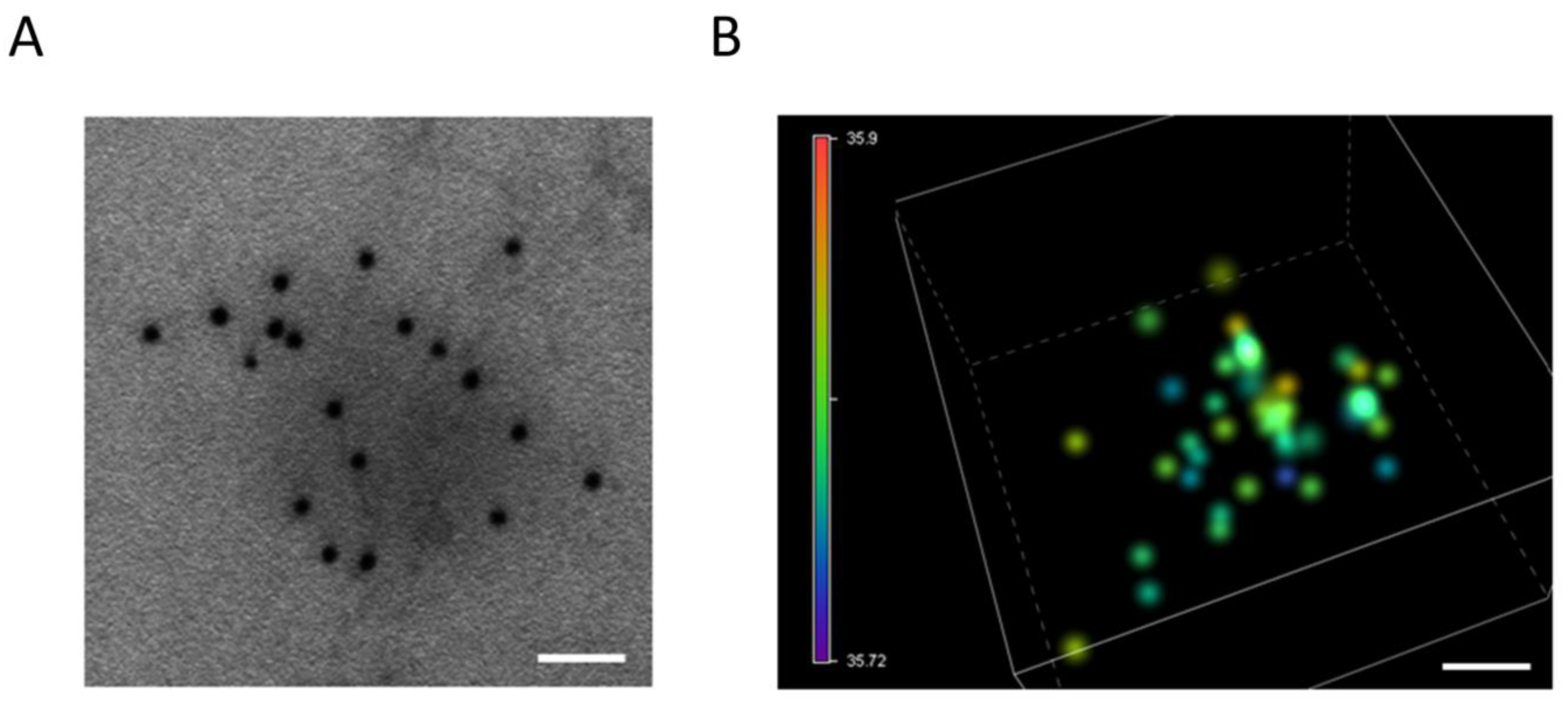
4.1. Electron Microscopy
4.2. Atomic Force Microscopy (AFM)
4.3. Single-Molecule Localization Microscopy (SMLM)
5. Combination Cancer Immunotherapy and New Immunomodulatory Targets
5.1. Selected Clinical Trials Providing Information on ICI Combination with Chemotherapy in Different Tumor Types (Table 1)
5.2. Selected Clinical Trials Providing Information on ICI Combination with PARP Inhibitors in Different Tumor Types (Table 2)
5.3. Selected Clinical Trials Providing Information on ICI Combination with Radionuclide in Different Tumor Types (Table 3)
5.4. Selected Clinical Trials Providing Information on ICI Combination with Vaccines in Different Tumor Types (Table 4)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Combined Therapy | Anti-PD-1/PD-L1 | Clinical Phase and Population | N of Patients | Result | Conclusion | NCT Number | Reference |
|---|---|---|---|---|---|---|---|---|
| LUNG non squamous | Pembrolizumab + platinum-pemetrexed | Pembrolizumab | III | 616 | Median OS: 22 m vs. 10.7 m (HR 0.56, CI95 [0.45 × 100.70]) ORR: 47.6% vs. 18.9% Subsequnet ICI: 53.9% | Long-lasting benefit in first-line setting | KEYNOTE 189 | [83] |
| LUNG advanced squamous NSCLC | Pembrolizumab/Carbo/Pacli or nab-Pac | Pembrolizumab | III; untreated metastatic | 559 | PFS (months) 6.4 HR (95%CI) 0.56 (0.45–0.70) OS (months) 15.9 HR (95%CI) 0.64 (0.49–0.85) | PFS and OS outcomes did not change based on taxane used | KEYNOTE 407 | [84] |
| LUNG non squamous | Atezolizumab + platinum-pemetrexed | Atezolizumab | III | 578 | Median PFS:7.6 m vs. 5.2 m (HR 0.596, CI95 [0.494 × 100.719], p < 0.0001) ORR: 46.9% vs. 32.2% Subsequnet ICI: 37.1% | Better ORR (47% vs. 32%) and longer median duration of response (10.5 m vs. 7.2 m) compared with chemotherapy alone. PFS benefit regardless of PD-L1 status | IMpower132 | [85] |
| LUNG advanced squamous NSCLC | Atezolizumab + carboplatin-nab-paclitaxel | Atezolizumab | III | 1021 | Median OS: 14.2 m vs. 13.5 m, NS Subsequnet ICI: 43.2% | Prolonged median OS in the PD-L1 high subgroup (23.4 m vs 10.2 m, HR 0.48, CI95 [0.29 × 100.81]) | IMpower 131 | [86] |
| BREAST | Atezolizumab PD-L1 + nab-PTX | Atezolizumab | III; previously untreated metastatic TNBC | 902 | PFS: 7.2 months in the atezolizumab + nab-PTX group vs. 5.5 months in the placebo + nab-PTX group. In PD-L1 + tumors, PFS: 7.5 and 5.0 months, respectively. OS: 21.3 months in the atezolizumab + nab-PTX group vs. 17.6 months in the placebo + nab-PTX group; In PD-L1 + tumors, OS: 25.0 and 15.5 months, respectively. | Clinically meaningful OS benefit observed in PD-L1+ pts (7.5-months median OS improvement). A + nP safe and tolerable | NCT02425891 (IMpassion130) | [88] |
| BREAST | Atezolizumab PD-L1 + nab-PTX-anthracycline as neoadjuvant for early-stage TNBC | Atezolizumab | III; Early TNBC | 455 | The pCR was documented with 58% in the atezolizumab + chemotherapy group versus 41% in the placebo + chemotherapy group; in the PD-L1 + population, the pCR was 69% and 49%, respectively. | Significantly improved pathological complete response rates with an acceptable safety profile. | NCT03197935 IMpassion031 | [89] |
| BREAST | Atezolizumab PD-L1 + cobimetinib and PTX | Atezolizumab | II; locally advanced or metastatic TNBC | 106 | median PFS 5.5 months in the cobimetinib + PTX group versus 3.8 months in the placebo + PTX group. | Cobimetinib +PTX increased PFS and ORR. A + cobi + taxane did not increase ORR. Safety profiles. | NCT02322814 (COLET) | [90] |
| BREAST | Durvalumab + Anthracycline/ taxane | Durvalumab | II | 117 | PCR (53.4%, Durvalumab; 44.2%, placebo), OR = 1.45. Durvalumab effect only in cohort (pCR 61.0% vs. 41.4%, OR = 2.22) 0.47% thyroid dysfunction. | The combination increases pCR rate in durvalumab alone. Increased pCR with higher sTILs. Trend for increased PCR rates in PD-L1C tumors. | NCT02685059 | [91] |
| BREAST HER-2-TARGETED | Pembrolizumab+ Trastuzumab | Pembrolizumab | I/II | 58 | OR (15%, PD-1+; 0%, PD-1−). fatigue 21% | Safe; with activity and durable clinical benefit in PD-L1+/HER2+, trastuzumab-resistant, advanced BC patients. | NCT02129556 PANACEA | [92] |
| PANCREAS | Pembrolizumab + Gemcitabine + Nab-paclitaxel | Pembrolizumab | Ib/II; Metastatic | 17 | PFS: 9.1 months. OS: 15.0 months | 70.6% (12/17) | NCT02331251 | [94] |
| PANCREAS | Ipilimumab + Gemcitabine | Ipilimumab | Ib | 21 | 21 ORR: 14% (3/21). PFS: 2.78 months. OS: 6.90 months | Safe and tolerable regimen for PDAC with a similar response rate to gemcitabine alone. 76.2% (16/21) elevated ALT, diarrhea, mostly hematologic toxicity | NCT01473940 | [95] |
| PANCREAS | Nivolumab + Gemcitabine + Nab-paclitaxel | I; Locally advanced or metastatic | 50 | ORR: 18%. PFS: 5.5 months. OS: 9.9 months | 36.0% (18/50; peripheral neuropathy, hypokalemia, diarrhea, increased AST/ALT, mostly hematologic toxicity) | NCT02309177 | [96] | |
| GASTRIC | Nivolumab + SOX and nivolumab + Cape/OX) | Nivolumab | II | 40 | median PFS 9.7 months and 10.6 months (5.6–12.5) | well tolerated. encouraging efficacy for unresectable advanced or recurrent HER2-negative G/GEJ cancer. | ATTRACTION-4 NCT02746796 | [97] |
| GASTRIC | Nivolumab + Cape/Oxa or Folfox | Nivolumab | III | 1581 | Significant improvements in OS (hazard ratio [HR] 0.71 [98.4% CI 0.59–0.86]; p < 0.0001) and PFS (HR 0.68 [98% CI 0.56–0.81] | Nivolumab is the first PD-1 inhibitor to show superior OS, PFS benefit; acceptable safety profile in combination | CheckMate 649 NCT02872116 | [98] |
| GASTRIC | Pembrolizumab + chemotherapy (DDP, 5FU or Cape) | Pembrolizumab | II; previously untreated advanced gastric/gastroesophageal junction adenocarcinoma | 56 | ORR 60.0% [95% confidence interval (CI), 38.7–78.9] (combination therapy) and 25.8% (95% CI 11.9–44.6) (monotherapy) | Pembrolizumab: antitumor activity; well tolerated. in combination with chemotherapy in patients | KEYNOTE-059 NCT02335411 | [99] |
| PROSTATE | Nivolumab + docetaxel | Nivolumab | II mCRPC in progression after second-generation hormonal therapy and CT-naïve | 41 | ORR was 36.8% in patients with measurable disease, PSA RR was 46.3%, and rPFS was 8.2 months | Clinical activity in patients with chemotherapy-naïve mCRPC. Safety consistent with the individual components. | CheckMate 9KD; NCT03338790 | [102] |
| PROSTATE | Pembrolizumab + docetaxel | Pembrolizumab | Ib/II | 104 mCRPC after second-generation hormonal therapy and CT-naïve | ORR was 18%, PSA RR was 28%, rPFS was 8.3 months, and OS was 20.4 months | KEYNOTE-365 | [103] | |
| PROSTATE | Pembrolizumab Plus Docetaxel and Prednisone | Pembrolizumab | Ib/II | 104 mCRPC | PSA response rate 34%; ORR (RECIST v1.1) was 23%. Median rPFS and OS 8.5 months and 20.2 months, respectively. | Antitumor activity in chemotherapy-naïve patients with mCRPC treated with abiraterone or enzalutamide for mCRPC. Safety consistent with individual agent profiles | KEYNOTE-365, (Cohort B); NCT02861573 | [104] |
| OVARIAN | Nivolumab + Bevacizumab (ICB + antiangiogenesis) | Nivolumab | II; Recurrent EOC (PSOC + PROC) PSOC PROC | 38 | ORR %: 40.0% in platinum-sensitive and 16.7% (95% CI 3.6–41.4%) platinum-resistant; mPFS, months: 8.1. | activity in patients with relapsed ovarian cancer; greater activity in platinum-sensitive setting, limited in platinum-resistant patients | NIVO-BEV | [105] |
