Simple Summary
Human colorectal cancer (CRC) is a global health burden, most notably for minority populations. While screening tools have helped improve disease detection and surveillance, CRC is a complex disease with a diverse set of molecular features that are linked to the location of the primary tumor. These features provide challenges to treatment and improving patient outcome. In addition, tumor metastases to liver and other organ systems are a main cause of CRC-related mortality and represent a substantial obstacle to improving patient outcomes. In this review, we summarize CRC tumor subgroups, their molecular features, treatments and outcomes with attention to health disparities and tumor metastases.
Abstract
Human colorectal cancer (CRC) is one of the most common malignancies in men and women across the globe, albeit CRC incidence and mortality shows a substantial racial and ethnic disparity, with the highest burden in African American patients. Even with effective screening tools such as colonoscopy and diagnostic detection assays, CRC remains a substantial health burden. In addition, primary tumors located in the proximal (right) or distal (left) sides of the colorectum have been shown to be unique tumor types that require unique treatment schema. Distal metastases in the liver and other organ systems are the major causes of mortality in CRC patients. Characterizing genomic, epigenomic, transcriptomic and proteomic (multi-omics) alterations has led to a better understanding of primary tumor biology, resulting in targeted therapeutic advancements. In this regard, molecular-based CRC subgroups have been developed that show correlations with patient outcomes. Molecular characterization of CRC metastases has highlighted similarities and differences between metastases and primary tumors; however, our understanding as to how to improve patient outcomes based on metastasis biology is lagging and remains a major obstacle to improving CRC patient outcomes. In this review, we will summarize the multi-omics features of primary CRC tumors and their metastases across racial and ethnic groups, the differences in proximal and distal tumor biology, molecular-based CRC subgroups, treatment strategies and challenges for improving patient outcomes.
1. Introduction
Human colon and rectal cancers are worldwide health burdens, with over 1.9 million new combined cases and 935,000 deaths in 2020, accounting for approximately 9–10% of all cancer incidence and mortality [1]. Colorectal cancer (CRC) is the third most diagnosed cancer and the second leading cause of cancer death [1], with an overall balance in numbers between males and females. Globally, CRC incidence is highest in Australia, Europe and the USA but lower in underdeveloped nations. CRC incidence is linked to diet/obesity, smoking, alcohol and lack of exercise, but genetic factors and family history are also influential in determining disease risk. In recent decades, CRC incidence rates for adults over 50 have decreased, mainly due to improved screening methods such as colonoscopy, imaging, and non-invasive biomarker tests. However, early-onset disease incidence for adults under 45 years old has increased in developed nations [1]. CRC mortality is mainly driven by metastases to distal tissues, mainly liver and brain tissues. Indeed, CRC metastases to liver tissues occur in 33–50% of all CRC patients [2,3] and reduce the overall survival (OS) to 3–11% if left untreated or given palliative care [2,4,5]. However, OS rates can climb up to 60% if the liver metastases are treated by removal of the affected liver tissue and thermal ablation of the tumor tissue [6,7].
2. Racial Disparities in CRC
As with every form of human cancer, racial disparities in incidence and outcome are present for CRC patients. African Americans followed by American Indians display increased CRC incidence, advanced disease stage and mortality as compared to Caucasian Americans [8]. African American patient CRC incidence is generally 3–7 years earlier than for Caucasian patients. African Americans are less likely to know their own family history of disease, to relay the detection of colorectal polyps to family members and in general, are distrustful of the US medical system [9]. The incidence disparity occurs even for early-onset patients under 45 years old. These findings suggest that several factors limit access to health care [8], including awareness and knowledge of recommended screenings; socioeconomics; healthcare access; limited, inadequate or no health insurance; or other barriers that ultimately limit preventative screening, treatment and follow-up health care [10,11,12,13]. From a socioeconomic perspective, individuals in lower socioeconomic populations also show lower participation in disease screening worldwide [14]. In general, racial disparities in patient outcomes are mainly related to disparities in CRC screening and clinical access and treatment differences [11].
With respect to treatment disparities, one report showed that while African American and Caucasian CRC patients were examined by oncologists at similar or equal rates, Caucasian patients were more likely to receive chemotherapy [11,15]. However, this is confounded by a recent study [16] showing that among stage III CC patients, a higher proportion of African American patients began adjuvant chemotherapy compared to Caucasians. It should be noted that duration, type, commitment and intensity of treatments were not included in the analyses and may be contributing factors to disparity [17]. African Americans have higher rates of recurrence and lower rates of successful therapy, further complicating the true sources of CRC health disparities [17].
Treatment disparities are also related to those physicians treating African American versus Caucasian patients, such that medical personnel treating African American patients are more likely to be trained outside of the United States and are less familiar with current screening methods, use older technologies, are understaffed, have overall less funding and have overall higher patient volumes. Since minority groups are generally geographically segregated, the access to local quality care is more limited, thereby resulting in critical overdemand of medical access that is inherently compromised [11]. Indeed, a report from Obrochta and colleagues showed that minority CRC patients were at an increased risk for delayed treatment or undertreatment compared to Caucasian patients, which is derived from neighborhood socioeconomic status [18].
A study led by Warren Anderson [19] investigated sociocultural factors, as well as environmental/dietary exposures and healthcare access among Caucasian and African American CRC patients and showed that increased screening and higher patient education were associated with lower CRC risk among African Americans [19,20]. In addition, CRC incidence disparity is not evident when correcting for screening, suggesting that screening is a key driver of CRC health disparities. In support of this, CRC incidence disparities were not explained fully by family history, healthcare access, environmental/dietary exposures or socioeconomic background [19,20].
Similar conclusions were reached by Kane et al., who showed that while all minority groups showed lower CRC screening rates compared to Caucasians, this disparity was reduced after adjustment for socioeconomic and behavioral factors [21]. Improved screening of both African Americans and Caucasians within a healthcare system over a 20-year period from 2000–2019 [22] and data from the USA National Cancer Institute (reviewed in [23]) have resulted in overall lower CRC incidence, as well as lower incidence of early- and late-stage disease and CRC mortality. Even though the incidence and death rates of African Americans are still higher than those of Caucasian patients, this shows that earlier detection and treatment are beneficial to improved patient outcomes across racial groups.
Early-onset CRC is becoming increasingly prevalent in the US population over the past 20 years, such that the recommended age for colonoscopy-based screening for polyps, adenomas and adenocarcinomas is now 45 years of age for the general population [24,25]. Early-onset CRC patients are more likely to present with advanced stage (stage IV) cancer and to be African American or Hispanic [26]. Specifically, early-onset African American CRC patients display worse OS compared to Caucasian patients across all income levels, even those patients who are more educated, have a higher income and have health insurance [24]. Petrick and colleagues also showed an increase in early-onset CRC among minority populations compared to Caucasian patients. Late-onset cases are most common in men, with the location in the left side of the colon, while women present with right-sided disease [27]. Interestingly, incidence of early-onset CRC was more prevalent in women [27]. Generally, CRCs are predominantly located in the distal (left) side of the colorectum, and African American patients show a bias towards primary tumors located in the proximal (right) area of the colon with microsatellite stable (MSS) disease. Interestingly, early-onset CRC patients present with left-sided primary tumors that have aggressive behavior [26], suggesting that there are also biological factors that help drive CRC disparities between racial and ethnic groups.
3. Genetic Progression of CRC
CRC progresses from the development of precancerous polyps from colonic epithelium to the formation of adenomas, followed by the development of adenocarcinomas that may ultimately invade the colonic epithelium and metastasize to liver and other tissues. Most CRCs develop sporadically, while only 5% of CRCs are derived from germline transmission of mutated driver genes. Among the heritable CRCs, Lynch syndrome represents less than 4% of all CRCs and occurs due to germline mutations in several mismatch repair genes, including MLH1, MSH2 and MSH6 [28].
CRC has been well studied with respect to the molecular alterations that drive tumor development and progression. Vogelstein originally proposed driver somatic mutation and deletion events that are key in the adenoma-carcinoma pathway that align with tumor cell selection and clonal expansion [29]. In this model, APC alterations (mutations or deletions) represent the first events in CRC tumorigenesis as normal colonic epithelial cells convert into hyperproliferative cells. APC alterations occur in up to 90% of all CRCs [30,31] and result in sustained Wnt signaling through stabilized β-catenin expression. Activating KRAS mutations (KRAS-mut), chromosome 18 deletions that include the DCC locus, and inactivation of the TGF-β response by SMAD2/SMAD4 changes are key steps in the development of adenomas. Finally, TP53 alterations (mutations and/or deletions) represent the gatekeeper events in the development of adenocarcinomas (Figure 1).
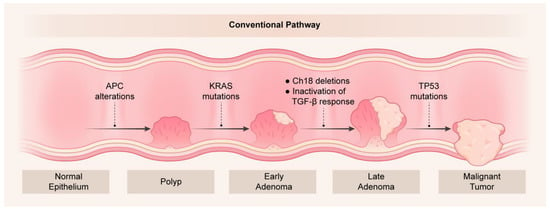
Figure 1.
Genetic changes as a function of colorectal tumorigenesis. Key molecular alterations are shown to occur with progression from normal epithelium to polyps and tumors in the adenoma-carcinoma sequence originally described by Vogelstein [29].
More recently, the Big Bang model [32] was developed and argues that tumors grow as a single expansion after an initial cellular transformation. The Big Bang model is supported by the fact that advanced tumors do not generally display clonal selection. When applied to metastases, the Vogelstein model assumes linearity in a stepwise fashion through novel mutations and their expansion by clonal selection [29]. Alternatively, the Big Bang model suggests that the metastatic potential of a tumor cell is defined early during tumorigenesis [32,33], in which a metastatic cell is selected based on growth advantage. In support of this, Hu and colleagues [33] performed WES on pairs of CRCs and liver metastases to show that driver mutations are formed early and that the mutation profiles between CRCs and their liver metastasis pair show strong convergence. In addition, metastases are seeded when the tumor is undetectable.
While approximately two thirds of CRCs develop from the traditional adenoma pathway, one third of CRCs develop from the serrated pathway [34,35]. These include hyperplastic polyps (HPPs), sessile serrated adenomas (SSAs) and traditional serrated adenomas (TSAs). HPPs are mainly located in the distal (left) side of the colon but do not develop past adenomas. TSAs are low-frequency serrated polyps that present in the distal colon with a sawtooth-like appearance and are enriched for KRAS mutations. SSAs are mainly located in the proximal (right) colon, display the BRAF (V600E) point mutation (BRAF-mut) and are underscored by increased size with a flattened and serrated appearance [34,35] (Figure 2).
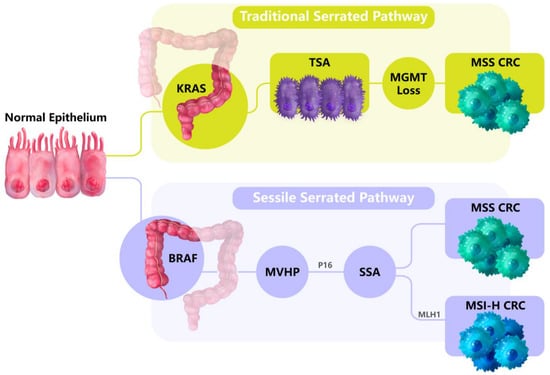
Figure 2.
The serrated pathways of CRC tumorigenesis. Traditional serrated pathways develop in the distal (left) side of the colorectum and are linked to KRAS mutations. Alternatively, sessile serrated pathways develop in the proximal (right) side of the colorectum and are enriched in the BRAF (V600E) point mutation. From the normal colon tissues, microvesicular hyperplastic polyps (MVHPs) form that are the precursors to sessile serrated adenomas (SSAs). SSAs then develop into CRCs, and display microsatellite instability (MSI) if MLH1 is epigenetically silenced.
These findings establish differences in tumor etiology and molecular features between CRCs that originate in the left (distal) versus the right (proximal) region of the colon. The normal colonic tissues on the left and right sides originate from unique developmental origins [36]. Most CRCs (85%) originate on the distal side of the colon, are enriched in KRAS, PIK3CA and TP53 mutations and display copy number variation (CNV) and chromosomal instability (CIN), in which key tumor suppressor genes are deleted and/or mutated. This contrasts with serrated CRCs located in the proximal colon, which are enriched in the BRAF mutation, are diploid (non-CIN) but are hypermutated, display microsatellite instability (MSI) due to MLH1 silencing by DNA methylation and are more prevalent in older women. Interestingly, proximal tumors present with a more diverse panel of somatic mutations compared to distal (non-hypermutated) tumors, namely with enrichment for mutations in ACVR2A, TGFBR2, the mismatch repair genes MSH3 and MSH6 and the DNA polymerase POLE [31]. From a treatment perspective, patients with proximal tumors showed worse OS and recurrence-free survival (RFS) than patients with left-sided primary tumors after partial hepatectomy or tumor ablation to remove liver metastases [2].
The Cancer Genome Atlas (TCGA) presented whole genome sequencing (WGS) and whole exome sequencing (WES) on nearly 300 CRCs to show mutations in APC, TP53, KRAS and PIK3CA in left-sided tumors, while mutations in ACVR2A, TGFBR2, the mismatch repair genes MSH3 and MSH6 and the DNA polymerase POLE were enriched in right-sided tumors. Giannakis and colleagues performed WES on 619 archived CRC tumors and normal-adjacent tissue pairs to identify novel mutations in BCL9L, CTCF, KLF5 and RBM10. In addition, the extent of somatic mutation correlates with the formation of neoantigen peptides that invoke anti-tumor immunity and is tied to lymphocytic infiltrations and patient survival [37].
3.1. Racial Differences in CRC Genetics
An analysis of genetic alterations in primary CRCs between African American and Caucasian patients showed elevated KRAS mutation rates in African American patients, as well as mutations of three genes (EPHA6, FLCN and HTR1F) only in African American CRC patients, although these mutations have a low frequency (3–6%) [8,38]. Guda and colleagues [38] performed next-generation sequencing of stage IV tumors with liver metastases from African American patients to identify mutations linked to metastatic disease. These data were analyzed to ultimately identify a panel of 15 genes that are more likely to be mutated in African Americans with CRC [38]. As a follow-up, Wang et al. then showed that stage I–III African American patients with these mutations were more prone to developing metastatic disease [39].
CNV analyses on primary CRCs from African American and Caucasian patients showed African American-specific aberrancies on chromosomes 11, 17p 20q and X [40,41,42], as well as elevated MSI. Moreover, an analysis of TP53 alterations showed that while TP53 mutation profiles are similar between African American and Caucasian CRC patients, the Pro/Pro polymorphism in codon 72 occurs at a higher frequency in African American patients and is linked to poor patient outcomes [43]. A comparison of somatic mutations among CRC patients showed that African American patients had a higher tumor mutation burden (TMB) than Caucasian patients in both early-onset and late-onset cases [44].
Early-onset African American patients also had increased ATRX mutations as well as significantly different mutation frequencies in ATRX, APC, FBXW7, LRP1B, PIK3CA and RNF43 across racial groups, suggesting that somatic mutation profiles occur as a function of race within early-onset tumors [44]. In contrast, a report from Xicola et al. [45] used WES in early-onset CRCs to demonstrate that African American patients had reduced TMB, CNV and mutation frequencies in APC and other known CRC driver genes compared to Caucasians but harbored loss-of-function mutations in BCL9L, a negative regulator of B-catenin. Interestingly, Wnt regulatory genes were silenced by promoter DNA hypermethylation in these tumors, suggesting distinct molecular routes for CRC tumorigenesis (discussed in Section 4: DNA Methylation Alterations) [45].
3.2. Genetic Features of CRC Metastases
Since CRC mortality is driven by metastasis, examining the genetic profiles of CRC metastases may identify novel molecular alterations to identify the changes that occur during metastasis and those that can be exploited for patient treatment and surveillance. Vermaat performed targeted sequencing of over 1200 cancer-related genes across 21 primary CRC/liver metastasis pairs to show that liver metastases are genetically unique from those of the primary tumor, with 817 coding region alterations exclusively in the liver metastases [46]. This analysis identified the most common gene mutations (KRAS, BRAF, PTEN, PIK3CA, etc.) as well as other mutations in genes in the EGFR and TGF-B pathways that influence treatment success [46]. An analysis of CNV in primary tumors from 349 metastatic CRC patients from the CAIRO and CAIRO2 phase III clinical trials [47] identified gains at chromosomes 1q, 7, 8q, 13 and 20 and the most common losses at 1p, 4, 8p, 14, 15, 17p and 18. In addition, gains of chromosomes 8p, 11q, 12p, 13q, 18q, 19q, 20q and X were present in patients who developed metastases, and variations of 194 chromosomal regions were associated with progression-free survival (PFS) [47]. Ishaque et al. [48] performed WGS on colorectal adenomas, carcinomas and metastases and identified mutations in AKT, BRCA2, FAT1, FGF1, KDR and NOTCH1 that may be therapeutic targets for disease treatment.
Yaeger et al. [49] analyzed sequencing data from 478 primary CRCs and 533 metastases from 979 patients with metastatic disease and 123 primary CRCs from patients with early-stage disease to find alterations related to metastasis. The mutation profiles of primary tumors and metastases were overall highly concordant. However, metastases harbored novel APC intron splicing alterations as well as CTNNB1 deletions. As shown by other reports, patients with right-sided primary metastatic disease were of older age at the time of diagnosis and had reduced survival, increased mutation burden, and enrichment of AKT1, KRAS, PIK3CA, PTEN, RNF43 and SMAD4 mutations. In contrast, left-sided tumors exhibit the following characteristics: (1) display mutations in APC, NRAS and TP53; (2) display amplification enrichment in receptor tyrosine kinase signaling genes ERBB2, ERBB3, EGFR and FGFR; (3) do not display mutations or CNV in genes related to cell division; and (4) may be more prone to fluctuations in intestinal microbiomes, suggesting that patients with left- and right-sided CRCs have unique molecular pathways of metastasis. Comparing these data to a separate data cohort of metastatic tumors, as well as to The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortia (ICGC) data for primary CRC mutations, Mendelaar et al. [50] identified the common mutations of left- and right-sided CRCs. Interestingly, the Mendelaar report also identified mutations in long non-coding RNA (lncRNA) genes, specifically noting that LINC00672 mutations are associated with treatment response. In addition, the presence of FBXW7 mutations signaled poor patient response to EGFR-targeted therapies.
4. DNA Methylation Alterations
Epigenetic alterations are hallmarks of CRC and every other tumor type. The main facets of epigenetics, DNA methylation, chromatin modifications of histone tail proteins, nucleosome occupancy and non-coding RNAs, namely microRNA (miRNA) and long non-coding RNA (lncRNA), are key for gene regulation and higher-order organization of chromatin. These epigenetic facets work in symphony to compose multiple levels of gene regulation. In human cancers, epigenetic aberrancies function to silence tumor suppressor genes and activate oncogenes. However, epigenetic marks are reversible via passive or active mechanisms, making them attractive therapeutic targets for disease treatment in reverting epigenome profiles to a more normal-like state. For the purposes of epigenetic alterations in CRC, we will focus on DNA methylation in this review.
DNA methylation in human and other mammalian tissues is generally defined as the addition of a methyl (-CH3) group to the C-5 position of cytosine nucleotides in the 5′-CG-3′ or CpG sequence context. Methyl marks are placed by the enzymatic activities of DNA methyltransferases (DNMTs) that use S-adenosylmethionine as a required co-factor. In human cells, DNMT1, DNMT3A and DNMT3B are largely responsible for DNA methylation catalysis, in which DNMT1 is involved in immediately copying the DNA methylation patterns from the parental strand to the nascent daughter strand during DNA replication, while DNMT3A and DNMT3B are responsible for placing methyl-marks at CpGs that were originally unmethylated. However, DNMT1 and DNMT3B are thought to work together to maintain DNA methylation profiles. DNMT3B also serves as an accessory scaffold protein in recruiting DNMT3A to specific CpG sites and is a key DNMT enzyme for catalyzing DNA methylation of gene body (transcribed) regions, in which DNMT3B is recognized by SETD2 for generating histone H3 lysine 36 trimethylation (H3K36me3) marks that are correlated with active gene expression.
While DNA methylation is pharmacologically reversible through the activities of DNMT inhibitors such as 5-azacytidine (Vidaza, AZA), 5-aza-2′-deoxycytidine (decitabine, DAC) and S110, enzymatic DNA demethylation is catalyzed in a stepwise manner through oxidative conversion of 5-methylcytosine (5mC) to 5-hydroxycytosince (5mC) by the TET family of enzymes. After TETs convert 5mC to 5hmC, 5hmC marks are further converted to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), after which both 5fC and 5caC marks are ultimately replaced with unmethylated cytosines through base excision repair [51,52,53].
DNA methylation aberrancies are common in CRC and all other cancer types in which gene-specific promoter DNA hypermethylation is concomitant with global DNA hypomethylation of non-CpG rich regions and repeat sequences. Promoter DNA hypermethylation may correlate with gene silencing, while gene body DNA methylation and CpG poor DNA hypomethylation may be positively associated with gene expression. Epigenetic driver events in CRC tumorigenesis are limited and include epigenetic silencing of MLH1, CDKN2A, MGMT, MLH1, RUNX3, the SFRP gene family, TPEF and VIM (reviewed in [35]). More recently, Xu et al. [54] analyzed TCGA CRC CNV, DNA methylation and gene expression data to identify LRRC26 and REP15 as prognostic CRC driver events in which high LRRC26 and REP15 expression was inversely correlated with the presence of metastases and tumor stage.
Most cancer-specific DNA methylation changes do not result in gene expression changes but are biomarkers of disease. DNA methylation-based biomarkers are highly effective in disease monitoring and predicting patient outcome and response to treatment. DNA methylation biomarkers can be found in tumor-derived cell-free DNA (cfDNA) in patient blood serum or plasma, urine and fecal matter. DNA methylation markers include ALX4, BMP3, CDH1, CDKN2A, MGMT, MLH1, NRDG4, PRIMA1, SDC2, SEPT9, SFPR2, TFPI2, TMEFF2 and VIM in blood, stool and/or urine specimens [55,56,57].
Two DNA methylation biomarker panels are FDA approved for early detection of CRC: (1) SEPT9 in tissue and blood plasma, marketed as Epi proColon, and (2) BMP3 and NDRG4 DNA methylation with KRAS mutation screening in stool, marketed as Cologuard. It should be noted that SDC2 DNA methylation testing in stool samples is approved in Korea (EarlyTect-Colon Cancer) and China (Colosafe) for CRC detection, and a yet unapproved test described as ColoDefense includes screening for both SDC2 and SEPT9 DNA methylation (reviewed in [58]). SEPT9 DNA methylation in plasma shows high sensitivity (60–80%) and specificity (80–99%) in identifying stage I and II disease and outperforms both carcinoembryonic antigen (CEA) and fecal occult blood tests for CRC detection. However, SEPT9 DNA methylation shows low sensitivity in detecting adenomas (summarized in [59]).
The large extent of DNA methylation changes in CRC has proven fruitful for the development of DNA methylation-based marker panels that predict patient outcome and response to treatment, as well as those that are linked to tumor stage and grade. Several reports have used DNA methylation data to build models related to patient outcome, which include the following: (1) Zhang and colleagues identified differential DNA methylation in B3GNT7, CHN2, MUC12 and TBX20 that is linked to gene expression and poor patient outcome [60]. (2) An analysis of Illumina Infinium DNA methylation array data from metastatic and non-metastatic CRCs revealed a 20-probe classifier with prognostic utility for non-metastatic cases [61]. This classifier included probes in BRD4, FBXL18, KCNQ1 and MET, among others, and the DNA methylation score of this panel was positively correlated with patient survival [61]. (3) Gong et al. mined TCGA CRC DNA methylation data to compile a three-gene (NR1H2, SCRIB and UACA) DNA methylation signature that correlated with patient OS irrespective of patient age and gender [62]. (4) Xie et al. [63] used tumor-derived cfDNA from blood plasma to evaluate the performance of a 14-gene panel (ARHGEF4, AV3, BMP3, CHST2, DOCK10, IKZF1, LRRC4, NDRG4, OPLAH, PPP2R5C, PDGFD, QKI, SFMBT2 and ZNF625). The panel detected metastatic CRC with 90% sensitivity and 90% sensitivity and is superior to CEA measurements. (5) Deng and colleagues [64] analyzed TCGA CRC DNA methylation data to develop a 23-probe panel that is associated with disease progression. (6) Cai et al. [65] evaluated a panel of nearly 600 cancer-specific DNA methylation markers on CRCs and normal colonic tissues to compile a panel of 150 differentially methylated regions (DMRs) that were subsequently evaluated on 328 blood plasma samples. From this analysis, 24 DMRs were subject to validation across CRCs, normal-colon and blood plasma samples to arrive at six top markers: BCAN, BCAT, IKZF1, VAV3 and two regions of SEPT9 that were predictive of disease recurrence and outperformed CEA expression to detect CRCs across all four disease stages. (7) Muthamilselvan et al. [66] surveyed TCGA CRC DNA methylation data to identify markers of CRC specific for stage: stage-I (FBN1), stage-II (FOXG1), stage-III (HCN1) and stage-IV (FAM123A, LAMA1, NELL1, ZNF135). (8) Chen and colleagues also analyzed TCGA DNA methylation data to identify a panel of five immune-related genes—FGF5, LTBP4, PIK3CD and PLXNC1 and SCTR—the composite DNA methylation signature of which correlated with prognosis for stage II and III CRC patients [67]. (9) As mentioned above, early-onset CRC incidence rates are rising [24,25], in contrast to lower incidence rates of late-onset disease seen in many parts of the developed world. DNA methylation profiles in early-onset CRCs display more extensive global DNA hypomethylation than late-onset cases [68], and a recent study from Joo and colleagues [69] used Illumina DNA methylation microarray profiling to identify 234 CpG regions with differential DNA methylation as well as accelerated cellular aging using epigenetic clocks (please see Section 4.3: Epigenetic Clocks) in early-onset cases.
4.1. CpG Island Methylator Phenotypes (CIMPs) in Colorectal Cancer
In addition to being used for predictive and prognostic purposes, cancer-specific DNA methylation profiles can be categorized to identify tumor subgroups. Indeed, Toyota and colleagues [70] first described a panel of cancer-specific DNA methylation events in a subgroup of CRC patients, termed CIMP. CIMP-specific loci remain unmethylated in non-CIMP tumors and normal-adjacent colonic mucosa, and patients with CIMP DNA methylation only represent 15–20% of all CRC cases. Weisenberger et al. [71] confirmed CIMP (now termed CIMP-high, CIMP-H) and developed a five-gene classification using real-time PCR technology: CACNA1G, IGF2, NEUROG1, RUNX3 and SOCS1. CIMP is highly concordant with BRAF (V600E) point mutations, MLH1 epigenetic silencing, MSI and a hypermutation profile but not TP53 mutations or chromosomal instability, and it occurs in right-sided tumors and is enriched in female patients with poor clinical outcome [31,71,72]. Ogino et al. [73] subsequently identified the CIMP-low (CIMP-L) subgroup of CRCs that display reduced DNA methylation at CIMP-specific loci, are enriched with KRAS mutations, chromosomal instability and microsatellite stability and are present in the right side of the colon in male patients [31,72,73]. Depending on the analysis, unsupervised clustering of CRCs unveils three to four DNA methylation-based subgroups CIMP-H, CIMP-L and non-CIMP, in which non-CIMP tumors are categorized as either one or two subgroups [31,72,74,75].
Stratifying CRCs by CIMP status and other molecular co-variates has been shown to result in a profound dissemination of CRC patient outcome and response to therapy. In pooling data for over 5000 CRC patients, Phipps et al. [76] categorized CRCs into five groups based on their molecular profiles: (1) CIMP-positive, BRAF mutant (BRAF-mut), KRAS wild-type (KRAS-wt), high microsatellite instability (MSI-H); (2) CIMP-positive, BRAF-mut, KRAS-wt, microsatellite stable (MSS) or low microsatellite instability (MSI-low); (3) CIMP-negative, BRAF-wt, KRAS-mut, MSS/MSI-low; (4) CIMP-negative, BRAF-wt, KRAS-wt, MSS/MSI-low; and (5) CIMP-negative, BRAF-wt, KRAS-wt, MSS/MSI-low. Group 2 patients displayed the worst outcome, while MSI-H status was significantly associated with improved patient outcome, especially in cases harboring CIMP and BRAF-mut. In addition, Group 1 and Group 2 patients had significant associations with disease-specific survival (DSS), while Group 3 patients had the worst DSS, most notably in Group 3 patients with stage I-III disease. Indeed, KRAS-mut patients showed poor survival overall, specifically in MSS/MSI-low cases. These findings were also shown by Murcia and colleagues [77], who performed similar molecular analyses on 878 CRC patients, in that CIMP-positive, BRAF-mut and MSS patients had the worst prognosis. This analysis also showed that 5-fluorouracil (5-FU)-based therapies demonstrated improved prognosis in patients with either CIMP-negative, KRAS-mut and MSS status or CIMP-negative, BRAF-wt, KRAS-wt and MSS status.
Given these complicated aspects of CRC biology, a set of four consensus molecular subgroups (CMS) were developed that include RNA expression signatures [78]: CMS1 (MSI immune), CMS2 (canonical), CMS3 (metabolic) and CMS4 (mesenchymal) (Figure 3). CMS1 tumors display CIMP-H, MSI, BRAF-mut, DNA hypermutation, immune infiltration and activation and are linked to poor patient survival after relapse. CMS2 tumors are non-CIMP, with extensive CNV and activated WNT and MYC signaling. CMS3 tumors display CIMP-L DNA methylation, MSI/MSS, nearly diploid copy number, KRAS-mut and metabolic dysregulation. CMS4 tumors are non-CIMP but display CNVs, stromal cell infiltration, activated TGF-β and angiogenic signaling, and CMS4 patients show poor survival. To simplify CMS classification, CMS1 status can be effectively determined by measuring MSI status, CMS2 and CMS3 subgroups can be stratified by differential DNA methylation of specific loci, and CMS4 tumors can be determined by immunohistochemistry and gene expression classifiers [79,80,81,82].
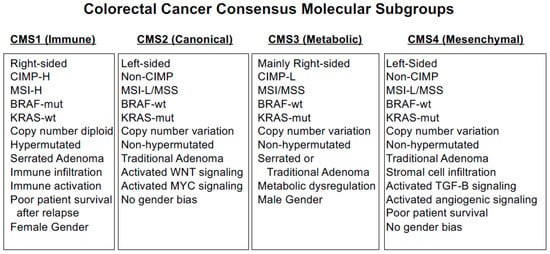
Figure 3.
CRC Consensus Molecular Subgroups.
4.2. DNA Methylation Profiles of Metastatic Disease
While molecular features of the primary tumor are expected to be represented in tumor metastases, there is an increasing body of evidence that metastases are characterized by novel molecular features that are not present in the corresponding primary tissue. Epigenetic profiling of liver metastases is limited compared to the primary tumor; however, several reports over the past decade have painted interesting portraits as to the role of DNA methylation changes in CRC. (1) Global DNA hypomethylation is common in CRC, in which non-CpG rich sequences in single copy loci and repetitive elements lose DNA methylation compared to normal tissues (reviewed in [35]). In comparing LINE-1 repetitive element DNA methylation levels between primary CRCs and liver metastases, Hur et al. [83] detected significantly lower levels of LINE-1 DNA methylation in metastases compared to primary CRCs, also resulting in the activation of proto-oncogenes, including MET, CHRM3 and RAB3IP. (2) Ili et al. [84] compared primary CRCs to normal-adjacent tissues and lymph node metastases using genome-scale bisulfite sequencing and found that non-CpG island DNA hypomethylation and CpG island hypermethylation were extensive in primary tumors but attenuated in lymph node metastases. However, there are additional CpG islands that display DNA hypermethylation in lymph node metastases over primary tumors. Finally, an analysis of CpG islands that show concordant DNA methylation between primary tumors and lymph nodes included BDNF, FIGN, HCN4, HTRA3 and STAC2, in which DNA methylation status of the panel was significantly associated with poor patient outcome. (3) Orjuela et al. [85] used methyl binding domain capture followed by sequencing to show that DNA hypermethylation of the primary tumor is well represented in liver metastases, although DNA hypomethylation was detected in the metastases compared to the primary tumors. (4) Yu and colleagues [86] used bisulfite sequencing to profile 30 CRC tumors and 19 lymph node metastases to ultimately identify DNA methylation of LBX2 as a biomarker of metastasis that outperforms CEA and imaging testing. (5) An analysis of DNA methylation in 59 early- and late-stage CRCs, normal-adjacent tissues and metastases identified DNA methylation signatures of metastases in early-stage tumors that predicted outcome and the possibility of metastasis [87]. (6) DNA hypermethylation of IMPA2 correlated with gene silencing in primary tumors and metastases and predicted poor patient outcome and advanced stage. (7) Bisulfite pyrosequencing of CDH1, CDH13, CDKN2A (p16), CDKN2A (p14), ESR1, HPP1, MGMT, MINT1, MINT2, MINT31, MLH1, THBS1 and TIMP3 across paired primary tumor-liver metastasis pairs, as well as CRCs with or without metastases, showed stochastic variability as a function of metastasis and suggest that cancer-specific DNA changes mostly occur prior to metastasis [88], which is in agreement with the other reports.
4.3. Epigenetic Clocks
Cancer is a disease of aging since increased age correlates with cancer risk, and age-related DNA methylation alterations have been described (reviewed in [89,90,91]). Although much less prevalent than cancer-associated DNA methylation alterations, global age-related DNA hypomethylation exists with regional DNA hypermethylation. In 1994, Issa and colleagues first identified age-related DNA hypermethylation of the human ESR1 CpG island [92]. Subsequently, age-related DNA hypermethylation changes were identified in human tissues [93,94,95,96]. As technologies to measure DNA methylation across the genome improved, so did the ability to identify age-specific DNA methylation markers that measure organismal age [97]. Using Illumina DNA methylation array data, Horvath identified 353 CpG sites that represented an epigenetic clock after analyses of data from over 6000 samples [98]. The Horvath clock was compatible with a large array of tissue and cell types. Alternatively, the Hannum clock [99] surveyed a disparate set of 71 CpGs to calculate cellular age in blood. Two other epigenetic clocks were subsequently developed for use with blood samples that utilize 3–513 CpGs [91,100,101].
4.4. Epigenetics of Racial Disparities in CRC and Disease Risk
Epigenetic clocks have been important contributors to better understanding age-related diseases such as Alzheimer’s disease, human cancer and cardiovascular disease, as well as how epigenetic age is linked to inherited syndromes and other diseases [91]. Epigenetic clocks help to identify factors that accelerate or decelerate epigenetic age. Since cancer is a disease of aging, it is understood that epigenetic age acceleration occurs in colorectal cancer and other human cancers. Moreover, it is known that African Americans are diagnosed with advanced stage disease on the right side of the colon at an earlier age compared to Caucasians. Devall and colleagues [102] generated Illumina EPIC DNA methylation microarray data on matched left- and right-sided normal colon tissues from European Americans (Caucasians) and African Americans to identify if epigenetic age acceleration is correlated with CRC risk and sidedness using the Horvath model. Interestingly, African Americans displayed age acceleration in the right (proximal) side of the colon compared to Caucasians, while Caucasians displayed age acceleration on the left (distal) colon tissue specimens. In general, more African Americans showed older right-sided colons than left-sided colons compared to Caucasians. In addition, an analysis of differentially methylated regions (DMRs) between the right and left sides of the colon showed an enrichment for DNA hypermethylation in the right-sided colon in African Americans—these regions were linked to colonic enhancers as well as CRC tumorigenesis and aging. Notably, in normal rectal tissues, European Americans displayed increased epigenetic age acceleration compared to African Americans; race-specific, age-associated DMRs were identified, and epigenetic drift of age-related DMRs was more pronounced in African Americans [103]. Age-related DMRs were represented across both racial groups, suggesting that age acceleration is a driver of disease risk [103].
Indeed, DNA methylation changes in ALU, LINE-1 and SAT2 repetitive elements have been linked to environmental exposures as well as education, occupation, social status, wealth and family income status (reviewed in [104]). With this in mind, Konradsen and colleagues [105] performed a meta-analysis of 27 studies that involved stage III CRC patients with socioeconomic markers that include education, employment, financial status, income level and health insurance and showed that patients with low socioeconomic status had significantly increased time to start treatment and lower odds of receiving adjuvant chemotherapy. Finally, Alonso et al. [106] identified MGMT and ADAMTS4 DNA hypermethylation in normal colonic mucosa, which are suggestive of an epigenetic field defect since these regions display DNA hypermethylation in CRC. Interestingly, DNA hypermethylation of these loci was more pronounced in the proximal colon in older African Americans and may serve as effective biomarkers for CRC risk in African Americans.
In contrast to cancer risk, there are only a handful of examples of CRC DNA methylation data that are linked to racial health disparities. A global meta-analysis of CIMP prevalence showed that it was highest in Africa and lowest in Asia, Europe and Australia [107]. In addition, a comparison of cancer-specific DNA methylation between African American and Caucasian CRC patients identified DNA hypermethylation of ARHGEF4, CHL1, CHL4, GDF, ITGA4, NELL and several microRNAs including miR-9-3p and miR-124-3p in African American CRC patients [108].
5. CRC Gene Expression Profiles
5.1. Gene Expression Subgroups and Functional Networks
Much like genetic (mutations and CNV) and epigenetic (DNA methylation) alterations, gene expression changes are common in CRC. From a tumor subgrouping perspective, CRC mRNA gene expression subgrouping generally follows the DNA methylation-based grouping with three subgroups: CIMP/MSI, invasive and chromosomal instability (CIN) [31]. In contrast, an unsupervised clustering analysis of mRNA gene expression data from 443 CRC patients revealed six subgroups: (C1) CIN with downregulated immune pathways, (C2) mismatch repair, (C3) KRAS-mut, (C4) cancer stem cell, (C5) CIN with Wnt pathway upregulation and (C6) CIN with a normal-like expression profile [109]. The C2 subgroup is linked to CIMP with BRAF-mut, but groups C2, C3 and C4 were enriched for CIMP and proximal primary tumor location. Alternatively, C1, C5 and C6 subgroups were enriched for CIN, non-CIMP and TP53-mut. The C4 subgroup was enriched for metastatic disease; however, there were no associations between subgroup and tumor staging. It should be noted that subgroups C4 and C6 were associated with shortened relapse-free survival.
Cancer-specific gene expression profiles are important in identifying functional networks important for cancer development and progression. Gene networks based on large expression data sets help to identify unique interactions within and between individual networks and expression pathways. Three recent examples examine this. First, Kim and colleagues analyzed CRC gene expression data sets [110] from unique regions of resected tumor tissues to extrapolate essential gene expression profiles in heterogeneous clinical tumor specimens and assign clinical specimens to specific subgroups. Notably, this analysis revealed that after stratifying the expression data by cancer specificity, residual expression data are present and unique for each patient and are cancer cell intrinsic. After clustering these residual gene expression profiles between patients, cancer cell-intrinsic subgroups are present and display novel prognostic value. Second, Emmert-Streib and colleagues [111] developed a gene regulatory network based on colon cancer gene expression data from the International Genomics Consortium with the goal of identifying co-regulated networks and the driver regions controlled by these networks at the chromosomal level to better understand CRC tumorigenesis. Of note, trans-chromosomal pairings are minor in CRC, with chromosome 22 genes mainly involved in these interactions. At a gene level, the main hub genes include OR7E104P, GATA1, NPHP3-AS1, POM121L12, ZNF843, NYX, SLC22A25, C20orf203 and SLC4A1, and the enriched pathways are related to cell cycle, mitosis, M phase, membrane targeting and translation. Third, Su et al. [112] mined the TCGA CRC gene expression data to develop CRC biomarker networks for cancer diagnosis and staging. After first identifying cancer-specific gene expression profiles, the group ultimately identified genes associated with tumor stage that include ADH1B, CLEC3B, GCNT2, GLDN, GLP2R, GREM2, JCHAIN, LYVE1, MAMDC2, PLP1, SCARA5 and TMEM100. In addition, individual expression levels of SULT1B1, PTGDR2, GPR15, BMP5 and CPT2 were significantly associated with OS. Pathway analyses showed enrichment of the curated expression panel for processes including metabolic processes and small molecule transport.
5.2. Gene Expression Specific to Race and Ethnicity
Based on whole transcriptome data, the Oncotype DX Colon Cancer Assay panel of 12 genes was developed to predict recurrence after resection of stage II/III disease [113,114,115,116]. The 12-gene panel consists of BGN, FAP, GADD45B, INHBA, MK167, MYBL2, MYC and five reference normalization genes [113]. Interestingly, a comparison of the 12-gene panel between Caucasians and African Americans with stage II CRC did not show any differences, nor were there any expression differences for each individual gene between the two groups [113]. These data indicate that although there are health disparities between Caucasian and African American CRC patients, they are not represented in this gene expression panel. However, comparison of African American and European American CRC disease using Agilent gene expression microarrays did reveal 95 genes with expression differences between the two groups [117]. This was reduced to a panel of 10 genes that can accurately predict ethnicity: ADAL, ANKRD36B, ARHGAP6, CRYBB2, C17orf81 PSPH, TRNT1, VSIG10L, WDR8 and ZNF835 [117].
5.3. Gene Expression Related to CRC Metastasis
Gene expression data can also be used in profiling CRC metastases. A meta-analysis of gene expression data sets of primary CRCs and liver metastases [118] unveiled a panel of 35 differentially expressed genes (DEGs), with the most dysregulated genes including AHSG, APOA2, CP, FGA, F2, HPX, HRG, ITIH2, PLG and SERPINC1. Among these, expression of ITIH2 was statistically significantly associated with survival, suggesting that it is important for CRC metastasis [118]. In comparison, Wu et al. examined whole transcriptome RNA expression (RNA-seq) and whole exome sequencing (WES) to examine gene expression and mutations/CNV, respectively, across 99 stage IV patients who underwent resection for only the primary tumor to identify correlates with stage IV disease [119]. Interestingly, among the clinical co-variates of tumor stage, gender, race, tumor location and tumor pathology, only tumor location was a significant determinant of outcome. In addition, mutations of key genes including APC, TP53, KRAS and PIK3CA did not correlate with patient outcome. The gene expression analyses, however, identified 97 genes with aberrant expression, including significant differences in DSCAML1, FGF18, NEUROD1, PLAC1, SAA2, SFTA2 and OTOP3 that were associated with patient outcome. FGF18 and DSCAML1 expression had a protective effect, while the expression of the other genes in the panel was positively associated with outcome risk [119].
6. Influence of the Tumor Microenvironment and Gut Microbiome on CRC Molecular Profiles
Increasing amounts of evidence indicate that the tumor microenvironment (TME) is a substantial contributor to CRC tumorigenesis and disease progression. The TME is composed of several cell types surrounding the colorectum, including adipocytes, endothelial cells, fibroblasts, immune cells and inflammatory cells [120,121]. Tumor cells and the TME communicate such that CRC tumor cells induce cytokine and growth factor production from the TME cell types to supplement tumor cell proliferation and immune escape and ultimately metastasis. [120,121]. From a molecular perspective in CRC, Wnt signaling is aberrant across 90% of all CRCs [31], and activated Wnt signaling correlates with the lack of T-cell infiltration [122]. In addition, CIMP-H tumors (CMS1, immune) with MSI-H display mutations, deletions and AXIN2 super-enhancer DNA hypomethylation in immune-modulating and antigen-presenting genes to provide MSI-H CIMP tumors with an advantage of immune escape [122].
It is becoming clear that immune-related gene expression differences play a role in CRC health disparities. For instance, African Americans display reduced antitumor cytotoxic immunity [123]. Second, gene expression differences in inflammatory and immune response pathways are present between African Americans and Caucasians [117]. An analysis of TCGA CRC data by Curran and colleagues [124] revealed that African American CRCs express reduced levels of CD8+ T cells and macrophages but overexpress B cells in comparison to European Americans, as was described by Basa et al. [125]. Gene expression profiling also showed that major histocompatibility complex genes as well as immune-inhibiting and immune-stimulating genes were differentially expressed between the two groups of patients [124]. Overall, these findings indicate that B and T cell expression difference may help address the health disparities in CRC patients.
The gut microbiome consists of microorganisms required for digestive health and is also influential in CRC progression [126], indicating crosstalk between the microbiome and the host colonic epithelium (reviewed in [127]). The microbiome is plentiful in CRC and hepatocellular tumors [128,129], and reports have highlighted the role of the microbiome in regulating cancer treatment response [130,131,132]. Poore and colleagues analyzed TCGA WGS and RNA-sequencing data from tissue and blood across all 33 cancer types profiled and found microbiomes present in most forms of human cancer [133]. Using these findings, Uriarte-Navarrete et al. [134] stratified CRC data into early- and late-stage groups and measured the interactions of each microorganism with each human gene. Interestingly, they found that due to how gene-microbiome expression networks are organized, early-stage CRC involves instituting structural features of the tumor cells, whereas late-stage disease displays advanced metabolic features and signaling [134]. Another report in which CRC mRNA expression data was integrated with microbiome information from several datasets revealed that 41 overexpressed genes in CRC (including AURKA, AURKB, BUB1, CCNA2, CDK1, CENPF, MAD2L1, TPX2 and UBE2C) are enriched in microtubule and tubule binding and were negatively associated with the presence of fusobacteria and positively associated with blautia, suggesting that these bacteria are interacting partners for modulating gene expression [135].
7. Drug Treatments and Therapeutic Strategies
Therapies using 5-fluorouracil (5-FU) have been the backbone of CRC treatments for several decades. Used as an inhibitor of DNA synthesis by preventing thymidine production, 5-FU has demonstrated efficacy in treating CRC patients and is now generally combined with (1) folinic acid (leucovorin) and oxaliplatin to form the FOLFOX chemotherapy regimen; (2) folinic acid and irinotecan to form the FOLFIRI regimen; (3) folinic acid, oxaliplatin and irinotecan in the FOXFOXIRI regimen [136]; or (4) XELOX, the combination of capecitabine and oxaliplatin.
Folinic acid is a vitamin B derivative that exacerbates the 5-FU cytotoxicity, irinotecan is a topoisomerase inhibitor that prevents DNA uncoiling and replication, and oxaliplatin is a platinum-based chemotherapy agent that inhibits DNA synthesis and DNA repair. Capecitabine (Xeloda) is an oral fluoropyrimidine that enters the cell and is converted to 5-FU in three steps [137], in which capecitabine is first converted to 5′deoxy-5-fluorocytidine by hepatic carboxylesterase, which is converted to 5′-deoxy-5-fluorouridine by cytidine deaminase. The substance 5′-deoxy-5-fluorouridine is converted to 5-FU by thymidine phosphorylase. Importantly, thymidine phosphorylase displays substantially higher activity in tumor tissues compared to normal tissues, thereby improving delivery of 5-FU specifically to tumor cells.
Most targeted CRC treatment strategies have focused on the epidermal growth factor receptor (EGFR) and vascular endothelial growth factor receptor (VEGFR) signaling pathways (summarized in [35]). The activation cascade of EGFR/KRAS/BRAF results in MEK1, MEK2 and ERK activation. ERK signaling activates oncogenic transcription factors that include ELK1, FOS, JUN and MYC. As a result, these transcription factors activate driver genes involved in cell proliferation, cell cycle progression and differentiation (reviewed in [35]). The VEGFR pathway also involves RAS and BRAF signaling but independently stimulates the PI3K/AKT/mTOR signaling cascade to promote cell growth, differentiation and angiogenesis.
Several monoclonal antibodies, such as cetuximab and panitumumab, have been developed that target the EGFR pathway by binding to the EGFR extracellular domain to block the binding of EGF ligands [138,139]. Two phase III clinical trials [140,141] showed that the addition of cetuximab to FOLFOX or FOLFIRI in first-line treatment of RAS-wt metastatic CRC patients resulted in improved patient OS and PFS. However, these agents are only effective in RAS-wt tumors. In fact, only RAS-mutation status and, to a lesser extent, BRAF-mutation status, guide CRC therapeutic decisions [142,143], Approximately 40% of RAS-wt tumors are resistant to cetuximab after earlier-line treatment failure with irinotecan-based regimens [142].
Other agents effective in treating CRC patients include nivolumab, a monoclonal antibody that targets the PD-1 receptor of lymphocytes. Ipilimumab is a monoclonal antibody that inhibits CTLA-4 and subsequently activates the immune response, while pembrolizumab is a monoclonal antibody that also targets PD-1 and is FDA approved for mismatch repair-deficient metastatic CRC [144]. Pertruzumab and trastuzumab are monoclonal antibodies that target ERBB2 in ERBB2-positive CRC and are widely used in breast cancer treatment [145]. ERBB2 amplification occurs in 3% of all metastatic CRCs and in 5% of CRC patients with RAS-wt tumors [146], thus representing an appreciably substantial patient subgroup that may benefit from ERBB2 inhibition. Bevacizumab is a monoclonal antibody that is directed towards the VEGF signaling pathway that is key for angiogenesis. Namely, bevacizumab inhibits tumor blood vessel growth and blood vessel recurrence and reverses permeability of the surviving vasculature. As a result, bevacizumab has been shown to be effective in improving CRC patient survival and is frequently used with 5-FU and other chemotherapies (reviewed in [147]).
Entrectinib is a small-molecule competitive inhibitor of tropomyosin neurotropic tyrosine receptor kinases A, B and C (TRKA, TRKB, TRKC), the proto-oncogene tyrosine protein kinase ROS1 and anaplastic lymphoma kinase (ALK) [148]. Similarly, larotrectinib is an inhibitor of TRKA, TRKB and TRKC. Specifically, TRKA activation induces phosphorylation of specific tyrosines across a series of proteins resulting in activation of multiple pathways that include PI3K/AKT and Ras [149,150,151]. TRKA is encoded by the neurotrophic tyrosine kinase receptor 1 (NTRK1) gene, and NTRK1 alterations have been observed in several human cancers, including colorectal cancer. Interestingly, NTRK1 is involved in multiple chromosomal rearrangements in human cancers that have oncogenic activity, including TPM3-TRK in colorectal cancer as well as MPRIP-NRTK1 and CD74 in non-small cell lung cancer [149]. The CRC TPM3-TRK fusion occurs at low frequencies (1–2%) in CRC [149] and co-occurs with APC and TP53 alterations as well as DNA hypermutation, MSI-H, enrichment in POLE/POLD1 mutations and location on the right side of the colorectum [152]. NTRK-altered CRCs are sensitive to both entrectinib and larotrectinib [68,153], suggesting that this subgroup of CRC patients may benefit from this personalized medicine approach.
7.1. Treatment Outcomes as a Function of Tumor Location and CIMP Status
CIMP-H tumors are generally found in the proximal (right) region of the colon; therefore, tumor location is an important determinant of CRC patient outcome (summarized in [35]). Even left and right colonic tissues have unique embryonic origins—right-sided colonic epithelial cells are derived from the midgut, while left-sided normal colonic mucosa is derived from the hindgut. In addition, colon crypt stem cell populations and embryonic gene expression differences begin in normal colonic tissues [154,155,156]. Left- and right-sided colon cancers display extensive differences in terms of genetic, epigenetic and transcriptomic profiles [154] as well as in clinical trials. A clinical trial of KRAS-wt metastatic colon cancer patients who were treated with either FOLFIRI or FOLFOX before treatment with bevacizumab or cetuximab showed significant differences in OS and PFS after stratification by tumor location [157,158], even though OS rates did not differ as a function of treatment arm. RAS-wt patients with left-sided tumors had improved PFS and OS compared to patients with right-sided tumors. Patients with left-sided RAS-wt tumors showed a significantly improved outcome when given cetuximab + FOLFIRI compared to right-sided, RAS-wt CRC patients [159]. Recently, Rossini et al. [160] performed a meta-analysis of five completed clinical trials (CAIRO5, FIRE-3, PARADIGM, PEAK and SWOG 80405) spanning over 2700 CRC patients to show that left-sided metastatic CRC patients showed improved OS after receiving anti-EGFR therapies compared to bevacizumab, while right-sided patients who received bevacizumab had longer progression-free survival but not significantly improved OS. Interestingly, there were significant correlations between tumor sidedness and treatment type with respect to PFS and OS, confirming the observations from other clinical trials.
From a molecular subtype perspective, CIMP-positive CRC patients have been evaluated in several clinical trials (summarized in [35,161]). In general, patients with right-sided tumors exhibit poorer OS and PFS and higher mortality rates than patients with left-sided tumors [162]; however, Min et al. and Van Rijnsoever et al. reported a survival benefit in stage II/III CIMP-positive patients when given 5-FU therapy [161,163,164]. CIMP-H CRC patients display worse DFS and OS than non-CIMP patients with microsatellite stable (MSS) or instable (MSI) status [165]. Similar findings were revealed after treatment with 5-FU in the FOLFOX setting, in which CIMP-positive patients generally showed no significant difference in DFS; however, OS rates improved. It should be noted that Zhang and colleagues did detect increased PFS and OS when CIMP-positive, metastatic CRC patients were given irinotecan followed by FOLFOX or when CIMP-positive patients received irinotecan, 5-FU and folinic acid [161,166,167]. CIMP-positive, KRAS-wt CRC patients who were given cetuximab demonstrated a shortened PFS compared to non-CIMP patients (reviewed in [161]). Finally, stratifying metastatic CRC patients by CMS gene expression subgrouping in the FIRE-3 clinical trial in which KRAS-wt patients received FOLFIRI + cetuximab or bevacizumab showed that CMS classification was significantly prognostic for PFS and OS [168]. Interestingly, CMS3 (right-sided, CIMP-L) and CMS4 (left-sided, non-CIMP) patients showed improved OS when given cetuximab compared to bevacizumab [168].
CIMP-positive tumors are highly enriched for the BRAF V600E point mutation and are resistant to EGFR-based therapies. In addition, treatments with BRAF inhibitors, including vemurafenib, as single agents have not demonstrated clinical efficacy [169,170]. Interestingly, clinical trials involving the combination of BRAF- and EGFR-inhibitors (vemurafenib and cetuximab) have shown improved performance for BRAF-mut CRCs [169,171,172,173]. Dabrafenib, another BRAF inhibitor, also showed an unexceptional response in treating BRAF-mut CRCs when combined with the MEK inhibitor trametinib, and response rates improved when dabrafenib (BRAF), trametinib (MEK) and panitumumab (EGFR) were combined. Similar findings were also found when combining vemurafenib (BRAF), cetuximab (EGFR) and irinotecan [169,174] or the combination of encorafenib (BRAF), cetuximab (EGFR) and binimetinib (MEK) (reviewed in [175]), supporting the observation that blocking EGFR signaling improves patient outcomes [169,176].
Clinical trials have also evaluated the efficacy of immune checkpoint inhibitors in treating BRAF-mut CRC patients. BRAF-mut and BRAF-wt CRC patients with microsatellite instability (MSI-H) given the anti-PD-1 antibody pembrolizumab showed improved outcomes [144,169]. Treatment-refractory, BRAF-mut, MSI-H CRC patients who were administered nivolumab (anti-PD-1) and ipilimumab (anti-T lymphocyte antigen-4) showed overall response rates similar to those for BRAF-wt CRC patients [169,177], although the duration of treatment response was shorter for BRAF-mut patients than for BRAF-wt patients. Finally, treatment of microsatellite-instable CRC patients with pembrolizumab (anti-PD-1) showed improved PFS compared to standard chemotherapy as a first-line approach [169,178].
7.2. Epigenetic Therapy in Colorectal Cancer
As described above, DNA methylation changes are pervasive in CRC and all other forms of human cancer and have roles in gene silencing and activation. DNA methylation is a dynamic and pharmacologically reversible epigenetic mark, making it an attractive therapeutic target summarized in [35]. Indeed, the global loss of DNA methylation in cancer cells, whether through genetic ablation or pharmacological intervention, results in dramatic cancer cell growth inhibition [179,180,181,182,183,184]. DNA methylation inhibitors, such as 5-aza-2′-deoxycytidine (decitabine, DAC), have anti-neoplastic effects on cancer cells through the reactivation of tumor suppressor genes [185,186] and downregulation of oncogenes [179]. DAC treatment also induces viral mimicry to stimulate an immune response and convert non-immune responsive tumors and/or those without T-cell infiltration (immune cold) to immune responsive tumors (immune hot) [180,181,182,184], which subsequently show increased responsiveness to chemotherapies [187]. This conversion starts with transcriptional reactivation of transposable elements (TEs) that include long terminal repeats (LTRs), short interspersed nuclear elements (SINEs) and long interspersed nuclear elements (LINEs) [180,181,182,183,184]. Therefore, DNA methylation inhibition combined with chemotherapeutic agents holds promise for cancer patient therapy.
7.3. Treatment of Metastatic CRC
Metastatic CRC represents a substantial therapeutic challenge, especially since the survival rates for patients with metastatic disease are dismal. Biller and Schrag [188] performed a meta-analysis of data from 222 clinical trials between 2014–2020 to better understand how molecular profiles affect treatment outcomes. Patients with BRAF-wt, KRAS-wt, NRAS-wt and metastatic CRCs have a median survival with treatment of 30 months [188]. However, stratifying patients by primary tumor location showed that right-sided patient median survival was 19 months compared to 34 months for patients with left-sided tumors [188]. These BRAF-wt, RAS-wt patients also show modest survival improvement of 2–4 additional months when given cetuximab and panitumumab with backbone chemotherapy compared to chemotherapy alone [188]. Left-sided metastatic CRC patients also demonstrated benefit from EGFR inhibition added to conventional chemotherapy when compared to bevacizumab and chemotherapy [188].
BRAF-mut patients saw a survival period of 9.3 months when given BRAF- and EGFR-based inhibitors compared to 5.9 months for those given standard chemotherapy [188]. Finally, patients with MSI or defective mismatch repair showed tremendous improvement in OS when given immunotherapy [188]. Patients with recurrent metastatic disease represent an especially challenging group, as there are no approved therapies that increase survival by more than three months, although treatment with regorafenib (a tyrosine kinase inhibitor), trifluridine (a thymidine analog) and tipiracil (a thymidine phosphorylase inhibitor) are FDA approved for third-line therapies [189] and show survival increases of 1–2 months [188].
8. Conclusions and Future Directions
In summary, CRC is a worldwide health burden, and CRC outcomes encapsulate socio-economic and racial disparities that affect patient diagnosis, treatment and outcome. From a biological standpoint, CRC is a heterogeneous disease in which biological covariates such as CIMP, KRAS/BRAF mutation, MSI and tumor location complicate treatment response and patient outcomes. CRC treatments historically target DNA synthesis and EGFR/VEGFR signaling; however, new treatment schemes have combined these with immunotherapies. Multi-omics profiling has provided new windows into CRC tumorigenesis and metastasis, but profiling minority populations with adenomas or adenocarcinomas is important for reducing health disparities and improving patient outcomes.
Author Contributions
Conceptualization, G.L. and T.L.; methodology, G.Y., X.Y. and D.J.W.; investigation, G.Y., X.Y. and D.J.W.; resources, G.Y., X.Y. and D.J.W.; writing—original draft preparation, G.Y., X.Y. and D.J.W.; writing—review and editing, D.J.W., T.L. and G.L.; visualization, G.Y., X.Y. and T.L.; supervision, G.L. and T.L.; project administration, G.L. and T.L.; funding acquisition, G.L. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Vicky Joseph Cancer Research Lab (to G.L.).
Conflicts of Interest
G.L. is a consultant for PANGEA Laboratory, Irvine, CA, USA. The remaining authors do not have conflict of interest to disclose.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, M.; Nieuwenhuizen, S.; Puijk, R.S.; Timmer, F.E.F.; Geboers, B.; Schouten, E.A.C.; Opperman, J.; Scheffer, H.J.; de Vries, J.J.J.; Versteeg, K.S.; et al. Primary Tumor Sidedness, RAS and BRAF Mutations and MSI Status as Prognostic Factors in Patients with Colorectal Liver Metastases Treated with Surgery and Thermal Ablation: Results from the Amsterdam Colorectal Liver Met Registry (AmCORE). Biomedicines 2021, 9, 962. [Google Scholar] [CrossRef] [PubMed]
- Vatandoust, S.; Price, T.J.; Karapetis, C.S. Colorectal cancer: Metastases to a single organ. World J. Gastroenterol. 2015, 21, 11767–11776. [Google Scholar] [CrossRef]
- Engstrand, J.; Nilsson, H.; Stromberg, C.; Jonas, E.; Freedman, J. Colorectal cancer liver metastases—A population-based study on incidence, management and survival. BMC Cancer 2018, 18, 78. [Google Scholar] [CrossRef]
- Meijerink, M.R.; Puijk, R.S.; van Tilborg, A.; Henningsen, K.H.; Fernandez, L.G.; Neyt, M.; Heymans, J.; Frankema, J.S.; de Jong, K.P.; Richel, D.J.; et al. Radiofrequency and Microwave Ablation Compared to Systemic Chemotherapy and to Partial Hepatectomy in the Treatment of Colorectal Liver Metastases: A Systematic Review and Meta-Analysis. Cardiovasc. Interv. Radiol. 2018, 41, 1189–1204. [Google Scholar] [CrossRef]
- Abdalla, E.K.; Vauthey, J.N.; Ellis, L.M.; Ellis, V.; Pollock, R.; Broglio, K.R.; Hess, K.; Curley, S.A. Recurrence and outcomes following hepatic resection, radiofrequency ablation, and combined resection/ablation for colorectal liver metastases. Ann. Surg. 2004, 239, 818–825; discussion 825–827. [Google Scholar] [CrossRef]
- Puijk, R.S.; Ruarus, A.H.; Vroomen, L.; van Tilborg, A.; Scheffer, H.J.; Nielsen, K.; de Jong, M.C.; de Vries, J.J.J.; Zonderhuis, B.M.; Eker, H.H.; et al. Colorectal liver metastases: Surgery versus thermal ablation (COLLISION)—A phase III single-blind prospective randomized controlled trial. BMC Cancer 2018, 18, 821. [Google Scholar] [CrossRef]
- Carethers, J.M. Racial and ethnic disparities in colorectal cancer incidence and mortality. Adv. Cancer Res. 2021, 151, 197–229. [Google Scholar] [CrossRef]
- Jackson, C.S.; Oman, M.; Patel, A.M.; Vega, K.J. Health disparities in colorectal cancer among racial and ethnic minorities in the United States. J. Gastrointest. Oncol. 2016, 7, S32–S43. [Google Scholar] [CrossRef]
- Carpten, J.D.; Fashoyin-Aje, L.; Garraway, L.A.; Winn, R. Making cancer research more inclusive. Nat. Rev. Cancer 2021, 21, 613–618. [Google Scholar] [CrossRef]
- Daniel, C.L.; Gilreath, K.; Keyes, D. Colorectal cancer disparities beyond biology: Screening, treatment, access. Front. Biosci. 2017, 22, 465–478. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, S.K.; Azim, S.; Ahmad, A.; Zubair, H.; Tyagi, N.; Srivastava, S.K.; Bhardwaj, A.; Singh, S.; Rocconi, R.P.; Singh, A.P. Biological basis of cancer health disparities: Resources and challenges for research. Am. J. Cancer Res. 2017, 7, 1–12. [Google Scholar] [PubMed]
- Williams, D.R.; Jackson, P.B. Social sources of racial disparities in health. Health Aff. 2005, 24, 325–334. [Google Scholar] [CrossRef] [PubMed]
- de Klerk, C.M.; Gupta, S.; Dekker, E.; Essink-Bot, M.L.; Expert Working Group ‘Coalition to reduce inequities in colorectal cancer screening’ of the World Endoscopy Organizaiton. Socioeconomic and ethnic inequities within organised colorectal cancer screening programmes worldwide. Gut 2018, 67, 679–687. [Google Scholar] [CrossRef]
- Baldwin, L.M.; Dobie, S.A.; Billingsley, K.; Cai, Y.; Wright, G.E.; Dominitz, J.A.; Barlow, W.; Warren, J.L.; Taplin, S.H. Explaining black-white differences in receipt of recommended colon cancer treatment. J. Natl. Cancer Inst. 2005, 97, 1211–1220. [Google Scholar] [CrossRef]
- Snyder, R.A.; Hu, C.Y.; Zafar, S.N.; Francescatti, A.; Chang, G.J. Racial Disparities in Recurrence and Overall Survival in Patients With Locoregional Colorectal Cancer. J. Natl. Cancer Inst. 2021, 113, 770–777. [Google Scholar] [CrossRef]
- Manz, C.R.; Schrag, D. Racial Disparities in Colorectal Cancer Recurrence and Mortality: Equitable Care, Inequitable Outcomes? J. Natl. Cancer Inst. 2021, 113, 656–657. [Google Scholar] [CrossRef]
- Obrochta, C.A.; Murphy, J.D.; Tsou, M.H.; Thompson, C.A. Disentangling Racial, Ethnic, and Socioeconomic Disparities in Treatment for Colorectal Cancer. Cancer Epidemiol. Biomarkers Prev. 2021, 30, 1546–1553. [Google Scholar] [CrossRef]
- Andersen, S.W.; Zheng, W.; Steinwandel, M.; Murff, H.J.; Lipworth, L.; Blot, W.J. Sociocultural Factors, Access to Healthcare, and Lifestyle: Multifactorial Indicators in Association with Colorectal Cancer Risk. Cancer Prev. Res. 2022, 15, 595–603. [Google Scholar] [CrossRef]
- Petrick, J.L.; Barber, L.E.; Rosenberg, L. What Are the Factors Underlying Colorectal Cancer Health Disparities? Cancer Prev. Res. 2022, 15, 561–563. [Google Scholar] [CrossRef]
- Kane, W.J.M.; Fleming, M.A.I.M.; Lynch, K.T.M.; Friel, C.M.M.; Williams, M.D.M.; Hedrick, T.L.M.; Yan, G.; Hoang, S.C.M. Associations of Race, Ethnicity, and Social Determinants of Health with Colorectal Cancer Screening. Dis. Colon. Rectum 2022. [Google Scholar] [CrossRef] [PubMed]
- Doubeni, C.A.; Corley, D.A.; Zhao, W.; Lau, Y.; Jensen, C.D.; Levin, T.R. Association between Improved Colorectal Screening and Racial Disparities. N. Engl. J. Med. 2022, 386, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Augustus, G.J.; Ellis, N.A. Colorectal Cancer Disparity in African Americans: Risk Factors and Carcinogenic Mechanisms. Am. J. Pathol. 2018, 188, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Kamath, S.D.; Torrejon, N.; Wei, W.; Tullio, K.; Nair, K.G.; Liska, D.; Krishnamurthi, S.S.; Khorana, A.A. Racial disparities negatively impact outcomes in early-onset colorectal cancer independent of socioeconomic status. Cancer Med. 2021, 10, 7542–7550. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.M.D.; Fontham, E.T.H.; Church, T.R.; Flowers, C.R.; Guerra, C.E.; LaMonte, S.J.; Etzioni, R.; McKenna, M.T.; Oeffinger, K.C.; Shih, Y.T.; et al. Colorectal cancer screening for average-risk adults: 2018 guideline update from the American Cancer Society. CA Cancer J. Clin. 2018, 68, 250–281. [Google Scholar] [CrossRef]
- Khan, S.A.; Morris, M.; Idrees, K.; Gimbel, M.I.; Rosenberg, S.; Zeng, Z.; Li, F.; Gan, G.; Shia, J.; LaQuaglia, M.P.; et al. Colorectal cancer in the very young: A comparative study of tumor markers, pathology and survival in early onset and adult onset patients. J. Pediatr. Surg. 2016, 51, 1812–1817. [Google Scholar] [CrossRef]
- Petrick, J.L.; Barber, L.E.; Andersen, S.W.; Florio, A.A.; Palmer, J.R.; Rosenberg, L. Racial Disparities and Sex Differences in Early- and Late-Onset Colorectal Cancer Incidence, 2001–2018. Front. Oncol. 2021, 11, 734998. [Google Scholar] [CrossRef]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Rev. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Becker, W.R.; Nevins, S.A.; Chen, D.C.; Chiu, R.; Horning, A.M.; Guha, T.K.; Laquindanum, R.; Mills, M.; Chaib, H.; Ladabaum, U.; et al. Single-cell analyses define a continuum of cell state and composition changes in the malignant transformation of polyps to colorectal cancer. Nat. Genet. 2022, 54, 985–995. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nat. Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Ding, J.; Ma, Z.; Sun, R.; Seoane, J.A.; Shaffer, J.S.; Suarez, C.J.; Berghoff, A.S.; Cremolini, C.; Falcone, A.; et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 2019, 51, 1113–1122. [Google Scholar] [CrossRef]
- Bettington, M.; Walker, N.; Clouston, A.; Brown, I.; Leggett, B.; Whitehall, V. The serrated pathway to colorectal carcinoma: Current concepts and challenges. Histopathology 2013, 62, 367–386. [Google Scholar] [CrossRef] [PubMed]
- Weisenberger, D.J.; Liang, G.; Lenz, H.J. DNA methylation aberrancies delineate clinically distinct subsets of colorectal cancer and provide novel targets for epigenetic therapies. Oncogene 2018, 37, 566–577. [Google Scholar] [CrossRef]
- Gervaz, P.; Bucher, P.; Morel, P. Two colons-two cancers: Paradigm shift and clinical implications. J. Surg. Oncol. 2004, 88, 261–266. [Google Scholar] [CrossRef]
- Giannakis, M.; Mu, X.J.; Shukla, S.A.; Qian, Z.R.; Cohen, O.; Nishihara, R.; Bahl, S.; Cao, Y.; Amin-Mansour, A.; Yamauchi, M.; et al. Genomic Correlates of Immune-Cell Infiltrates in Colorectal Carcinoma. Cell Rep. 2016, 17, 1206. [Google Scholar] [CrossRef]
- Guda, K.; Veigl, M.L.; Varadan, V.; Nosrati, A.; Ravi, L.; Lutterbaugh, J.; Beard, L.; Willson, J.K.; Sedwick, W.D.; Wang, Z.J.; et al. Novel recurrently mutated genes in African American colon cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 1149–1154. [Google Scholar] [CrossRef]
- Wang, Z.; Li, L.; Guda, K.; Chen, Z.; Barnholtz-Sloan, J.; Park, Y.S.; Markowitz, S.D.; Willis, J. Adverse Clinical Outcome Associated With Mutations That Typify African American Colorectal Cancers. J. Natl. Cancer Inst. 2016, 108, djw164. [Google Scholar] [CrossRef]
- Brim, H.; Lee, E.; Abu-Asab, M.S.; Chaouchi, M.; Razjouyan, H.; Namin, H.; Goel, A.; Schaffer, A.A.; Ashktorab, H. Genomic aberrations in an African American colorectal cancer cohort reveals a MSI-specific profile and chromosome X amplification in male patients. PLoS ONE 2012, 7, e40392. [Google Scholar] [CrossRef]
- Manne, U.; Jadhav, T.; Putcha, B.K.; Samuel, T.; Soni, S.; Shanmugam, C.; Suswam, E.A. Molecular Biomarkers of Colorectal Cancer and Cancer Disparities: Current Status and Perspective. Curr. Color. Cancer Rep. 2016, 12, 332–344. [Google Scholar] [CrossRef] [PubMed]
- Nayani, R.; Ashktorab, H.; Brim, H.; Laiyemo, A.O. Genetic Basis for Colorectal Cancer Disparities. Curr. Color. Cancer Rep. 2015, 11, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Katkoori, V.R.; Jia, X.; Shanmugam, C.; Wan, W.; Meleth, S.; Bumpers, H.; Grizzle, W.E.; Manne, U. Prognostic significance of p53 codon 72 polymorphism differs with race in colorectal adenocarcinoma. Clin. Cancer Res. 2009, 15, 2406–2416. [Google Scholar] [CrossRef] [PubMed]
- Holowatyj, A.N.; Wen, W.; Gibbs, T.; Seagle, H.M.; Keller, S.R.; Edwards, D.R.V.; Washington, M.K.; Eng, C.; Perea, J.; Zheng, W.; et al. Racial/ethnic and sex differences in somatic cancer gene mutations among patients with early-onset colorectal cancer. Cancer Discov. 2022, 13, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Xicola, R.M.; Manojlovic, Z.; Augustus, G.J.; Kupfer, S.S.; Emmadi, R.; Alagiozian-Angelova, V.; Triche, T., Jr.; Salhia, B.; Carpten, J.; Llor, X.; et al. Lack of APC somatic mutation is associated with early-onset colorectal cancer in African Americans. Carcinogenesis 2018, 39, 1331–1341. [Google Scholar] [CrossRef] [PubMed]
- Vermaat, J.S.; Nijman, I.J.; Koudijs, M.J.; Gerritse, F.L.; Scherer, S.J.; Mokry, M.; Roessingh, W.M.; Lansu, N.; de Bruijn, E.; van Hillegersberg, R.; et al. Primary colorectal cancers and their subsequent hepatic metastases are genetically different: Implications for selection of patients for targeted treatment. Clin. Cancer Res. 2012, 18, 688–699. [Google Scholar] [CrossRef]
- Haan, J.C.; Labots, M.; Rausch, C.; Koopman, M.; Tol, J.; Mekenkamp, L.J.; van de Wiel, M.A.; Israeli, D.; van Essen, H.F.; van Grieken, N.C.; et al. Genomic landscape of metastatic colorectal cancer. Nat. Commun. 2014, 5, 5457. [Google Scholar] [CrossRef] [PubMed]
- Ishaque, N.; Abba, M.L.; Hauser, C.; Patil, N.; Paramasivam, N.; Huebschmann, D.; Leupold, J.H.; Balasubramanian, G.P.; Kleinheinz, K.; Toprak, U.H.; et al. Whole genome sequencing puts forward hypotheses on metastasis evolution and therapy in colorectal cancer. Nat. Commun. 2018, 9, 4782. [Google Scholar] [CrossRef]
- Yaeger, R.; Chatila, W.K.; Lipsyc, M.D.; Hechtman, J.F.; Cercek, A.; Sanchez-Vega, F.; Jayakumaran, G.; Middha, S.; Zehir, A.; Donoghue, M.T.A.; et al. Clinical Sequencing Defines the Genomic Landscape of Metastatic Colorectal Cancer. Cancer Cell. 2018, 33, 125–136.e3. [Google Scholar] [CrossRef]
- Mendelaar, P.A.J.; Smid, M.; van Riet, J.; Angus, L.; Labots, M.; Steeghs, N.; Hendriks, M.P.; Cirkel, G.A.; van Rooijen, J.M.; Tije, A.J.T.; et al. Whole genome sequencing of metastatic colorectal cancer reveals prior treatment effects and specific metastasis features. Nat. Commun. 2021, 12, 574. [Google Scholar] [CrossRef]
- Booth, M.J.; Branco, M.R.; Ficz, G.; Oxley, D.; Krueger, F.; Reik, W.; Balasubramanian, S. Quantitative sequencing of 5-methylcytosine and 5-hydroxymethylcytosine at single-base resolution. Science 2012, 336, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Shen, L.; Dai, Q.; Wu, S.C.; Collins, L.B.; Swenberg, J.A.; He, C.; Zhang, Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 2011, 333, 1300–1303. [Google Scholar] [CrossRef] [PubMed]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Gong, C.; Wang, Y.; Hu, Y.; Liu, H.; Fang, Z. Multi-omics analysis to identify driving factors in colorectal cancer. Epigenomics 2020, 12, 1633–1650. [Google Scholar] [CrossRef]
- Bach, S.; Paulis, I.; Sluiter, N.R.; Tibbesma, M.; Martin, I.; van de Wiel, M.A.; Tuynman, J.B.; Bahce, I.; Kazemier, G.; Steenbergen, R.D.M. Detection of colorectal cancer in urine using DNA methylation analysis. Sci. Rep. 2021, 11, 2363. [Google Scholar] [CrossRef]
- Fatemi, N.; Tierling, S.; Es, H.A.; Varkiani, M.; Mojarad, E.N.; Aghdaei, H.A.; Walter, J.; Totonchi, M. DNA methylation biomarkers in colorectal cancer: Clinical applications for precision medicine. Int. J. Cancer 2022, 151, 2068–2081. [Google Scholar] [CrossRef]
- Muller, D.; Gyorffy, B. DNA methylation-based diagnostic, prognostic, and predictive biomarkers in colorectal cancer. Biochim. Biophys. Acta Rev. Cancer 2022, 1877, 188722. [Google Scholar] [CrossRef]
- Anghel, S.A.; Ionita-Mindrican, C.B.; Luca, I.; Pop, A.L. Promising Epigenetic Biomarkers for the Early Detection of Colorectal Cancer: A Systematic Review. Cancers 2021, 13, 4965. [Google Scholar] [CrossRef]
- Jung, G.; Hernandez-Illan, E.; Moreira, L.; Balaguer, F.; Goel, A. Epigenetics of colorectal cancer: Biomarker and therapeutic potential. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 111–130. [Google Scholar] [CrossRef]
- Zhang, L.; Li, D.; Du, F.; Huang, H.; Yuan, C.; Fu, J.; Sun, S.; Tian, T.; Liu, X.; Sun, H.; et al. A panel of differentially methylated regions enable prognosis prediction for colorectal cancer. Genomics 2021, 113, 3285–3293. [Google Scholar] [CrossRef]
- Gundert, M.; Edelmann, D.; Benner, A.; Jansen, L.; Jia, M.; Walter, V.; Knebel, P.; Herpel, E.; Chang-Claude, J.; Hoffmeister, M.; et al. Genome-wide DNA methylation analysis reveals a prognostic classifier for non-metastatic colorectal cancer (ProMCol classifier). Gut 2019, 68, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Ye, W.; Liu, T.; Jian, S.; Liu, W. The Development of Three-DNA Methylation Signature as a Novel Prognostic Biomarker in Patients with Colorectal Cancer. Biomed. Res. Int. 2020, 2020, 3497810. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Mahoney, D.W.; Foote, P.H.; Burger, K.N.; Doering, K.A.; Taylor, W.R.; Then, S.S.; Cao, X.; McGlinch, M.; Berger, C.K.; et al. Novel Methylated DNA Markers in the Surveillance of Colorectal Cancer Recurrence. Clin. Cancer Res. 2021, 27, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wan, H.; Tian, J.; Cheng, X.; Rao, M.; Li, J.; Zhang, H.; Zhang, M.; Cai, Y.; Lu, Z.; et al. CpG-methylation-based risk score predicts progression in colorectal cancer. Epigenomics 2020, 12, 605–615. [Google Scholar] [CrossRef]
- Cai, G.; Cai, M.; Feng, Z.; Liu, R.; Liang, L.; Zhou, P.; ColonAi, Q.G.; Zhu, B.; Mo, S.; Wang, H.; et al. A Multilocus Blood-Based Assay Targeting Circulating Tumor DNA Methylation Enables Early Detection and Early Relapse Prediction of Colorectal Cancer. Gastroenterology 2021, 161, 2053–2056.e2. [Google Scholar] [CrossRef]
- Muthamilselvan, S.; Raghavendran, A.; Palaniappan, A. Stage-differentiated ensemble modeling of DNA methylation landscapes uncovers salient biomarkers and prognostic signatures in colorectal cancer progression. PLoS ONE 2022, 17, e0249151. [Google Scholar] [CrossRef]
- Chen, F.; Pei, L.; Liu, S.; Lin, Y.; Han, X.; Meng, E.; Wang, X.; Hong, S.; Wang, D.; Liu, F.; et al. Identification of a Novel Immune-Related CpG Methylation Signature to Predict Prognosis in Stage II/III Colorectal Cancer. Front. Genet. 2021, 12, 684349. [Google Scholar] [CrossRef]
- Antelo, M.; Balaguer, F.; Shia, J.; Shen, Y.; Hur, K.; Moreira, L.; Cuatrecasas, M.; Bujanda, L.; Giraldez, M.D.; Takahashi, M.; et al. A high degree of LINE-1 hypomethylation is a unique feature of early-onset colorectal cancer. PLoS ONE 2012, 7, e45357. [Google Scholar] [CrossRef]
- Joo, J.E.; Clendenning, M.; Wong, E.M.; Rosty, C.; Mahmood, K.; Georgeson, P.; Winship, I.M.; Preston, S.G.; Win, A.K.; Dugue, P.A.; et al. DNA Methylation Signatures and the Contribution of Age-Associated Methylomic Drift to Carcinogenesis in Early-Onset Colorectal Cancer. Cancers 2021, 13, 2589. [Google Scholar] [CrossRef]
- Toyota, M.; Ahuja, N.; Ohe-Toyota, M.; Herman, J.G.; Baylin, S.B.; Issa, J.P. CpG island methylator phenotype in colorectal cancer. Proc. Natl. Acad. Sci. USA 1999, 96, 8681–8686. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Siegmund, K.D.; Campan, M.; Young, J.; Long, T.I.; Faasse, M.A.; Kang, G.H.; Widschwendter, M.; Weener, D.; Buchanan, D.; et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat. Genet. 2006, 38, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Hinoue, T.; Weisenberger, D.J.; Lange, C.P.; Shen, H.; Byun, H.M.; Van Den Berg, D.; Malik, S.; Pan, F.; Noushmehr, H.; van Dijk, C.M.; et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012, 22, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Kawasaki, T.; Kirkner, G.J.; Loda, M.; Fuchs, C.S. CpG island methylator phenotype-low (CIMP-low) in colorectal cancer: Possible associations with male sex and KRAS mutations. J. Mol. Diagn. 2006, 8, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Toyota, M.; Kondo, Y.; Lin, E.; Zhang, L.; Guo, Y.; Hernandez, N.S.; Chen, X.; Ahmed, S.; Konishi, K.; et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 18654–18659. [Google Scholar] [CrossRef]
- Yagi, K.; Akagi, K.; Hayashi, H.; Nagae, G.; Tsuji, S.; Isagawa, T.; Midorikawa, Y.; Nishimura, Y.; Sakamoto, H.; Seto, Y.; et al. Three DNA methylation epigenotypes in human colorectal cancer. Clin. Cancer Res. 2010, 16, 21–33. [Google Scholar] [CrossRef]
- Phipps, A.I.; Alwers, E.; Harrison, T.; Banbury, B.; Brenner, H.; Campbell, P.T.; Chang-Claude, J.; Buchanan, D.; Chan, A.T.; Farris, A.B.; et al. Association Between Molecular Subtypes of Colorectal Tumors and Patient Survival, Based on Pooled Analysis of 7 International Studies. Gastroenterology 2020, 158, 2158–2168 e4. [Google Scholar] [CrossRef]
- Murcia, O.; Juarez, M.; Rodriguez-Soler, M.; Hernandez-Illan, E.; Giner-Calabuig, M.; Alustiza, M.; Egoavil, C.; Castillejo, A.; Alenda, C.; Barbera, V.; et al. Colorectal cancer molecular classification using BRAF, KRAS, microsatellite instability and CIMP status: Prognostic implications and response to chemotherapy. PLoS ONE 2018, 13, e0203051. [Google Scholar] [CrossRef]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef]
- Lindor, N.M.; Burgart, L.J.; Leontovich, O.; Goldberg, R.M.; Cunningham, J.M.; Sargent, D.J.; Walsh-Vockley, C.; Petersen, G.M.; Walsh, M.D.; Leggett, B.A.; et al. Immunohistochemistry versus microsatellite instability testing in phenotyping colorectal tumors. J. Clin. Oncol. 2002, 20, 1043–1048. [Google Scholar] [CrossRef]
- Trinh, A.; Trumpi, K.; Melo, F.D.S.E.; Wang, X.; de Jong, J.H.; Fessler, E.; Kuppen, P.J.; Reimers, M.S.; Swets, M.; Koopman, M.; et al. Practical and Robust Identification of Molecular Subtypes in Colorectal Cancer by Immunohistochemistry. Clin. Cancer Res. 2017, 23, 387–398. [Google Scholar] [CrossRef]
- Ubink, I.; Elias, S.G.; Moelans, C.B.; Lacle, M.M.; van Grevenstein, W.M.U.; van Diest, P.J.; Rinkes, I.H.M.V.B.; Kranenburg, O. A Novel Diagnostic Tool for Selecting Patients With Mesenchymal-Type Colon Cancer Reveals Intratumor Subtype Heterogeneity. J. Natl. Cancer Inst. 2017, 109, djw303. [Google Scholar] [CrossRef] [PubMed]
- Berg, I.V.D.; Smid, M.; Braak, R.R.J.C.V.D.; van de Wiel, M.A.; van Deurzen, C.H.; de Weerd, V.; Martens, J.W.M.; Ijzermans, J.N.M.; Wilting, S.M. A panel of DNA methylation markers for the classification of consensus molecular subtypes 2 and 3 in patients with colorectal cancer. Mol. Oncol. 2021, 15, 3348–3362. [Google Scholar] [CrossRef] [PubMed]
- Hur, K.; Cejas, P.; Feliu, J.; Moreno-Rubio, J.; Burgos, E.; Boland, C.R.; Goel, A. Hypomethylation of long interspersed nuclear element-1 (LINE-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut 2014, 63, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Ili, C.; Buchegger, K.; Demond, H.; Castillo-Fernandez, J.; Kelsey, G.; Zanella, L.; Abanto, M.; Riquelme, I.; Lopez, J.; Viscarra, T.; et al. Landscape of Genome-Wide DNA Methylation of Colorectal Cancer Metastasis. Cancers 2020, 12, 2710. [Google Scholar] [CrossRef] [PubMed]
- Orjuela, S.; Menigatti, M.; Schraml, P.; Kambakamba, P.; Robinson, M.D.; Marra, G. The DNA hypermethylation phenotype of colorectal cancer liver metastases resembles that of the primary colorectal cancers. BMC Cancer 2020, 20, 290. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Xue, W.; Liu, Z.; Chen, S.; Wang, J.; Peng, Q.; Xu, L.; Liu, X.; Cui, C.; Fan, J.B. A novel DNA methylation marker to identify lymph node metastasis of colorectal cancer. Front. Oncol. 2022, 12, 1000823. [Google Scholar] [CrossRef]
- Li, W.; Guo, L.; Tang, W.; Ma, Y.; Wang, X.; Shao, Y.; Zhao, H.; Ying, J. Identification of DNA methylation biomarkers for risk of liver metastasis in early-stage colorectal cancer. Clin. Epigenetics 2021, 13, 126. [Google Scholar] [CrossRef]
- Konishi, K.; Watanabe, Y.; Shen, L.; Guo, Y.; Castoro, R.J.; Kondo, K.; Chung, W.; Ahmed, S.; Jelinek, J.; Boumber, Y.A.; et al. DNA methylation profiles of primary colorectal carcinoma and matched liver metastasis. PLoS ONE 2011, 6, e27889. [Google Scholar] [CrossRef]
- Issa, J.P. Aging, DNA methylation and cancer. Crit. Rev. Oncol. Hematol. 1999, 32, 31–43. [Google Scholar] [CrossRef]
- Jung, M.; Pfeifer, G.P. Aging and DNA methylation. BMC Biol. 2015, 13, 7. [Google Scholar] [CrossRef]
- Unnikrishnan, A.; Freeman, W.M.; Jackson, J.; Wren, J.D.; Porter, H.; Richardson, A. The role of DNA methylation in epigenetics of aging. Pharmacol. Ther. 2019, 195, 172–185. [Google Scholar] [CrossRef] [PubMed]
- Issa, J.P.; Ottaviano, Y.L.; Celano, P.; Hamilton, S.R.; Davidson, N.E.; Baylin, S.B. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat. Genet. 1994, 7, 536–540. [Google Scholar] [CrossRef] [PubMed]
- Bjornsson, H.T.; Sigurdsson, M.I.; Fallin, M.D.; Irizarry, R.A.; Aspelund, T.; Cui, H.; Yu, W.; Rongione, M.A.; Ekstrom, T.J.; Harris, T.B.; et al. Intra-individual change over time in DNA methylation with familial clustering. JAMA 2008, 299, 2877–2883. [Google Scholar] [CrossRef] [PubMed]
- Bocklandt, S.; Lin, W.; Sehl, M.E.; Sanchez, F.J.; Sinsheimer, J.S.; Horvath, S.; Vilain, E. Epigenetic predictor of age. PLoS ONE 2011, 6, e14821. [Google Scholar] [CrossRef] [PubMed]
- Christensen, B.C.; Houseman, E.A.; Marsit, C.J.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.H.; Karagas, M.R.; Padbury, J.F.; Bueno, R.; et al. Aging and environmental exposures alter tissue-specific DNA methylation dependent upon CpG island context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.G.; Maxwell, A.P.; et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Weidner, C.I.; Lin, Q.; Koch, C.M.; Eisele, L.; Beier, F.; Ziegler, P.; Bauerschlag, D.O.; Jockel, K.H.; Erbel, R.; Muhleisen, T.W.; et al. Aging of blood can be tracked by DNA methylation changes at just three CpG sites. Genome Biol. 2014, 15, R24. [Google Scholar] [CrossRef] [PubMed]
- Devall, M.; Sun, X.; Yuan, F.; Cooper, G.S.; Willis, J.; Weisenberger, D.J.; Casey, G.; Li, L. Racial Disparities in Epigenetic Aging of the Right vs Left Colon. J. Natl. Cancer Inst. 2021, 113, 1779–1782. [Google Scholar] [CrossRef] [PubMed]
- Devall, M.A.; Sun, X.; Eaton, S.; Cooper, G.S.; Willis, J.E.; Weisenberger, D.J.; Casey, G.; Li, L. A Race-Specific, DNA Methylation Analysis of Aging in Normal Rectum: Implications for the Biology of Aging and Its Relationship to Rectal Cancer. Cancers 2022, 15, 45. [Google Scholar] [CrossRef] [PubMed]
- Rajaprakash, M.; Dean, L.T.; Palmore, M.; Johnson, S.B.; Kaufman, J.; Fallin, D.M.; Ladd-Acosta, C. DNA methylation signatures as biomarkers of socioeconomic position. Environ. Epigenetics 2023, 9, dvac027. [Google Scholar] [CrossRef] [PubMed]
- Konradsen, A.A.; Lund, C.M.; Vistisen, K.K.; Albieri, V.; Dalton, S.O.; Nielsen, D.L. The influence of socioeconomic position on adjuvant treatment of stage III colon cancer: A systematic review and meta-analysis. Acta Oncol. 2020, 59, 1291–1299. [Google Scholar] [CrossRef]
- Alonso, S.; Dai, Y.; Yamashita, K.; Horiuchi, S.; Dai, T.; Matsunaga, A.; Sanchez-Munoz, R.; Bilbao-Sieyro, C.; Diaz-Chico, J.C.; Chernov, A.V.; et al. Methylation of MGMT and ADAMTS14 in normal colon mucosa: Biomarkers of a field defect for cancerization preferentially targeting elder African-Americans. Oncotarget 2015, 6, 3420–3431. [Google Scholar] [CrossRef]
- Advani, S.M.; Advani, P.S.; Brown, D.W.; DeSantis, S.M.; Korphaisarn, K.; VonVille, H.M.; Bressler, J.; Lopez, D.S.; Davis, J.S.; Daniel, C.R.; et al. Global differences in the prevalence of the CpG island methylator phenotype of colorectal cancer. BMC Cancer 2019, 19, 964. [Google Scholar] [CrossRef]
- Wang, X.; Ji, P.; Zhang, Y.; LaComb, J.F.; Tian, X.; Li, E.; Williams, J.L. Aberrant DNA Methylation: Implications in Racial Health Disparity. PLoS ONE 2016, 11, e0153125. [Google Scholar] [CrossRef]
- Marisa, L.; de Reynies, A.; Duval, A.; Selves, J.; Gaub, M.P.; Vescovo, L.; Etienne-Grimaldi, M.C.; Schiappa, R.; Guenot, D.; Ayadi, M.; et al. Gene expression classification of colon cancer into molecular subtypes: Characterization, validation, and prognostic value. PLoS Med. 2013, 10, e1001453. [Google Scholar] [CrossRef]
- Kim, D.; Cho, K.H. Hidden patterns of gene expression provide prognostic insight for colorectal cancer. Cancer Gene Ther. 2023, 30, 11–21. [Google Scholar] [CrossRef]
- Emmert-Streib, F.; Simoes, R.d.M.; Glazko, G.; McDade, S.; Haibe-Kains, B.; Holzinger, A.; Dehmer, M.; Campbell, F.C. Functional and genetic analysis of the colon cancer network. BMC Bioinform. 2014, 15 (Suppl. 6), S6. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Tian, X.; Gao, R.; Guo, W.; Chen, C.; Jia, D.; Li, H.; Lv, X. Colon cancer diagnosis and staging classification based on machine learning and bioinformatics analysis. Comput. Biol. Med. 2022, 145, 105409. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, R.; Posey, J.; Chao, C.Y.; Lu, R.; Jadhav, T.; Javed, A.Y.; Javed, A.; Mahmoud, F.A.; Osarogiagbon, R.U.; Manne, U. A comparison of 12-gene colon cancer assay gene expression in African American and Caucasian patients with stage II colon cancer. BMC Cancer 2016, 16, 368. [Google Scholar] [CrossRef]
- Gray, R.G.; Quirke, P.; Handley, K.; Lopatin, M.; Magill, L.; Baehner, F.L.; Beaumont, C.; Clark-Langone, K.M.; Yoshizawa, C.N.; Lee, M.; et al. Validation study of a quantitative multigene reverse transcriptase-polymerase chain reaction assay for assessment of recurrence risk in patients with stage II colon cancer. J. Clin. Oncol. 2011, 29, 4611–4619. [Google Scholar] [CrossRef] [PubMed]
- Venook, A.P.; Niedzwiecki, D.; Lopatin, M.; Ye, X.; Lee, M.; Friedman, P.N.; Frankel, W.; Clark-Langone, K.; Millward, C.; Shak, S.; et al. Biologic determinants of tumor recurrence in stage II colon cancer: Validation study of the 12-gene recurrence score in cancer and leukemia group B (CALGB) 9581. J. Clin. Oncol. 2013, 31, 1775–1781. [Google Scholar] [CrossRef]
- Yothers, G.; O’Connell, M.J.; Lee, M.; Lopatin, M.; Clark-Langone, K.M.; Millward, C.; Paik, S.; Sharif, S.; Shak, S.; Wolmark, N. Validation of the 12-gene colon cancer recurrence score in NSABP C-07 as a predictor of recurrence in patients with stage II and III colon cancer treated with fluorouracil and leucovorin (FU/LV) and FU/LV plus oxaliplatin. J. Clin. Oncol. 2013, 31, 4512–4519. [Google Scholar] [CrossRef]
- Jovov, B.; Araujo-Perez, F.; Sigel, C.S.; Stratford, J.K.; McCoy, A.N.; Yeh, J.J.; Keku, T. Differential gene expression between African American and European American colorectal cancer patients. PLoS ONE 2012, 7, e30168. [Google Scholar] [CrossRef]
- Niu, L.; Gao, C.; Li, Y. Identification of potential core genes in colorectal carcinoma and key genes in colorectal cancer liver metastasis using bioinformatics analysis. Sci. Rep. 2021, 11, 23938. [Google Scholar] [CrossRef]
- Wu, B.; Yang, J.; Qin, Z.; Yang, H.; Shao, J.; Shang, Y. Prognosis prediction of stage IV colorectal cancer patients by mRNA transcriptional profile. Cancer Med. 2022, 11, 4900–4912. [Google Scholar] [CrossRef]
- Hinshaw, D.C.; Shevde, L.A. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019, 79, 4557–4566. [Google Scholar] [CrossRef]
- Zheng, X.; Ma, Y.; Bai, Y.; Huang, T.; Lv, X.; Deng, J.; Wang, Z.; Lian, W.; Tong, Y.; Zhang, X.; et al. Identification and validation of immunotherapy for four novel clusters of colorectal cancer based on the tumor microenvironment. Front. Immunol. 2022, 13, 984480. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Giannakis, M.; Wells, D.K.; Hamada, T.; Mu, X.J.; Quist, M.; Nowak, J.A.; Nishihara, R.; Qian, Z.R.; Inamura, K.; et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018, 8, 730–749. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.; Lewin, D.N.; Sun, S.; Spiceland, C.M.; Rockey, D.C.; Alekseyenko, A.V.; Wu, J.D.; Baron, J.A.; Alberg, A.J.; Hill, E.G. Tumor-Infiltrating Lymphocytes and Colorectal Cancer Survival in African American and Caucasian Patients. Cancer Epidemiol. Biomark. Prev. 2018, 27, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Curran, T.; Sun, Z.; Gerry, B.; Findlay, V.J.; Wallace, K.; Li, Z.; Paulos, C.; Ford, M.; Rubinstein, M.P.; Chung, D.; et al. Differential immune signatures in the tumor microenvironment are associated with colon cancer racial disparities. Cancer Med. 2021, 10, 1805–1814. [Google Scholar] [CrossRef] [PubMed]
- Basa, R.C.; Davies, V.; Li, X.; Murali, B.; Shah, J.; Yang, B.; Li, S.; Khan, M.W.; Tian, M.; Tejada, R.; et al. Decreased Anti-Tumor Cytotoxic Immunity among Microsatellite-Stable Colon Cancers from African Americans. PLoS ONE 2016, 11, e0156660. [Google Scholar] [CrossRef]
- Yu, M.R.; Kim, H.J.; Park, H.R. Fusobacterium nucleatum Accelerates the Progression of Colitis-Associated Colorectal Cancer by Promoting EMT. Cancers 2020, 12, 2728. [Google Scholar] [CrossRef]
- Yang, Q.; Wang, Y.; Jia, A.; Wang, Y.; Bi, Y.; Liu, G. The crosstalk between gut bacteria and host immunity in intestinal inflammation. J. Cell. Physiol. 2021, 236, 2239–2254. [Google Scholar] [CrossRef]
- Bullman, S.; Pedamallu, C.S.; Sicinska, E.; Clancy, T.E.; Zhang, X.; Cai, D.; Neuberg, D.; Huang, K.; Guevara, F.; Nelson, T.; et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 2017, 358, 1443–1448. [Google Scholar] [CrossRef]
- Ma, C.; Han, M.; Heinrich, B.; Fu, Q.; Zhang, Q.; Sandhu, M.; Agdashian, D.; Terabe, M.; Berzofsky, J.A.; Fako, V.; et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018, 360, eaan5931. [Google Scholar] [CrossRef]
- Geller, L.T.; Barzily-Rokni, M.; Danino, T.; Jonas, O.H.; Shental, N.; Nejman, D.; Gavert, N.; Zwang, Y.; Cooper, Z.A.; Shee, K.; et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 2017, 357, 1156–1160. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillere, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Poore, G.D.; Kopylova, E.; Zhu, Q.; Carpenter, C.; Fraraccio, S.; Wandro, S.; Kosciolek, T.; Janssen, S.; Metcalf, J.; Song, S.J.; et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature 2020, 579, 567–574. [Google Scholar] [CrossRef]
- Uriarte-Navarrete, I.; Hernandez-Lemus, E.; de Anda-Jauregui, G. Gene-Microbiome Co-expression Networks in Colon Cancer. Front. Genet. 2021, 12, 617505. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Si, H.; Bao, B.; Li, S.; Teng, D.; Yan, Y.; Hu, S.; Xu, Y.; Du, X. Integrated analysis of intestinal microbiota and host gene expression in colorectal cancer patients. J. Med. Microbiol. 2022, 71, 001596. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.P.; Duarte, G.S.; Sterrantino, C.; Pais, H.L.; Quintela, A.; Martins, A.P.; Costa, J. Triplet (FOLFOXIRI) versus doublet (FOLFOX or FOLFIRI) backbone chemotherapy as first-line treatment of metastatic colorectal cancer: A systematic review and meta-analysis. Crit. Rev. Oncol. Hematol. 2017, 118, 54–62. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Twelves, C.; Cassidy, J.; Allman, D.; Bajetta, E.; Boyer, M.; Bugat, R.; Findlay, M.; Frings, S.; Jahn, M.; et al. Oral capecitabine compared with intravenous fluorouracil plus leucovorin in patients with metastatic colorectal cancer: Results of a large phase III study. J. Clin. Oncol. 2001, 19, 4097–4106. [Google Scholar] [CrossRef]
- Bhattacharya, S. An empirical review on the resistance mechanisms of epidermal growth factor receptor inhibitors and predictive molecular biomarkers in colorectal cancer. Crit. Rev. Oncol. Hematol. 2023, 183, 103916. [Google Scholar] [CrossRef]
- Dassonville, O.; Bozec, A.; Fischel, J.L.; Milano, G. EGFR targeting therapies: Monoclonal antibodies versus tyrosine kinase inhibitors. Similarities and differences. Crit. Rev. Oncol. Hematol. 2007, 62, 53–61. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Lenz, H.J.; Kohne, C.H.; Heinemann, V.; Tejpar, S.; Melezinek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef]
- Bokemeyer, C.; Kohne, C.H.; Ciardiello, F.; Lenz, H.J.; Heinemann, V.; Klinkhardt, U.; Beier, F.; Duecker, K.; van Krieken, J.H.; Tejpar, S. FOLFOX4 plus cetuximab treatment and RAS mutations in colorectal cancer. Eur. J. Cancer 2015, 51, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Lievre, A.; Bachet, J.B.; Boige, V.; Cayre, A.; Le Corre, D.; Buc, E.; Ychou, M.; Bouche, O.; Landi, B.; Louvet, C.; et al. KRAS mutations as an independent prognostic factor in patients with advanced colorectal cancer treated with cetuximab. J. Clin. Oncol. 2008, 26, 374–379. [Google Scholar] [CrossRef] [PubMed]
- Punt, C.J.; Koopman, M.; Vermeulen, L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Kim, T.W.; Van Cutsem, E.; Geva, R.; Jager, D.; Hara, H.; Burge, M.; O’Neil, B.; Kavan, P.; Yoshino, T.; et al. Phase II Open-Label Study of Pembrolizumab in Treatment-Refractory, Microsatellite Instability-High/Mismatch Repair-Deficient Metastatic Colorectal Cancer: KEYNOTE-164. J. Clin. Oncol. 2020, 38, 11–19. [Google Scholar] [CrossRef]
- Bando, H.; Ohtsu, A.; Yoshino, T. Therapeutic landscape and future direction of metastatic colorectal cancer. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 306–32210. [Google Scholar] [CrossRef]
- Strickler, J.H.; Yoshino, T.; Graham, R.P.; Siena, S.; Bekaii-Saab, T. Diagnosis and Treatment of ERBB2-Positive Metastatic Colorectal Cancer: A Review. JAMA Oncol. 2022, 8, 760–769. [Google Scholar] [CrossRef]
- Yeung, Y.; Tebbutt, N.C. Bevacizumab in colorectal cancer: Current and future directions. Expert Rev. Anticancer. Ther. 2012, 12, 1263–1273. [Google Scholar] [CrossRef]
- Frampton, J.E. Entrectinib: A Review in NTRK+ Solid Tumours and ROS1+ NSCLC. Drugs 2021, 81, 697–708. [Google Scholar] [CrossRef]
- Ardini, E.; Bosotti, R.; Borgia, A.L.; De Ponti, C.; Somaschini, A.; Cammarota, R.; Amboldi, N.; Raddrizzani, L.; Milani, A.; Magnaghi, P.; et al. The TPM3-NTRK1 rearrangement is a recurring event in colorectal carcinoma and is associated with tumor sensitivity to TRKA kinase inhibition. Mol. Oncol. 2014, 8, 1495–1507. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Miller, F.D. Signal transduction by the neurotrophin receptors. Curr. Opin. Cell. Biol. 1997, 9, 213–221. [Google Scholar] [CrossRef]
- Kaplan, D.R.; Miller, F.D. Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol. 2000, 10, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.W.; Ou, Q.; Wu, X.; Nagasaka, M.; Shao, Y.; Ou, S.I.; Yang, Y. NTRK fusion positive colorectal cancer is a unique subset of CRC with high TMB and microsatellite instability. Cancer Med. 2022, 11, 2541–2549. [Google Scholar] [CrossRef] [PubMed]
- Rolfo, C.; Ruiz, R.; Giovannetti, E.; Gil-Bazo, I.; Russo, A.; Passiglia, F.; Giallombardo, M.; Peeters, M.; Raez, L. Entrectinib: A potent new TRK, ROS1, and ALK inhibitor. Expert Opin. Investig. Drugs 2015, 24, 1493–1500. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Jacobs, B.; D’Ario, G.; Di Narzo, A.F.; Soneson, C.; Budinska, E.; Popovici, V.; Vecchione, L.; Gerster, S.; Yan, P.; et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann. Oncol. 2014, 25, 1995–2001. [Google Scholar] [CrossRef] [PubMed]
- Powell, A.E.; Vlacich, G.; Zhao, Z.Y.; McKinley, E.T.; Washington, M.K.; Manning, H.C.; Coffey, R.J. Inducible loss of one Apc allele in Lrig1-expressing progenitor cells results in multiple distal colonic tumors with features of familial adenomatous polyposis. Am. J. Physiol. Liver Physiol. 2014, 307, G16–G23. [Google Scholar] [CrossRef]
- Wang, Y.; Poulin, E.J.; Coffey, R.J. LRIG1 is a triple threat: ERBB negative regulator, intestinal stem cell marker and tumour suppressor. Br. J. Cancer 2013, 108, 1765–1770. [Google Scholar] [CrossRef]
- Venook, A.P.; Niedzwiecki, D.; Innocenti, F.; Fruth, B.; Greene, C.; O’Neil, B.H.; Shaw, J.E.; Atkins, J.N.; Horvath, L.E.; Polite, B.N.; et al. Impact of primary (1º) tumor location on overall survival (OS) and progression-free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 80405 (Alliance). J. Clin. Oncol. 2016, 34, 3504. [Google Scholar] [CrossRef]
- Venook, A.P.; Niedzwiecki, D.; Lenz, H.-J.; Innocenti, F.; Mahoney, M.R.; O’Neil, B.H.; Shaw, J.E.; Polite, B.N.; Hochster, H.S.; Atkins, J.N.; et al. CALGB/SWOG 80405: Phase III trial of irinotecan/5-FU/leucovorin (FOLFIRI) or oxaliplatin/5-FU/leucovorin (mFOLFOX6) with bevacizumab (BV) or cetuximab (CET) for patients (pts) with KRAS wild-type (wt) untreated metastatic adenocarcinoma of the colon or rectum (MCRC). J. Clin. Oncol. 2014, 32, 15. [Google Scholar]
- Tejpar, S.; Stintzing, S.; Ciardiello, F.; Tabernero, J.; Van Cutsem, E.; Beier, F.; Esser, R.; Lenz, H.J.; Heinemann, V. Prognostic and Predictive Relevance of Primary Tumor Location in Patients With RAS Wild-Type Metastatic Colorectal Cancer: Retrospective Analyses of the CRYSTAL and FIRE-3 Trials. JAMA Oncol. 2016, 3, 194–201. [Google Scholar] [CrossRef]
- Rossini, D.; Boccaccino, A.; Carullo, M.; Antoniotti, C.; Dima, G.; Ciraci, P.; Marmorino, F.; Moretto, R.; Masi, G.; Cremolini, C. Primary tumour side as a driver for treatment choice in RAS wild-type metastatic colorectal cancer patients: A systematic review and pooled analysis of randomised trials. Eur. J. Cancer 2023, 184, 106–116. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, W.; Cao, P. Advances in CpG Island Methylator Phenotype Colorectal Cancer Therapies. Front. Oncol. 2021, 11, 629390. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Yang, D.; Yau, L.; Feng, S.; Cremolini, C.; Zhang, W.; Maus, M.K.; Antoniotti, C.; Langer, C.; Scherer, S.J.; et al. Primary tumor location as a prognostic factor in metastatic colorectal cancer. J. Natl. Cancer Inst. 2015, 107, dju427. [Google Scholar] [CrossRef] [PubMed]
- Min, B.H.; Bae, J.M.; Lee, E.J.; Yu, H.S.; Kim, Y.H.; Chang, D.K.; Kim, H.C.; Park, C.K.; Lee, S.H.; Kim, K.M.; et al. The CpG island methylator phenotype may confer a survival benefit in patients with stage II or III colorectal carcinomas receiving fluoropyrimidine-based adjuvant chemotherapy. BMC Cancer 2011, 11, 344. [Google Scholar] [CrossRef] [PubMed]
- Van Rijnsoever, M.; Elsaleh, H.; Joseph, D.; McCaul, K.; Iacopetta, B. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin. Cancer Res. 2003, 9, 2898–2903. [Google Scholar] [PubMed]
- Juo, Y.Y.; Johnston, F.M.; Zhang, D.Y.; Juo, H.H.; Wang, H.; Pappou, E.P.; Yu, T.; Easwaran, H.; Baylin, S.; van Engeland, M.; et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: A systematic review and meta-analysis. Ann. Oncol. 2014, 25, 2314–2327. [Google Scholar] [CrossRef]
- Shiovitz, S.; Bertagnolli, M.M.; Renfro, L.A.; Nam, E.; Foster, N.R.; Dzieciatkowski, S.; Luo, Y.; Lao, V.V.; Monnat, R.J., Jr.; Emond, M.J.; et al. CpG island methylator phenotype is associated with response to adjuvant irinotecan-based therapy for stage III colon cancer. Gastroenterology 2014, 147, 637–645. [Google Scholar] [CrossRef]
- Zhang, X.; Shimodaira, H.; Soeda, H.; Komine, K.; Takahashi, H.; Ouchi, K.; Inoue, M.; Takahashi, M.; Takahashi, S.; Ishioka, C. CpG island methylator phenotype is associated with the efficacy of sequential oxaliplatin- and irinotecan-based chemotherapy and EGFR-related gene mutation in Japanese patients with metastatic colorectal cancer. Int. J. Clin. Oncol. 2016, 21, 1091–1101. [Google Scholar] [CrossRef]
- Stintzing, S.; Wirapati, P.; Lenz, H.J.; Neureiter, D.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Kaiser, F.; Al-Batran, S.; Heintges, T.; et al. Consensus molecular subgroups (CMS) of colorectal cancer (CRC) and first-line efficacy of FOLFIRI plus cetuximab or bevacizumab in the FIRE3 (AIO KRK-0306) trial. Ann. Oncol. 2019, 30, 1796–1803. [Google Scholar] [CrossRef]
- Sahin, I.H.; Klostergaard, J. BRAF Mutations as Actionable Targets: A Paradigm Shift in the Management of Colorectal Cancer and Novel Avenues. JCO Oncol. Pract. 2021, 17, 723–730. [Google Scholar] [CrossRef]
- Yokota, T.; Ura, T.; Shibata, N.; Takahari, D.; Shitara, K.; Nomura, M.; Kondo, C.; Mizota, A.; Utsunomiya, S.; Muro, K.; et al. BRAF mutation is a powerful prognostic factor in advanced and recurrent colorectal cancer. Br. J. Cancer 2011, 104, 856–862. [Google Scholar] [CrossRef]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients With Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Grothey, A.; Yaeger, R.; Van Cutsem, E.; Desai, J.; Yoshino, T.; Wasan, H.; Ciardiello, F.; Loupakis, F.; Hong, Y.S.; et al. Encorafenib, Binimetinib, and Cetuximab in BRAF V600E-Mutated Colorectal Cancer. N. Engl. J. Med. 2019, 381, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Guthrie, K.A.; Morris, V.K.; Lenz, H.J.; Magliocco, A.M.; Maru, D.; Yan, Y.; Lanman, R.; Manyam, G.; Hong, D.S.; et al. Randomized Trial of Irinotecan and Cetuximab With or Without Vemurafenib in BRAF-Mutant Metastatic Colorectal Cancer (SWOG S1406). J. Clin. Oncol. 2021, 39, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.S.; Morris, V.K.; El Osta, B.; Sorokin, A.V.; Janku, F.; Fu, S.; Overman, M.J.; Piha-Paul, S.; Subbiah, V.; Kee, B.; et al. Phase IB Study of Vemurafenib in Combination with Irinotecan and Cetuximab in Patients with Metastatic Colorectal Cancer with BRAFV600E Mutation. Cancer Discov. 2016, 6, 1352–1365. [Google Scholar] [CrossRef] [PubMed]
- Ros, J.; Baraibar, I.; Sardo, E.; Mulet, N.; Salva, F.; Argiles, G.; Martini, G.; Ciardiello, D.; Cuadra, J.L.; Tabernero, J.; et al. BRAF, MEK and EGFR inhibition as treatment strategies in BRAF V600E metastatic colorectal cancer. Ther. Adv. Med. Oncol. 2021, 13, 1758835921992974. [Google Scholar] [CrossRef]
- Corcoran, R.B.; Andre, T.; Atreya, C.E.; Schellens, J.H.M.; Yoshino, T.; Bendell, J.C.; Hollebecque, A.; McRee, A.J.; Siena, S.; Middleton, G.; et al. Combined BRAF, EGFR, and MEK Inhibition in Patients with BRAF(V600E)-Mutant Colorectal Cancer. Cancer Discov. 2018, 8, 428–443. [Google Scholar] [CrossRef] [PubMed]
- Overman, M.J.; Lonardi, S.; Wong, K.Y.M.; Lenz, H.J.; Gelsomino, F.; Aglietta, M.; Morse, M.A.; Van Cutsem, E.; McDermott, R.; Hill, A.; et al. Durable Clinical Benefit With Nivolumab Plus Ipilimumab in DNA Mismatch Repair-Deficient/Microsatellite Instability-High Metastatic Colorectal Cancer. J. Clin. Oncol. 2018, 36, 773–779. [Google Scholar] [CrossRef]
- Andre, T.; Shiu, K.K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef]
- Chiappinelli, K.B.; Strissel, P.L.; Desrichard, A.; Li, H.; Henke, C.; Akman, B.; Hein, A.; Rote, N.S.; Cope, L.M.; Snyder, A.; et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2015, 162, 974–986. [Google Scholar] [CrossRef]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Mehdipour, P.; Marhon, S.A.; Ettayebi, I.; Chakravarthy, A.; Hosseini, A.; Wang, Y.; de Castro, F.A.; Yau, H.L.; Ishak, C.; Abelson, S.; et al. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 2020, 588, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.Z.; Qiu, Z.; Wu, Q.; Finlay, D.; Garcia, G.; Sun, D.; Rantala, J.; Barshop, W.; Hope, J.L.; Gimple, R.C.; et al. FBXO44 promotes DNA replication-coupled repetitive element silencing in cancer cells. Cell. 2020, 184, 352–369.e23. [Google Scholar] [CrossRef]
- Tunbak, H.; Enriquez-Gasca, R.; Tie, C.H.C.; Gould, P.A.; Mlcochova, P.; Gupta, R.K.; Fernandes, L.; Holt, J.; van der Veen, A.G.; Giampazolias, E.; et al. The HUSH complex is a gatekeeper of type I interferon through epigenetic regulation of LINE-1s. Nat. Commun. 2020, 11, 5387. [Google Scholar] [CrossRef]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2009, 31, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. A decade of exploring the cancer epigenome—Biological and translational implications. Nat. Rev. Cancer 2011, 11, 726–734. [Google Scholar] [CrossRef]
- Guler, G.D.; Tindell, C.A.; Pitti, R.; Wilson, C.; Nichols, K.; Cheung, T.K.; Kim, H.J.; Wongchenko, M.; Yan, Y.; Haley, B.; et al. Repression of Stress-Induced LINE-1 Expression Protects Cancer Cell Subpopulations from Lethal Drug Exposure. Cancer Cell 2017, 32, 221–237 e13. [Google Scholar] [CrossRef]
- Biller, L.H.; Schrag, D. A Review of the Diagnosis and Treatment of Metastatic Colorectal Cancer-Reply. JAMA 2021, 325, 2405. [Google Scholar] [CrossRef]
- Burness, C.B.; Duggan, S.T. Trifluridine/Tipiracil: A Review in Metastatic Colorectal Cancer. Drugs 2016, 76, 1393–1402. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).