
Crosstalk between Noncoding RNAs and the Epigenetics Machinery in Pediatric Tumors and Their Microenvironment

Abstract
Simple Summary
Abstract
1. Introduction
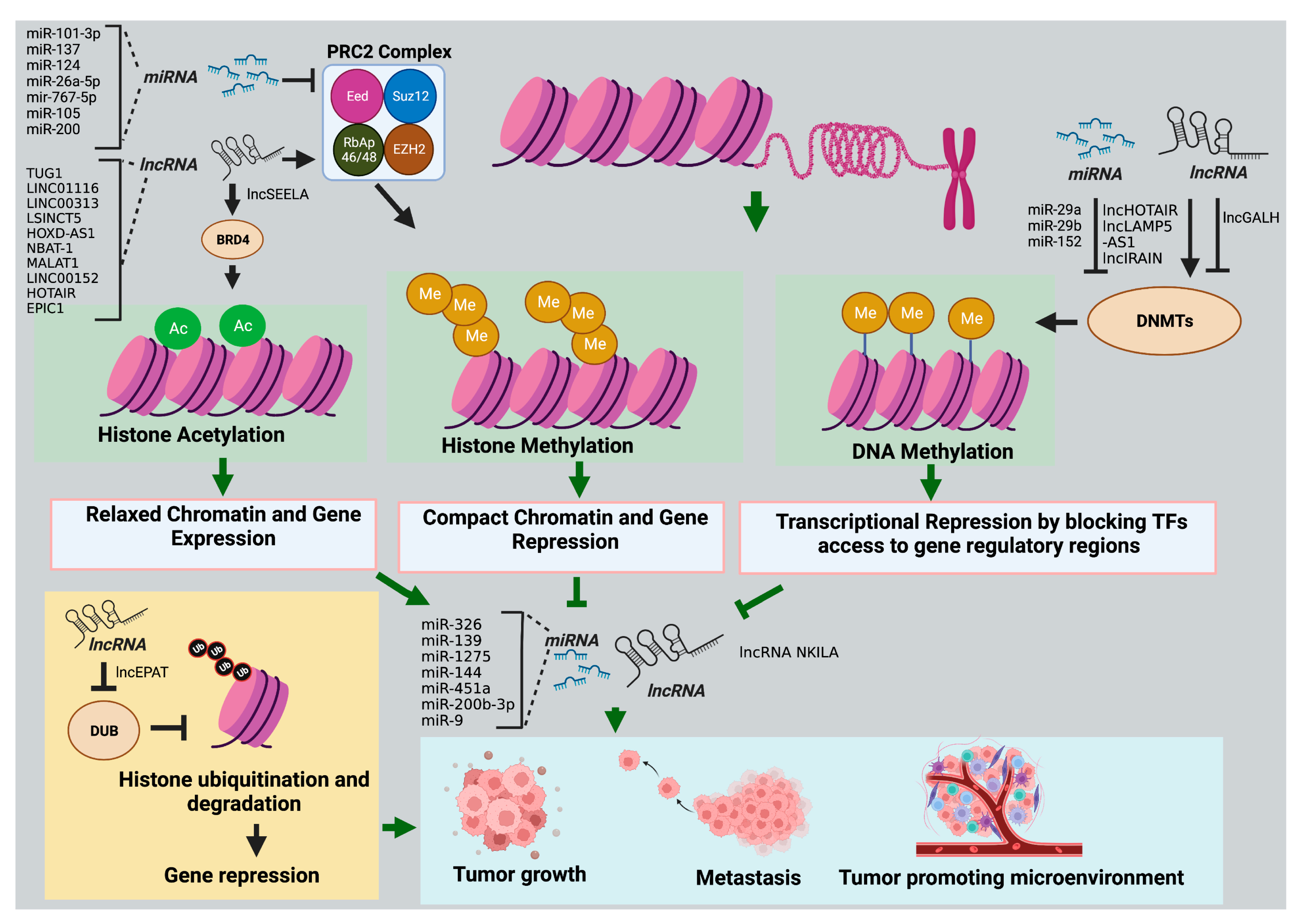
2. Role of ncRNAS in the Epigenetic Regulation of Pediatric Tumors
2.1. Histone Modifications
2.1.1. miRNAs
2.1.2. LncRNAs
2.2. DNA Methylation
2.2.1. miRNAs
2.2.2. LncRNAs
3. Role of ncRNAS in the Epigenetic Regulation of the TME
3.1. miRNAs
3.2. LncRNAs
3.3. PiRNAs and snRNAs

{kind=link}
{kind=link}
| LncRNAs | Cancer Type | Target Epigenetic Regulator | Mechanism of Action | Role as Potential Biomarker |
|---|---|---|---|---|
| HOX antisense intergenic RNA (HOTAIR) [178] | Cutaneous squamous cell carcinoma (CSCC) | Transcription factor Sp1 | HOTAIR interacts and upregulates Sp1, promoting Sp1-induced DNMT1-mediated promoter methylation and repression of miR-199a. Downregulation of miR-199a promotes CSCC cell stemness and tumor progression. | Upregulated in CSCC tissues compared to normal adjacent cells and associated with worse patient prognosis. |
| LncRNA IRAIN [179,180] | RCC | Dnmt1, Dnmt3a, and Dnmt3b | IRAIN recruits Dnmt1, Dnmt3a, and Dnmt3b to the VEGFA promoter, inhibiting its expression. VEGFA downregulation inhibits ECs recruitment, tumor angiogenesis, and growth. | IRAIN has lower expression in RC tissues than in healthy renal tissues. |
| LINC00152 [181] | Gastric cancer | EZH2 | LINC00152 recruits EZH2 to CXCL9 and CXCL10 promoters and epigenetically silences them. LINC00152 inhibition upregulates CXCL9, CXCL10, and CXCR3, which promotes intratumoral cytotoxic CD8+ T-cell infiltration. | LINC00152 is highly expressed in gastric cancer patients than normal counterparts. |
| Nuclear paraspeckle assembly transcript 1 (NEAT1) [182,183] | Glioblastoma | RNA-binding protein SFPQ | NEAT1 promotes paraspeckle assembly and relocates transcriptional repressor SFPQ from the CXCL8 promoter to paraspeckles, upregulating its protein product IL8 expression. IL8 secretion from tumor cells facilitates TAM recruitment and immunosuppression. | GBM tissues have higher expression of NEAT1 than low-grade glioma and normal brain tissues. |
| Colorectal neoplasia differentially expressed (CRNDE) [184,185] | HCC | p300/YY1 complex | CRNDE stabilizes the p300/YY1 complex and enhances histone H3K9 and H3K27 acetylation at the EGFR promoter, upregulating its expression. Exosomal EGFR is known to modulate the liver microenvironment to facilitate liver metastases. | CRNDE is highly expressed in human HCC compared to normal liver cells. |
| MIAT [186,187,188] | Thyroid cancer | EZH2 | MIAT sponges miR-150 activity and upregulates its target EZH2, promoting tumor cell proliferation, migration, and invasion. MIAT upregulation is associated with immune suppression in cancer. | MIAT is overexpressed in thyroid cancer patients |
| HOTAIR [189] | AML | EZH2 | HOTAIR recruits EZH2 to the p15 promoter, inducing its H3K27me3 and silencing gene expression. p15 downregulation is associated with the enhanced self-renewal capacity of leukemia stem cells, promoting leukemogenesis. | HOTAIR expression is significantly upregulated in AML patients. |
| NcRNA Types | Cancer Type | Target Epigenetic Regulator | Mechanism of Action |
|---|---|---|---|
| piRNA-823 [190] | Multiple myeloma | DNMT3A and 3B | piRNA-823 overexpression upregulates DNMT3A and DNMT3B levels and increases global DNA methylation. PiRNA-823 silencing reexpress methylation-silenced tumor suppressor, p16INK4A, decreases tumor angiogenesis, and inhibits tumor growth. |
| piR_011186 [191] | AML | DNMT1, Suv39H1 and/or EZH2 | piR_011186 promotes DNA and histone H3 methylation of the CDKN2B promoter, which downregulates its expression and is associated with enhanced cell proliferation. |
| PIWI-like 4 (piRNA associated protein) [192] | Glioma | H3K27me3 demethylase UTX | PIWIL4 interacts with UTX, which removes transcriptionally repressive H3K27me3 marks on neuronal genes, promoting neuronal differentiation and activity. The upregulation of neuronal genes due to PIWIL4–UTX interaction can further modify the glioma microenvironment and promote glioma cell proliferation. |
| piRNA-823 [193] | Multiple myeloma | DNMT3B | G-MDSCs induce piRNA-823 expression in multiple myeloma cells, which in turn activates DNMT3B expression and increases global DNA methylation. These changes are associated with enhanced stemness of multiple myeloma stem cells and tumor growth. |
| piRNA-30473 [194] | DLBCL | m6A mRNA methylase WTAP | piRNA-30473 stabilizes WTAP mRNA, upregulating global m6A levels in DLBCL cells. This increases hexokinase 2 (HK2) expression, which is associated with increased cell proliferation and tumorigenicity of DLBCL cells. |
| sdnRNA-3 [176] | Melanoma | Chromatin-remodeling regulator Mi-2β | sdnRNA-3 promotes the enrichment of chromodomain-helicase-DNA-binding protein 4 (CHD4), also known as Mi-2β, to the Nos2 promoter. This induces H3K27me3 modification of the Nos2 gene and represses the transcription of its gene product, inducible nitric oxide synthase (iNOS). The decrease in sdnRNA-3 expression in TAMs increases iNOS transcription and inhibits tumor growth. |
| RN7SK [177] | Multiple cancers including lung, liver, colon, and gastric | m6A readers | M6A readers recognize and interact with m6A-modified RN7SK, which facilitates the formation of RN7SK secondary structures and stabilizes its expression. RN7SK prevents the mRNA degradation of m6A readers by exonucleases, increasing their expression. The upregulation of m6A readers such as EWSR1 and KHDRBS1 promotes Wnt/β-catenin signaling and tumorigenesis by suppressing ubiquitin protein Cullin1 in various tumor types. |
| U1 snRNP [195] | Lung and breast cancer | Proximal polyadenylation signals (PASs) in introns and exons | U1 snRNP inhibits proximal polyadenylation signals (PASs) in introns and last exons, preventing premature transcription termination and mRNA shortening of target genes. U1 snRNP inhibition in cancer cells prompts the removal of 3ʹUTR miRNA target sites from many oncogenic mRNAs, resulting in their upregulation. The upregulated genes are involved in signaling pathways that control cell-cycle progression (CDC25A, CCNB1, and BRCA1), apoptosis (BCL6), cell growth (FGFR1, EGFR, and BRAF), cell migration (FGFR1, FYN, and TIMP2), extracellular matrix remodeling (TIMP2), DNA replication (APC), and transcription (EWSR1). |
| Database Name | Used for | Link |
|---|---|---|
| miRBase [196] | This database contains information about miRNAs including sequences, annotations, and expression data. It also provides tools for predicting miRNA targets. | https://www.mirbase.org (accessed on 26 April 2023) |
| TargetScan [197] | This provides computational predictions of miRNA targets based on the matching of miRNA seed sequence with complementary mRNA sequences. | www.targetscan.org (accessed on 26 April 2023) |
| miRTarBase [198] | This database provides information about experimentally validated miRNA–target mRNA interactions. | www.mirtarbase.cuhk.edu.cn/ (accessed on 26 April 2023) |
| miRWalk [199] | miRWalk provides predictions of miRNA–target interactions based on several existing miRNA–target prediction programs, including TargetScan, miRanda, miRBase, and miRDB4. It also integrates these predictions with experimentally validated interactions from other databases, such as miRTarBase. | https://mirtarbase.cuhk.edu.cn/ (accessed on 26 April 2023) |
| NONCODE [200] | This comprehensive database contains information about expression and functions of lncRNA. | www.noncode.org/introduce.php (accessed on 26 April 2023) |
| lncRNAdb [201] | This database includes lncRNA annotations, functions, and interactions with other molecules. | https://ngdc.cncb.ac.cn/ (accessed on 26 April 2023) |
| LNCipedia [202] | It includes comprehensive information on lncRNA structure, sequence, expression, and functional annotation. | https://lncipedia.org/ (accessed on 26 April 2023) |