| Tumor Type | Combined Therapy | Anti-PD-1/PD-L1 | Clinical Phase and Population | N of Patients | Result | Conclusion | NCT Number | Reference |
|---|---|---|---|---|---|---|---|---|
| PROSTATE | Pembrolizumab + Olaparib (Anti-PD1/anti-PDL1 + PARPi) | Pembrolizumab | Ib/II mCRPC progressed to docetaxel and second-generation hormonal therapies | 84 | PSA RR of 9%, ORR of 8.3%, rPFS of 4 months and OS of 14 months | Additional follow-up: combination still active in docetaxel-pretreated pts. Combination safety consistent with individual profiles of each agent. | KEYNOTE-365 (cohort A) | [106] |
| PROSTATE | Durvalumab + Olaparib (Anti-PD1/anti-PDL1 + PARPi) | Durvalumab | II Metastatic mCRPC after progression to abiraterone and/or enzalutamide | 17 | rPFS 16.1 months with 53% serological or radiographic response. rPFS 16.1 months in DNA repair genes alterations and ORR of 83%. Biomarkers: MLH1, PMS2, MSH2, MSH6 | Acceptable toxicity and efficacy, particularly in men with DDR abnormalities. Identified biomarkers: MLH1, PMS2, MSH2, MSH6 | NCT02484404 | [107] |
| PROSTATE | Nivolumab + ipilimumab (Anti-PD1 + anti-CTLA4) | Nivolumab + ipilimumab | II Metastatic mCRPC | 7/44 (16%) | 25% ORR in pre-chemotherapy cohort 1 and 10% ORR in post-chemotherapy cohort2. 5.5 and 3.8 months median rPFS and 19.0 and 15.2 months median OS. | Identified potential biomarkers of response: BRCA2, FANCA, ATRX, ERCC3, MLH1, XRCC2 | NCT02985957 | [108] |
| BREAST | Pembrolizumab PD-1 + niraparib | Pembrolizumab | II advanced or metastatic TNBC | 55 | ORR 21% in the pembrolizumab + niraparib group; 47%. in BRCA -mutated tumors. | higher response rates in BC with tumor BRCA mutations | NCT02657889 | [109] |
| BREAST | Camrelizumab + apatinib | Camrelizumab | II advanced TNBC | 40 | ORR 43.3% in the continuous dosing cohort, no objective response in the intermittent dosing cohort. | ORR dramatically higher than previously reported ORR by anti-PD-1/PD-L1 antibody or apatinib monotherapy. Favorable therapeutic effects and a manageable safety profile. | NCT03394287 | [110] |
| OVARIAN | Anti-PD1 (pembrolizumab) + ICB + PARPi (niraparib) | Pembrolizumab | Metastatic OC I/II | 20/39 (51%) | Mutational signature 3 correlates with clinical benefit. mutations assessed in BRCA1, BRCA2 | Response dependent on interactions of exhausted CD8 + T cells and PD-L1 + macrophages and PD-L1 + tumor cells | NCT02657889 | [112] |
| OVARIAN | ICB + PARPi Pembrolizumab + Niraparib | Pembrolizumab | I-II | 62, PR-ROC | ORR 25% DCR 68% In BRCAm: ORR 45%, DCR 73% | Grade 3 TRAE occurred in 16 patients (30%), anemia in 21%, thrombocytopenia (9%) | TOPACIO/Keynote-162 NCT02657889 | [11] |
| OVARIAN | ICB + PARPi Durvalumab and olaparib | Durvalumab | II An uMbrella Study of BIomarker-driven Targeted Therapy | 70 PR-ROC | 77% overall response rate (ORR) in gBRCA-mutant patients and a 34% ORR in platinum- sensitive BRCA- wild- type patients (n = 23/32) | Clinical utility with biomarker-driven targeted therapy. All treatment combinations were manageable, and without unexpected toxicities. | NCT03699449 (AMBITION) | [114] |
| OVARIAN | ICB + PARPi + anti-angiogenesis. Triplet combination Durvalumab + bevacizumab + olaparib | Durvalumab | I/II Recurrent PSOC: BRCAwt BRCAm | 32 PS ROC with a germline BRCA1/2 mutation | ORR%: BRCAwt: 31.3 BRCAm: 72 DCR of 77.4% (90% CI 61.7–88.9) versus 28.1% (90% CI 15.5–43.9), respectively. The ORR was 87.1% (95% CI 70.2–96.4) versus 34.4% (95% CI 18.6–53.2). The median PFS was 14.7 months (95% CI 10.0–18.1) | Triplet superior over the doublet for all the endpoints. Genomic instability did not correlate with response | MEDIOLA NCT02734004 | [115] |
| OVARIAN | Dostarlimab + niraparib + bevacizumab (Triplet combination) | Dostarlimab | II | 41 Patients PR ROC | PFS months (95% CI) 7.6 (4.2–10.6) ORR (%): 17.9 Partial responce | ORR of 47.5% and a clinical benefit rate of 95.0%. Durable responses of longer than 12 months were observed in 25% | OPAL trial NCT03574779 | [116] |
| Tumor Type | Combined Therapy | Anti-PD-1/PD-L1 | Clinical Phase and Population | N of Patients | Result | Conclusion | NCT Number | Reference |
|---|---|---|---|---|---|---|---|---|
| PROSTATE | Ipilimumab ± radiotherapy | Ipilimumab | Phase I/II, completed mCRPC | 71 | 16% patients (8/50) had about 50% PSA decline and 1/28 complete response. Grade 3–4 colitis and hepatitis and 1 treatment-related death. | Clinical antitumor activity with disease control and manageable AEs | NCT00323882 | [117] |
| PROSTATE | Atezolizumab + Radium 223 | Atezolizumab | Ib mCRPC | 44 | ORR of 6.8%, PSA RR of 4.5%, and rPFS of 3 months | Low clinical response | NCT02814669 | [118] |
| GMB | Durvalumab and/or bevacizumab + radiotherapy | Durvalumab | II | 158 Newly diagnosed MGMT methylated Glioblastoma and recurrent GMB | cohort A (n = 40, treatment well-tolerated. mOS:15.1 months, 8 still alive (with survival ranging SR 15.7–34.9 months). cohort B (n = 30): OS 59.0% and 44.4% for 6, and 12 months. Post-treatment:partial response in 13.3% of the cohort population (n = 4) and none of the patients experienced high-grade treatment-related adverse events (grade 4) | Treatment with RT was well-tolerated. Adding durva to BEV did not improve the outcome of durvalumab alone. | NCT02336165 | [119] |
| Type of Tumor | Combined Therapy | Anti-PD-1/PD-L1 | Clinical Phase and Population | N of Patients | Result | Conclusion | NCT Number | Reference |
|---|---|---|---|---|---|---|---|---|
| PANCREAS | GVAX + Ipilimumab after FOLFIRINOX vs. FOLFIRINOX continuation | Ipilimumab | II Metastatic | 40 vs. 42 | PFS: 2.4 mo vs. 5.55 mo. OS: 9.38 mo vs. 14.7 mo) 41.0% (16/39; adrenal insufficiency, hypophysitis, rash, diarrhea) | GVAX and ipilimumab maintenance therapy did not improve OS over continuation of chemotherapy and resulted in a numerically inferior survival in metastatic PDAC | [120] | |
| PANCREAS | Cy/GVAX + CRS-207 vs. CRS-207 vs. Single-agent chemotherapy | Cy/GVAX | IIb Metastatic, previously treated | 213 (73 vs. 68 vs. 72) | Median OS in the primary cohort (n = 213) was 3.7 (2.9–5.3), 5.4 (4.2–6.4), and 4.6 (4.2–5.7) months in arms A, B, and C, respectively, showing no significant difference between arm A and arm C [p = not significant (NS), HR 1.17; 95% CI, 0.84–1.64] | The combination of Cy/GVAX + CRS-207 did not improve survival over chemotherapy | NCT02004262 | [121] |
| PROSTATE | Atezolizumab+ Sipuleucel T | Atezolizumab | Ib | 37 mCRPC | ORR 8% after 6 months DCR 41%. rPFS: 8.2 months in arm 1 vs. 5.8 months in arm 2 | [122] | ||
| PROSTATE | Ipilimumab+ Sipuleucel T | Ipilimumab | III | 50 mCRPC | No alteration of antigen-specific responses. Lower baseline frequencies of CTLA-4 expressing T cells and a history of RT. | Modest clinical activity. | NCT01804465 | [123] |
| OVARIAN | Durvalumab + Anti-FRα vaccine | Durvalumab | II | 27 Recurrent PROC | Robust FRα-specific T cell responses in all patients | Safe and tolerable. Unexpectedly durable survival in heavily pretreated population. | NCT02764333 | [124] |
| OVARIAN | TILs + ipilimumab + nivolumab | Ipilimumab + nivolumab | I/II | Recurrent EOC | One patient achieved a partial response and 5 others experienced disease stabilization for up to 12 months | improved T cell fold expansion, increased CD8 T cell tumor reactivity, and favorably affect the T cell phenotype | NCT03287674 | [125] |
