
WNT-Conditioned Mechanism of Exit from Postchemotherapy Shock of Differentiated Tumour Cells

 , , ,
, , ,  and
and
Abstract
Simple Summary
Abstract

1. Introduction
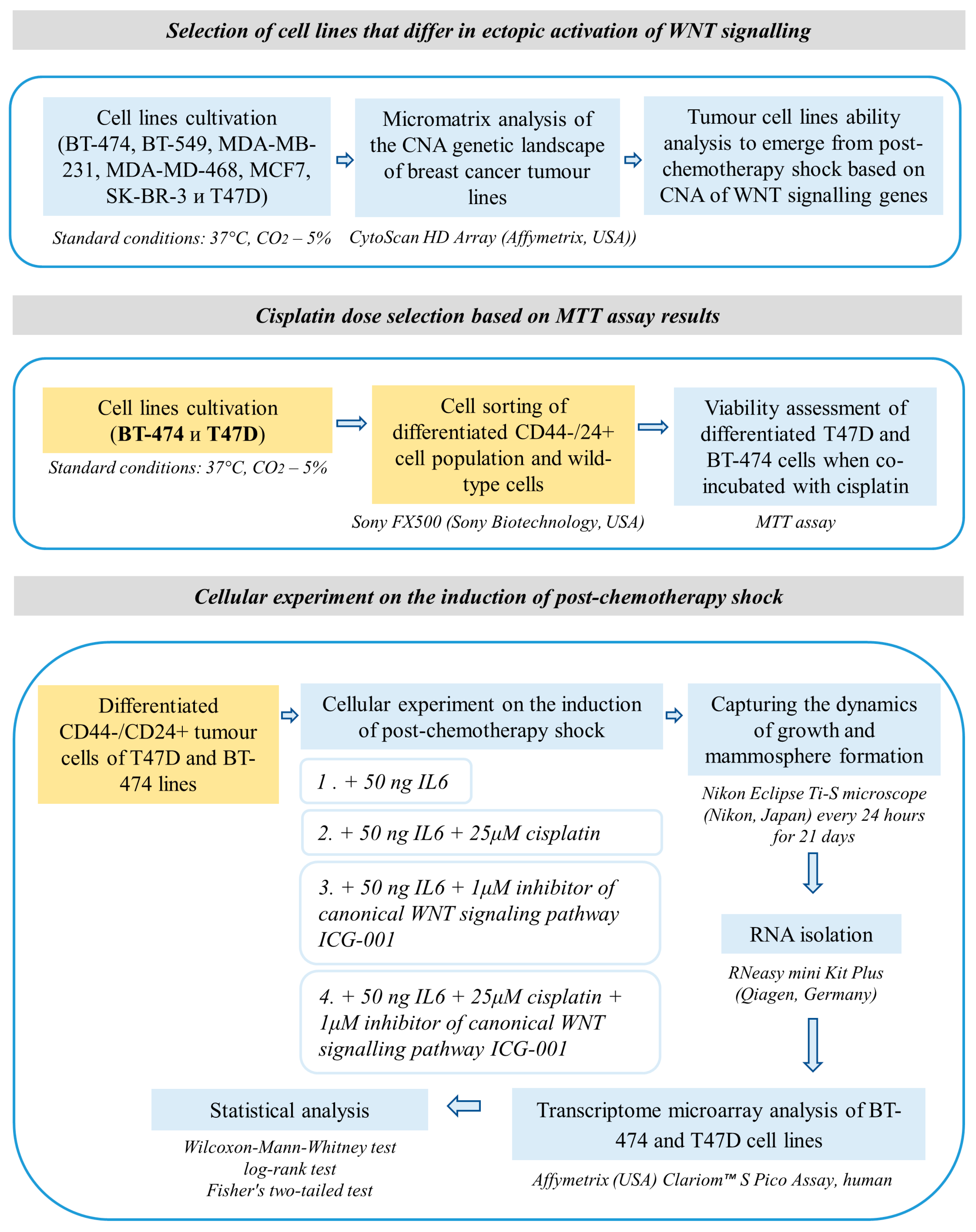
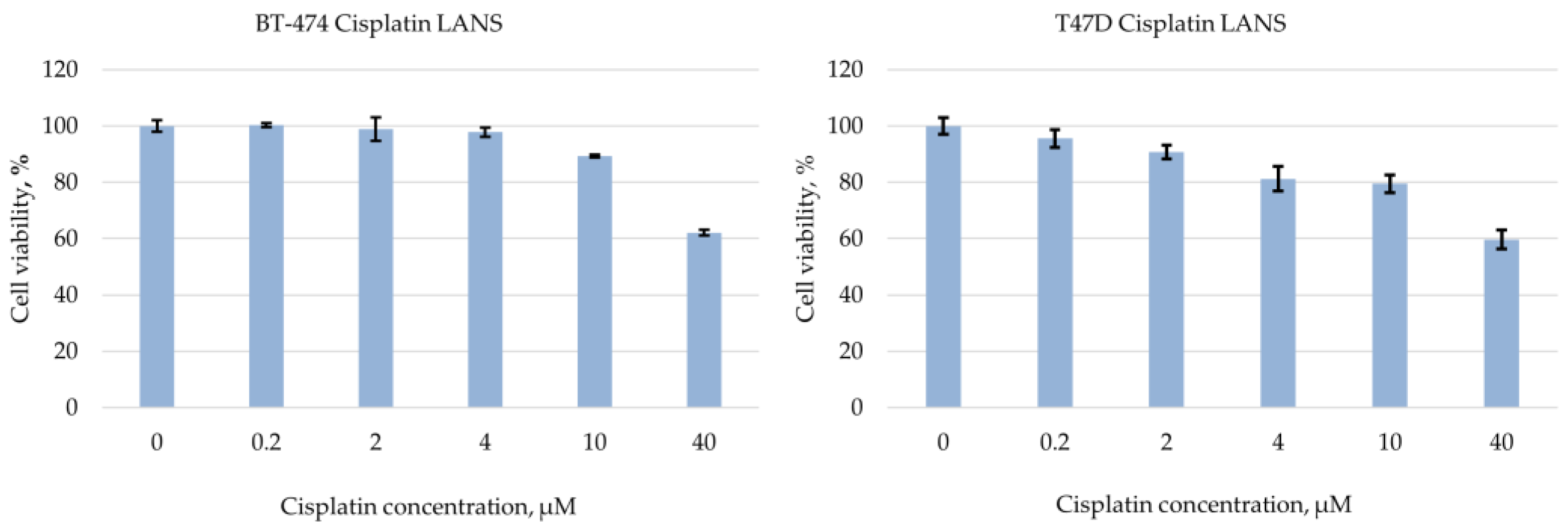
2. Materials and Methods
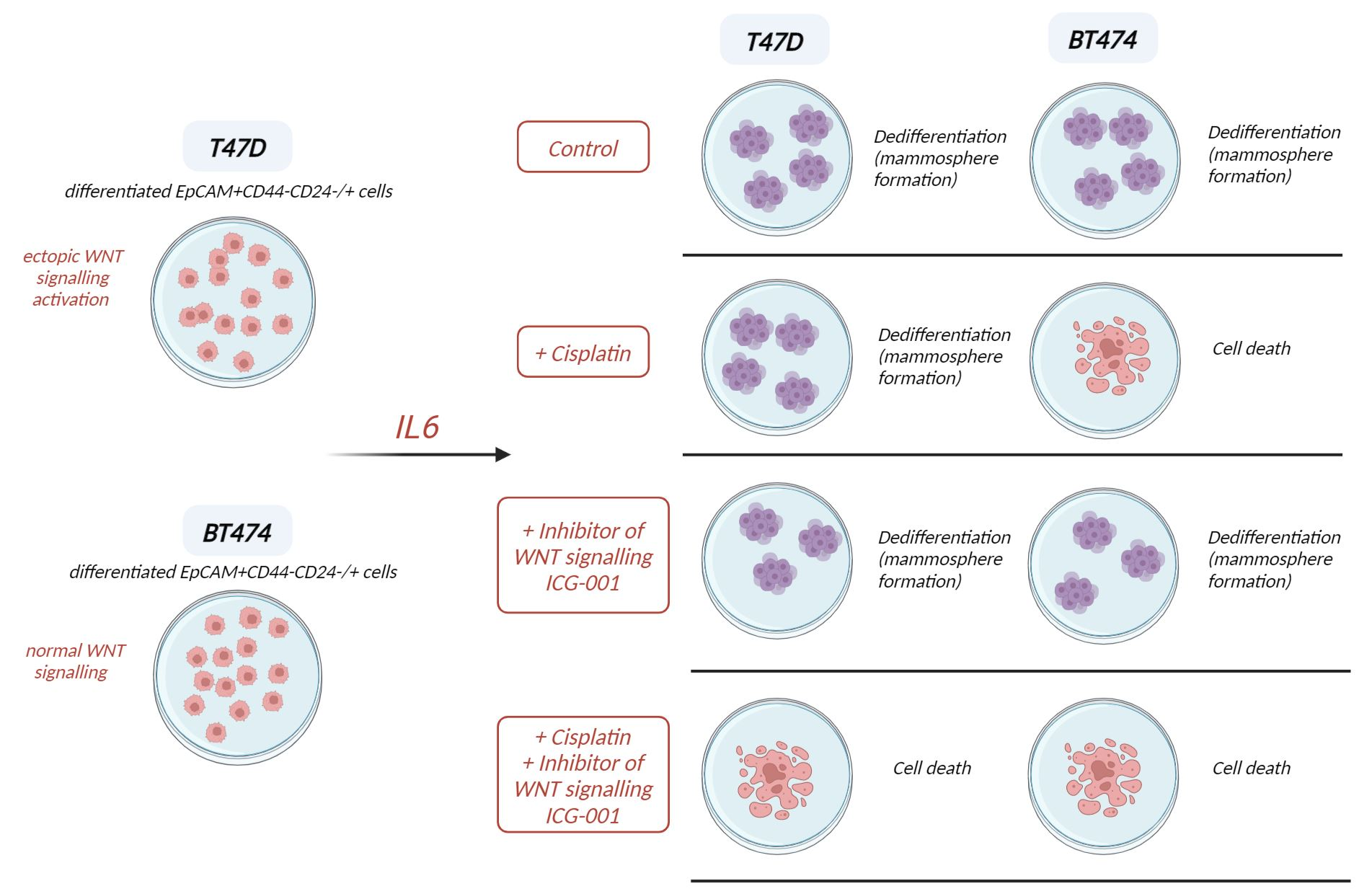
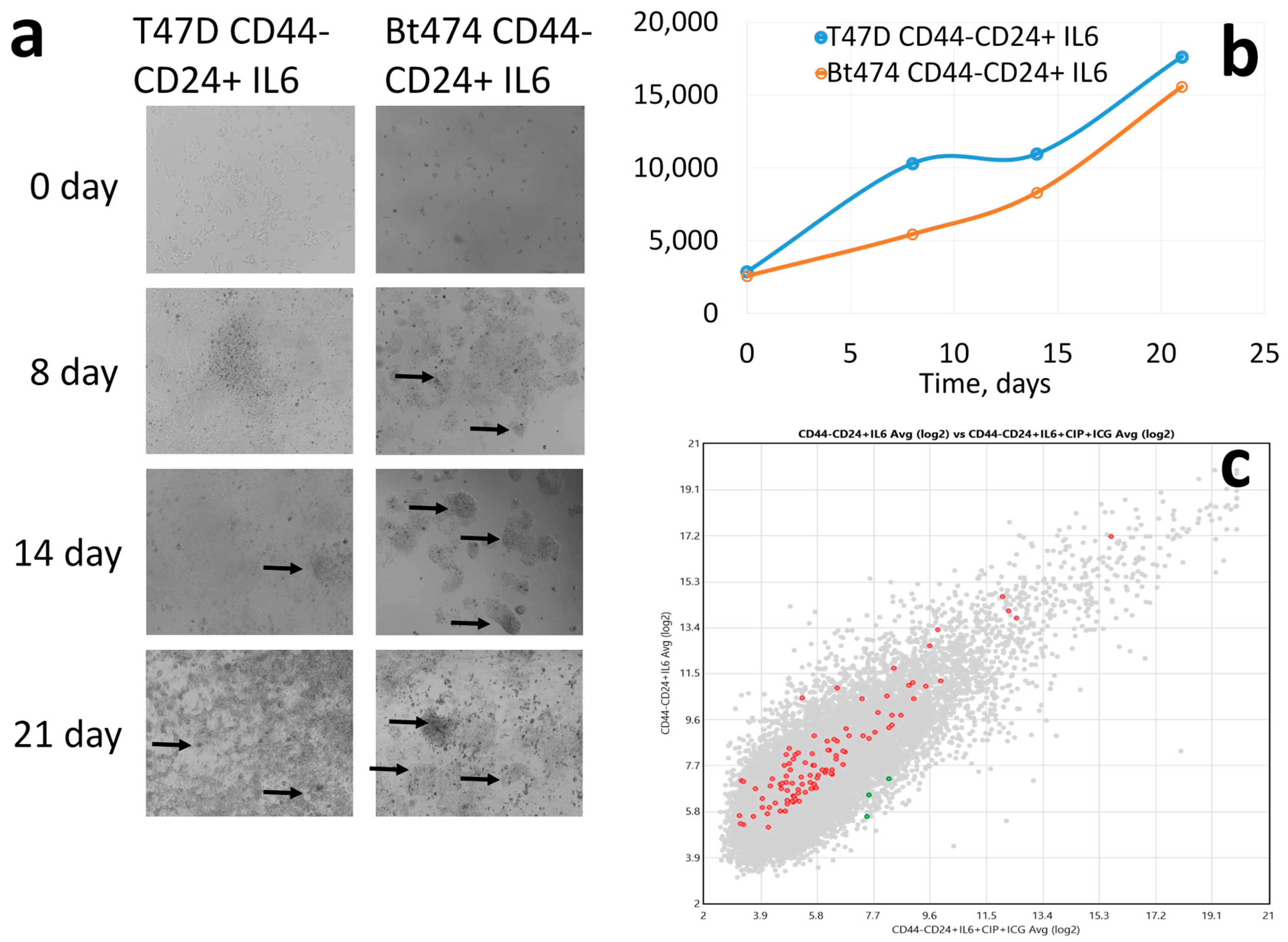
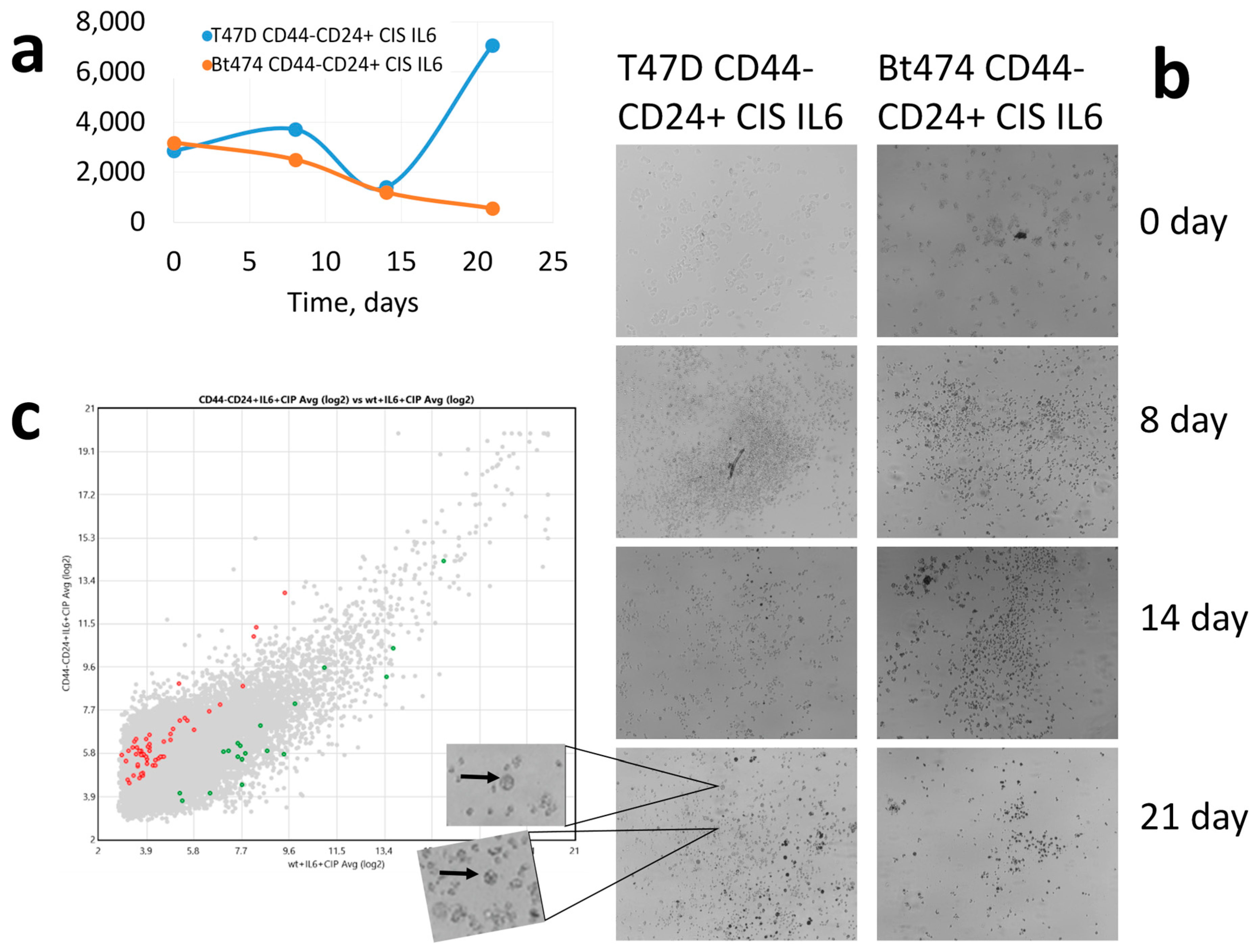
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
References
- Pohl, S.-G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef] [PubMed]
- Koni, M.; Pinnarò, V.; Brizzi, M.F. The Wnt Signalling Pathway: A Tailored Target in Cancer. Int. J. Mol. Sci. 2020, 21, 7697. [Google Scholar] [CrossRef]
- de Oliveira, W.A.A.; El Laithy, Y.; Bruna, A.; Annibali, D.; Lluis, F. Wnt Signaling in the Breast: From Development to Disease. Front. Cell Dev. Biol. 2022, 10. [Google Scholar] [CrossRef]