| LncBook [203] | It is a comprehensive dataset for studying the functions and mechanisms of lncRNAs. It integrates multi-omics data from expression, methylation, genome variation, and lncRNA–miRNA interactions, providing a more complete picture of the lncRNA molecular networks. | https://ngdc.cncb.ac.cn/lncbook/ (accessed on 26 April 2023) |
| LncATLAS [204] and LncLocator [205] | LncATLAS is a database of lncRNA subcellular localization, whereas LncLocator can predict the subcellular localization of the lncRNAs. | https://lncatlas.crg.eu/ (accessed on 26 April 2023) http://www.csbio.sjtu.edu.cn/bioinf/lncLocator/ (accessed on 26 April 2023) |
| piRNABank [206] | It provides information on piRNA sequences, genomic locations, expression patterns, and potential targets. | http://pirnabank.ibab.ac.in/ (accessed on 26 April 2023) |
| piRBase [207] | The database comprises piRNA sequences, genomic locations, expression patterns, targets, and functions. | http://bigdata.ibp.ac.cn/piRBase/ (accessed on 26 April 2023) |
| Rfam [208] | This database includes a variety of RNA families, including small nuclear RNAs. It provides annotation and alignment data, secondary structure predictions, and functional information for each family. | https://rfam.org/ (accessed on 26 April 2023) |
| snOPY [209] | It provides comprehensive information about small nucleolar RNA (snoRNAs), snoRNA gene loci, and target RNAs. | http://snoopy.med.miyazaki-u.ac.jp/snorna_db.cgi (accessed on 26 April 2023) |
| RNAcentral [210] | It is a comprehensive database that provides a single access point to a large and diverse collection of RNA sequences and their functions. | https://rnacentral.org/ (accessed on 26 April 2023) |
| starBase [211] | This database can be used to identify the RNA–RNA and protein–RNA interaction networks. | http://starbase.sysu.edu.cn/ (accessed on 26 April 2023) |
| RSEM [212] | This tool can be used for quantifying gene and transcript expression levels from RNA-Seq data. | software package |
| miRDeep2 [213] | This tool can be used to identify novel and known miRNAs in deep sequencing data. | software package |
| ANNOVAR [214] | This tool can be used to functionally annotate genetic variants, including noncoding regions detected from diverse genomes. | https://annovar.openbioinformatics.org/ (accessed on 26 April 2023) |

4. Conclusions
Funding
Conflicts of Interest
References
- Gröbner, S.N.; Worst, B.C.; Weischenfeldt, J.; Buchhalter, I.; Kleinheinz, K.; Rudneva, V.A.; Johann, P.D.; Balasubramanian, G.P.; Segura-Wang, M.; Brabetz, S.; et al. The landscape of genomic alterations across childhood cancers. Nature 2018, 555, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, E.R.; Thiele, C.J. Epigenetic changes in pediatric solid tumors: Promising new targets. Clin. Cancer Res. 2012, 18, 2768–2779. [Google Scholar] [CrossRef] [PubMed]
- Panditharatna, E.; Filbin, M.G. The growing role of epigenetics in childhood cancers. Curr. Opin. Pediatr. 2020, 32, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Drozak, P.; Brylinski, L.; Zawitkowska, J. A Comprehensive Overview of Recent Advances in Epigenetics in Pediatric Acute Lymphoblastic Leukemia. Cancers 2022, 14, 5384. [Google Scholar] [CrossRef]
- Angione, S.D.A.; Akalu, A.Y.; Gartrell, J.; Fletcher, E.P.; Burckart, G.J.; Reaman, G.H.; Leong, R.; Stewart, C.F. Fusion Oncoproteins in Childhood Cancers: Potential Role in Targeted Therapy. J. Pediatr. Pharmacol. Ther. 2021, 26, 541–555. [Google Scholar] [CrossRef]
- Powers, M.P. The ever-changing world of gene fusions in cancer: A secondary gene fusion and progression. Oncogene 2019, 38, 7197–7199. [Google Scholar] [CrossRef]
- Terry, R.L.; Meyran, D.; Ziegler, D.S.; Haber, M.; Ekert, P.G.; Trapani, J.A.; Neeson, P.J. Immune profiling of pediatric solid tumors. J. Clin. Investig. 2020, 130, 3391–3402. [Google Scholar] [CrossRef]
- Vakkila, J.; Jaffe, R.; Michelow, M.; Lotze, M.T. Pediatric cancers are infiltrated predominantly by macrophages and contain a paucity of dendritic cells: A major nosologic difference with adult tumors. Clin. Cancer Res. 2006, 12 Pt 1, 2049–2054. [Google Scholar] [CrossRef]
- Koo, J.; Hayashi, M.; Verneris, M.R.; Lee-Sherick, A.B. Targeting Tumor-Associated Macrophages in the Pediatric Sarcoma Tumor Microenvironment. Front. Oncol. 2020, 10, 581107. [Google Scholar] [CrossRef]
- Pauli, A.; Rinn, J.L.; Schier, A.F. Non-coding RNAs as regulators of embryogenesis. Nat. Rev. Genet. 2011, 12, 136–149. [Google Scholar] [CrossRef]
- Lopez-Ramirez, M.A.; Nicoli, S. Role of miRNAs and epigenetics in neural stem cell fate determination. Epigenetics 2014, 9, 90–100. [Google Scholar] [CrossRef]
- Han, C.; Sun, L.Y.; Wang, W.T.; Sun, Y.M.; Chen, Y.Q. Non-coding RNAs in cancers with chromosomal rearrangements: The signatures, causes, functions and implications. J. Mol. Cell Biol. 2019, 11, 886–898. [Google Scholar] [CrossRef]
- Di Agostino, S.; Vahabi, M.; Turco, C.; Fontemaggi, G. Secreted Non-Coding RNAs: Functional Impact on the Tumor Microenvironment and Clinical Relevance in Triple-Negative Breast Cancer. Noncoding RNA 2022, 8, 5. [Google Scholar] [CrossRef]
- Smith, C.M.; Catchpoole, D.; Hutvagner, G. Non-Coding RNAs in Pediatric Solid Tumors. Front. Genet. 2019, 10, 798. [Google Scholar] [CrossRef]
- Dong, A.; Preusch, C.B.; So, W.K.; Lin, K.; Luan, S.; Yi, R.; Wong, J.W.; Wu, Z.; Cheung, T.H. A long noncoding RNA, LncMyoD, modulates chromatin accessibility to regulate muscle stem cell myogenic lineage progression. Proc. Natl. Acad. Sci. USA 2020, 117, 32464–32475. [Google Scholar] [CrossRef]
- Vierbuchen, T.; Agarwal, S.; Johnson, J.L.; Galia, L.; Lei, X.; Stein, K.; Olagnier, D.; Gaede, K.I.; Herzmann, C.; Holm, C.K.; et al. The lncRNA LUCAT1 is elevated in inflammatory disease and restrains inflammation by regulating the splicing and stability of NR4A2. Proc. Natl. Acad. Sci. USA 2023, 120, e2213715120. [Google Scholar] [CrossRef]
- Sauvageau, M. Diverging RNPs: Toward Understanding lncRNA-Protein Interactions and Functions. Adv. Exp. Med. Biol. 2019, 1203, 285–312. [Google Scholar] [CrossRef]
- Ninomiya, K.; Adachi, S.; Natsume, T.; Iwakiri, J.; Terai, G.; Asai, K.; Hirose, T. LncRNA-dependent nuclear stress bodies promote intron retention through SR protein phosphorylation. EMBO J. 2020, 39, e102729. [Google Scholar] [CrossRef]
- Taniue, K.; Kurimoto, A.; Sugimasa, H.; Nasu, E.; Takeda, Y.; Iwasaki, K.; Nagashima, T.; Okada-Hatakeyama, M.; Oyama, M.; Kozuka-Hata, H.; et al. Long noncoding RNA UPAT promotes colon tumorigenesis by inhibiting degradation of UHRF1. Proc. Natl. Acad. Sci. USA 2016, 113, 1273–1278. [Google Scholar] [CrossRef]
- Fatica, A.; Bozzoni, I. Long non-coding RNAs: New players in cell differentiation and development. Nat. Rev. Genet. 2014, 15, 7–21. [Google Scholar] [CrossRef]
- Bianchi, M.; Renzini, A.; Adamo, S.; Moresi, V. Coordinated Actions of MicroRNAs with other Epigenetic Factors Regulate Skeletal Muscle Development and Adaptation. Int. J. Mol. Sci. 2017, 18, 840. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Yu, L.Y.; Huang, X.Q.; Zhao, Z.H.; Liu, J. Epigenetic regulation by long noncoding RNAs in osteo-/adipogenic differentiation of mesenchymal stromal cells and degenerative bone diseases. World J. Stem Cells 2022, 14, 92–103. [Google Scholar] [CrossRef]
- Ponnusamy, M.; Yan, K.W.; Liu, C.Y.; Li, P.F.; Wang, K. PIWI family emerging as a decisive factor of cell fate: An overview. Eur. J. Cell Biol. 2017, 96, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Salvatori, B.; Biscarini, S.; Morlando, M. Non-coding RNAs in Nervous System Development and Disease. Front. Cell Dev. Biol. 2020, 8, 273. [Google Scholar] [CrossRef] [PubMed]
- Stavast, C.J.; van Zuijen, I.; Karkoulia, E.; Ozcelik, A.; van Hoven-Beijen, A.; Leon, L.G.; Voerman, J.S.A.; Janssen, G.M.C.; van Veelen, P.A.; Burocziova, M.; et al. The tumor suppressor MIR139 is silenced by POLR2M to promote AML oncogenesis. Leukemia 2022, 36, 687–700. [Google Scholar] [CrossRef]
- Pandey, G.K.; Mitra, S.; Subhash, S.; Hertwig, F.; Kanduri, M.; Mishra, K.; Fransson, S.; Ganeshram, A.; Mondal, T.; Bandaru, S.; et al. The risk-associated long noncoding RNA NBAT-1 controls neuroblastoma progression by regulating cell proliferation and neuronal differentiation. Cancer Cell 2014, 26, 722–737. [Google Scholar] [CrossRef]
- Fernandez-Diaz, D.; Rodriguez-Vidal, C.; Silva-Rodriguez, P.; Paniagua, L.; Blanco-Teijeiro, M.J.; Pardo, M.; Pineiro, A.; Bande, M. Applications of Non-Coding RNAs in Patients with Retinoblastoma. Front. Genet. 2022, 13, 842509. [Google Scholar] [CrossRef]
- Esperanza-Cebollada, E.; Gomez-Gonzalez, S.; Perez-Jaume, S.; Vega-Garcia, N.; Vicente-Garces, C.; Richarte-Franques, M.; Rives, S.; Catala, A.; Torrebadell, M.; Camos, M. A miRNA signature related to stemness identifies high-risk patients in paediatric acute myeloid leukaemia. Br. J. Haematol. 2023. [Google Scholar] [CrossRef]
- Audia, J.E.; Campbell, R.M. Histone Modifications and Cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019521. [Google Scholar] [CrossRef]
- Porazzi, P.; Petruk, S.; Pagliaroli, L.; De Dominici, M.; Deming, D., 2nd; Puccetti, M.V.; Kushinsky, S.; Kumar, G.; Minieri, V.; Barbieri, E.; et al. Targeting Chemotherapy to Decondensed H3K27me3-Marked Chromatin of AML Cells Enhances Leukemia Suppression. Cancer Res. 2022, 82, 458–471. [Google Scholar] [CrossRef]
- Gollner, S.; Oellerich, T.; Agrawal-Singh, S.; Schenk, T.; Klein, H.U.; Rohde, C.; Pabst, C.; Sauer, T.; Lerdrup, M.; Tavor, S.; et al. Loss of the histone methyltransferase EZH2 induces resistance to multiple drugs in acute myeloid leukemia. Nat. Med. 2017, 23, 69–78. [Google Scholar] [CrossRef]
- Shen, J.K.; Cote, G.M.; Gao, Y.; Choy, E.; Mankin, H.J.; Hornicek, F.J.; Duan, Z. Targeting EZH2-mediated methylation of H3K27 inhibits proliferation and migration of Synovial Sarcoma in vitro. Sci. Rep. 2016, 6, 25239. [Google Scholar] [CrossRef]
- Wang, Z.; Dai, J.; Yan, J.; Zhang, Y.; Yin, Z. Targeting EZH2 as a novel therapeutic strategy for sorafenib-resistant thyroid carcinoma. J. Cell Mol. Med. 2019, 23, 4770–4778. [Google Scholar] [CrossRef]
- Chase, A.; Cross, N.C. Aberrations of EZH2 in cancer. Clin. Cancer Res. 2011, 17, 2613–2618. [Google Scholar] [CrossRef]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; Van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef]
- Collinson, A.; Collier, A.J.; Morgan, N.P.; Sienerth, A.R.; Chandra, T.; Andrews, S.; Rugg-Gunn, P.J. Deletion of the Polycomb-Group Protein EZH2 Leads to Compromised Self-Renewal and Differentiation Defects in Human Embryonic Stem Cells. Cell Rep. 2016, 17, 2700–2714. [Google Scholar] [CrossRef]
- Erhardt, S.; Su, I.H.; Schneider, R.; Barton, S.; Bannister, A.J.; Perez-Burgos, L.; Jenuwein, T.; Kouzarides, T.; Tarakhovsky, A.; Surani, M.A. Consequences of the depletion of zygotic and embryonic enhancer of zeste 2 during preimplantation mouse development. Development 2003, 130, 4235–4248. [Google Scholar] [CrossRef]
- Laugesen, A.; Hojfeldt, J.W.; Helin, K. Molecular Mechanisms Directing PRC2 Recruitment and H3K27 Methylation. Mol. Cell 2019, 74, 8–18. [Google Scholar] [CrossRef]
- Xue, P.; Huang, S.; Han, X.; Zhang, C.; Yang, L.; Xiao, W.; Fu, J.; Li, H.; Zhou, Y. Exosomal miR-101-3p and miR-423-5p inhibit medulloblastoma tumorigenesis through targeting FOXP4 and EZH2. Cell Death Differ. 2022, 29, 82–95. [Google Scholar] [CrossRef]
- Ren, X.; Bai, X.; Zhang, X.; Li, Z.; Tang, L.; Zhao, X.; Li, Z.; Ren, Y.; Wei, S.; Wang, Q.; et al. Quantitative nuclear proteomics identifies that miR-137-mediated EZH2 reduction regulates resveratrol-induced apoptosis of neuroblastoma cells. Mol. Cell. Proteom. MCP 2015, 14, 316–328. [Google Scholar] [CrossRef]
- Zeineldin, M.; Patel, A.G.; Dyer, M.A. Neuroblastoma: When differentiation goes awry. Neuron 2022, 110, 2916–2928. [Google Scholar] [CrossRef] [PubMed]
- Neo, W.H.; Yap, K.; Lee, S.H.; Looi, L.S.; Khandelia, P.; Neo, S.X.; Makeyev, E.V.; Su, I.H. MicroRNA miR-124 controls the choice between neuronal and astrocyte differentiation by fine-tuning Ezh2 expression. J. Biol. Chem. 2014, 289, 20788–20801. [Google Scholar] [CrossRef] [PubMed]
- Akpa, M.M.; Iglesias, D.; Chu, L.; Thiebaut, A.; Jentoft, I.; Hammond, L.; Torban, E.; Goodyer, P.R. Wilms Tumor Suppressor, WT1, Cooperates with MicroRNA-26a and MicroRNA-101 to Suppress Translation of the Polycomb Protein, EZH2, in Mesenchymal Stem Cells. J. Biol. Chem. 2016, 291, 3785–3795. [Google Scholar] [CrossRef]
- Wu, M.K.; Sabbaghian, N.; Xu, B.; Addidou-Kalucki, S.; Bernard, C.; Zou, D.; Reeve, A.E.; Eccles, M.R.; Cole, C.; Choong, C.S.; et al. Biallelic DICER1 mutations occur in Wilms tumours. J. Pathol. 2013, 230, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Torrezan, G.T.; Ferreira, E.N.; Nakahata, A.M.; Barros, B.D.; Castro, M.T.; Correa, B.R.; Krepischi, A.C.; Olivieri, E.H.; Cunha, I.W.; Tabori, U.; et al. Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat. Commun. 2014, 5, 4039. [Google Scholar] [CrossRef]
- Zakrzewska, M.; Fendler, W.; Zakrzewski, K.; Sikorska, B.; Grajkowska, W.; Dembowska-Baginska, B.; Filipek, I.; Stefanczyk, L.; Liberski, P.P. Altered MicroRNA Expression Is Associated with Tumor Grade, Molecular Background and Outcome in Childhood Infratentorial Ependymoma. PLoS ONE 2016, 11, e0158464. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, X.; Jia, L.T.; Hu, S.J.; Zhao, J.; Yang, J.D.; Wen, W.H.; Wang, Z.; Wang, T.; Zhao, J.; et al. c-Myc-mediated epigenetic silencing of MicroRNA-101 contributes to dysregulation of multiple pathways in hepatocellular carcinoma. Hepatology 2014, 59, 1850–1863. [Google Scholar] [CrossRef]
- Chiang, C.W.; Huang, Y.; Leong, K.W.; Chen, L.C.; Chen, H.C.; Chen, S.J.; Chou, C.K. PKCalpha mediated induction of miR-101 in human hepatoma HepG2 cells. J. Biomed. Sci. 2010, 17, 35. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, S.; Xu, J.; Li, Y.; Zhang, J.; Zhang, J.; Lu, X. miR-767-5p inhibits glioma proliferation and metastasis by targeting SUZ12. Oncol. Rep. 2019, 42, 55–66. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Lindahl-Allen, M.; Polytarchou, C.; Hirsch, H.A.; Tsichlis, P.N.; Struhl, K. Loss of miR-200 inhibition of Suz12 leads to polycomb-mediated repression required for the formation and maintenance of cancer stem cells. Mol. Cell 2010, 39, 761–772. [Google Scholar] [CrossRef]
- Liu, X.; Lei, Q.; Yu, Z.; Xu, G.; Tang, H.; Wang, W.; Wang, Z.; Li, G.; Wu, M. MiR-101 reverses the hypomethylation of the LMO3 promoter in glioma cells. Oncotarget 2015, 6, 7930–7943. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, W.; Xu, S.; Zhang, J.; Zhang, J.; Yu, Q.; Jiao, Y.; Wang, Y.; Lu, A.; You, Y.; et al. MicroRNA-105 inhibits human glioma cell malignancy by directly targeting SUZ12. Tumour Biol. 2017, 39, 1010428317705766. [Google Scholar] [CrossRef]
- Farmakovskaya, M.; Khromova, N.; Rybko, V.; Dugina, V.; Kopnin, B.; Kopnin, P. E-Cadherin repression increases amount of cancer stem cells in human A549 lung adenocarcinoma and stimulates tumor growth. Cell Cycle 2016, 15, 1084–1092. [Google Scholar] [CrossRef]
- Miele, E.; Po, A.; Mastronuzzi, A.; Carai, A.; Besharat, Z.M.; Pediconi, N.; Abballe, L.; Catanzaro, G.; Sabato, C.; De Smaele, E.; et al. Downregulation of miR-326 and its host gene beta-arrestin1 induces pro-survival activity of E2F1 and promotes medulloblastoma growth. Mol. Oncol. 2021, 15, 523–542. [Google Scholar] [CrossRef]
- Winters, A.C.; Bernt, K.M. MLL-Rearranged Leukemias-An Update on Science and Clinical Approaches. Front. Pediatr. 2017, 5, 4. [Google Scholar] [CrossRef]
- Mai, J.; Gu, J.; Liu, Y.; Liu, X.; Sai, K.; Chen, Z.; Lu, W.; Yang, X.; Wang, J.; Guo, C.; et al. Negative regulation of miR-1275 by H3K27me3 is critical for glial induction of glioblastoma cells. Mol. Oncol. 2019, 13, 1589–1604. [Google Scholar] [CrossRef]
- Li, Q.; Song, W.; Wang, J. TUG1 confers Adriamycin resistance in acute myeloid leukemia by epigenetically suppressing miR-34a expression via EZH2. Biomed. Pharmacother. 2019, 109, 1793–1801. [Google Scholar] [CrossRef]
- Li, R.; Ruan, Q.; Zheng, J.; Zhang, B.; Yang, H. LINC01116 Promotes Doxorubicin Resistance in Osteosarcoma by Epigenetically Silencing miR-424-5p and Inducing Epithelial-Mesenchymal Transition. Front. Pharmacol. 2021, 12, 632206. [Google Scholar] [CrossRef]
- Singh, A.P.; Luo, H.; Matur, M.; Eshelman, M.A.; Hamamoto, K.; Sharma, A.; Lesperance, J.; Huang, S. A coordinated function of lncRNA HOTTIP and miRNA-196b underpinning leukemogenesis by targeting FAS signaling. Oncogene 2022, 41, 718–731. [Google Scholar] [CrossRef]
- Xing, C.Y.; Zhang, Y.Z.; Hu, W.; Zhao, L.Y. LINC00313 facilitates osteosarcoma carcinogenesis and metastasis through enhancing EZH2 mRNA stability and EZH2-mediated silence of PTEN expression. Cell. Mol. Life Sci. 2022, 79, 382. [Google Scholar] [CrossRef]
- Kong, D.; Li, C.; Yang, Q.; Wei, B.; Wang, L.; Peng, C. Long noncoding RNA LSINCT5 acts as an oncogene via increasing EZH2-induced inhibition of APC expression in osteosarcoma. Biochem. Biophys. Res. Commun. 2018, 507, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Zhang, E.; Song, L.; Tu, L.; Wang, Z.; Tian, F.; Aikenmu, K.; Chu, G.; Zhao, J. Long noncoding RNA HOXD-AS1 aggravates osteosarcoma carcinogenesis through epigenetically inhibiting p57 via EZH2. Biomed. Pharmacother. 2018, 106, 890–895. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Yin, C.F.; Chang, Y.W.; Fan, Y.C.; Lin, S.H.; Wu, Y.C.; Huang, H.C.; Juan, H.F. LncRNA SNHG1 regulates neuroblastoma cell fate via interactions with HDAC1/2. Cell Death Dis. 2022, 13, 809. [Google Scholar] [CrossRef] [PubMed]
- Durbin, A.D.; Zimmerman, M.W.; Dharia, N.V.; Abraham, B.J.; Iniguez, A.B.; Weichert-Leahey, N.; He, S.; Krill-Burger, J.M.; Root, D.E.; Vazquez, F.; et al. Selective gene dependencies in MYCN-amplified neuroblastoma include the core transcriptional regulatory circuitry. Nat. Genet. 2018, 50, 1240–1246. [Google Scholar] [CrossRef]
- Bernard, L.D.; Dubois, A.; Heurtier, V.; Fischer, V.; Gonzalez, I.; Chervova, A.; Tachtsidi, A.; Gil, N.; Owens, N.; Bates, L.E.; et al. OCT4 activates a Suv39h1-repressive antisense lncRNA to couple histone H3 Lysine 9 methylation to pluripotency. Nucleic Acids Res. 2022, 50, 7367–7379. [Google Scholar] [CrossRef]
- Monferrer, E.; Burgos-Panadero, R.; Blanquer-Maceiras, M.; Canete, A.; Navarro, S.; Noguera, R. High Oct4 expression: Implications in the pathogenesis of neuroblastic tumours. BMC Cancer 2019, 19, 1. [Google Scholar] [CrossRef]
- Pezzolo, A.; Parodi, F.; Marimpietri, D.; Raffaghello, L.; Cocco, C.; Pistorio, A.; Mosconi, M.; Gambini, C.; Cilli, M.; Deaglio, S.; et al. Oct-4+/Tenascin C+ neuroblastoma cells serve as progenitors of tumor-derived endothelial cells. Cell Res. 2011, 21, 1470–1486. [Google Scholar] [CrossRef]
- Kim, D.K.; Song, B.; Han, S.; Jang, H.; Bae, S.H.; Kim, H.Y.; Lee, S.H.; Lee, S.; Kim, J.K.; Kim, H.S.; et al. Phosphorylation of OCT4 Serine 236 Inhibits Germ Cell Tumor Growth by Inducing Differentiation. Cancers 2020, 12, 2601. [Google Scholar] [CrossRef]
- Park, C.S.; Lewis, A.; Chen, T.; Lacorazza, D. Concise Review: Regulation of Self-Renewal in Normal and Malignant Hematopoietic Stem Cells by Kruppel-Like Factor 4. Stem Cells Transl. Med. 2019, 8, 568–574. [Google Scholar] [CrossRef]
- Khateb, A.; Deshpande, A.; Feng, Y.; Finlay, D.; Lee, J.S.; Lazar, I.; Fabre, B.; Li, Y.; Fujita, Y.; Zhang, T.; et al. The ubiquitin ligase RNF5 determines acute myeloid leukemia growth and susceptibility to histone deacetylase inhibitors. Nat. Commun. 2021, 12, 5397. [Google Scholar] [CrossRef]
- Yadav, P.; Subbarayalu, P.; Medina, D.; Nirzhor, S.; Timilsina, S.; Rajamanickam, S.; Eedunuri, V.K.; Gupta, Y.; Zheng, S.; Abdelfattah, N.; et al. M6A RNA Methylation Regulates Histone Ubiquitination to Support Cancer Growth and Progression. Cancer Res. 2022, 82, 1872–1889. [Google Scholar] [CrossRef] [PubMed]
- Tamburri, S.; Conway, E.; Pasini, D. Polycomb-dependent histone H2A ubiquitination links developmental disorders with cancer. Trends Genet. 2022, 38, 333–352. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.; Kawaoka, S.; Yu, M.; Shi, J.; Ni, T.; Yang, W.; Zhu, J.; Roeder, R.G.; Vakoc, C.R. Histone H2B ubiquitin ligase RNF20 is required for MLL-rearranged leukemia. Proc. Natl. Acad. Sci. USA 2013, 110, 3901–3906. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zhou, A.; Wei, Y.; Liu, F.; Li, P.; Fang, R.; Ma, L.; Zhang, S.; Wang, L.; Liu, J.; et al. Critical role of lncEPAT in coupling dysregulated EGFR pathway and histone H2A deubiquitination during glioblastoma tumorigenesis. Sci. Adv. 2022, 8, eabn2571. [Google Scholar] [CrossRef] [PubMed]
- Fang, K.; Huang, W.; Sun, Y.M.; Chen, T.Q.; Zeng, Z.C.; Yang, Q.Q.; Pan, Q.; Han, C.; Sun, L.Y.; Luo, X.Q.; et al. Cis-acting lnc-eRNA SEELA directly binds histone H4 to promote histone recognition and leukemia progression. Genome Biol. 2020, 21, 269. [Google Scholar] [CrossRef]
- Alharbi, R.A.; Pettengell, R.; Pandha, H.S.; Morgan, R. The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia 2013, 27, 1000–1008. [Google Scholar] [CrossRef]
- Quentmeier, H.; Dirks, W.G.; Macleod, R.A.; Reinhardt, J.; Zaborski, M.; Drexler, H.G. Expression of HOX genes in acute leukemia cell lines with and without MLL translocations. Leuk. Lymphoma 2004, 45, 567–574. [Google Scholar] [CrossRef]
- Adriaens, C.; Standaert, L.; Barra, J.; Latil, M.; Verfaillie, A.; Kalev, P.; Boeckx, B.; Wijnhoven, P.W.; Radaelli, E.; Vermi, W.; et al. p53 induces formation of NEAT1 lncRNA-containing paraspeckles that modulate replication stress response and chemosensitivity. Nat. Med. 2016, 22, 861–868. [Google Scholar] [CrossRef]
- Unfried, J.P.; Marin-Baquero, M.; Rivera-Calzada, A.; Razquin, N.; Martin-Cuevas, E.M.; de Braganca, S.; Aicart-Ramos, C.; McCoy, C.; Prats-Mari, L.; Arribas-Bosacoma, R.; et al. Long Noncoding RNA NIHCOLE Promotes Ligation Efficiency of DNA Double-Strand Breaks in Hepatocellular Carcinoma. Cancer Res. 2021, 81, 4910–4925. [Google Scholar] [CrossRef]
- Chen, K.Y.; Zhu, S.G.; He, J.W.; Duan, X.P. LncRNA CRNDE is involved in radiation resistance in hepatocellular carcinoma via modulating the SP1/PDK1 axis. Neoplasma 2022, 69, 918–930. [Google Scholar] [CrossRef]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Chen, Y.L.; Zhang, Z.X.; Shou, L.H.; Di, J.Y. Regulation of DNA methylation and tumor suppression gene expression by miR-29b in leukemia patients and related mechanisms. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 158–165. [Google Scholar] [CrossRef]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009, 113, 6411–6418. [Google Scholar] [CrossRef]
- Das, S.; Foley, N.; Bryan, K.; Watters, K.M.; Bray, I.; Murphy, D.M.; Buckley, P.G.; Stallings, R.L. MicroRNA mediates DNA demethylation events triggered by retinoic acid during neuroblastoma cell differentiation. Cancer Res. 2010, 70, 7874–7881. [Google Scholar] [CrossRef]
- Gong, H.L.; Tao, Y.; Mao, X.Z.; Song, D.Y.; You, D.; Ni, J.D. MicroRNA-29a suppresses the invasion and migration of osteosarcoma cells by regulating the SOCS1/NF-kappaB signalling pathway through negatively targeting DNMT3B. Int. J. Mol. Med. 2019, 44, 1219–1232. [Google Scholar] [CrossRef]
- Chen, C.Y.; Tsay, W.; Tang, J.L.; Shen, H.L.; Lin, S.W.; Huang, S.Y.; Yao, M.; Chen, Y.C.; Shen, M.C.; Wang, C.H.; et al. SOCS1 methylation in patients with newly diagnosed acute myeloid leukemia. Genes Chromosom. Cancer 2003, 37, 300–305. [Google Scholar] [CrossRef]
- Zhou, H.; Miki, R.; Eeva, M.; Fike, F.M.; Seligson, D.; Yang, L.; Yoshimura, A.; Teitell, M.A.; Jamieson, C.A.; Cacalano, N.A. Reciprocal regulation of SOCS 1 and SOCS3 enhances resistance to ionizing radiation in glioblastoma multiforme. Clin. Cancer Res. 2007, 13, 2344–2353. [Google Scholar] [CrossRef]
- Martini, M.; Pallini, R.; Luongo, G.; Cenci, T.; Lucantoni, C.; Larocca, L.M. Prognostic relevance of SOCS3 hypermethylation in patients with glioblastoma multiforme. Int. J. Cancer 2008, 123, 2955–2960. [Google Scholar] [CrossRef]
- Khan, A.W.; Ziemann, M.; Rafehi, H.; Maxwell, S.; Ciccotosto, G.D.; El-Osta, A. MeCP2 interacts with chromosomal microRNAs in brain. Epigenetics 2017, 12, 1028–1037. [Google Scholar] [CrossRef]
- Chen, Y.J.; Luo, J.; Yang, G.Y.; Yang, K.; Wen, S.Q.; Zou, S.Q. Mutual regulation between microRNA-373 and methyl-CpG-binding domain protein 2 in hilar cholangiocarcinoma. World J. Gastroenterol. 2012, 18, 3849–3861. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Liu, Q.L.; Zhai, T.S.; Lu, J.; Dong, Y.Z.; Xu, Y.F. Silencing miR-454 suppresses cell proliferation, migration and invasion via directly targeting MECP2 in renal cell carcinoma. Am. J. Transl. Res. 2020, 12, 4277–4289. [Google Scholar] [PubMed]
- Cui, S.; Liu, L.; Wan, T.; Jiang, L.; Shi, Y.; Luo, L. MiR-520b inhibits the development of glioma by directly targeting MBD2. Am. J. Cancer Res. 2017, 7, 1528–1539. [Google Scholar] [PubMed]
- He, L.; Li, H.; Wu, A.; Peng, Y.; Shu, G.; Yin, G. Functions of N6-methyladenosine and its role in cancer. Mol. Cancer 2019, 18, 176. [Google Scholar] [CrossRef]
- Kang, H.; Lee, S.; Kim, K.; Jeon, J.; Kang, S.G.; Youn, H.; Kim, H.Y.; Youn, B. Downregulated CLIP3 induces radioresistance by enhancing stemness and glycolytic flux in glioblastoma. J. Exp. Clin. Cancer Res. 2021, 40, 282. [Google Scholar] [CrossRef]
- Zepecki, J.P.; Karambizi, D.; Fajardo, J.E.; Snyder, K.M.; Guetta-Terrier, C.; Tang, O.Y.; Chen, J.S.; Sarkar, A.; Fiser, A.; Toms, S.A.; et al. miRNA-mediated loss of m6A increases nascent translation in glioblastoma. PLoS Genet. 2021, 17, e1009086. [Google Scholar] [CrossRef]
- Wong, K.Y.; So, C.C.; Loong, F.; Chung, L.P.; Lam, W.W.; Liang, R.; Li, G.K.; Jin, D.Y.; Chim, C.S. Epigenetic inactivation of the miR-124-1 in haematological malignancies. PLoS ONE 2011, 6, e19027. [Google Scholar] [CrossRef]
- Namlos, H.M.; Skarn, M.; Ahmed, D.; Grad, I.; Andresen, K.; Kresse, S.H.; Munthe, E.; Serra, M.; Scotlandi, K.; Llombart-Bosch, A.; et al. miR-486-5p expression is regulated by DNA methylation in osteosarcoma. BMC Genom. 2022, 23, 142. [Google Scholar] [CrossRef]
- Li, Q.; Li, H.; Zhao, X.; Wang, B.; Zhang, L.; Zhang, C.; Zhang, F. DNA Methylation Mediated Downregulation of miR-449c Controls Osteosarcoma Cell Cycle Progression by Directly Targeting Oncogene c-Myc. Int. J. Biol. Sci. 2017, 13, 1038–1050. [Google Scholar] [CrossRef]
- Bi, L.; Zhou, B.; Li, H.; He, L.; Wang, C.; Wang, Z.; Zhu, L.; Chen, M.; Gao, S. A novel miR-375-HOXB3-CDCA3/DNMT3B regulatory circuitry contributes to leukemogenesis in acute myeloid leukemia. BMC Cancer 2018, 18, 182. [Google Scholar] [CrossRef]
- Stumpel, D.J.; Schotte, D.; Lange-Turenhout, E.A.; Schneider, P.; Seslija, L.; de Menezes, R.X.; Marquez, V.E.; Pieters, R.; den Boer, M.L.; Stam, R.W. Hypermethylation of specific microRNA genes in MLL-rearranged infant acute lymphoblastic leukemia: Major matters at a micro scale. Leukemia 2011, 25, 429–439. [Google Scholar] [CrossRef]
- Nishi, M.; Eguchi-Ishimae, M.; Wu, Z.; Gao, W.; Iwabuki, H.; Kawakami, S.; Tauchi, H.; Inukai, T.; Sugita, K.; Hamasaki, Y.; et al. Suppression of the let-7b microRNA pathway by DNA hypermethylation in infant acute lymphoblastic leukemia with MLL gene rearrangements. Leukemia 2013, 27, 389–397. [Google Scholar] [CrossRef]
- Cheray, M.; Etcheverry, A.; Jacques, C.; Pacaud, R.; Bougras-Cartron, G.; Aubry, M.; Denoual, F.; Peterlongo, P.; Nadaradjane, A.; Briand, J.; et al. Cytosine methylation of mature microRNAs inhibits their functions and is associated with poor prognosis in glioblastoma multiforme. Mol. Cancer 2020, 19, 36. [Google Scholar] [CrossRef]
- Lovat, F.; Nigita, G.; Distefano, R.; Nakamura, T.; Gasparini, P.; Tomasello, L.; Fadda, P.; Ibrahimova, N.; Catricala, S.; Palamarchuk, A.; et al. Combined loss of function of two different loci of miR-15/16 drives the pathogenesis of acute myeloid leukemia. Proc. Natl. Acad. Sci. USA 2020, 117, 12332–12340. [Google Scholar] [CrossRef]
- Wang, S.L.; Huang, Y.; Su, R.; Yu, Y.Y. Silencing long non-coding RNA HOTAIR exerts anti-oncogenic effect on human acute myeloid leukemia via demethylation of HOXA5 by inhibiting Dnmt3b. Cancer Cell Int. 2019, 19, 114. [Google Scholar] [CrossRef]
- Wang, W.T.; Chen, T.Q.; Zeng, Z.C.; Pan, Q.; Huang, W.; Han, C.; Fang, K.; Sun, L.Y.; Yang, Q.Q.; Wang, D.; et al. The lncRNA LAMP5-AS1 drives leukemia cell stemness by directly modulating DOT1L methyltransferase activity in MLL leukemia. J. Hematol. Oncol. 2020, 13, 78. [Google Scholar] [CrossRef]
- Xu, X.; Lou, Y.; Tang, J.; Teng, Y.; Zhang, Z.; Yin, Y.; Zhuo, H.; Tan, Z. The long non-coding RNA Linc-GALH promotes hepatocellular carcinoma metastasis via epigenetically regulating Gankyrin. Cell Death Dis. 2019, 10, 86. [Google Scholar] [CrossRef]
- Cheng, D.; Deng, J.; Zhang, B.; He, X.; Meng, Z.; Li, G.; Ye, H.; Zheng, S.; Wei, L.; Deng, X.; et al. LncRNA HOTAIR epigenetically suppresses miR-122 expression in hepatocellular carcinoma via DNA methylation. EBioMedicine 2018, 36, 159–170. [Google Scholar] [CrossRef]
- Zhang, M.Y.; Calin, G.; Deng, M.D.; Au-Yeung, R.K.H.; Wang, L.Q.; Chim, C.S. Epigenetic Silencing of Tumor Suppressor lncRNA NKILA: Implication on NF-kappaB Signaling in Non-Hodgkin’s Lymphoma. Genes 2022, 13, 128. [Google Scholar] [CrossRef]
- Lwin, T.; Zhao, X.; Cheng, F.; Zhang, X.; Huang, A.; Shah, B.; Zhang, Y.; Moscinski, L.C.; Choi, Y.S.; Kozikowski, A.P.; et al. A microenvironment-mediated c-Myc/miR-548m/HDAC6 amplification loop in non-Hodgkin B cell lymphomas. J. Clin. Investig. 2013, 123, 4612–4626. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Tang, F.; Hu, P.; Wang, Y.; Gong, J.; Sun, S.; Xie, C. HDAC6 promotes cell proliferation and confers resistance to gefitinib in lung adenocarcinoma. Oncol. Rep. 2016, 36, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.W.; Shin, D.H.; Lee, D.H.; Choi, J.; Han, G.; Lee, K.Y.; Kwon, S.H. HDAC6 deacetylates p53 at lysines 381/382 and differentially coordinates p53-induced apoptosis. Cancer Lett. 2017, 391, 162–171. [Google Scholar] [CrossRef]
- Pham, T.Q.; Robinson, K.; Xu, L.; Pavlova, M.N.; Skapek, S.X.; Chen, E.Y. HDAC6 promotes growth, migration/invasion, and self-renewal of rhabdomyosarcoma. Oncogene 2021, 40, 578–591. [Google Scholar] [CrossRef]
- Banik, D.; Noonepalle, S.; Hadley, M.; Palmer, E.; Gracia-Hernandez, M.; Zevallos-Delgado, C.; Manhas, N.; Simonyan, H.; Young, C.N.; Popratiloff, A.; et al. HDAC6 Plays a Noncanonical Role in the Regulation of Antitumor Immune Responses, Dissemination, and Invasiveness of Breast Cancer. Cancer Res. 2020, 80, 3649–3662. [Google Scholar] [CrossRef]
- Song, L.; Luan, B.; Xu, Q.; Shi, R.; Wang, X. microRNA-155-3p delivered by M2 macrophages-derived exosomes enhances the progression of medulloblastoma through regulation of WDR82. J. Transl. Med. 2022, 20, 13. [Google Scholar] [CrossRef]
- Youn, G.S.; Park, J.K.; Lee, C.Y.; Jang, J.H.; Yun, S.H.; Kwon, H.Y.; Choi, S.Y.; Park, J. MicroRNA-22 negatively regulates LPS-induced inflammatory responses by targeting HDAC6 in macrophages. BMB Rep. 2020, 53, 223–228. [Google Scholar] [CrossRef]
- Zhong, C.; Tao, B.; Yang, F.; Xia, K.; Yang, X.; Chen, L.; Peng, T.; Xia, X.; Li, X.; Peng, L. Histone demethylase JMJD1C promotes the polarization of M1 macrophages to prevent glioma by upregulating miR-302a. Clin. Transl. Med. 2021, 11, e424. [Google Scholar] [CrossRef]
- Yang, H.D.; Kim, H.S.; Kim, S.Y.; Na, M.J.; Yang, G.; Eun, J.W.; Wang, H.J.; Cheong, J.Y.; Park, W.S.; Nam, S.W. HDAC6 Suppresses Let-7i-5p to Elicit TSP1/CD47-Mediated Anti-Tumorigenesis and Phagocytosis of Hepatocellular Carcinoma. Hepatology 2019, 70, 1262–1279. [Google Scholar] [CrossRef]
- Zhao, J.; Li, H.; Zhao, S.; Wang, E.; Zhu, J.; Feng, D.; Zhu, Y.; Dou, W.; Fan, Q.; Hu, J.; et al. Epigenetic silencing of miR-144/451a cluster contributes to HCC progression via paracrine HGF/MIF-mediated TAM remodeling. Mol. Cancer 2021, 20, 46. [Google Scholar] [CrossRef]
- Winkler, I.; Bitter, C.; Winkler, S.; Weichenhan, D.; Thavamani, A.; Hengstler, J.G.; Borkham-Kamphorst, E.; Kohlbacher, O.; Plass, C.; Geffers, R.; et al. Identification of Ppargamma-modulated miRNA hubs that target the fibrotic tumor microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 454–463. [Google Scholar] [CrossRef]
- Kurashige, J.; Mima, K.; Sawada, G.; Takahashi, Y.; Eguchi, H.; Sugimachi, K.; Mori, M.; Yanagihara, K.; Yashiro, M.; Hirakawa, K.; et al. Epigenetic modulation and repression of miR-200b by cancer-associated fibroblasts contribute to cancer invasion and peritoneal dissemination in gastric cancer. Carcinogenesis 2015, 36, 133–141. [Google Scholar] [CrossRef]
- Li, X.; Fan, Q.; Li, J.; Song, J.; Gu, Y. MiR-124 down-regulation is critical for cancer associated fibroblasts-enhanced tumor growth of oral carcinoma. Exp. Cell Res. 2017, 351, 100–108. [Google Scholar] [CrossRef]
- Dudley, A.C. Tumor endothelial cells. Cold Spring Harb. Perspect. Med. 2012, 2, a006536. [Google Scholar] [CrossRef]
- Lucero, R.; Zappulli, V.; Sammarco, A.; Murillo, O.D.; Cheah, P.S.; Srinivasan, S.; Tai, E.; Ting, D.T.; Wei, Z.; Roth, M.E.; et al. Glioma-Derived miRNA-Containing Extracellular Vesicles Induce Angiogenesis by Reprogramming Brain Endothelial Cells. Cell Rep. 2020, 30, 2065–2074.e4. [Google Scholar] [CrossRef]
- Bahrami, A.; Jafari, A.; Ferns, G.A. The dual role of microRNA-9 in gastrointestinal cancers: oncomiR or tumor suppressor? Biomed. Pharmacother. 2022, 145, 112394. [Google Scholar] [CrossRef]
- Minor, J.; Wang, X.; Zhang, F.; Song, J.; Jimeno, A.; Wang, X.J.; Lu, X.; Gross, N.; Kulesz-Martin, M.; Wang, D.; et al. Methylation of microRNA-9 is a specific and sensitive biomarker for oral and oropharyngeal squamous cell carcinomas. Oral Oncol. 2012, 48, 73–78. [Google Scholar] [CrossRef]
- Ji, Y.; Fioravanti, J.; Zhu, W.; Wang, H.; Wu, T.; Hu, J.; Lacey, N.E.; Gautam, S.; Le Gall, J.B.; Yang, X.; et al. miR-155 harnesses Phf19 to potentiate cancer immunotherapy through epigenetic reprogramming of CD8(+) T cell fate. Nat. Commun. 2019, 10, 2157. [Google Scholar] [CrossRef]
- Liu, L.; Yi, H.; Wang, C.; He, H.; Li, P.; Pan, H.; Sheng, N.; Ji, M.; Cai, L.; Ma, Y. Integrated Nanovaccine with MicroRNA-148a Inhibition Reprograms Tumor-Associated Dendritic Cells by Modulating miR-148a/DNMT1/SOCS1 Axis. J. Immunol. 2016, 197, 1231–1241. [Google Scholar] [CrossRef]
- Schiavoni, G.; Munitz, A.; Strid, J. Editorial: Emerging Roles for Type 2-Associated Cells and Cytokines in Cancer Immunity. Front. Immunol. 2021, 12, 811125. [Google Scholar] [CrossRef] [PubMed]
- Koyasu, S.; Moro, K. Type 2 innate immune responses and the natural helper cell. Immunology 2011, 132, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Xiang, Y.; Eyers, F.; Young, I.G.; Rosenberg, H.F.; Foster, P.S.; Yang, M. Identification of microRNAs regulating the developmental pathways of bone marrow derived mast cells. PLoS ONE 2014, 9, e98139. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Lee, S.S. The Interplay among miRNAs, Major Cytokines, and Cancer-Related Inflammation. Mol. Ther. Nucleic Acids 2020, 20, 606–620. [Google Scholar] [CrossRef]
- Yamada, Y.; Kosaka, K.; Miyazawa, T.; Kurata-Miura, K.; Yoshida, T. miR-142-3p enhances FcepsilonRI-mediated degranulation in mast cells. Biochem. Biophys. Res. Commun. 2014, 443, 980–986. [Google Scholar] [CrossRef]
- Kwon, Y.; Kim, Y.; Eom, S.; Kim, M.; Park, D.; Kim, H.; Noh, K.; Lee, H.; Lee, Y.S.; Choe, J.; et al. MicroRNA-26a/-26b-COX-2-MIP-2 Loop Regulates Allergic Inflammation and Allergic Inflammation-promoted Enhanced Tumorigenic and Metastatic Potential of Cancer Cells. J. Biol. Chem. 2015, 290, 14245–14266. [Google Scholar] [CrossRef]
- Eom, S.; Kim, Y.; Kim, M.; Park, D.; Lee, H.; Lee, Y.S.; Choe, J.; Kim, Y.M.; Jeoung, D. Transglutaminase II/microRNA-218/-181a loop regulates positive feedback relationship between allergic inflammation and tumor metastasis. J. Biol. Chem. 2014, 289, 29483–29505. [Google Scholar] [CrossRef]
- Knolle, M.D.; Chin, S.B.; Rana, B.M.J.; Englezakis, A.; Nakagawa, R.; Fallon, P.G.; Git, A.; McKenzie, A.N.J. MicroRNA-155 Protects Group 2 Innate Lymphoid Cells From Apoptosis to Promote Type-2 Immunity. Front. Immunol. 2018, 9, 2232. [Google Scholar] [CrossRef]
- Roberts, L.B.; Jowett, G.M.; Read, E.; Zabinski, T.; Berkachy, R.; Selkirk, M.E.; Jackson, I.; Niazi, U.; Anandagoda, N.; Araki, M.; et al. MicroRNA-142 Critically Regulates Group 2 Innate Lymphoid Cell Homeostasis and Function. J. Immunol. 2021, 206, 2725–2739. [Google Scholar] [CrossRef]
- Johansson, K.; Weidner, J.; Radinger, M. MicroRNAs in type 2 immunity. Cancer Lett. 2018, 425, 116–124. [Google Scholar] [CrossRef]
- Chen, C.; He, W.; Huang, J.; Wang, B.; Li, H.; Cai, Q.; Su, F.; Bi, J.; Liu, H.; Zhang, B.; et al. LNMAT1 promotes lymphatic metastasis of bladder cancer via CCL2 dependent macrophage recruitment. Nat. Commun. 2018, 9, 3826. [Google Scholar] [CrossRef]
- Han, X.; Huang, S.; Xue, P.; Fu, J.; Liu, L.; Zhang, C.; Yang, L.; Xia, L.; Sun, L.; Huang, S.K.; et al. LncRNA PTPRE-AS1 modulates M2 macrophage activation and inflammatory diseases by epigenetic promotion of PTPRE. Sci. Adv. 2019, 5, eaax9230. [Google Scholar] [CrossRef]
- de Groot, A.E.; Myers, K.V.; Krueger, T.E.G.; Brennen, W.N.; Amend, S.R.; Pienta, K.J. Targeting interleukin 4 receptor alpha on tumor-associated macrophages reduces the pro-tumor macrophage phenotype. Neoplasia 2022, 32, 100830. [Google Scholar] [CrossRef]
- Li, L.; Xu, F.; Xie, P.; Yuan, L.; Zhou, M. PTPRT Could Be a Treatment Predictive and Prognostic Biomarker for Breast Cancer. Biomed. Res. Int. 2021, 2021, 3301402. [Google Scholar] [CrossRef]
- Zhang, P.; Becka, S.; Craig, S.E.; Lodowski, D.T.; Brady-Kalnay, S.M.; Wang, Z. Cancer-derived mutations in the fibronectin III repeats of PTPRT/PTPrho inhibit cell-cell aggregation. Cell Commun. Adhes. 2009, 16, 146–153. [Google Scholar] [CrossRef]
- Peng, C.; Zhang, C.; Yu, W.; Li, L.; Zhang, Z.; Liu, T.; Zhang, Y.; Fan, G.; Huangfu, H. Receptor Type Protein Tyrosine Phosphatase Epsilon (PTPRE) Plays an Oncogenic Role in Thyroid Carcinoma by Activating the AKT and ERK1/2 Signaling Pathway. Curr. Cancer Drug Targets 2023, 23, 471–481. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, J.; Yao, Q.; Wang, Y.; Liu, Z.; Zhang, L. LncRNA SNHG6 Promotes Wilms’ Tumor Progression Through Regulating miR-429/FRS2 Axis. Cancer Biother. Radiopharm. 2021. [Google Scholar] [CrossRef]
- Zhang, Y.; An, J.; Pei, Y. LncRNA SNHG6 promotes LMO3 expression by sponging miR-543 in glioma. Mol. Cell. Biochem. 2020, 472, 9–17. [Google Scholar] [CrossRef]
- Ruan, J.; Zheng, L.; Hu, N.; Guan, G.; Chen, J.; Zhou, X.; Li, M. Long noncoding RNA SNHG6 promotes osteosarcoma cell proliferation through regulating p21 and KLF2. Arch. Biochem. Biophys. 2018, 646, 128–136. [Google Scholar] [CrossRef]
- Liu, F.; Tian, T.; Zhang, Z.; Xie, S.; Yang, J.; Zhu, L.; Wang, W.; Shi, C.; Sang, L.; Guo, K.; et al. Long non-coding RNA SNHG6 couples cholesterol sensing with mTORC1 activation in hepatocellular carcinoma. Nat. Metab. 2022, 4, 1022–1040. [Google Scholar] [CrossRef]
- Kanbar, J.N.; Ma, S.; Kim, E.S.; Kurd, N.S.; Tsai, M.S.; Tysl, T.; Widjaja, C.E.; Limary, A.E.; Yee, B.; He, Z.; et al. The long noncoding RNA Malat1 regulates CD8+ T cell differentiation by mediating epigenetic repression. J. Exp. Med. 2022, 219, e20211756. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.M.; Lian, G.Y.; Song, Y.; Huang, Y.F.; Gong, Y. LncRNA MALAT1 promotes tumorigenesis and immune escape of diffuse large B cell lymphoma by sponging miR-195. Life Sci. 2019, 231, 116335. [Google Scholar] [CrossRef]
- Guo, W.; Wang, Y.; Yang, M.; Wang, Z.; Wang, Y.; Chaurasia, S.; Wu, Z.; Zhang, M.; Yadav, G.S.; Rathod, S.; et al. LincRNA-immunity landscape analysis identifies EPIC1 as a regulator of tumor immune evasion and immunotherapy resistance. Sci. Adv. 2021, 7, eabb3555. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Li, Y.; Zhang, J.; Zhang, B. PD-L1 expression increased by IFN-gamma via JAK2-STAT1 signaling and predicts a poor survival in colorectal cancer. Oncol. Lett. 2020, 20, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.W.; Sriswasdi, S.; Kinugasa, Y.; Adachi, J.; Horikoshi, Y.; Shibuya, A.; Iwasaki, W.; Tashiro, S.; Tomonaga, T.; Siomi, H. Piwi-piRNA complexes induce stepwise changes in nuclear architecture at target loci. EMBO J. 2021, 40, e108345. [Google Scholar] [CrossRef]
- Thomson, T.; Lin, H. The biogenesis and function of PIWI proteins and piRNAs: Progress and prospect. Annu. Rev. Cell Dev. Biol. 2009, 25, 355–376. [Google Scholar] [CrossRef]
- Aravin, A.A.; Sachidanandam, R.; Girard, A.; Fejes-Toth, K.; Hannon, G.J. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science 2007, 316, 744–747. [Google Scholar] [CrossRef]
- Brennecke, J.; Aravin, A.A.; Stark, A.; Dus, M.; Kellis, M.; Sachidanandam, R.; Hannon, G.J. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 2007, 128, 1089–1103. [Google Scholar] [CrossRef]
- Ishizu, H.; Kinoshita, T.; Hirakata, S.; Komatsuzaki, C.; Siomi, M.C. Distinct and Collaborative Functions of Yb and Armitage in Transposon-Targeting piRNA Biogenesis. Cell Rep. 2019, 27, 1822–1835.e8. [Google Scholar] [CrossRef]
- Rogers, A.K.; Situ, K.; Perkins, E.M.; Toth, K.F. Zucchini-dependent piRNA processing is triggered by recruitment to the cytoplasmic processing machinery. Genes Dev. 2017, 31, 1858–1869. [Google Scholar] [CrossRef]
- Czech, B.; Hannon, G.J. One Loop to Rule Them All: The Ping-Pong Cycle and piRNA-Guided Silencing. Trends Biochem. Sci. 2016, 41, 324–337. [Google Scholar] [CrossRef]
- Pandey, R.R.; Homolka, D.; Chen, K.M.; Sachidanandam, R.; Fauvarque, M.O.; Pillai, R.S. Recruitment of Armitage and Yb to a transcript triggers its phased processing into primary piRNAs in Drosophila ovaries. PLoS Genet. 2017, 13, e1006956. [Google Scholar] [CrossRef]
- Houwing, S.; Kamminga, L.M.; Berezikov, E.; Cronembold, D.; Girard, A.; van den Elst, H.; Filippov, D.V.; Blaser, H.; Raz, E.; Moens, C.B.; et al. A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell 2007, 129, 69–82. [Google Scholar] [CrossRef]
- Kawaoka, S.; Izumi, N.; Katsuma, S.; Tomari, Y. 3′ end formation of PIWI-interacting RNAs in vitro. Mol. Cell 2011, 43, 1015–1022. [Google Scholar] [CrossRef]
- Horwich, M.D.; Li, C.; Matranga, C.; Vagin, V.; Farley, G.; Wang, P.; Zamore, P.D. The Drosophila RNA methyltransferase, DmHen1, modifies germline piRNAs and single-stranded siRNAs in RISC. Curr. Biol. CB 2007, 17, 1265–1272. [Google Scholar] [CrossRef]
- Kirino, Y.; Mourelatos, Z. The mouse homolog of HEN1 is a potential methylase for Piwi-interacting RNAs. RNA 2007, 13, 1397–1401. [Google Scholar] [CrossRef]
- Nagao, A.; Mituyama, T.; Huang, H.; Chen, D.; Siomi, M.C.; Siomi, H. Biogenesis pathways of piRNAs loaded onto AGO3 in the Drosophila testis. RNA 2010, 16, 2503–2515. [Google Scholar] [CrossRef]
- Li, C.; Vagin, V.V.; Lee, S.; Xu, J.; Ma, S.; Xi, H.; Seitz, H.; Horwich, M.D.; Syrzycka, M.; Honda, B.M.; et al. Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell 2009, 137, 509–521. [Google Scholar] [CrossRef]
- Halic, M.; Moazed, D. Transposon silencing by piRNAs. Cell 2009, 138, 1058–1060. [Google Scholar] [CrossRef]
- Cornes, E.; Bourdon, L.; Singh, M.; Mueller, F.; Quarato, P.; Wernersson, E.; Bienko, M.; Li, B.; Cecere, G. piRNAs initiate transcriptional silencing of spermatogenic genes during C. elegans germline development. Dev. Cell 2022, 57, 180–196.e7. [Google Scholar] [CrossRef]
- Le Thomas, A.; Rogers, A.K.; Webster, A.; Marinov, G.K.; Liao, S.E.; Perkins, E.M.; Hur, J.K.; Aravin, A.A.; Toth, K.F. Piwi induces piRNA-guided transcriptional silencing and establishment of a repressive chromatin state. Genes Dev. 2013, 27, 390–399. [Google Scholar] [CrossRef] [PubMed]
- Morais, P.; Adachi, H.; Yu, Y.T. Spliceosomal snRNA Epitranscriptomics. Front. Genet. 2021, 12, 652129. [Google Scholar] [CrossRef]
- Valadkhan, S.; Gunawardane, L.S. Role of small nuclear RNAs in eukaryotic gene expression. Essays Biochem. 2013, 54, 79–90. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.A.; Sachidanandam, R.; Bourc’his, D.; Schaefer, C.; Pezic, D.; Toth, K.F.; Bestor, T.; Hannon, G.J. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol. Cell 2008, 31, 785–799. [Google Scholar] [CrossRef] [PubMed]
- Pezic, D.; Manakov, S.A.; Sachidanandam, R.; Aravin, A.A. piRNA pathway targets active LINE1 elements to establish the repressive H3K9me3 mark in germ cells. Genes Dev. 2014, 28, 1410–1428. [Google Scholar] [CrossRef]
- Shi, Y.; Shi, Q.; Shen, Q.; Zhang, Q.; Cao, X. Dicer-independent snRNA/snoRNA-derived nuclear RNA 3 regulates tumor-associated macrophage function by epigenetically repressing inducible nitric oxide synthase transcription. Cancer Commun. 2021, 41, 140–153. [Google Scholar] [CrossRef]
- Xu, X.; Ma, L.; Zhang, X.; Guo, S.; Guo, W.; Wang, Y.; Qiu, S.; Tian, X.; Miao, Y.; Yu, Y.; et al. A positive feedback circuit between RN7SK snRNA and m(6)A readers is essential for tumorigenesis. Mol. Ther. 2022. [Google Scholar] [CrossRef]
- Chen, J.; Hou, S.F.; Tang, F.J.; Liu, D.S.; Chen, Z.Z.; Zhang, H.L.; Wang, S.H. HOTAIR/Sp1/miR-199a critically regulates cancer stemness and malignant progression of cutaneous squamous cell carcinoma. Oncogene 2022, 41, 99–111. [Google Scholar] [CrossRef]
- Li, Y.; Luo, Q.; Li, Z.; Wang, Y.; Zhu, C.; Li, T.; Li, X. Long Non-coding RNA IRAIN Inhibits VEGFA Expression via Enhancing Its DNA Methylation Leading to Tumor Suppression in Renal Carcinoma. Front. Oncol. 2020, 10, 1082. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, H.; Chong, Y.; Guan, B.; Guo, P. YAP Promotes VEGFA Expression and Tumor Angiogenesis Though Gli2 in Human Renal Cell Carcinoma. Arch. Med. Res. 2019, 50, 225–233. [Google Scholar] [CrossRef]
- Ou, J.; Lei, P.; Yang, Z.; Yang, M.; Luo, L.; Mo, H.; Luo, G.; He, J. LINC00152 mediates CD8(+) T-cell infiltration in gastric cancer through binding to EZH2 and regulating the CXCL9, 10/CXCR3 axis. J. Mol. Histol. 2021, 52, 611–620. [Google Scholar] [CrossRef]
- Dong, F.; Qin, X.; Wang, B.; Li, Q.; Hu, J.; Cheng, X.; Guo, D.; Cheng, F.; Fang, C.; Tan, Y.; et al. ALKBH5 Facilitates Hypoxia-Induced Paraspeckle Assembly and IL8 Secretion to Generate an Immunosuppressive Tumor Microenvironment. Cancer Res. 2021, 81, 5876–5888. [Google Scholar] [CrossRef]
- Liang, J.; Liu, C.; Xu, D.; Xie, K.; Li, A. LncRNA NEAT1 facilitates glioma progression via stabilizing PGK1. J. Transl. Med. 2022, 20, 80. [Google Scholar] [CrossRef]
- Liu, Y.C.; Lin, Y.H.; Chi, H.C.; Huang, P.S.; Liao, C.J.; Liou, Y.S.; Lin, C.C.; Yu, C.J.; Yeh, C.T.; Huang, Y.H.; et al. CRNDE acts as an epigenetic modulator of the p300/YY1 complex to promote HCC progression and therapeutic resistance. Clin. Epigenet. 2022, 14, 106. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Bai, M.; Zhou, L.; Wang, X.; Li, S.; Wang, X.; Yang, H.; Li, J.; et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat. Commun. 2017, 8, 15016. [Google Scholar] [CrossRef]
- Guo, K.; Qian, K.; Shi, Y.; Sun, T.; Wang, Z. LncRNA-MIAT promotes thyroid cancer progression and function as ceRNA to target EZH2 by sponging miR-150-5p. Cell Death Dis. 2021, 12, 1097. [Google Scholar] [CrossRef]
- Liu, W.; Wang, Z.; Wang, C.; Ai, Z. Long non-coding RNA MIAT promotes papillary thyroid cancer progression through upregulating LASP1. Cancer Cell Int. 2019, 19, 194. [Google Scholar] [CrossRef]
- Zhang, X.; Pan, B.; Qiu, J.; Ke, X.; Shen, S.; Wang, X.; Tang, N. lncRNA MIAT targets miR-411-5p/STAT3/PD-L1 axis mediating hepatocellular carcinoma immune response. Int. J. Exp. Pathol. 2022, 103, 102–111. [Google Scholar] [CrossRef]
- Gao, S.; Zhou, B.; Li, H.; Huang, X.; Wu, Y.; Xing, C.; Yu, X.; Ji, Y. Long noncoding RNA HOTAIR promotes the self-renewal of leukemia stem cells through epigenetic silencing of p15. Exp. Hematol. 2018, 67, 32–40.e3. [Google Scholar] [CrossRef]
- Yan, H.; Wu, Q.L.; Sun, C.Y.; Ai, L.S.; Deng, J.; Zhang, L.; Chen, L.; Chu, Z.B.; Tang, B.; Wang, K.; et al. piRNA-823 contributes to tumorigenesis by regulating de novo DNA methylation and angiogenesis in multiple myeloma. Leukemia 2015, 29, 196–206. [Google Scholar] [CrossRef]
- Wu, D.; Fu, H.; Zhou, H.; Su, J.; Zhang, F.; Shen, J. Effects of Novel ncRNA Molecules, p15-piRNAs, on the Methylation of DNA and Histone H3 of the CDKN2B Promoter Region in U937 Cells. J. Cell. Biochem. 2015, 116, 2744–2754. [Google Scholar] [CrossRef] [PubMed]
- Subhramanyam, C.S.; Cao, Q.; Wang, C.; Heng, Z.S.L.; Zhou, Z.; Hu, Q. Role of PIWI-like 4 in modulating neuronal differentiation from human embryonal carcinoma cells. RNA Biol. 2020, 17, 1613–1624. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Mu, S.; Sun, C.; Fan, F.; Yan, H.; Qin, Y.; Cui, G.; Wang, Y.; Guo, T.; Mei, H.; et al. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol. Cancer 2019, 18, 88. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Fan, G.; Song, S.; Jiang, Y.; Qian, C.; Zhang, W.; Su, Q.; Xue, X.; Zhuang, W.; Li, B. piRNA-30473 contributes to tumorigenesis and poor prognosis by regulating m6A RNA methylation in DLBCL. Blood 2021, 137, 1603–1614. [Google Scholar] [CrossRef]
- Oh, J.M.; Venters, C.C.; Di, C.; Pinto, A.M.; Wan, L.; Younis, I.; Cai, Z.; Arai, C.; So, B.R.; Duan, J.; et al. U1 snRNP regulates cancer cell migration and invasion in vitro. Nat. Commun. 2020, 11, 1. [Google Scholar] [CrossRef]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. miRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Huang, H.Y.; Lin, Y.C.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase update 2022: An informative resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. miRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef]
- Bu, D.; Yu, K.; Sun, S.; Xie, C.; Skogerbo, G.; Miao, R.; Xiao, H.; Liao, Q.; Luo, H.; Zhao, G.; et al. NONCODE v3.0: Integrative annotation of long noncoding RNAs. Nucleic Acids Res. 2012, 40, D210–D215. [Google Scholar] [CrossRef]
- Quek, X.C.; Thomson, D.W.; Maag, J.L.; Bartonicek, N.; Signal, B.; Clark, M.B.; Gloss, B.S.; Dinger, M.E. lncRNAdb v2.0: Expanding the reference database for functional long noncoding RNAs. Nucleic Acids Res. 2015, 43, D168–D173. [Google Scholar] [CrossRef]
- Volders, P.J.; Anckaert, J.; Verheggen, K.; Nuytens, J.; Martens, L.; Mestdagh, P.; Vandesompele, J. LNCipedia 5: Towards a reference set of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, D135–D139. [Google Scholar] [CrossRef]
- Ma, L.; Cao, J.; Liu, L.; Du, Q.; Li, Z.; Zou, D.; Bajic, V.B.; Zhang, Z. LncBook: A curated knowledgebase of human long non-coding RNAs. Nucleic Acids Res. 2019, 47, 2699. [Google Scholar] [CrossRef]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Hermoso Pulido, T.; Guigo, R.; Johnson, R. LncATLAS database for subcellular localization of long noncoding RNAs. RNA 2017, 23, 1080–1087. [Google Scholar] [CrossRef]
- Lin, Y.; Pan, X.; Shen, H.B. lncLocator 2.0: A cell-line-specific subcellular localization predictor for long non-coding RNAs with interpretable deep learning. Bioinformatics 2021, 37, 2308–2316. [Google Scholar] [CrossRef]
- Sai Lakshmi, S.; Agrawal, S. piRNABank: A web resource on classified and clustered Piwi-interacting RNAs. Nucleic Acids Res. 2008, 36, D173–D177. [Google Scholar] [CrossRef]
- Wang, J.; Zhang, P.; Lu, Y.; Li, Y.; Zheng, Y.; Kan, Y.; Chen, R.; He, S. piRBase: A comprehensive database of piRNA sequences. Nucleic Acids Res. 2019, 47, D175–D180. [Google Scholar] [CrossRef]
- Kalvari, I.; Nawrocki, E.P.; Argasinska, J.; Quinones-Olvera, N.; Finn, R.D.; Bateman, A.; Petrov, A.I. Non-Coding RNA Analysis Using the Rfam Database. Curr. Protoc. Bioinform. 2018, 62, e51. [Google Scholar] [CrossRef]
- Yoshihama, M.; Nakao, A.; Kenmochi, N. snOPY: A small nucleolar RNA orthological gene database. BMC Res. Notes 2013, 6, 426. [Google Scholar] [CrossRef]
- The, R.C.; Petrov, A.I.; Kay, S.J.E.; Kalvari, I.; Howe, K.L.; Gray, K.A.; Bruford, E.A.; Kersey, P.J.; Cochrane, G.; Finn, R.D.; et al. RNAcentral: A comprehensive database of non-coding RNA sequences. Nucleic Acids Res. 2017, 45, D128–D134. [Google Scholar] [CrossRef]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. starBase v2.0: Decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef]
- Gu, Y.; Niu, S.; Wang, Y.; Duan, L.; Pan, Y.; Tong, Z.; Zhang, X.; Yang, Z.; Peng, B.; Wang, X.; et al. DMDRMR-Mediated Regulation of m(6)A-Modified CDK4 by m(6)A Reader IGF2BP3 Drives ccRCC Progression. Cancer Res. 2021, 81, 923–934. [Google Scholar] [CrossRef]
- Hu, J.J.; Song, W.; Zhang, S.D.; Shen, X.H.; Qiu, X.M.; Wu, H.Z.; Gong, P.H.; Lu, S.; Zhao, Z.J.; He, M.L.; et al. HBx-upregulated lncRNA UCA1 promotes cell growth and tumorigenesis by recruiting EZH2 and repressing p27Kip1/CDK2 signaling. Sci. Rep. 2016, 6, 23521. [Google Scholar] [CrossRef]
- Yoneda, R.; Ueda, N.; Uranishi, K.; Hirasaki, M.; Kurokawa, R. Long noncoding RNA pncRNA-D reduces cyclin D1 gene expression and arrests cell cycle through RNA m(6)A modification. J. Biol. Chem. 2020, 295, 5626–5639. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Zhang, J.; Mao, L. E6 hijacks KDM5C/lnc_000231/miR-497-5p/CCNE1 axis to promote cervical cancer progression. J. Cell. Mol. Med. 2020, 24, 11422–11433. [Google Scholar] [CrossRef]
- Huang, Z.F.; Tang, Y.L.; Shen, Z.L.; Yang, K.Y.; Gao, K. UXT, a novel DNMT3b-binding protein, promotes breast cancer progression via negatively modulating lncRNA MEG3/p53 axis. Mol. Ther. Oncolytics 2022, 24, 497–506. [Google Scholar] [CrossRef]
- Song, W.; Fei, F.; Qiao, F.; Weng, Z.; Yang, Y.; Cao, B.; Yue, J.; Xu, J.; Zheng, M.; Li, J. ALKBH5-mediated N(6)-methyladenosine modification of TRERNA1 promotes DLBCL proliferation via p21 downregulation. Cell Death Discov. 2022, 8, 25. [Google Scholar] [CrossRef]
- Ahn, E.; Araki, K.; Hashimoto, M.; Li, W.; Riley, J.L.; Cheung, J.; Sharpe, A.H.; Freeman, G.J.; Irving, B.A.; Ahmed, R. Role of PD-1 during effector CD8 T cell differentiation. Proc. Natl. Acad. Sci. USA 2018, 115, 4749–4754. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Hsu, J.M.; Yang, W.H.; Hung, M.C. Mechanisms regulating PD-L1 expression in cancers and associated opportunities for novel small-molecule therapeutics. Nat. Rev. Clin. Oncol. 2022, 19, 287–305. [Google Scholar] [CrossRef]
- Wang, Q.; Li, G.; Ma, X.; Liu, L.; Liu, J.; Yin, Y.; Li, H.; Chen, Y.; Zhang, X.; Zhang, L.; et al. LncRNA TINCR impairs the efficacy of immunotherapy against breast cancer by recruiting DNMT1 and downregulating MiR-199a-5p via the STAT1-TINCR-USP20-PD-L1 axis. Cell Death Dis. 2023, 14, 76. [Google Scholar] [CrossRef]
- Padmanaban, V.; Krol, I.; Suhail, Y.; Szczerba, B.M.; Aceto, N.; Bader, J.S.; Ewald, A.J. E-cadherin is required for metastasis in multiple models of breast cancer. Nature 2019, 573, 439–444. [Google Scholar] [CrossRef]
- Li, Y.; Cheng, C. Long noncoding RNA NEAT1 promotes the metastasis of osteosarcoma via interaction with the G9a-DNMT1-Snail complex. Am. J. Cancer Res. 2018, 8, 81–90. [Google Scholar]
- Carbone, C.; Piro, G.; Merz, V.; Simionato, F.; Santoro, R.; Zecchetto, C.; Tortora, G.; Melisi, D. Angiopoietin-Like Proteins in Angiogenesis, Inflammation and Cancer. Int. J. Mol. Sci. 2018, 19, 431. [Google Scholar] [CrossRef]
- Yu, Y.; Zhang, M.; Liu, J.; Xu, B.; Yang, J.; Wang, N.; Yan, S.; Wang, F.; He, X.; Ji, G.; et al. Long Non-coding RNA PVT1 Promotes Cell Proliferation and Migration by Silencing ANGPTL4 Expression in Cholangiocarcinoma. Mol. Ther. Nucleic Acids 2018, 13, 503–513. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, L.; Li, H.; Sun, T.; Wen, X.; Li, X.; Meng, Y.; Li, Y.; Liu, M.; Liu, S.; et al. Nuclear-Encoded lncRNA MALAT1 Epigenetically Controls Metabolic Reprogramming in HCC Cells through the Mitophagy Pathway. Mol. Ther. Nucleic Acids 2021, 23, 264–276. [Google Scholar] [CrossRef]
- Arunkumar, G.; Baek, S.; Sturgill, D.; Bui, M.; Dalal, Y. Oncogenic lncRNAs alter epigenetic memory at a fragile chromosomal site in human cancer cells. Sci. Adv. 2022, 8, eabl5621. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pathania, A.S. Crosstalk between Noncoding RNAs and the Epigenetics Machinery in Pediatric Tumors and Their Microenvironment. Cancers 2023, 15, 2833. https://doi.org/10.3390/cancers15102833
Pathania AS. Crosstalk between Noncoding RNAs and the Epigenetics Machinery in Pediatric Tumors and Their Microenvironment. Cancers. 2023; 15(10):2833. https://doi.org/10.3390/cancers15102833
Chicago/Turabian StylePathania, Anup S. 2023. "Crosstalk between Noncoding RNAs and the Epigenetics Machinery in Pediatric Tumors and Their Microenvironment" Cancers 15, no. 10: 2833. https://doi.org/10.3390/cancers15102833
APA StylePathania, A. S. (2023). Crosstalk between Noncoding RNAs and the Epigenetics Machinery in Pediatric Tumors and Their Microenvironment. Cancers, 15(10), 2833. https://doi.org/10.3390/cancers15102833