5.5. Selected Clinical Trials Providing Information on ICI Combination with Novel Hormonal Therapies in Different Tumor Types (Table 5)
5.6. Selected Clinical Trials Providing Information on ICI Combination with CAR-T Cell Immunotherapy in Different Tumor Types (Table 6)
5.7. Selected Clinical Trials Providing Information on ICI Combination with Viral and other Therapies in Different Tumor Types (Table 7)
| Tumor Type | Combined Therapy | Anti-PD-1/PD-L1 | Clinical Phase and Population | N of Patients | Result | Conclusion | NCT Number | Reference |
|---|---|---|---|---|---|---|---|---|
| PROSTATE | Pembrolizumab (immune checkpoint blockade) + enzalutamide (androgen receptor inhibitor) | Pembrolizumab | mCRPC refractory to enzalutamide | 126 | In cohort 4: 12% had a response, 51% disease control rate (DCR). In cohort 5, 51% DCR. rPFS: 4 months in both cohorts. All Grade (C4 75%, C5 69%) and Grade 3–5 (C4 26%, C5 24%) adverse events (AEs) were similar as compared to cohorts 1–3 but numerically more frequent | Data demonstrate clinical support the addition of enzalutamide to with pembrolizumab. | KEYNOTE-199 | [127] |
| PROSTATE | Pembrolizumab + enzalutamide | Pembrolizumab | Ib/II mCRPC that had progressed to abiraterone | 103 | PSA RR 22%, ORR 12% DCR of 32% | Pembro + enza continued to show activity in pts with abi-pretreated mCRPC. Safety was consistent with the known profiles of pembro and enza. | KEYNOTE-365 | [128] |
| Tumor Type | Combined Therapy | Anti-PD-1/PD-L1 | Clinical Phase and Population | N of Patients | Result | Conclusion | NCT Number | Reference |
|---|---|---|---|---|---|---|---|---|
| PANCREAS | Mesothelin-specific | I | 6 Metastatic | SD: stabilized disease: 2 patients (33%) with PFS of 3.8 and 5.4 mo No severe TRAEs | Evidence for the potential antitumor activity of messenger RNA CARTmeso cells, as well as PDAC resistance to the immune response | [129] | ||
| PANCREAS OVARIAN | Knocked-out PD-1, mesothelin-directed CAR Ts (GC008t) | Mesothelin-positive solid tumors including EOC | I Mesothelin-positive solid tumors including EOC | 9 Mesothelin-positive solid tumors including EOC | The best response of the 7 evaluable patients was stable disease in 4 and partial response in 2 patients (dosed ≥ 1 × 107/kg) with PFS of 80 and 160 days. | genetic inactivation of PD-1 in CAR-T cells by CRISPR is feasible and safe | NCT03747965 | [130] |
| Solid tumors | PD-1 and T cell receptor (TCR) deficient mesothelin-specific CAR-T (MPTK-CAR-T | Mesothelin-positive solid tumors | I | 15 | The best overall response was stable disease (2/15 patients). No dose-limiting toxicity or unexpected adverse events were observed in any of the 15 patients | Feasibility and safety of CRISPR-engineered CAR-T cells with PD-1 disruption | [131] |
| Tumor Type | Combined Therapy | Anti-PD-1/PD-L1 | Clinical Phase and Population | N of Patients | Result | Conclusion | NCT Number | Reference |
|---|---|---|---|---|---|---|---|---|
| GMB | Pembrolizumab+ DNX-2401 oncolytic adenovirus. Intratumoral administration of DNX-2401 and sequential, adjuvant pembrolizumab b | Pembrolizumab | II Open label single group study | 49 Recurrent glioblastoma or gliosarcoma | >94% tumor regression.12 months mOS in 48 patients | safe without any dose-limiting conditions | NCT02798406 (CAPTIVE/KEYNOTE-192) | [132] |
| Targeting ROS | ||||||||
| PROSTATE | Docetaxel + OV Docetaxel + Radiotherapy + OV | II | 25 Hormone refractory | Oncoxin-Viusid (OV) −75 mL/day Suppresses ROS production PFS 59% OS rate 64% at 1 year | Clinical and humoral response, high survival rates, delayed appearance of signs of disease progression. | NCT03543670 | [133] | |
6. The Immunotherapy Side Effects: Focus on Cardiac Toxicity
7. Challenges in Immunotherapy
8. Conclusions
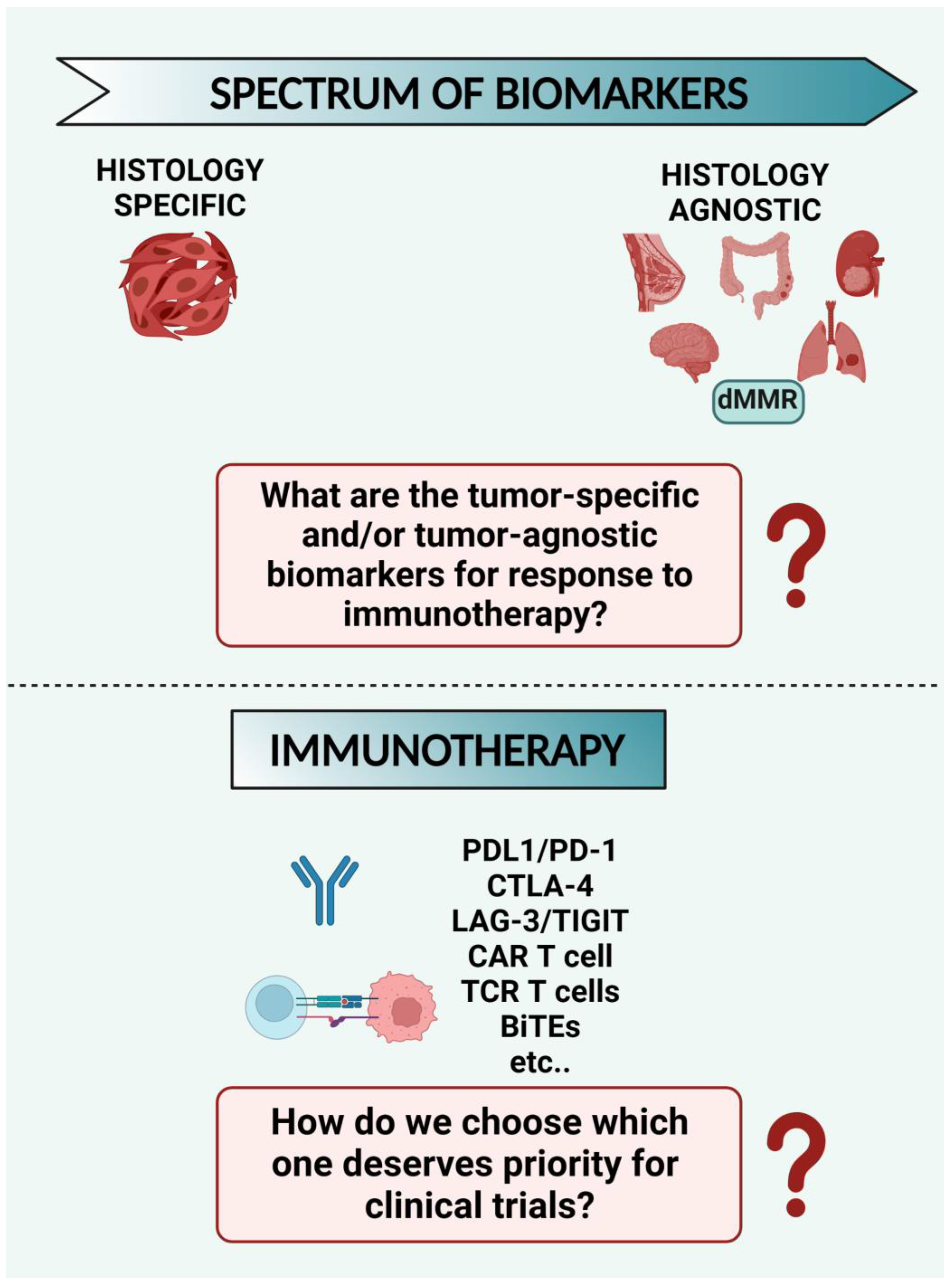
8.1. What Are the Tumor-Specific and/or Tumor-Agnostic Biomarkers for Response to Immunotherapy? Despite the Many Options Available for Immunotherapy Strategies, How do We Choose which One Deserves Priority for Clinical Trials? (Figure 3)

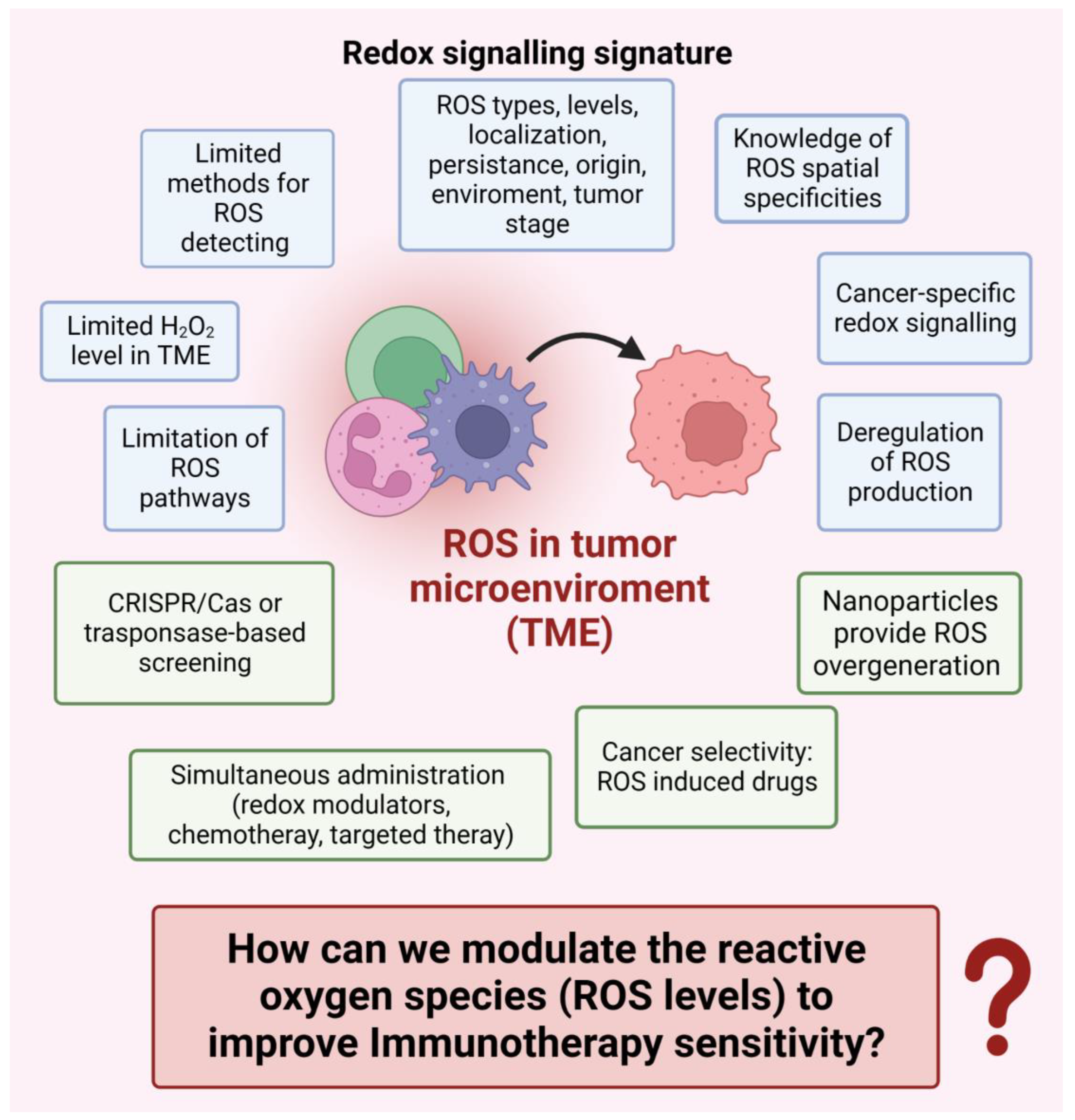
8.2. How can we Modulate the Reactive Oxygen Species (ROS Levels) to Improve Immunotherapy Sensitivity? (Figure 4)

8.3. What Effect may the Ubiquitin Signaling Modulation have on Immunotherapy Sensitivity? (Figure 5)

8.4. How can Imaging Techniques Be Used in the Diagnostic and Therapeutic Immunotherapy Field? (Figure 6)

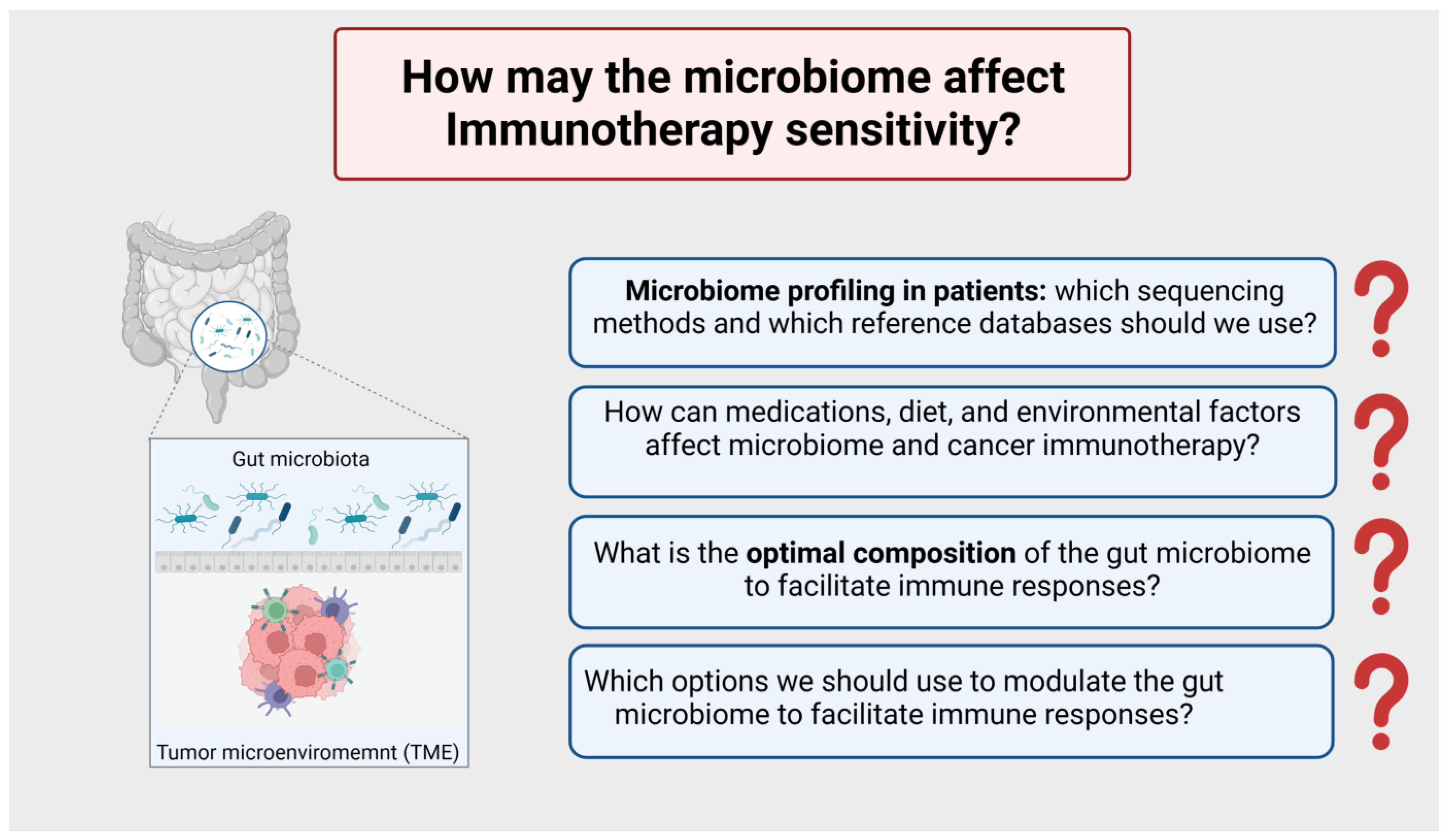
8.5. How may the Microbiome Affect Immunotherapy Sensitivity? (Figure 7)

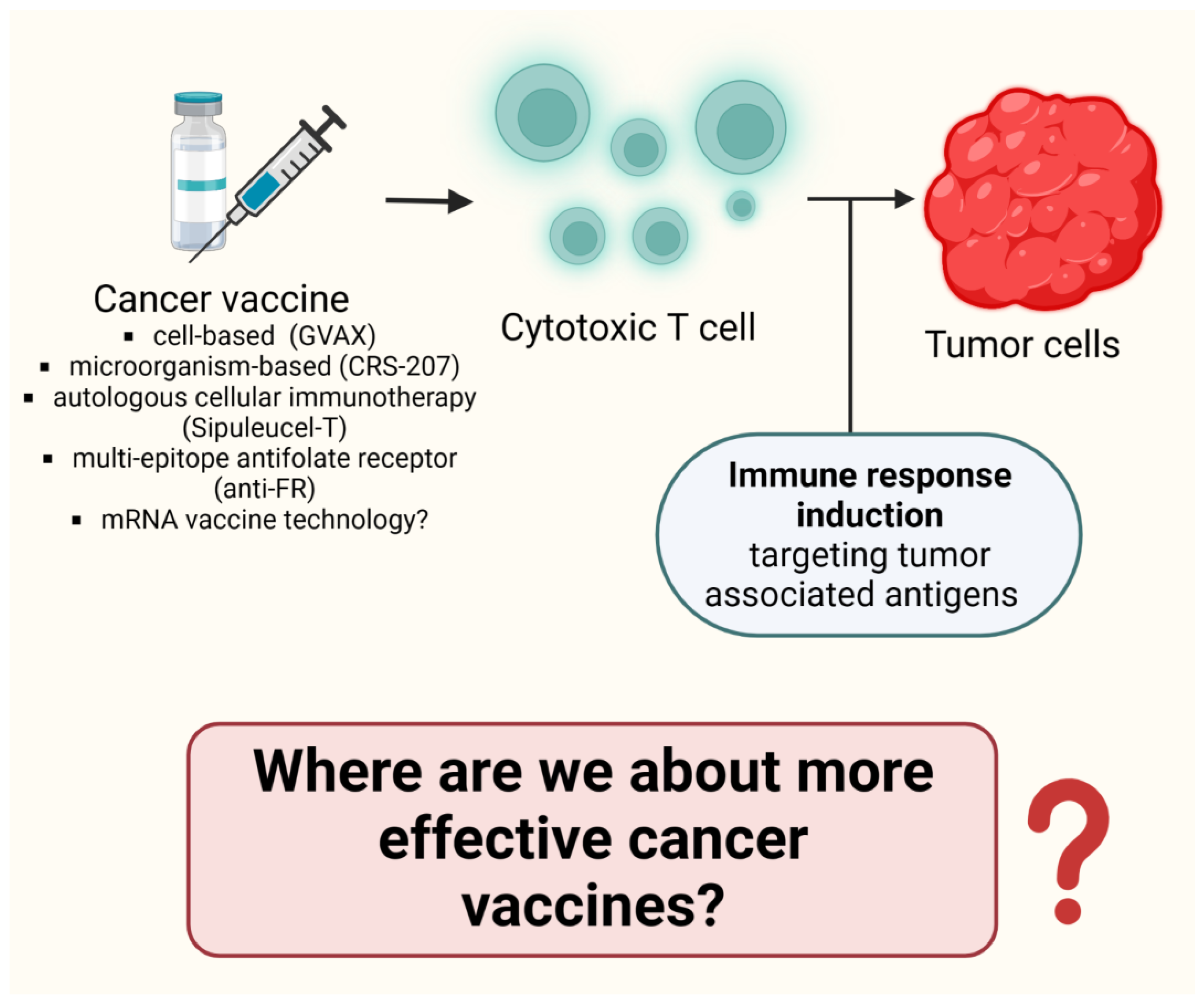
8.6. Where Are We about more Effective Cancer Vaccines? (Figure 8)

Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Finck, A.; Gill, S.-I.; June, C.H. Cancer immunotherapy comes of age and looks for maturity. Nat. Commun. 2020, 11, 3325. [Google Scholar] [PubMed]
- Darvishi, M.; Tosan, F.; Nakhaei, P.; Manjili, D.A.; Kharkouei, S.A.; Alizadeh, A.; Ilkhani, S.; Khalafi, F.; Zadeh, F.A.; Shafagh, S.G. Recent progress in cancer immunotherapy: Overview of current status and challenges. Pathol. Res. Pract. 2023, 241, 15424. [Google Scholar]
- Haslam, A.; Prasad, V. Estimation of the percentage of US patients with cancer who are eligible for and respond to checkpoint inhibitor immunotherapy drugs. JAMA Netw. Open 2019, 2, e192535. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Shao, X.; Zhang, Y.; Zhu, M.; Wang, F.X.C.; Mu, J.; Li, J.; Yao, H.; Chen, K. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med. 2023. [Google Scholar] [CrossRef] [PubMed]
- Peña-Romero, A.C.; Orenes-Piñero, E. Dual Effect of Immune Cells within Tumour Microenvironment: Pro- and Anti-Tumour Effects and Their Triggers. Cancers 2022, 14, 1681. [Google Scholar] [CrossRef]
- Jin, M.-Z.; Jin, W.-L. The updated landscape of tumor microenvironment and drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 166. [Google Scholar] [PubMed]
- Lu, C.; Li, Y.; Ali, N.M.; Zhang, B.; Cui, X. The role of innate immune cells in the tumor microenvironment and research progress in anti-tumor therapy. Front. Immunol. 2023, 13, 1039260. [Google Scholar] [CrossRef]
- Deng, L.; Meng, T.; Chen, L.; Wei, W.; Wang, P. The Role of Ubiquitination in Tumorigenesis and Targeted Drug Discovery. Signal Transduct. Target. Ther. 2020, 5, 11. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, S.-C. Targeting Ubiquitin Signaling for Cancer Immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 16. [Google Scholar] [CrossRef]
- Hou, B.; Chen, T.; Zhang, H.; Li, J.; Wang, P.; Shang, G. The E3 ubiquitin ligases regulate PD-1/PD-L1 protein levels in tumor microenvironment to improve immunotherapy. Front. Immunol. 2023, 14, 1123244. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-M.; Zhao, Z.-Y.; Yu, X.; Xia, Q.-D.; Zhou, P.; Wang, S.-G.; Wu, H.-L.; Hu, J. Exploiting E3 ubiquitin ligases to reeducate the tumor microenvironment for cancer therapy. Exp. Hematol. Oncol. 2023, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.-W.; Qiu, S.-Q.; Zhang, G.-J. Molecular and functional imaging in cancer-targeted therapy: Current applications and future directions. Signal Transduct. Target. Ther. 2023, 8, 89. [Google Scholar] [CrossRef] [PubMed]
- Crișan, G.; Moldovean-Cioroianu, N.S.; Timaru, D.-G.; Andrieș, G.; Călin Căinap, G.; Vasile Chiș, G. Radiopharmaceuticals for PET and SPECT Imaging: A Literature Review over the Last Decade. Int. J. Mol. Sci. 2022, 23, 5023. [Google Scholar] [CrossRef] [PubMed]
- Calvo, V.; Izquierdo, M. T Lymphocyte and CAR-T Cell-Derived Extracellular Vesicles and Their Applications in Cancer Therapy. Cells 2022, 11, 790. [Google Scholar] [CrossRef] [PubMed]
- Parisse, P.; Rago, I.; Ulloa Severino, L.; Perissinotto, F.; Ambrosetti, E.; Paoletti, P.; Ricci, M.; Beltrami, A.P.; Cesselli, D.; Casalis, L. Atomic force microscopy analysis of extracellular vesicles. Eur. Biophys. J. 2017, 46, 813–820. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Wang, M.; Xu, T.; Zhang, M.; Dai, H.; Wang, C.; Ding, D.; Zhong, Z. Reactive oxygen species-powered cancer immunotherapy: Current status and challenges. J. Control. Rel. 2023, 356, 623–648. [Google Scholar] [CrossRef]
- Kennel, K.B.; Greten, F.R. Immune cell—Produced ROS and their impact on tumor growth and metastasis. Redox Biol. 2021, 42, 101891. [Google Scholar] [CrossRef]
- Liu, R.; Peng, L.; Zhou, L.; Huang, Z.; Zhou, C.; Huang, C. Oxidative Stress in Cancer Immunotherapy: Molecular Mechanisms and Potential Applications. Antioxidants 2022, 11, 853. [Google Scholar] [CrossRef]
- Arfin, S.; Jha, N.K.; Jha, S.K.; Kesari, K.K.; Ruokolainen, J.; Roychoudhury, S.; Rathi, B.; Kumar, D. Oxidative Stress in Cancer Cell Metabolism. Antioxidants 2021, 10, 642. [Google Scholar] [CrossRef]
- Kuo, C.-L.; Babuharisankar, A.P.; Lin, Y.-C.; Lien, H.-W.; Lo, Y.-K.; Chou, H.-Y.; Tangeda, V.; Cheng, L.-C.; Cheng, A.N.; Lee, A.N.L. Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: Foe or friend? J. Biomed. Sci. 2022, 29, 74. [Google Scholar] [CrossRef] [PubMed]
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Aboelella, N.S.; Brandle, C.; Kim, T.; Ding, Z.C.; Zhou, G. Oxidative Stress in the Tumor Microenvironment and Its Relevance to Cancer Immunotherapy. Cancers 2021, 13, 986. [Google Scholar] [CrossRef]
- Wang, J.; Liu, N.; Jiang, H.; Li, Q.; Xing, D. Reactive Oxygen Species in Anticancer Immunity: A Double-Edged Sword. Front. Bioeng. Biotechnol. 2021, 9, 784612. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.; Ngiow, S.F.; Ribas, A.; Smyth, M.J. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015, 75, 2139–2145. [Google Scholar] [CrossRef] [PubMed]
- Hildeman, D.A.; Mitchell, T.; Teague, T.K.; Henson, P.; Day, B.J.; Kappler, J.; Marrack, P.C. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity 1999, 10, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Kotsafti, A.; Scarpa, M.; Castagliuolo, I.; Scarpa, M. Reactive Oxygen Species and Antitumor Immunity—From Surveillance to Evasion. Cancers 2020, 12, 1748. [Google Scholar] [CrossRef]
- Deng, H.; Yang, W.; Zhou, Z.; Tian, R.; Lin, L.; Ma, Y.; Song, J.; Chen, X. Targeted scavenging of extracellular ROS relieves suppressive immunogenic cell death. Nat. Commun. 2020, 11, 4951. [Google Scholar] [CrossRef]
- Hayward, S.W. Immunotherapeutic Response in Tumors Is Affected by Microenvironmental ROS. Cancer Res. 2020, 80, 1799–1800. [Google Scholar] [CrossRef]
- Wei, C.; Ma, Y.; Wang, F.; Liao, Y.; Chen, Y.; Zhao, B.; Zhao, Q.; Wang, D.; Tang, D. Igniting Hope for Tumor Immunotherapy: Promoting the “Hot and Cold” Tumor Transition. Oncology 2022, 16, 11795549221120708. [Google Scholar] [CrossRef]
- Bailly, C. Regulation of PD-L1 expression on cancer cells with ROS modulating drugs. Life Sci. 2020, 246, 117403. [Google Scholar] [CrossRef] [PubMed]
- Van Loenhout, J.; Peeters, M.; Bogaerts, A.; Smits, E.; Deben, C. Oxidative Stress-Inducing Anticancer Therapies: Taking a Closer Look at Their Immunomodulating Effects. Antioxidants 2020, 9, 1188. [Google Scholar] [CrossRef]
- Magrì, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chilà, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin c enhances cancer immunotherapy. Sci. Trans. Med. 2020, 12, eaay8707. [Google Scholar] [CrossRef] [PubMed]
- Zaher, A.; Stephens, L.M.; Miller, A.M.; Hartwig, S.M.; Stolwijk, J.M.; Petronek, M.S.; Zacharias, Z.R.; Wadas, T.J.; Monga, V.; Cullen, J.J.; et al. Pharmacological ascorbate as a novel therapeutic strategy to enhance cancer immunotherapy. Front. Immunol. 2022, 13, 989000. [Google Scholar] [CrossRef] [PubMed]
- Bedhiafi, T.; Inchakalody, V.P.; Fernandes, Q.; Mestiri, S.; Billa, N.; Uddin, S.; Merhi, M.; Dermime, S. The potential role of vitamin C in empowering cancer immunotherapy. Biomed. Pharmacother. 2022, 146, 112553. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Gu, X.; Han, F.; Hea, Z.; Wang, Y. Emerging role of natural products in cancer immunotherapy. Acta Pharm. Sin. B 2022, 12, 1163–1185. [Google Scholar] [CrossRef]
- Weichselbaum, R.R.; Liang, H.; Deng, L.; Fu, Y.-X. Radiotherapy and immunotherapy: A beneficial liaison? Nat. Rev. Clin. Oncol. 2017, 14, 365–379. [Google Scholar] [CrossRef]
- Hwang, W.L.; Pike, L.R.; Royce, T.J.; Mahal, B.A.; Loeffler, J.S. Safety of combining radiotherapy with immune-checkpoint inhibition. Nat. Rev. Clin. Oncol. 2018, 15, 477–494. [Google Scholar] [CrossRef]
- Pointer, K.B.; Pitroda, S.P.; Weichselbaum, R.R. Radiotherapy and immunotherapy: Open questions and future strategies. Trends Cancer 2022, 8, 9–20. [Google Scholar] [CrossRef]
- Cramer, G.M.; Moon, E.K.; Cengel, K.A.; Busch, T.M. Photodynamic Therapy and Immune Checkpoint Blockade. Photochem. Photobiol. 2020, 96, 954–961. [Google Scholar] [CrossRef]
- Jiang, W.; Liang, M.; Guangzhi, L.Q.; Wu, S. The Current Status of Photodynamic Therapy in Cancer Treatment. Cancers 2023, 15, 585. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Chen, X. Combined Photodynamic and Photothermal Therapy and Immunotherapy for Cancer Treatment: A Review. Int. J. Nanomed. 2022, 17, 6427–6446. [Google Scholar] [CrossRef] [PubMed]
- Son, S.; Kim, J.H.; Wang, X.; Zhang, C.; Yoon, S.A.; Shin, J.; Sharma, A.; Lee, M.H.; Cheng, L.; Wu, J.; et al. Multifunctional sonosensitizers in sonodynamic cancer therapy. Chem. Soc. Rev. 2020, 49, 3244–3261. [Google Scholar] [PubMed]
- Sharma, A.; Sharma, L.; Nandy, S.K.; Payal, N.; Yadav, S.; Vargas-De-La-Cruz, C.; Anwer, M.K.; Khan, H.; Behl, T.; Bungau, S.G. Molecular Aspects and Therapeutic Implications of Herbal Compounds Targeting Different Types of Cancer. Molecules 2023, 11, 750. [Google Scholar] [CrossRef]
- Chan, W.J.; Adiwidjaja, J.; McLachlan, A.J.; Boddy, A.V.; Harnett, J.E. Interactions between natural products and cancer treatments: Underlying mechanisms and clinical importance. Cancer Chemother. Pharmacol. 2023, 91, 103–119. [Google Scholar] [CrossRef]
- Yang, C.; Li, D.; Ko, C.N.; Wang, K.; Wang, H. Active ingredients of traditional Chinese medicine for enhancing the effect of tumor immunotherapy. Front. Immunol. 2023, 10, 1133050. [Google Scholar] [CrossRef]
- Xu, H.; Hu, M.; Liu, M.; An, S.; Guan, K.; Wang, M.; Li, L.; Zhang, J.; Li, J. Leaf Huang. Nano-puerarin regulates tumor microenvironment and facilitates chemo- and immunotherapy in murine triple negative breast cancer model. Biomaterials 2020, 235, 119769. [Google Scholar] [CrossRef]
- Vo, H.; Cartwright, C.; Song, I.-W.; Karp, D.D.; Nogueras Gonzalez, G.M. Ipilimumab, Pembrolizumab, or Nivolumab in Combination with BBI608 in Patients with Advanced Cancers Treated at MD Anderson Cancer Center Henry. J. Clin. Oncol. 2022, 40, 127–137. [Google Scholar]
- Jing, B.; Gu, F.; Rui, A.R.; Gao, Y.; Li, Y.; Xie, Y.; Wang, J.; Chen, Y.; Li, H.; Gao, T.; et al. Apoptotic tumor cell-derived microparticles loading Napabucasin inhibit CSCs and synergistic immune therapy. J. Nanobiotec. 2023, 21, 37. [Google Scholar]