- Parsons, M.J.; Tammela, T.; Dow, L.E. WNT as a Driver and Dependency in CancerWNT as a Driver and Dependency in Cancer. Cancer Discov. 2021, 11, 2413–2429. [Google Scholar] [CrossRef]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef]
- Katoh, M.; Katoh, M. WNT signaling and cancer stemness. Essays Biochem. 2022, 66, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.-G.; Polotskaia, A.; Xiao, G.; Di, L.; Zhao, Y.; Hu, W.; Philip, J.; Hendrickson, R.C.; Bargonetti, J. Identification, validation, and targeting of the mutant p53-PARP-MCM chromatin axis in triple negative breast cancer. Npj Breast Cancer 2017, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Bojko, A.; Czarnecka-Herok, J.; Charzynska, A.; Dabrowski, M.; Sikora, E. Diversity of the Senescence Phenotype of Cancer Cells Treated with Chemotherapeutic Agents. Cells 2019, 8, 1501. [Google Scholar] [CrossRef] [PubMed]
- Mikuła-Pietrasik, J.; Niklas, A.; Uruski, P.; Tykarski, A.; Książek, K. Mechanisms and significance of therapy-induced and spontaneous senescence of cancer cells. Cell. Mol. Life Sci. 2019, 77, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-induced senescence in cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [PubMed]
- Mikuła-Pietrasik, J.; Witucka, A.; Pakuła, M.; Uruski, P.; Begier-Krasińska, B.; Niklas, A.; Tykarski, A.; Książek, K. Comprehensive review on how platinum- and taxane-based chemotherapy of ovarian cancer affects biology of normal cells. Cell. Mol. Life Sci. 2019, 76, 681–697. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Bloukh, S.; Carpenter, V.J.; Alwohoush, E.; Bakeer, J.; Darwish, S.; Azab, B.; Gewirtz, D.A. Therapy-Induced Senescence: An “Old” Friend Becomes the Enemy. Cancers 2020, 12, 822. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, W.; Dong, H.; Li, Y.; Li, L.; Han, L.; Han, Z.; Wang, S.; Ma, D.; Wang, H. Cisplatin-induced senescence in ovarian cancer cells is mediated by GRP78. Oncol. Rep. 2014, 31, 2525–2534. [Google Scholar] [CrossRef]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.-Y.J.; Wu, D.Y. Escape from Therapy-Induced Accelerated Cellular Senescence in p53-Null Lung Cancer Cells and in Human Lung Cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small molecule compounds that induce cellular senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef] [PubMed]
- Vesel, M.; Rapp, J.; Feller, D.; Kiss, E.; Jaromi, L.; Meggyes, M.; Miskei, G.; Duga, B.; Smuk, G.; Laszlo, T.; et al. ABCB1 and ABCG2 drug transporters are differentially expressed in non-small cell lung cancers (NSCLC) and expression is modified by cisplatin treatment via altered Wnt signaling. Respir. Res. 2017, 18, 52. [Google Scholar] [CrossRef] [PubMed]
- De Blander, H.; Morel, A.-P.; Senaratne, A.P.; Ouzounova, M.; Puisieux, A. Cellular Plasticity: A Route to Senescence Exit and Tumorigenesis. Cancers 2021, 13, 4561. [Google Scholar] [CrossRef]
- Otero-Albiol, D.; Carnero, A. Cellular senescence or stemness: Hypoxia flips the coin. J. Exp. Clin. Cancer Res. 2021, 40, 243. [Google Scholar] [CrossRef]
- Kumari, R.; Jat, P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front. Cell Dev. Biol. 2021, 9, 645593. [Google Scholar] [CrossRef]
- Liu, H.; Zhao, H.; Sun, Y. Tumor microenvironment and cellular senescence: Understanding therapeutic resistance and harnessing strategies. Semin. Cancer Biol. 2021, 86, 769–781. [Google Scholar] [CrossRef]
- Karabicici, M.; Alptekin, S.; Fırtına Karagonlar, Z.; Erdal, E. Doxorubicin-induced senescence promotes stemness and tumorigenicity in EpCAM−/CD133− nonstem cell population in hepatocellular carcinoma cell line, HuH-7. Mol. Oncol. 2021, 15, 2185–2202. [Google Scholar] [CrossRef]
- Milanovic, M.; Fan, D.N.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Barbosa, I.A.M. Senescence-associated reprogramming promotes cancer stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Litviakov, N.; Ibragimova, M.; Tsyganov, M.; Kazantseva, P.; Deryusheva, I.; Pevzner, A.; Doroshenko, A.; Garbukov, E.; Tarabanovskaya, N.; Slonimskaya, E. Amplifications of stemness genes and the capacity of breast tumors for metastasis. Oncotarget 2020, 11, 1988–2001. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef]
- Kumar, S.M.; Liu, S.; Lu, H.; Zhang, H.; