
Molecular Biology of Pediatric and Adult Ovarian Germ Cell Tumors: A Review
 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
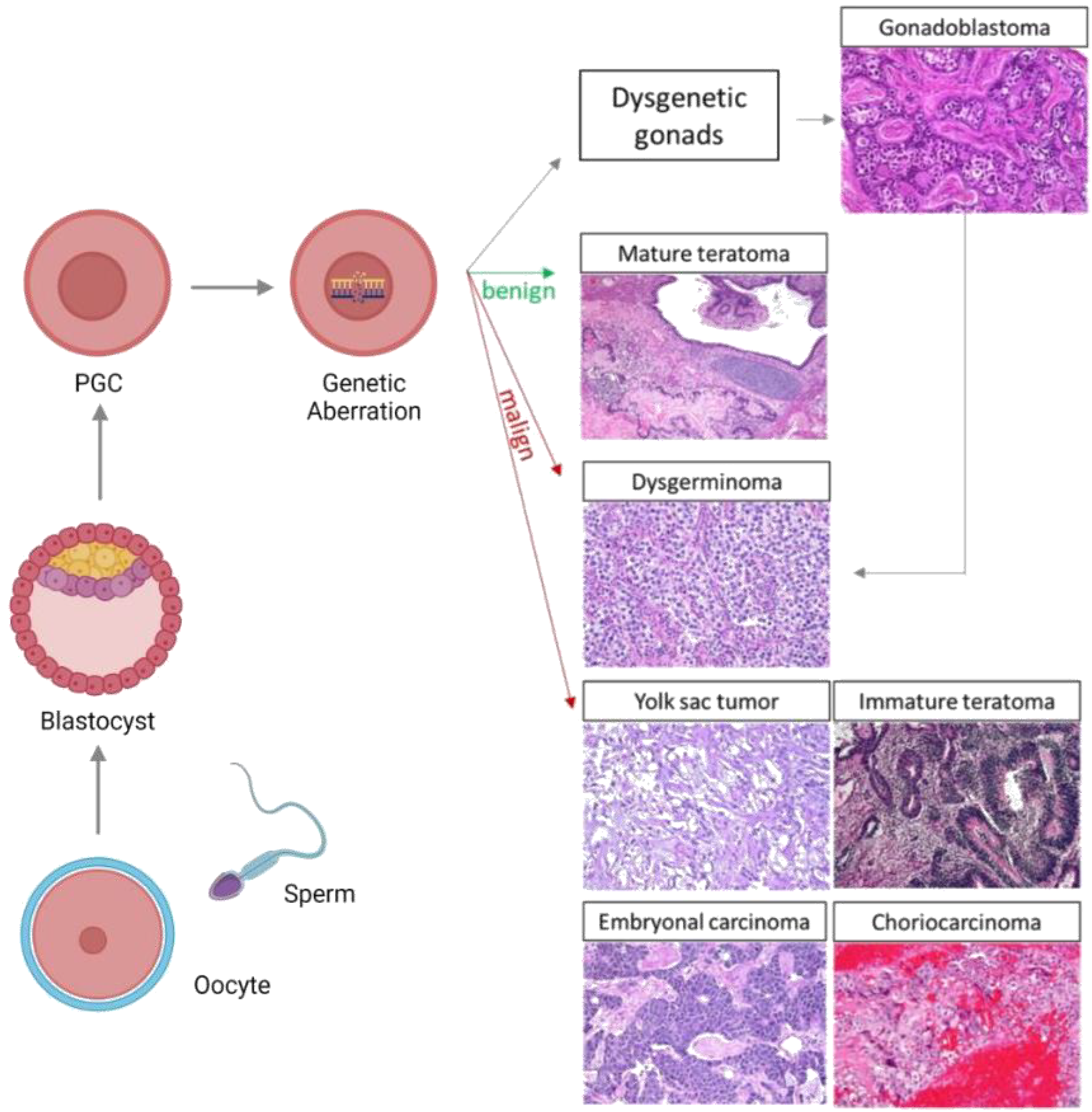
2. Etiopathogenesis of Ovarian Germ Cell Tumors
2.1. Gonadal Dysgenesis
2.2. General Features of Embryonic and Gonadal Development
2.3. OGCT Development
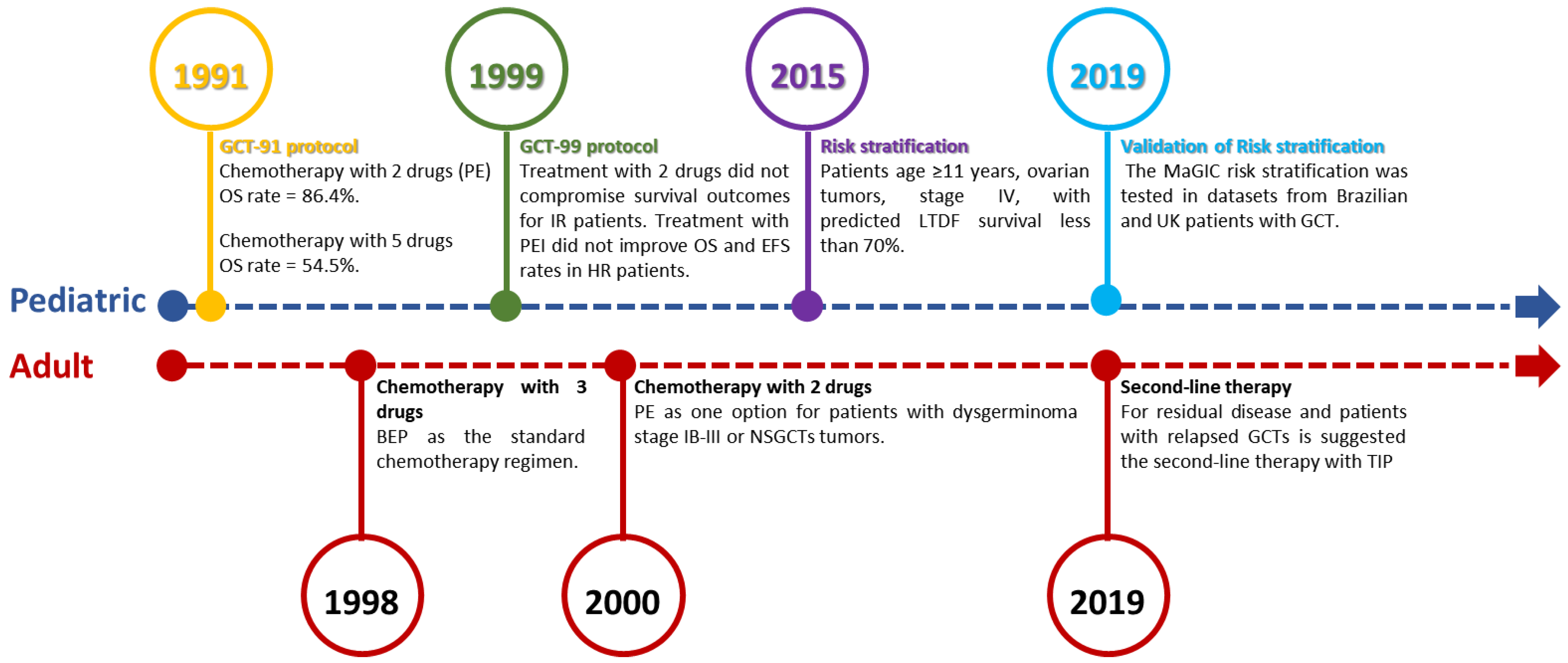
3. Search for Better Therapeutic Approaches

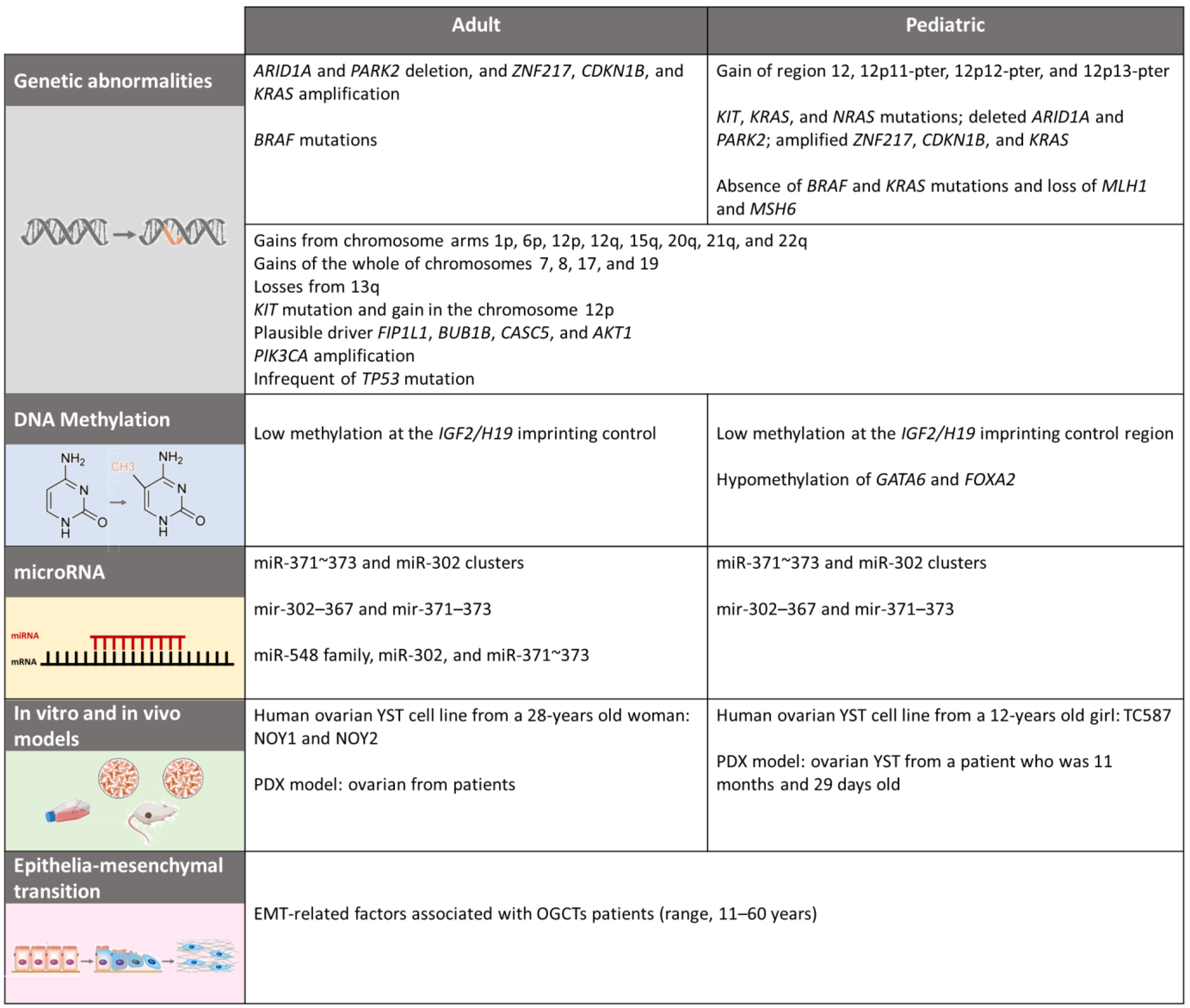
4. Molecular Biology
4.1. Genetics and Citogenetics
4.2. Epigenetics
4.2.1. DNA Methylation
4.2.2. MicroRNA
5. In Vitro and In Vivo Models
6. Molecular Implications of Treatment Resistance
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sessa, C.; Schneider, D.T.; Planchamp, F.; Baust, K.; Braicu, E.I.; Concin, N.; Godzinski, J.; McCluggage, W.G.; Orbach, D.; Pautier, P.; et al. ESGO–SIOPE Guidelines for the Management of Adolescents and Young Adults with Non-Epithelial Ovarian Cancers. Lancet Oncol. 2020, 21, e360–e368. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer Statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Morice, P.; Lorusso, D.; Prat, J.; Oaknin, A.; Pautier, P.; Colombo, N. Non-Epithelial Ovarian Cancer: ESMO Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2018, 29, iv1–iv18. [Google Scholar] [CrossRef]
- Maoz, A.; Matsuo, K.; Ciccone, M.A.; Matsuzaki, S.; Klar, M.; Roman, L.D.; Sood, A.K.; Gershenson, D.M. Molecular Pathways and Targeted Therapies for Malignant Ovarian Germ Cell Tumors and Sex Cord–Stromal Tumors: A Contemporary Review. Cancers 2020, 12, 1398. [Google Scholar] [CrossRef]
- Kraggerud, S.M.; Hoei-Hansen, C.E.; Alagaratnam, S.; Skotheim, R.I.; Abeler, V.M.; Rajpert-De Meyts, E.; Lothe, R.A. Molecular Characteristics of Malignant Ovarian Germ Cell Tumors and Comparison with Testicular Counterparts: Implications for Pathogenesis. Endocr. Rev. 2013, 34, 339–376. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Sakamoto, K.; Murakami, K.; Iwasaki, Y.; Kamataki, A.; Kurose, A. Genetic Zygosity of Mature Ovarian Teratomas, Struma Ovarii, and Ovarian Carcinoids. Virchows Arch. 2018, 473, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Van Nieuwenhuysen, E.; Busschaert, P.; Neven, P.; Han, S.N.; Moerman, P.; Liontos, M.; Papaspirou, M.; Kupryjanczyk, J.; Hogdall, C.; Hogdall, E.; et al. The Genetic Landscape of 87 Ovarian Germ Cell Tumors. Gynecol. Oncol. 2018, 151, 61–68. [Google Scholar] [CrossRef]
- Kubota, Y.; Seki, M.; Kawai, T.; Isobe, T.; Yoshida, M.; Sekiguchi, M.; Kimura, S.; Watanabe, K.; Sato-Otsubo, A.; Yoshida, K.; et al. Comprehensive Genetic Analysis of Pediatric Germ Cell Tumors Identifies Potential Drug Targets. Commun. Biol. 2020, 3, 544. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.D.; Foster, N.A.; Vowler, S.L.; Roberts, I.; Thornton, C.M.; Hale, J.P.; Schneider, D.T.; Nicholson, J.C.; Coleman, N. Malignant Germ Cell Tumours of Childhood: New Associations of Genomic Imbalance. Br. J. Cancer 2007, 96, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Faure-Conter, C.; Orbach, D.; Fresneau, B.; Verité, C.; Bonneau, J.; Thebaud, E.; Poirée, M.; Thouvenin, S.; Pluchart, C.; Mure, P.Y.; et al. Disorder of Sex Development with Germ Cell Tumors: Which Is Uncovered First? Pediatr. Blood Cancer 2020, 67. [Google Scholar] [CrossRef]
- Hughes, I.A. Consensus Statement on Management of Intersex Disorders. Arch. Dis. Child. 2005, 91, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Piazza, M.J.; Urbanetz, A.A. Germ Cell Tumors in Dysgenetic Gonads. Clinics 2019, 74, e408. [Google Scholar] [CrossRef] [PubMed]
- Hennes, E.; Zahn, S.; Lopes, L.; Schönberger, S.; Leuschner, I.; Göbel, U.; Calaminus, G.; Schneider, D. Molecular Genetic Analysis of Bilateral Ovarian Germ Cell Tumors. Klin. Padiatr. 2012, 224, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Duhil de Bénazé, G.; Pacquement, H.; Faure-Conter, C.; Patte, C.; Orbach, D.; Corradini, N.; Berger, C.; Sudour-Bonnange, H.; Vérité, C.; Martelli, H.; et al. Paediatric Dysgerminoma: Results of Three Consecutive French Germ Cell Tumours Clinical Studies (TGM-85/90/95) with Late Effects Study. Eur. J. Cancer 2018, 91, 30–37. [Google Scholar] [CrossRef]
- Capito, C.; Leclair, M.-D.; Arnaud, A.; David, A.; Baron, S.; Corradini, N.; Héloury, Y. 46,XY Pure Gonadal Dysgenesis: Clinical Presentations and Management of the Tumor Risk. J. Pediatr. Urol. 2011, 7, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.Y.; Bryant, S.; Miller, D.S.; Kehoe, S.M.; Richardson, D.L.; Lea, J.S. Malignant Ovarian Germ Cell Tumor—Role of Surgical Staging and Gonadal Dysgenesis. Gynecol. Oncol. 2014, 134, 84–89. [Google Scholar] [CrossRef]
- Scully, R.E. Gonadoblastoma. A Review of 74 Cases. Cancer 1970, 25, 1340–1356. [Google Scholar] [CrossRef]
- Hersmus, R.; Stoop, H.; van de Geijn, G.J.; Eini, R.; Biermann, K.; Oosterhuis, J.W.; DHooge, C.; Schneider, D.T.; Meijssen, I.C.; Dinjens, W.N.M.; et al. Prevalence of C-KIT Mutations in Gonadoblastoma and Dysgerminomas of Patients with Disorders of Sex Development (DSD) and Ovarian Dysgerminomas. PLoS ONE 2012, 7, e43952. [Google Scholar] [CrossRef]
- Cools, M.; Stoop, H.; Kersemaekers, A.-M.F.; Drop, S.L.S.; Wolffenbuttel, K.P.; Bourguignon, J.-P.; Slowikowska-Hilczer, J.; Kula, K.; Faradz, S.M.H.; Oosterhuis, J.W.; et al. Gonadoblastoma Arising in Undifferentiated Gonadal Tissue within Dysgenetic Gonads. J. Clin. Endocrinol. Metab. 2006, 91, 2404–2413. [Google Scholar] [CrossRef]
- Looijenga, L.H.J.; Stoop, H.; de Leeuw, H.P.J.C.; de Gouveia Brazao, C.A.; Gillis, A.J.M.; van Roozendaal, K.E.P.; van Zoelen, E.J.J.; Weber, R.F.A.; Wolffenbuttel, K.P.; van Dekken, H.; et al. POU5F1 (OCT3/4) Identifies Cells with Pluripotent Potential in Human Germ Cell Tumors. Cancer Res. 2003, 63, 2244–2250. [Google Scholar]
- Dolci, S.; Campolo, F.; De Felici, M. Gonadal Development and Germ Cell Tumors in Mouse and Humans. Semin. Cell. Dev. Biol. 2015, 45, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, J.W.; Looijenga, L.H.J. Human Germ Cell Tumours from a Developmental Perspective. Nat. Rev. Cancer 2019, 19, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Hackett, J.A.; Surani, M.A. Regulatory Principles of Pluripotency: From the Ground State Up. Cell Stem Cell 2014, 15, 416–430. [Google Scholar] [CrossRef] [PubMed]
- Runyan, C.; Schaible, K.; Molyneaux, K.; Wang, Z.; Levin, L.; Wylie, C. Steel Factor Controls Midline Cell Death of Primordial Germ Cells and Is Essential for Their Normal Proliferation and Migration. Development 2006, 133, 4861–4869. [Google Scholar] [CrossRef]
- Anderson, R.A.; Fulton, N.; Cowan, G.; Coutts, S.; Saunders, P.T. Conserved and Divergent Patterns of Expression of DAZL, VASA and OCT4 in the Germ Cells of the Human Fetal Ovary and Testis. BMC Dev. Biol 2007, 7, 136. [Google Scholar] [CrossRef]
- Schneider, D.T.; Schuster, A.E.; Fritsch, M.K.; Hu, J.; Olson, T.; Lauer, S.; Göbel, U.; Perlman, E.J. Multipoint Imprinting Analysis Indicates a Common Precursor Cell for Gonadal and Nongonadal Pediatric Germ Cell Tumors. Cancer Res. 2001, 61, 7268–7276. [Google Scholar]
- Ross, J.A.; Schmidt, P.T.; Perentesis, J.P.; Davies, S.M. Genomic Imprinting of H19 and Insulin-like Growth Factor-2 in Pediatric Germ Cell Tumors. Cancer 1999, 85, 1389–1394. [Google Scholar] [CrossRef]
- Teilum, G. Classification of Endodermal Sinus Tumour (Mesoblastoma Vitellinum) and so-called “Embryonal Carcinoma” of the Ovary. Acta Pathol. Microbiol. Scand. 1965, 64, 407–429. [Google Scholar] [CrossRef]
- Shaikh, F.; Murray, M.J.; Amatruda, J.F.; Coleman, N.; Nicholson, J.C.; Hale, J.P.; Pashankar, F.; Stoneham, S.J.; Poynter, J.N.; Olson, T.A.; et al. Paediatric Extracranial Germ-Cell Tumours. Lancet Oncol. 2016, 17, e149–e162. [Google Scholar] [CrossRef]
- Bryant, C.S.; Kumar, S.; Shah, J.P.; Mahdi, H.; Ali-Fehmi, R.; Munkarah, A.R.; Deppe, G.; Morris, R.T. Racial Disparities in Survival among Patients with Germ Cell Tumors of the Ovary—United States. Gynecol. Oncol. 2009, 114, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.O.; Berwick, M.; Verschraegen, C.F.; Wiggins, C.; Lansing, L.; Muller, C.Y.; Qualls, C.R. Incidence and Survival Rates for Female Malignant Germ Cell Tumors. Obstet. Gynecol. 2006, 107, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Euscher, E.D. Germ Cell Tumors of the Female Genital Tract. Surg. Pathol. Clin. 2019, 12, 621–649. [Google Scholar] [CrossRef]
- Veneris, J.T.; Mahajan, P.; Frazier, A.L. Contemporary Management of Ovarian Germ Cell Tumors and Remaining Controversies. Gynecol. Oncol. 2020, 158, 467–475. [Google Scholar] [CrossRef] [PubMed]
- Heskett, M.B.; Sanborn, J.Z.; Boniface, C.; Goode, B.; Chapman, J.; Garg, K.; Rabban, J.T.; Zaloudek, C.; Benz, S.C.; Spellman, P.T.; et al. Multiregion Exome Sequencing of Ovarian Immature Teratomas Reveals 2N Near-Diploid Genomes, Paucity of Somatic Mutations, and Extensive Allelic Imbalances Shared across Mature, Immature, and Disseminated Components. Mod. Pathol. 2020, 33, 1193–1206. [Google Scholar] [CrossRef]
- Pen-in, L.C.; Low, J.; Nicklin, J.L.; Ward, B.G.; Crandon, A.J. Fertility and Ovarian Function After Conservative Surgery for Germ Cell Tumours of the Ovary. Aust. N. Z. J. Obs. Gynaecol. 1999, 39, 243–245. [Google Scholar] [CrossRef] [PubMed]
- Meisel, J.L.; Woo, K.M.; Sudarsan, N.; Eng, J.; Patil, S.; Jacobsen, E.P.; Murali, R.; Gardner, G.J.; Bosl, G.J.; Aghajanian, C.; et al. Development of a Risk Stratification System to Guide Treatment for Female Germ Cell Tumors. Gynecol. Oncol. 2015, 138, 566–572. [Google Scholar] [CrossRef]
- Williams, S.D.; Blessing, J.A.; DiSaia, P.J.; Major, F.J.; Ball, H.G.; Liao, S.-Y. Second-Look Laparotomy in Ovarian Germ Cell Tumors: The Gynecologic Oncology Group Experience. Gynecol. Oncol. 1994, 52, 287–291. [Google Scholar] [CrossRef]
- De Giorgi, U.; Casadei, C.; Bergamini, A.; Attademo, L.; Cormio, G.; Lorusso, D.; Pignata, S.; Mangili, G. Therapeutic Challenges for Cisplatin-Resistant Ovarian Germ Cell Tumors. Cancers 2019, 11, 1584. [Google Scholar] [CrossRef]
- Lopes, L.F.; Macedo, C.R.P.; Pontes, E.M.; dos Santos Aguiar, S.; Mastellaro, M.J.; Melaragno, R.; Vianna, S.M.R.; Lopes, P.A.A.; Mendonça, N.; de Assis Almeida, M.T.; et al. Cisplatin and Etoposide in Childhood Germ Cell Tumor: Brazilian Pediatric Oncology Society Protocol GCT-91. J. Clin. Oncol. 2009, 27, 1297–1303. [Google Scholar] [CrossRef]
- Lopes, L.F.; Macedo, C.R.P.D.; Aguiar, S.d.S.; Barreto, J.H.S.; Martins, G.E.; Sonaglio, V.; Milone, M.; Lima, E.R.; Almeida, M.T.d.A.; Lopes, P.M.A.A.; et al. Lowered Cisplatin Dose and No Bleomycin in the Treatment of Pediatric Germ Cell Tumors: Results of the GCT-99 Protocol from the Brazilian Germ Cell Pediatric Oncology Cooperative Group. J. Clin. Oncol. 2016, 34, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Frazier, A.L.; Hale, J.P.; Rodriguez-Galindo, C.; Dang, H.; Olson, T.; Murray, M.J.; Amatruda, J.F.; Thornton, C.; Arul, G.S.; Billmire, D.; et al. Revised Risk Classification for Pediatric Extracranial Germ Cell Tumors Based on 25 Years of Clinical Trial Data from the United Kingdom and United States. J. Clin. Oncol. 2015, 33, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Mangili, G.; Giorda, G.; Ferrandina, G.; Cormio, G.; Cassani, C.; Savarese, A.; Danese, S.; Carnelli, M.; Vasta, F.M.; Perrone, A.M.; et al. Surveillance Alone in Stage I Malignant Ovarian Germ Cell Tumors: A MITO (Multicenter Italian Trials in Ovarian Cancer) Prospective Observational Study. Int. J. Gynecol. Cancer 2021, 31, 1242–1247. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.D.; Blessing, J.A.; Hatch, K.D.; Homesley, H.D. Chemotherapy of Advanced Dysgerminoma: Trials of the Gynecologic Oncology Group. J. Clin. Oncol. 1991, 9, 1950–1955. [Google Scholar] [CrossRef]
- Vicus, D.; Beiner, M.E.; Clarke, B.; Klachook, S.; Le, L.W.; Laframboise, S.; Mackay, H. Ovarian Immature Teratoma: Treatment and Outcome in a Single Institutional Cohort. Gynecol. Oncol. 2011, 123, 50–53. [Google Scholar] [CrossRef]
- Mangili, G.; Scarfone, G.; Gadducci, A.; Sigismondi, C.; Ferrandina, G.; Scibilia, G.; Viganò, R.; Tateo, S.; Villa, A.; Lorusso, D. Is Adjuvant Chemotherapy Indicated in Stage I Pure Immature Ovarian Teratoma (IT)? A Multicentre Italian Trial in Ovarian Cancer (MITO-9). Gynecol. Oncol. 2010, 119, 48–52. [Google Scholar] [CrossRef]
- Rogers, P.C.; Olson, T.A.; Cullen, J.W.; Billmire, D.F.; Marina, N.; Rescorla, F.; Davis, M.M.; London, W.B.; Lauer, S.J.; Giller, R.H.; et al. Treatment of Children and Adolescents with Stage II Testicular and Stages I and II Ovarian Malignant Germ Cell Tumors: A Pediatric Intergroup Study—Pediatric Oncology Group 9048 and Children’s Cancer Group 8891. J. Clin. Oncol. 2004, 22, 3563–3569. [Google Scholar] [CrossRef]
- Dimopoulos, M.A.; Papadopoulou, M.; Andreopoulou, E.; Papadimitriou, C.; Pavlidis, N.; Aravantinos, G.; Aspropotamitis, A.; Anagnostopoulos, A.; Fountzilas, G.; Michalas, S.; et al. Favorable Outcome of Ovarian Germ Cell Malignancies Treated with Cisplatin or Carboplatin-Based Chemotherapy: A Hellenic Cooperative Oncology Group Study. Gynecol. Oncol. 1998, 70, 70–74. [Google Scholar] [CrossRef]
- Williams, S.D.; Kauderer, J.; Burnett, A.F.; Lentz, S.S.; Aghajanian, C.; Armstrong, D.K. Adjuvant Therapy of Completely Resected Dysgerminoma with Carboplatin and Etoposide: A Trial of the Gynecologic Oncology Group. Gynecol. Oncol. 2004, 95, 496–499. [Google Scholar] [CrossRef]
- Mann, J.R.; Raafat, F.; Robinson, K.; Imeson, J.; Gornall, P.; Sokal, M.; Gray, E.; McKeever, P.; Hale, J.; Bailey, S.; et al. The United Kingdom Children’s Cancer Study Group’s Second Germ Cell Tumor Study: Carboplatin, Etoposide, and Bleomycin Are Effective Treatment for Children With Malignant Extracranial Germ Cell Tumors, With Acceptable Toxicity. J. Clin. Oncol. 2000, 18, 3809–3818. [Google Scholar] [CrossRef]
- Lu, Y.; Yang, J.; Cao, D.; Huang, H.; Wu, M.; You, Y.; Chen, J.; Lang, J.; Shen, K. Role of Neoadjuvant Chemotherapy in the Management of Advanced Ovarian Yolk Sac Tumor. Gynecol. Oncol. 2014, 134, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, D.K.; Alvarez, R.D.; Bakkum-Gamez, J.N.; Barroilhet, L.; Behbakht, K.; Berchuck, A.; Chen, L.; Cristea, M.; DeRosa, M.; Eisenhauer, E.L.; et al. Ovarian Cancer, Version 2.2020, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2021, 19, 191–226. [Google Scholar] [CrossRef] [PubMed]
- Lindsay Frazier, A.; Faure-Conter, C.; Fresneau, B.; Lopes, L.F.; Pashankar, F.; Shaikh, F.; O’Neill, A.F.; Amatruda, J.F.; Krailo, M.; Klosterkemper, L.; et al. Validation of the MaGIC Paediatric Germ Cell Tumour Risk Stratification. Eur. Urol. Suppl. 2019, 18, 20. [Google Scholar] [CrossRef]
- Newton, C.; Murali, K.; Ahmad, A.; Hockings, H.; Graham, R.; Liberale, V.; Sarker, S.-J.; Ledermann, J.; Berney, D.M.; Shamash, J.; et al. A Multicentre Retrospective Cohort Study of Ovarian Germ Cell Tumours: Evidence for Chemotherapy de-Escalation and Alignment of Paediatric and Adult Practice. Eur. J. Cancer 2019, 113, 19–27. [Google Scholar] [CrossRef]
- Pinto, M.T.; Cárcano, F.M.; Vieira, A.G.S.; Cabral, E.R.M.; Lopes, L.F. Molecular Biology of Pediatric and Adult Male Germ Cell Tumors. Cancers 2021, 13, 2349. [Google Scholar] [CrossRef]
- Dharia, N.V.; Kugener, G.; Guenther, L.M.; Malone, C.F.; Durbin, A.D.; Hong, A.L.; Howard, T.P.; Bandopadhayay, P.; Wechsler, C.S.; Fung, I.; et al. A First-Generation Pediatric Cancer Dependency Map. Nat. Genet. 2021, 53, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Masqué-Soler, N.; Szczepanowski, M.; Leuschner, I.; Vokuhl, C.; Haag, J.; Calaminus, G.; Klapper, W. Absence of BRAF Mutation in Pediatric and Adolescent Germ Cell Tumors Indicate Biological Differences to Adult Tumors. Pediatr. Blood Cancer 2012, 59, 732–735. [Google Scholar] [CrossRef]
- Göbel, U.; Schneider, D.T.; Calaminus, G.; Haas, R.J.; Schmidt, P.; Harms, D. Germ-Cell Tumors in Childhood and Adolescence. Ann. Oncol. 2000, 11, 263–272. [Google Scholar] [CrossRef]
- Oosterhuis, J.W.; Looijenga, L.H.J. Testicular Germ-Cell Tumours in a Broader Perspective. Nat. Rev. Cancer 2005, 5, 210–222. [Google Scholar] [CrossRef]
- Mosbech, C.H.; Rechnitzer, C.; Brok, J.S.; Rajpert-De Meyts, E.; Hoei-Hansen, C.E. Recent Advances in Understanding the Etiology and Pathogenesis of Pediatric Germ Cell Tumors. J. Pediatr. Hematol. Oncol. 2014, 36, 263–270. [Google Scholar] [CrossRef]
- Riopel, M.A.; Spellerberg, A.; Griffin, C.A.; Perlman, E.J. Genetic Analysis of Ovarian Germ Cell Tumors by Comparative Genomic Hybridization. Cancer Res. 1998, 58, 3105–3110. [Google Scholar]
- Kraggerud, S.M.; Szymanska, J.; Abeler, V.M.; Kaern, J.; Eknaes, M.; Heim, S.; Teixeira, M.R.; Tropé, C.G.; Peltomäki, P.; Lothe, R.A. DNA Copy Number Changes in Malignant Ovarian Germ Cell Tumors. Cancer Res. 2000, 60, 3025–3030. [Google Scholar]
- Cheng, L.; Zhang, S.; MacLennan, G.T.; Poulos, C.K.; Sung, M.-T.; Beck, S.D.; Foster, R.S. Interphase Fluorescence In Situ Hybridization Analysis of Chromosome 12p Abnormalities Is Useful for Distinguishing Epidermoid Cysts of the Testis from Pure Mature Teratoma. Clin. Cancer Res. 2006, 12, 5668–5672. [Google Scholar] [CrossRef]
- Kernek, K.M.; Brunelli, M.; Ulbright, T.M.; Eble, J.N.; Martignoni, G.; Zhang, S.; Michael, H.; Cummings, O.W.; Cheng, L. Fluorescence in Situ Hybridization Analysis of Chromosome 12p in Paraffin-Embedded Tissue Is Useful for Establishing Germ Cell Origin of Metastatic Tumors. Mod. Pathol. 2004, 17, 1309–1313. [Google Scholar] [CrossRef]
- Zong, X.; Zhang, Y.; Peng, X.; Cao, D.; Yu, M.; Wang, J.; Li, H.; Guo, X.; Liang, H.; Yang, J. Analysis of the Genomic Landscape of Yolk Sac Tumors Reveals Mechanisms of Evolution and Chemoresistance. Nat. Commun. 2021, 12, 3579. [Google Scholar] [CrossRef]
- André, F.; Arnedos, M.; Baras, A.S.; Baselga, J.; Bedard, P.L.; Berger, M.F.; Bierkens, M.; Calvo, F.; Cerami, E.; Chakravarty, D.; et al. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef]
- Morfouace, M.; Stevovic, A.; Vinches, M.; Golfinopoulos, V.; Jin, D.X.; Holmes, O.; Erlich, R.; Fayette, J.; Croce, S.; Ray-Coquard, I.; et al. First Results of the EORTC-SPECTA/Arcagen Study Exploring the Genomics of Rare Cancers in Collaboration with the European Reference Network EURACAN. ESMO Open 2020, 5, e001075. [Google Scholar] [CrossRef] [PubMed]
- Korn, W.M.; Weghuis, D.E.M.O.; Suijkerbuijk, R.F.; Schmidt, U.; Otto, T.; du Manoir, S.; van Kessel, A.G.; Harstrick, A.; Seeber, S.; Becher, R. Detection of Chromosomal DNA Gains and Losses in Testicular Germ Cell Tumors by Comparative Genomic Hybridization. Genes Chromosomes Cancer 1996, 17, 78–87. [Google Scholar] [CrossRef]
- Fritsch, M.K.; Schneider, D.T.; Schuster, A.E.; Murdoch, F.E.; Perlman, E.J. Activation of Wnt/β-Catenin Signaling in Distinct Histologic Subtypes of Human Germ Cell Tumors. Pediatr. Dev. Pathol. 2006, 9, 115–131. [Google Scholar] [CrossRef]
- Fustino, N.; Rakheja, D.; Ateek, C.S.; Neumann, J.C.; Amatruda, J.F. Bone Morphogenetic Protein Signalling Activity Distinguishes Histological Subsets of Paediatric Germ Cell Tumours. Int. J. Androl. 2011, 34, e218–e233. [Google Scholar] [CrossRef] [PubMed]
- Honecker, F.; Wermann, H.; Mayer, F.; Gillis, A.J.M.; Stoop, H.; van Gurp, R.J.L.M.; Oechsle, K.; Steyerberg, E.; Hartmann, J.T.; Dinjens, W.N.M.; et al. Microsatellite Instability, Mismatch Repair Deficiency, and BRAF Mutation in Treatment-Resistant Germ Cell Tumors. J. Clin. Oncol. 2009, 27, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-C.; Baylin, S.B. Cancer Epigenetics: Linking Basic Biology to Clinical Medicine. Cell Res. 2011, 21, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Lo, P.-K.; Sukumar, S. Epigenomics and Breast Cancer. Pharmacogenomics 2008, 9, 1879–1902. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of Epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef]
- Rodenhiser, D. Epigenetics and Human Disease: Translating Basic Biology into Clinical Applications. Can. Med. Assoc. J. 2006, 174, 341–348. [Google Scholar] [CrossRef]
- Esteller, M. Epigenetics in Cancer. N. Engl. J. Med. 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Amatruda, J.F.; Ross, J.A.; Christensen, B.; Fustino, N.J.; Chen, K.S.; Hooten, A.J.; Nelson, H.; Kuriger, J.K.; Rakheja, D.; Frazier, A.L.; et al. DNA Methylation Analysis Reveals Distinct Methylation Signatures in Pediatric Germ Cell Tumors. BMC Cancer 2013, 13, 313. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.A.; Mills, L.; Hooten, A.J.; Langer, E.; Roesler, M.; Frazier, A.L.; Krailo, M.; Nelson, H.H.; Bestrashniy, J.; Amatruda, J.F.; et al. Differences in DNA Methylation Profiles by Histologic Subtype of Paediatric Germ Cell Tumours: A Report from the Children’s Oncology Group. Br. J. Cancer 2018, 119, 864–872. [Google Scholar] [CrossRef]
- Wermann, H.; Stoop, H.; Gillis, A.J.; Honecker, F.; van Gurp, R.J.; Ammerpohl, O.; Richter, J.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H. Global DNA Methylation in Fetal Human Germ Cells and Germ Cell Tumours: Association with Differentiation and Cisplatin Resistance. J. Pathol. 2010, 221, 433–442. [Google Scholar] [CrossRef]
- Yu, X.; Han, Y.; Liu, S.; Jiang, W.; Song, Y.; Tong, J.; Qiao, T.; Lv, Z.; Li, D. Analysis of Genetic Alterations Related to DNA Methylation in Testicular Germ Cell Tumors Based on Data Mining. Cytogenet. Genome Res. 2021, 161, 382–394. [Google Scholar] [CrossRef]
- Koul, S.; Houldsworth, J.; Mansukhani, M.M.; Donadio, A.; McKiernan, J.M.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S.; Murty, V.V. Characteristic Promoter Hypermethylation Signatures in Male Germ Cell Tumors. Mol. Cancer 2002, 1, 8. [Google Scholar] [CrossRef]
- Rainier, S.; Dobry, C.J.; Feinberg, A.P. Loss of Imprinting in Hepatoblastoma. Cancer Res. 1995, 55, 1836–1838. [Google Scholar] [PubMed]
- Sievers, S.; Alemazkour, K.; Zahn, S.; Perlman, E.J.; Gillis, A.J.M.; Looijenga, L.H.J.; Göbel, U.; Schneider, D.T. IGF2/H19 Imprinting Analysis of Human Germ Cell Tumors (GCTs) Using the Methylation-Sensitive Single-Nucleotide Primer Extension Method Reflects the Origin of GCTs in Different Stages of Primordial Germ Cell Development. Genes Chromosomes Cancer 2005, 44, 256–264. [Google Scholar] [CrossRef]
- Shivdasani, R.A. MicroRNAs: Regulators of Gene Expression and Cell Differentiation. Blood 2006, 108, 3646–3653. [Google Scholar] [CrossRef] [PubMed]
- Dykxhoorn, D.M. MicroRNAs and Metastasis: Little RNAs Go a Long Way. Cancer Res. 2010, 70, 6401–6406. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Das, M.K.; Evensen, H.S.F.; Furu, K.; Haugen, T.B. MiRNA-302s May Act as Oncogenes in Human Testicular Germ Cell Tumours. Sci. Rep. 2019, 9, 9189. [Google Scholar] [CrossRef]