- Mussa, A.; Idris, R.A.M.; Ahmed, N.; Ahmad, S.; Murtadha, A.H.; Tengku Din, T.A.D.A.A.; Yean Yean, C.Y.; Rahman, W.F.A.; Lazim, N.M.; Uskokovi’c, V.; et al. High-Dose Vitamin C for Cancer Therapy. Pharmaceuticals 2022, 15, 711. [Google Scholar] [CrossRef]
- Yang, B.; Gao, J.; Pei, Q.; Xu, H.; Yu, H. Engineering prodrug nanomedicine for cancer immunotherapy. Adv. Sci. 2020, 7, 2002365. [Google Scholar]
- Peng, S.; Xiao, F.; Chen, M.; Gao, H. Tumor-Microenvironment-Responsive Nanomedicine for Enhanced Cancer Immunotherapy. Adv. Sci. 2022, 9, 2103836. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Chen, Y.; Shi, J. Reactive Oxygen Species (ROS)-Based Nanomedicine. Chem. Rev. 2019, 119, 4881–4985. [Google Scholar] [CrossRef] [PubMed]
- Saravanakumar, G.; Kim, J.; Kim, W.J. Reactive-Oxygen-Species-Responsive Drug Delivery Systems: Promises and Challenges. Adv. Sci. 2016, 4, 1600124. [Google Scholar] [CrossRef]
- Yuniati, L.; Lauriola, A.; Gerritsen, M.; Abreu, S.; Ni, E.; Tesoriero, C.; Onireti, J.O.; Low, T.Y.; Heck, A.J.R.; Vettori, A.; et al. Ubiquitylation of the ER-Shaping Protein Lunapark via the CRL3KLHL12 Ubiquitin Ligase Complex. Cell Rep. 2020, 31, 107664. [Google Scholar] [CrossRef]
- Zinngrebe, J.; Montinaro, A.; Peltzer, N.; Walczak, H. Ubiquitin in the Immune System. EMBO Rep. 2014, 15, 28–45. [Google Scholar] [CrossRef]
- Fujita, Y.; Tinoco, R.; Li, Y.; Senft, D.; Ronai, Z.A. Ubiquitin Ligases in Cancer Immunotherapy—Balancing Antitumor and Autoimmunity. Trends Mol. Med. 2019, 25, 428–443. [Google Scholar]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. N. Engl. J. Med. 2016, 375, 1767–1778. [Google Scholar] [CrossRef]
- Ding, L.; Chen, X.; Zhang, W.; Dai, X.; Guo, H.; Pan, X.; Xu, Y.; Feng, J.; Yuan, M.; Gao, X.; et al. Canagliflozin Primes Antitumor Immunity by Triggering PD-L1 Degradation in Endocytic Recycling. J. Clin. Investig. 2023, 133, e154754. [Google Scholar]
- Jiang, C.; He, L.; Xiao, S.; Wu, W.; Zhao, Q.; Liu, F. E3 Ubiquitin Ligase RNF125 Suppresses Immune Escape in Head and Neck Squamous Cell Carcinoma by Regulating PD-L1 Expression. Mol. Biotechnol. 2022, 65, 891–903. [Google Scholar]
- Yang, H.; Zhang, X.; Lao, M.; Sun, K.; He, L.; Xu, J.; Duan, Y.; Chen, Y.; Ying, H.; Li, M.; et al. Targeting Ubiquitin-Specific Protease 8 Sensitizes Anti-Programmed Death-Ligand 1 Immunotherapy of Pancreatic Cancer. Cell Death Differ. 2022, 30, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Shi, D.; Wu, X.; Jian, Y.; Wang, J.; Huang, C.; Mo, S.; Li, Y.; Li, F.; Zhang, C.; Zhang, D.; et al. USP14 Promotes Tryptophan Metabolism and Immune Suppression by Stabilizing IDO1 in Colorectal Cancer. Nat. Commun. 2022, 13, 5644. [Google Scholar] [CrossRef]
- Wang, X.; Tokheim, C.; Gu, S.S.; Wang, B.; Tang, Q.; Li, Y.; Traugh, N.; Zeng, Z.; Zhang, Y.; Li, Z.; et al. In Vivo CRISPR Screens Identify the E3 Ligase Cop1 as a Modulator of Macrophage Infiltration and Cancer Immunotherapy Target. Cell 2021, 184, 5357–5374.e22. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, J.; Liu, Z.; Zhang, H. MARCH1 as a Novel Immune-Related Prognostic Biomarker That Shapes an Inflamed Tumor Microenvironment in Lung Adenocarcinoma. Front. Oncol. 2022, 12, 1008753. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, X.; Sun, Y.; Chu, Y.; Liu, F.; Chen, C. TRIM44 Regulates Tumor Immunity in Gastric Cancer through LOXL2-Dependent Extracellular Matrix Remodeling. Cell Oncol. 2022, 46, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Hargadon, K.M.; Johnson, C.E.; Williams, C.J. Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 2018, 62, 29–39. [Google Scholar] [CrossRef]
- Lv, G.; Sun, X.; Qiu, L.; Sun, Y.; Li, K.; Liu, Q.; Zhao, Q.; Qin, S.; Lin, J. PET imaging of tumor PD-L1 expression with a highly specific nonblocking single-domain antibody. J. Nucl. Med. 2020, 61, 117–122. [Google Scholar] [CrossRef]
- Broos, K.; Lecocq, Q.; Raes, G.; Devoogdt, N.; Keyaerts, M.; Breckpot, K. Noninvasive imaging of the PD-1:PD-L1 immune checkpoint: Embracing nuclear medicine for the benefit of personalized immunotherapy. Theranostics 2018, 8, 3559–3570. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: Mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef]
- Bensch, F.; van der Veen, V.; Lub-de Hooge, M.N.; Jorritsma-Smit, A.; Boellaard, R.; Kok, I.C.; Oosting, S.F.; Schröder, C.P.; Hiltermann, T.J.N.; van der Wekken, A.J.; et al. 9Zr-atezolizumab imaging as a non-invasive approach to assess clinical response to PD-L1 blockade in cancer. Nat. Med. 2018, 24, 1852–1858. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Shalabi, A.; Hubbard-Lucey, V.M. Comprehensive analysis of the clinical immuno-oncology landscape. Ann. Oncol. 2018, 29, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Moses, W.W. Fundamental Limits of Spatial Resolution in PET. Nucl. Instrum. Methods Phys. Res. A 2011, 648, S236–S240. [Google Scholar] [CrossRef] [PubMed]
- Krekorian, M.; Fruhwirth, G.O.; Srinivas, M.; Figdor, C.G.; Heskamp, S.; Witney, T.H.; Aarntzen, E.H.J.G. Imaging of T-cells and their responses during anti-cancer immunotherapy. Theranostics 2019, 9, 7924–7947. [Google Scholar] [CrossRef] [PubMed]
- Dudley, C.V.; Baer, B.; Simons, R.M. Utilization of Chimeric Antigen Receptor T-cell Therapy in Adults. Semin. Oncol. Nurs. 2019, 35, 150930. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. Car t cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [PubMed]
- Valiullina, A.K.; Zmievskaya, E.A.; Ganeeva, I.A.; Zhuravleva, M.N.; Garanina, E.E.; Rizvanov, A.A.; Petukhov, A.V.; Bulatov, E.R. Evaluation of CAR-T Cells’ Cytotoxicity against Modified Solid Tumor Cell Lines. Biomedicines 2023, 11, 626. [Google Scholar] [CrossRef]
- Yang, P.; Cao, X.; Cai, H.; Feng, P.; Chen, X.; Zhu, Y.; Yang, Y.; An, W.; Yang, Y.; Jie, J. The exosomes derived from CAR-T cell efficiently target mesothelin and reduce triple-negative breast cancer growth. Cell Immunol. 2021, 360, 104262. [Google Scholar] [CrossRef]
- Momen-Heravi, F.; Balaj, L.; Alian, S. Alternative methods for characterization of extracellular vesicles. Front. Physiol. 2012, 3, 354. [Google Scholar] [CrossRef]
- Nizamudeen, Z.; Markus, R.; Lodge, R.; Parmenter, C.; Platt, M.; Chakrabarti, L.; Sottile, V. Rapid and accurate analysis of stem cell-derived extracellular vesicles with super resolution microscopy and live imaging. Biochim. Biophys. Acta Mol. Cell Res. 2018, 1865, 1891–1900. [Google Scholar] [CrossRef]
- Gautron, J.; Stapane, L.; Le Roy, N.; Nys, Y.; Rodriguez-Navarro, A.B.; Hincke, M.T. Avian eggshell biomineralization: An update on its structure, mineralogy and protein tool kit. BMC Mol. Cell Biol. 2021, 22, 11. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Bates, M.; Zhuang, X. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 2006, 3, 793–796. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S.; Rodríguez-Abreu, D.; Speranza, G.; Esteban, E.; Felip, E.; Dómine, M.; Hui, R.; Hochmair, M.J.; Clingan, P.; Powell, S.F.; et al. Updated analysis from KEYNOTE-189: Pembrolizumab or placebo plus pemetrexed and platinum for previously untreated metastatic nonsquamous non-small-cell lung cancer. J. Clin. Oncol. 2020, 38, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus chemotherapy for squamous non-small-cell lung cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Nishio, M.; Barlesi, F.; West, H.; Ball, S.; Bordoni, R.; Cobo, M.; Longeras, P.D.; Goldschmidt, J.; Novello, S.; Orlandi, F.; et al. Atezolizumab plus chemotherapy for first-line treatment of nonsquamous NSCLC: Results from the randomized phase 3 IMpower132 trial. J. Thorac. Oncol. 2021, 16, 653–664. [Google Scholar] [CrossRef]
- Jotte, R.; Cappuzzo, F.; Vynnychenko, I.; Stroyakovskiy, D.; Rodrıguez-Abreu, D.; Hussein, M.; Soo, R.; Conter, H.J.; Kozuki, T.; Huang, K.-C.; et al. Atezolizumab in combination with carboplatin and nab-paclitaxel in advanced squamous NSCLC (IMpower131): Results from a randomized phase III trial. J. Thorac. Oncol. 2020, 15, 1351–1360. [Google Scholar] [CrossRef]
- Luen, S.; Virassamy, B.; Savas, P.; Salgado, R.; Loi, S. The genomic landscape of breast cancer and its interaction with host immunity. Breast 2016, 29, 241–250. [Google Scholar] [CrossRef]
- Emens, L.A.; Adams, S.; Barrios, C.H.; Dieras, V.C.; Iwata, H.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. LBA16 IMpassion130: Final OS analysis from the pivotal phase III study of atezolizumab + nab-paclitaxel vs placebo + nab-paclitaxel in previously untreated locally advanced or metastatic triple-negative breast cancer. Ann. Oncol. 2020, 31, S1148. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): A randomised, double-blind, phase 3 trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef]
- Brufsky, A.; Kim, S.B.; Zvirbule, Z.; Dirix, L.Y.; Eniu, A.E.; Carabantes, F.; Izarzugaza, Y.; Mebis, J.; Sohn, J.; Wongchenko, M.; et al. Phase II COLET study: Atezolizumab (A) + cobimetinib (C) + paclitaxel (P)/nab-paclitaxel (nP) as first-line (1L) treatment (tx) for patients (pts) with locally advanced or metastatic triple-negative breast cancer (mTNBC). J. Clin. Oncol. 2019, 37, 1013. [Google Scholar] [CrossRef]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.-U.; Grischke, E.-M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple negative breast cancer—Clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Giobbie-Hurder, A.; Gombos, A.; Bachelot, T.; Hui, R.; Curigliano, G.; Campone, M.; Biganzoli, L.; Bonnefoi, H.; Jerusalem, G.; et al. Pembrolizumab plus trastuzumab in trastuzumab-resistant, advanced, HER2-positive breast cancer (PANACEA): A single-arm, multicentre, phase 1b–2 trial. Lancet Oncol. 2019, 20, 371–382. [Google Scholar] [CrossRef]
- Yoon, J.H.; Jung, Y.-J.; Moon, S.-H. Immunotherapy for pancreatic cancer. World J. Clin. Cases 2021, 9, 2969–2982. [Google Scholar] [PubMed]
- Weiss, G.J.; Blaydorn, L.; Beck, J.; Bornemann-Kolatzki, K.; Urnovitz, H.; Schütz, E.; Khemka, V. Phase Ib/II study of gemcitabine, nab-paclitaxel, and pembrolizumab in metastatic pancreatic adenocarcinoma. Investig. New Drugs 2018, 36, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Kamath, S.D.; Kalyan, A.; Kircher, S.; Nimeiri, H.; Fought, A.J.; Benson, A., 3rd; Mulcahy, M. Ipilimumab and Gemcitabine for Advanced Pancreatic Cancer: A Phase Ib Study. Oncologist 2020, 25, e808–e815. [Google Scholar] [CrossRef]
- Wainberg, Z.A.; Hochster, H.S.; Kim, E.J.; George, B.; Kaylan, A.; Chiorean, E.G.; Waterhouse, D.M.; Guiterrez, M.; Parikh, A.; Jain, R.; et al. Open-label, Phase I Study of Nivolumab Combined with nab-Paclitaxel Plus Gemcitabine in Advanced Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 4814–4822. [Google Scholar] [CrossRef]
- Boku, N.; Ryu, M.H.; Kato, K.; Chung, H.C.; Minashi, K.; Lee, K.W.; Cho, H.; Kang, W.K.; Komatsu, Y.; Tsuda, M.; et al. Safety and efficacy of nivolumab in combination with S-1/capecitabine plus oxaliplatin in patients with previously untreated, unresectable, advanced, or recurrent gastric/gastroesophageal junction cancer: Interim results of a randomized, phase II trial (ATTRACTION-4). Ann. Oncol. 2019, 30, 250–258. [Google Scholar]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Bragagnoli, A.C.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): A randomised, open-label, phase 3 trial. Lancet 2021, 398, 27–40. [Google Scholar] [CrossRef]
- Bang, Y.-J.; Kang, Y.-K.; Catenacci, D.V.; Muro, K.; Fuchs, C.S.; Geva, R.; Hara, H.; Golan, T.; Garrido, M.; Jalal, S.I.; et al. Pembrolizumab alone or in combination with chemotherapy as first-line therapy for patients with advanced gastric or gastroesophageal junction adenocarcinoma: Results from the phase II nonrandomized KEYNOTE-059 study. Gastric Cancer 2019, 22, 828–837. [Google Scholar] [CrossRef]
- Bausart, M.; Preat, V.; Malfanti, A. Immunotherapy for glioblastoma: The promise of combination strategies. J. Exp. Clin. Cancer Res. 2022, 41, 35. [Google Scholar]
- López-Campos, F.; Gajate, P.; Romero-Laorden, N.; Zafra-Martín, J.; Juan, M.; Hernando Polo, S.; Conde Moreno, A.; Couñago, F. Immunotherapy in Advanced Prostate Cancer: Current Knowledge and Future Directions. Biomedicines 2022, 10, 537. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Gonzalez Mella, P.; Castellano, D.; Minatta, J.N.; Rezazadeh Kalebasty, A.; Shaffer, D.; Vazquez-Limon, J.C.; Armstrong, A.J.; Sanchez-Lopez, H.M.; Sharkey, B.; et al. Efficacy and safety of nivolumab in combination with docetaxel in men with metastatic castration-resistant prostate cancer in CheckMate 9KD. Ann. Oncol. 2019, 30, 885–886. [Google Scholar]
- Sridhar, S.S.; Kolinsky, M.P.; Gravis, G.; Mourey, L.; Piulats Rodriguez, J.M.M.; Romano, E.; Berry, W.R.; Gurney, H.; Retz, M.; Appleman, L.J.; et al. Pembrolizumab (pembro) plus docetaxel and prednisone in patients (pts) with abiraterone acetate (abi) or enzalutamide (enza)-pretreated metastatic castration-resistant prostate cancer (mCRPC): KEYNOTE-365 cohort B efficacy, safety and, biomarker results. J. Clin. Oncol. 2020, 38, 5550. [Google Scholar]
- Yu, E.Y.; Kolinsky, M.P.; Berry, W.R.; Retz, M.; Mourey, L.; Piulats, J.M.; Appleman, L.J.; Romano, E.; Gravis, G.; Gurney, H.; et al. Pembrolizumab Plus Docetaxel and Prednisone in Patients with Metastatic Castration-resistant Prostate Cancer: Long-term Results from the Phase 1b/2 KEYNOTE-365 Cohort B Study. Eur. Urol. 2022, 82, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.F.; Herold, C.; Gray, K.P.; Penson, R.T.; Horowitz, N.; Konstantinopoulos, P.A.; Castro, C.M.; Hill, S.J.; Curtis, J.; Luo, W.; et al. Assessment of combined nivolumab and bevacizumab in relapsed ovarian cancer: A phase 2 clinical trial. JAMA Oncol. 2019, 5, 1731–1738. [Google Scholar] [CrossRef]
- Yu, E.Y.; Piulats, J.M.; Gravis, G.; Laguerre, B.; Arranz Arija, J.A.; Oudard, S.; Fong, P.C.C.; Kolinsky, M.P.; Augustin, M.; Feyerabend, S.; et al. KEYNOTE-365 cohort A updated results: Pembrolizumab (pembro) plus olaparib in docetaxel-pretreated patients (pts) with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, 100. [Google Scholar] [CrossRef]
- Karzai, F.; Vanderweele, D.; Madan, R.A.; Owens, H.; Cordes, L.M.; Hankin, A.; Couvillon, A.; Nichols, E.; Bilusic, M.; Beshiri, M.L. Activity of durvalumab plus olaparib in metastatic castration-resistant prostate cancer in men with and without DNA damage repair mutations 11 Medical and Health Sciences 1112 Oncology and Carcinogenesis. J. Immunother. Cancer 2018, 6, 141. [Google Scholar] [CrossRef]
- Sharma, P.; Pachynski, R.K.; Narayan, V.; Hu, Y.; Han, G.C.; Fizaz, K. Nivolumab plus ipilimumab for metastatic castration- resistant prostate cancer: Preliminary analysis of patients in the checkmate 650 trial. Cell 2020, 38, 489–499. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-Label Clinical Trial of Niraparib Combined with Pembrolizumab for Treatment of Advanced or Metastatic Triple- Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef]
- Liu, J.; Liu, Q.; Li, Y.; Li, Q.; Su, F.; Yao, H.; Su, S.; Wang, Q.; Jin, L.; Wang, Y.; et al. Efficacy and safety of camrelizumab combined with apatinib in advanced triple-negative breast cancer: An open-label phase II trial. J. Immunother. Cancer 2020, 8, e000696. [Google Scholar] [CrossRef]
- Porter, R.; Matulonis, U.A. Immunotherapy for Ovarian Cancer. Clin. Adv. Hematol. Oncol. 2022, 20, 240–253. [Google Scholar]
- Färkkilä, A.; Gulhan, D.C.; Casado, J.; Jacobson, C.A.; Nguyen, H.; Kochupurakkal, B.; Maliga, Z.; Yapp, C.; Chen, Y.-A.; Schapiro, D.; et al. Immunogenomic profiling determines responses to combined PARP and PD-1 inhibition in ovarian cancer. Nat. Commun. 2020, 11, 1459. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; Van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. TOPACIO/Keynote-162 (NCT02657889): A phase 1/2 study of niraparib + pembrolizumab in patients (pts) with advanced triple-negative breast cancer or recurrent ovarian cancer (ROC): Results from ROC cohort. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Kim, B.-G.; Kim, J.-W.; Lee, J.B.; Park, F.; Joung, J.-G.; Kim, S.; Choi, C.H.; Kim, H.S. Biomarker-guided targeted therapy in platinum-resistant ovarian cancer (AMBITION. KGOG 3045): A multicentre, open-label, five-arm, uncontrolled, umbrella trial. Gynecol. Oncol. 2022, 33, e45. [Google Scholar] [CrossRef] [PubMed]
- Drew, Y.; Penson, R.T.; O’malley, D.M.; Kim, J.W.; Zimmermann, S.; Roxburgh, P.; Sohn, J.; Stemmer, S.M.; Bastian, S.; Ferguson, M.; et al. Phase II study of olaparib (O) plus durvalumab (D) and bevacizumab (B) (MEDIOLA): Initial results in patients (pts) with non-germline BRCA-mutated (non-gBRCAm) platinum sensitive relapsed (PSR) ovarian cancer (OC) [ESMO abstract 814MO]. Ann. Oncol. 2020, 31, S615–S616. [Google Scholar]
- Liu, J.; Gaillard, S.; Hendrickson, A.W.; Moroney, J.; Yeku, O.; Diver, E.; Gunderson, C.; Arend, R.; Ratner, E.; Samnotra, V.; et al. An open-label phase II study of dostarlimab (TSR-042), bevacizumab (bev), and niraparib combination in patients (pts) with platinum-resistant ovarian cancer (PROC): Cohort A of the OPAL trial. Gynecol. Oncol. 2021, 162, S17–S18. [Google Scholar]
- Slovin, S.F.; Higano, C.S.; Hamid, O.; Tejwani, S.; Harzstark, A.; Alumkal, J.J.; Scher, H.I.; Chin, K.; Gagnier, P.; McHenry, M.B.; et al. Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: Results from an open-label, multicenter phase I/II study. Ann. Oncol. 2013, 24, 1813–1821. [Google Scholar]
- Morris, M.J.; Fong, L.; Petrylak, D.P.; Sartor, A.O.; Higano, C.S.; Pagliaro, L.C.; Alva, A.S.; Appleman, L.J.; Tan, W.; Vaishampayan, U.N.; et al. Safety and clinical activity of atezolizumab (atezo) + radium-223 dichloride (r-223) in 2L metastatic castration-resistant prostate cancer (mCRPC): Results from a phase Ib clinical trial. J. Clin. Oncol. 2020, 38, 5565. [Google Scholar] [CrossRef]
- Reardon, D.; Kaley, T.; Dietrich, J.; Clarke, J.; Dunn, G.; Lim, M.; Cloughesy, T.; Gan, H.; Park, A.; Schwarzenberger, P.; et al. Atim-38. Phase 2 study to evaluate the clinical efficacy and safety of Medi4736 (Durvalumab, Durva) + bevacizumab (Bev) in Bev-naïve patients with recurrent glioblastoma (Gbm). Neuro-Oncology 2018, 20, vi10. [Google Scholar] [CrossRef]
- Wu, A.A.; Bever, K.M.; Ho, W.J.; Fertig, E.J.; Niu, N.; Zheng, L.; Parkinson, R.M.; Durham, J.N.; Onners, B.; Ferguson, A.K.; et al. A Phase II Study of Allogeneic GM-CSF-Transfected Pancreatic Tumor Vaccine (GVAX) with Ipilimumab as Maintenance Treatment for Metastatic Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 5129–5139. [Google Scholar] [CrossRef]
- Le, D.T.; Picozzi, V.J.; Ko, A.H.; Wainberg, Z.A.; Kindler, H.; Wang-Gillam, A.; Oberstein, P.; Morse, M.A.; Zeh, H.J., 3rd; Weekes, C.; et al. Results from a Phase IIb, Randomized, Multicenter Study of GVAX Pancreas and CRS-207 Compared with Chemotherapy in Adults with Previously Treated Metastatic Pancreatic Adenocarcinoma (ECLIPSE Study). Clin. Cancer Res. 2019, 25, 5493–5502. [Google Scholar] [CrossRef] [PubMed]
- Rosser, C.J.; Hirasawa, Y.; Acoba, J.D.; Tamura, D.J.; Pal, S.K.; Huang, J.; Scholz, M.C.; Dorffet, T.B. Phase Ib study assessing different sequencing regimens of atezolizumab (anti-PD-L1) and sipuleucel-T (SipT)in patients who have asymptomatic or minimally symptomatic metastatic castrate resistant prostate cancer. J. Clin. Oncol. 2020, 38, e17564. [Google Scholar] [CrossRef]
- Sinha, M.; Zhang, L.; Subudhi, S.; Chen, B.; Marquez, J.; Liu, E.V.; Allaire, K.; Cheung, A.; Ng, S.; Nguyen, C.; et al. Preexisting immune status associated with response to combination of sipuleucel-T and ipilimumab in patients with metastatic castration-resistant prostate cancer. J. Immunother. Cancer 2021, 9, e002254. [Google Scholar] [CrossRef]
- Zamarin, D.; Walderich, S.; Holland, A.; Zhou, Q.; Iasonos, A.E.; Torrisi, J.M.; Merghoub, T.; Chesebrough, L.F.; Mcdonnell, A.S.; Gallagher, J.M.; et al. Safety, immunogenicity, and clinical efficacy of durvalumab in combination with folate receptor alpha vaccine TPIV200 in patients with advanced ovarian cancer: A phase II trial. J. Immunother. Cancer 2020, 8, e000829. [Google Scholar] [CrossRef]
- Kverneland, A.H.; Pedersen, M.; Westergaard, M.C.W.; Nielsen, M.; Borch, T.H.; Olsen, L.R.; Aasbjerg, G.; Santegoets, S.J.; van der Burg, S.H.; Milne, K.; et al. Adoptive cell therapy in combination with checkpoint inhibitors in ovarian cancer. Oncotarget 2020, 11, 2092–2105. [Google Scholar] [CrossRef]
- Akhuba, L.; Tigai, Z.; Shek, D. Where Do We Stand with Immunotherapy for Advanced Pancreatic Ductal Adenocarcinoma: A Synopsis of Clinical Outcomes. Biomedicines 2022, 10, 3196. [Google Scholar] [CrossRef]
- Hoimes, C.J.; Graff, J.N.; Tagawa, S.T.; Hwang, C.; Kilari, D.; Ten Tije, A.J.; Omlin, A.U.; McDermott, R.S.; Vaishampayan, U.N.; Tony Elliott, T.; et al. KEYNOTE-199 cohorts (C) 4 and 5: Phase II study of pembrolizumab (pembro) plus enzalutamide (enza) for enza-resistant metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2020, 38, 5543. [Google Scholar] [CrossRef]
- Conter, H.J.; Shore, N.D.; Berry, W.R.; Fong, P.C.C.; Piulats Rodriguez, J.M.M.; Appleman, L.J.; Todenhöfer, T.; Gravis, G.; Laguerre, B.; Gurney, H.; et al. Pembrolizumab (pembro) plus enzalutamide (enza) in patients (pts) with abiraterone acetate (abi)-pretreated metastatic castration-resistant prostate cancer (mCRPC): KEYNOTE-365 cohort C efficacy, safety, and biomarker results. J. Clin. Oncol. 2020, 38, 5545. [Google Scholar] [CrossRef]
- Beatty, G.L.; O’Hara, M.H.; Lacey, S.F.; Torigian, D.A.; Nazimuddin, F.; Chen, F.; Kulikovskaya, I.M.; Soulen, M.C.; McGarvey, M.; Nelson, A.M.; et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology 2018, 155, 29–32. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J. Phase I study of CRISPR-engineered CAR-T cells with PD-1 inactivation in treating mesothelin-positive solid tumors. J. Clin. Oncol. 2020, 38 (Suppl. S15), 3038. [Google Scholar]
- Wang, Z.; Li, N.; Feng, K.; Chen, M.; Zhang, Y.; Liu, Y.; Yang, Q.; Nie, J.; Tang, N.; Zhang, X.; et al. Phase I study of CAR-T cells with PD-1 and TCR disruption in mesothelin-positive solid tumors. Cell. Mol. Immunol. 2021, 18, 2188–2198. [Google Scholar] [PubMed]
- Aiken, R.; Chen, C.; Cloughesy, T.; Colman, H.; Daras, M.; Groves, M.; Khagi, S.; Kumthekar, P.; Lang, F.; Nassiri, F.; et al. Atim-33. Interim results of a phase II multi-center study of oncolytic adenovirus Dnx-2401 with pembrolizumab for recurrent glioblastoma; captive study (Keynote-192). Neuro-Oncology 2019, 21, vi8–vi9. [Google Scholar] [CrossRef]
- Fundora Ramos, M.I.; Maden, L.B.; Casanova, F.O.; Cruz, F.H.; Reyes, C.S.; Gato, A.H.; Lyncon, I.B.; González, E.V.; Morales, K.P.; Lence, J.J.; et al. Oncoxin-Viusid(®) may improve quality of life and survival in patients with hormone-refractory prostate cancer undergoing oncospecific treatments. Mol. Clin. Oncol. 2021, 14, 5. [Google Scholar] [PubMed]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. JCO 2018, 36, 1714–1768. [Google Scholar]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy. N. Engl. J. Med. 2018, 379, 64–73. [Google Scholar] [CrossRef]
- U.S. Department of Health and Human Services; National Institute of Health; National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) Version 5.0. Available online: https://Ctep.Cancer.Gov/Protocoldevelopment/Electronic_applications/Ctc.Htm (accessed on 27 November 2017).
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric Antigen Receptor T-Cell Therapy—Assessment and Management of Toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar]
- Alvi, R.M.; Frigault, M.J.; Fradley, M.G.; Jain, M.D.; Mahmood, S.S.; Awadalla, M.; Lee, D.H.; Zlotoff, D.A.; Zhang, L.; Drobni, Z.D.; et al. Cardiovascular Events Among Adults Treated with Chimeric Antigen Receptor T-Cells (CAR-T). J. Am. Coll. Cardiol. 2019, 74, 3099–3108. [Google Scholar] [CrossRef]
- Lefebvre, B.; Kang, Y.; Smith, A.M.; Frey, N.V.; Carver, J.R.; Scherrer-Crosbie, M. Cardiovascular Effects of CAR T Cell Therapy. JACC CardioOncol. 2020, 2, 193–203. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Chen, D.H.; Guha, A.; Mackenzie, S.; Walker, J.M.; Roddie, C. CAR T Cell Therapy–Related Cardiovascular Outcomes and Management. JACC CardioOncol. 2020, 2, 97–109. [Google Scholar] [CrossRef]
- Ganatra, S.; Redd, R.; Hayek, S.S.; Parikh, R.; Azam, T.; Yanik, G.A.; Spendley, L.; Nikiforow, S.; Jacobson, C.; Nohria, A. Chimeric Antigen Receptor T-Cell Therapy–Associated Cardiomyopathy in Patients With Refractory or Relapsed Non-Hodgkin Lymphoma. Circulation 2020, 142, 1687–1690. [Google Scholar] [CrossRef] [PubMed]
- Pathan, N.; Hemingway, C.A.; Alizadeh, A.A.; Stephens, A.C.; Boldrick, J.C.; Oragui, E.E.; McCabe, C.; Welch, S.B.; Whitney, A.; O’Gara, P.; et al. Role of Interleukin 6 in Myocardial Dysfunction of Meningococcal Septic Shock. Lancet 2004, 363, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Totzeck, M.; Michel, L.; Lin, Y.; Herrmann, J.; Rassaf, T. Cardiotoxicity from Chimeric Antigen Receptor-T Cell Therapy for Advanced Malignancies. Eur. Heart J. 2022, 43, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Wang, V.; Gauthier, M.; Decot, V.; Reppel, L.; Bensoussan, D. Systematic Review on CAR-T Cell Clinical Trials Up to 2022: Academic Center Input. Cancers 2023, 15, 1003. [Google Scholar] [CrossRef]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries on the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Larroquette, M.; Guegan, J.P.; Besse, B.; Cousin, S.; Brunet, M.; Le Moulec, S.; Le Loarer, F.; Rey, C.; Soria, J.C.; Barlesi, F.; et al. Spatial transcriptomics of macrophage infiltration in non- small cell lung cancer reveals determinants of sensitivity and resistance to anti- PD1/ PD- L1 antibodies. J. ImmunoTher. Cancer 2022, 10, e003890. [Google Scholar] [CrossRef]
- Chai, L.F.; Prince, E.; Pillarisetty, V.G.; Katz, S.C. Challenges in assessing solid tumor responses to immunotherapy. Cancer Gene Ther. 2020, 7–8, 528–538. [Google Scholar] [CrossRef]
- Bai, R.; Cui, J. Development of Immunotherapy Strategies Targeting Tumor Microenvironment Is Fiercely Ongoing. Front. Immunol. 2022, 13, 890166. [Google Scholar] [CrossRef]
- Bader, J.E.; Voss, K.; Rathmell, J.C. Targeting Metabolism to Improve the Tumor Microenvironment for Cancer Immunotherapy. Mol. Cell. 2020, 78, 1019–1033. [Google Scholar] [CrossRef]
- Murciano-Goroff, Y.R.; Warner, A.B.; Wolchok, J.D. The future of cancer immunotherapy: Microenvironment targeting combinations. Cell Res. 2020, 30, 507–519. [Google Scholar] [CrossRef]
- Tan, A.C.; Bagley, S.J.; Wen, P.Y.; Lim, M.; Platten, M.; Colman, H.; Ashley, D.M.; Wick, W.; Chang, S.M.; Galanis, E.; et al. Systematic review of combinations of targeted or immunotherapy in advanced solid tumors. J. Immunol. Ther. Cancer 2021, 9, e002459. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Parkes, E.E.; Peng, W.; Moyers, J.T.; Curran, M.A.; Tawbi, H.A. Development of Immunotherapy Combination Strategies in Cancer. Cancer Discov. 2021, 11, 1368–1397. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.J.; Svensson-Arvelund, J.; Lubitz, G.S.; Marabelle, A.; Melero, I.; Brown, B.D.; Brody, J.D. Cancer vaccines: The next immunotherapy frontier. Nat. Cancer 2022, 3, 911–926. [Google Scholar] [CrossRef] [PubMed]
- Baraniuk, C. When will the world get cancer vaccines? BMJ 2023, 6, o3041. [Google Scholar] [CrossRef] [PubMed]
- Alburquerque-González, B.; López-Abellán, M.D.; Luengo-Gil, G.; Montoro-García, S.; Conesa-Zamora, P. Design of Personalized Neoantigen RNA Vaccines Against Cancer Based on Next-Generation Sequencing Data. In Pharmacogenomics in Drug Discovery and Development; Part of the Methods in Molecular Biology Book Series; Springer: New York, NY, USA, 2022; Volume 2547, pp. 165–185. [Google Scholar]
- Wang, D.R.; Wu, X.L.; Sun, Y.L. Therapeutic targets and biomarkers of tumor immunotherapy: Response versus non-response. Signal Trans. Target Ther. 2022, 7, 331. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Le, D.T. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 373, 1979. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Gopalakrishnan, V.; Helmink, B.A.; Spencer, C.N.; Reuben, A.; Wargo, J.A. The Influence of the Gut Microbiome on Cancer, Immunity, and Cancer Immunotherapy. Cancer Cell 2018, 33, 570–580. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lauriola, A.; Davalli, P.; Marverti, G.; Santi, S.; Caporali, A.; D’Arca, D. Targeting the Interplay of Independent Cellular Pathways and Immunity: A Challenge in Cancer Immunotherapy. Cancers 2023, 15, 3009. https://doi.org/10.3390/cancers15113009
Lauriola A, Davalli P, Marverti G, Santi S, Caporali A, D’Arca D. Targeting the Interplay of Independent Cellular Pathways and Immunity: A Challenge in Cancer Immunotherapy. Cancers. 2023; 15(11):3009. https://doi.org/10.3390/cancers15113009
Chicago/Turabian StyleLauriola, Angela, Pierpaola Davalli, Gaetano Marverti, Spartaco Santi, Andrea Caporali, and Domenico D’Arca. 2023. "Targeting the Interplay of Independent Cellular Pathways and Immunity: A Challenge in Cancer Immunotherapy" Cancers 15, no. 11: 3009. https://doi.org/10.3390/cancers15113009
APA StyleLauriola, A., Davalli, P., Marverti, G., Santi, S., Caporali, A., & D’Arca, D. (2023). Targeting the Interplay of Independent Cellular Pathways and Immunity: A Challenge in Cancer Immunotherapy. Cancers, 15(11), 3009. https://doi.org/10.3390/cancers15113009