- Sedaghat, N.; Fathy, M.; Modarressi, M.H.; Shojaie, A. Identifying Functional Cancer-Specific MiRNA–MRNA Interactions in Testicular Germ Cell Tumor. J. Theor. Biol. 2016, 404, 82–96. [Google Scholar] [CrossRef]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.M.; Stoop, H.; Nagel, R.; Liu, Y.-P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A Genetic Screen Implicates MiRNA-372 and MiRNA-373 as Oncogenes in Testicular Germ Cell Tumors. In Advances in Molecular Oncology; d’Adda di Fagagna, F., Chiocca, S., McBlane, F., Cavallaro, U., Eds.; Springer: New York, NY, USA, 2007; pp. 17–46. [Google Scholar]
- Palmer, R.D.; Murray, M.J.; Saini, H.K.; van Dongen, S.; Abreu-Goodger, C.; Muralidhar, B.; Pett, M.R.; Thornton, C.M.; Nicholson, J.C.; Enright, A.J.; et al. Malignant Germ Cell Tumors Display Common MicroRNA Profiles Resulting in Global Changes in Expression of Messenger RNA Targets. Cancer Res. 2010, 70, 2911–2923. [Google Scholar] [CrossRef]
- Syring, I.; Bartels, J.; Holdenrieder, S.; Kristiansen, G.; Müller, S.C.; Ellinger, J. Circulating Serum MiRNA (MiR-367-3p, MiR-371a-3p, MiR-372-3p and MiR-373-3p) as Biomarkers in Patients with Testicular Germ Cell Cancer. J. Urol. 2015, 193, 331–337. [Google Scholar] [CrossRef]
- Dieckmann, K.-P.; Radtke, A.; Geczi, L.; Matthies, C.; Anheuser, P.; Eckardt, U.; Sommer, J.; Zengerling, F.; Trenti, E.; Pichler, R.; et al. Serum Levels of MicroRNA-371a-3p (M371 Test) as a New Biomarker of Testicular Germ Cell Tumors: Results of a Prospective Multicentric Study. J. Clin. Oncol. 2019, 37, 1412–1423. [Google Scholar] [CrossRef]
- Murray, M.J.; Halsall, D.J.; Hook, C.E.; Williams, D.M.; Nicholson, J.C.; Coleman, N. Identification of MicroRNAs from the MiR-371–373 and MiR-302 Clusters as Potential Serum Biomarkers of Malignant Germ Cell Tumors. Am. J. Clin. Pathol. 2011, 135, 119–125. [Google Scholar] [CrossRef]
- Chang, R.; Li, X.; Mu, N.; Hrydziuszko, O.; Garcia-Majano, B.; Larsson, C.; Lui, W.-O. MicroRNA Expression Profiles in Non-epithelial Ovarian Tumors. Int. J. Oncol. 2017. [Google Scholar] [CrossRef]
- Port, M.; Glaesener, S.; Ruf, C.; Riecke, A.; Bokemeyer, C.; Meineke, V.; Honecker, F.; Abend, M. Micro-RNA Expression in Cisplatin Resistant Germ Cell Tumor Cell Lines. Mol. Cancer 2011, 10, 52. [Google Scholar] [CrossRef]
- Gillis, A.; Stoop, H.; Hersmus, R.; Oosterhuis, J.; Sun, Y.; Chen, C.; Guenther, S.; Sherlock, J.; Veltman, I.; Baeten, J.; et al. High-Throughput MicroRNAome Analysis in Human Germ Cell Tumours. J. Pathol. 2007, 213, 319–328. [Google Scholar] [CrossRef]
- Murray, M.J.; Saini, H.K.; van Dongen, S.; Palmer, R.D.; Muralidhar, B.; Pett, M.R.; Piipari, M.; Thornton, C.M.; Nicholson, J.C.; Enright, A.J.; et al. The Two Most Common Histological Subtypes of Malignant Germ Cell Tumour Are Distinguished by Global MicroRNA Profiles, Associated with Differential Transcription Factor Expression. Mol. Cancer 2010, 9, 290. [Google Scholar] [CrossRef]
- Palmer, R.D.; Barbosa-Morais, N.L.; Gooding, E.L.; Muralidhar, B.; Thornton, C.M.; Pett, M.R.; Roberts, I.; Schneider, D.T.; Thorne, N.; Tavaré, S.; et al. Pediatric Malignant Germ Cell Tumors Show Characteristic Transcriptome Profiles. Cancer Res. 2008, 68, 4239–4247. [Google Scholar] [CrossRef] [PubMed]
- Masters, J.R.W. Human Cancer Cell Lines: Fact and Fantasy. Nat. Rev. Mol. Cell Biol. 2000, 1, 233–236. [Google Scholar] [CrossRef]
- Sinha, R.; Luna, A.; Schultz, N.; Sander, C. A Pan-Cancer Survey of Cell Line Tumor Similarity by Feature-Weighted Molecular Profiles. Cell Rep. Methods 2021, 1, 100039. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Kajiyama, H.; Yamamoto, E.; Terauchi, M.; Ino, K.; Nawa, A.; Kikkawa, F. Establishment and Characterization of an Ovarian Yolk Sac Tumor Cell Line Reveals Possible Involvement of Nkx2.5 in Tumor Development. Oncology 2008, 74, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Shibata, K.; Umezu, T.; Sakurai, M.; Kajiyama, H.; Yamamoto, E.; Ino, K.; Nawa, A.; Kikkawa, F. Establishment of Cisplatin-Resistant Ovarian Yolk Sac Tumor Cells and Investigation of the Mechanism of Cisplatin Resistance Using This Cell Line. Gynecol. Obs. Investig. 2011, 71, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Schmidtova, S.; Dorssers, L.C.J.; Kalavska, K.; Gillis, A.J.M.; Oosterhuis, J.W.; Stoop, H.; Miklikova, S.; Kozovska, Z.; Burikova, M.; Gercakova, K.; et al. Napabucasin Overcomes Cisplatin Resistance in Ovarian Germ Cell Tumor-Derived Cell Line by Inhibiting Cancer Stemness. Cancer Cell. Int. 2020, 20, 364. [Google Scholar] [CrossRef]
- Mitsui, H.; Shibata, K.; Suzuki, S.; Umezu, T.; Mizuno, M.; Kajiyama, H.; Kikkawa, F. Functional Interaction between Peritoneal Mesothelial Cells and Stem Cells of Ovarian Yolk Sac Tumor (SC-OYST) in Peritoneal Dissemination. Gynecol. Oncol. 2012, 124, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Kohashi, K.; Ohno, M.; Taguchi, T.; Oda, Y. Establishment and Characterization of a Novel Primitive Yolk Sac Tumour Cell Line, TC587. Anticancer Res. 2020, 40, 759–766. [Google Scholar] [CrossRef]
- Piulats, J.M.; Vidal, A.; García-Rodríguez, F.J.; Muñoz, C.; Nadal, M.; Moutinho, C.; Martínez-Iniesta, M.; Mora, J.; Figueras, A.; Guinó, E.; et al. Orthoxenografts of Testicular Germ Cell Tumors Demonstrate Genomic Changes Associated with Cisplatin Resistance and Identify PDMP as a Resensitizing Agent. Clin. Cancer Res. 2018, 24, 3755–3766. [Google Scholar] [CrossRef] [PubMed]
- Kameya, T.; Shimosato, Y.; Tumuraya, M.; Ohsawa, N.; Nomura, T. Human Gastric Choriocarcinoma Serially Transplanted in Nude Mice2. J. Natl. Cancer Inst. 1976, 56, 325–332. [Google Scholar] [CrossRef]
- Shimosato, Y.; Kameya, T.; Nagai, K.; Hirohashi, S.; Koide, T.; Hayashi, H.; Nomura, T. Transplantation of Human Tumors in Nude Mice23. J. Natl. Cancer Inst. 1976, 56, 1251–1260. [Google Scholar] [CrossRef]
- Tamauchi, S.; Suzuki, S.; Xuboya, C.; Yoshihara, M.; Yoshida, K.; Ikeda, Y.; Yoshikawa, N.; Kajiyama, H.; Kikkawa, F. Establishment of a Patient-derived Xenograft Model and Cell Line of Malignant Transformation of Mature Cystic Teratoma of the Ovary. J. Obstet. Gynaecol. Res. 2021, 47, 713–719. [Google Scholar] [CrossRef]
- Weroha, S.J.; Becker, M.A.; Enderica-Gonzalez, S.; Harrington, S.C.; Oberg, A.L.; Maurer, M.J.; Perkins, S.E.; AlHilli, M.; Butler, K.A.; McKinstry, S.; et al. Tumorgrafts as In Vivo Surrogates for Women with Ovarian Cancer. Clin. Cancer Res. 2014, 20, 1288–1297. [Google Scholar] [CrossRef]
- Ricci, F.; Bizzaro, F.; Cesca, M.; Guffanti, F.; Ganzinelli, M.; Decio, A.; Ghilardi, C.; Perego, P.; Fruscio, R.; Buda, A.; et al. Patient-Derived Ovarian Tumor Xenografts Recapitulate Human Clinicopathology and Genetic Alterations. Cancer Res. 2014, 74, 6980–6990. [Google Scholar] [CrossRef]
- Heo, E.J.; Cho, Y.J.; Cho, W.C.; Hong, J.E.; Jeon, H.-K.; Oh, D.-Y.; Choi, Y.-L.; Song, S.Y.; Choi, J.-J.; Bae, D.-S.; et al. Patient-Derived Xenograft Models of Epithelial Ovarian Cancer for Preclinical Studies. Cancer Res. Treat. 2017, 49, 915–926. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; He, Y.; Xie, B.; Li, S.; Gan, F.; Zhang, S.; Luo, P. Establishment and Characterization of an Ovarian Yolk Sac Tumor Patient-Derived Xenograft Model. Pediatr. Surg. Int. 2021, 37, 1031–1040. [Google Scholar] [CrossRef] [PubMed]
- Prestayko, A.W.; D’Aoust, J.C.; Issell, B.F.; Crooke, S.T. Cisplatin (Cis-Diamminedichloroplatinum II). Cancer Treat. Rev. 1979, 6, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Lebwohl, D.; Canetta, R. Clinical Development of Platinum Complexes in Cancer Therapy: An Historical Perspective and an Update. Eur. J. Cancer 1998, 34, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Galanski, M. Recent Developments in the Field of Anticancer Platinum Complexes. Recent Patterns Anti-Cancer Drug Discov. 2006, 1, 285–295. [Google Scholar] [CrossRef]
- Ozols, R.F. Ovarian Cancer: New Clinical Approaches. Cancer Treat. Rev. 1991, 18, 77–83. [Google Scholar] [CrossRef]
- Giaccone, G. Clinical Perspectives on Platinum Resistance. Drugs 2000, 59, 9–17. [Google Scholar] [CrossRef]
- Köberle, B.; Tomicic, M.T.; Usanova, S.; Kaina, B. Cisplatin Resistance: Preclinical Findings and Clinical Implications. Biochim. Biophys. Acta BBA Rev. Cancer 2010, 1806, 172–182. [Google Scholar] [CrossRef]
- Rocha, C.R.R.; Silva, M.M.; Quinet, A.; Cabral-Neto, J.B.; Menck, C.F.M. DNA Repair Pathways and Cisplatin Resistance: An Intimate Relationship. Clinics 2018, 73, e478s. [Google Scholar] [CrossRef]
- Brasseur, K.; Gévry, N.; Asselin, E. Chemoresistance and Targeted Therapies in Ovarian and Endometrial Cancers. Oncotarget 2017, 8, 4008–4042. [Google Scholar] [CrossRef]
- Duan, M.; Ulibarri, J.; Liu, K.J.; Mao, P. Role of Nucleotide Excision Repair in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 9248. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease: Old Views and New Perspectives. Int. J. Dev. Biol. 2009, 53, 1541–1547. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R. A Review Series the Basics of Epithelial-Mesenchymal Transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and BHLH Factors in Tumour Progression: An Alliance against the Epithelial Phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Palma, C.d.S.; Grassi, M.L.; Thomé, C.H.; Ferreira, G.A.; Albuquerque, D.; Pinto, M.T.; Ferreira Melo, F.U.; Kashima, S.; Covas, D.T.; Pitteri, S.J.; et al. Proteomic Analysis of Epithelial to Mesenchymal Transition (EMT) Reveals Cross-Talk between SNAIL and HDAC1 Proteins in Breast Cancer Cells. Mol. Cell Proteom. 2016, 15, 906–917. [Google Scholar] [CrossRef]
- Guo, W.; Keckesova, Z.; Donaher, J.L.; Shibue, T.; Tischler, V.; Reinhardt, F.; Itzkovitz, S.; Noske, A.; Zürrer-Härdi, U.; Bell, G.; et al. Slug and Sox9 Cooperatively Determine the Mammary Stem Cell State. Cell 2012, 148, 1015–1028. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Becker, A.; Zimmer, A.; Lu, J.; Buettner, R.; Kirfel, J. SNAI1-Mediated Epithelial-Mesenchymal Transition Confers Chemoresistance and Cellular Plasticity by Regulating Genes Involved in Cell Death and Stem Cell Maintenance. PLoS ONE 2013, 8, e66558. [Google Scholar] [CrossRef]
- Pinto, F.; Pértega-Gomes, N.; Vizcaíno, J.R.; Andrade, R.P.; Cárcano, F.M.; Reis, R.M. Brachyury as a Potential Modulator of Androgen Receptor Activity and a Key Player in Therapy Resistance in Prostate Cancer. Oncotarget 2016, 7. [Google Scholar] [CrossRef]
- Solheim, O.; Førsund, M.; Tropé, C.G.; Kraggerud, S.M.; Nesland, J.M.; Davidson, B. Epithelial-Mesenchymal Transition Markers in Malignant Ovarian Germ Cell Tumors. J. Pathol. Microbiol. Immunol. 2017, 125, 781–786. [Google Scholar] [CrossRef]
- Vousden, K.H.; Lane, D.P. P53 in Health and Disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 275–283. [Google Scholar] [CrossRef]
- Kirsch, D.G.; Kastan, M.B. Tumor-Suppressor P53: Implications for Tumor Development and Prognosis. J. Clin. Oncol. 1998, 16, 3158–3168. [Google Scholar] [CrossRef] [PubMed]
- Heidenreich, A.; Schenkman, N.S.; Sesterhenn, I.A.; Mostofi, K.F.; Moul, J.W.; Srivastava, S.; Engelmann, U.H. Immunohistochemical and Mutational Analysis of the P53 Tumour Suppressor Gene and the Bcl-2 Oncogene in Primary Testicular Germ Cell Tumours. J. Pathol. Microbiol. Immunol. 1998, 106, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Guillou, L.; Estreicher, A.; Chaubert, P.; Hurlimann, J.; Kurt, A.M.; Metthez, G.; Iggo, R.; Gray, A.C.; Jichlinski, P.; Leisinger, H.J.; et al. Germ Cell Tumors of the Testis Overexpress Wild-Type P53. Am. J. Pathol. 1996, 149, 1221–1228. [Google Scholar] [PubMed]
- Houldsworth, J.; Xiao, H.; Murty, V.; Chen, W.; Ray, B.; Reuter, V.E.; Bosl, G.J.; Chaganti, R. Human Male Germ Cell Tumor Resistance to Cisplatin Is Linked to TP53 Gene Mutation. Oncogene 1998, 16, 2345–2349. [Google Scholar] [CrossRef] [PubMed]
- Gadducci, A.; Cosio, S.; Muraca, S.; Genazzani, A.R. Molecular Mechanisms of Apoptosis and Chemosensitivity to Platinum and Paclitaxel in Ovarian Cancer: Biological Data and Clinical Implications. Eur. J. Gynaecol. Oncol. 2002, 23, 390–396. [Google Scholar]
- Feldman, D.R. Medical Treatment of Advanced Testicular Cancer. JAMA 2008, 299, 672. [Google Scholar] [CrossRef]
- Kersemaekers, A.-M.F.; Mayer, F.; Molier, M.; van Weeren, P.C.; Oosterhuis, J.W.; Bokemeyer, C.; Looijenga, L.H.J. Role of P53 and MDM2 in Treatment Response of Human Germ Cell Tumors. J. Clin. Oncol. 2002, 20, 1551–1561. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, M.T.; Eiras Martins, G.; Vieira, A.G.S.; Galvão, J.M.S.; de Pádua Souza, C.; Macedo, C.R.P.D.; Lopes, L.F. Molecular Biology of Pediatric and Adult Ovarian Germ Cell Tumors: A Review. Cancers 2023, 15, 2990. https://doi.org/10.3390/cancers15112990
Pinto MT, Eiras Martins G, Vieira AGS, Galvão JMS, de Pádua Souza C, Macedo CRPD, Lopes LF. Molecular Biology of Pediatric and Adult Ovarian Germ Cell Tumors: A Review. Cancers. 2023; 15(11):2990. https://doi.org/10.3390/cancers15112990
Chicago/Turabian StylePinto, Mariana Tomazini, Gisele Eiras Martins, Ana Glenda Santarosa Vieira, Janaina Mello Soares Galvão, Cristiano de Pádua Souza, Carla Renata Pacheco Donato Macedo, and Luiz Fernando Lopes. 2023. "Molecular Biology of Pediatric and Adult Ovarian Germ Cell Tumors: A Review" Cancers 15, no. 11: 2990. https://doi.org/10.3390/cancers15112990
APA StylePinto, M. T., Eiras Martins, G., Vieira, A. G. S., Galvão, J. M. S., de Pádua Souza, C., Macedo, C. R. P. D., & Lopes, L. F. (2023). Molecular Biology of Pediatric and Adult Ovarian Germ Cell Tumors: A Review. Cancers, 15(11), 2990. https://doi.org/10.3390/cancers15112990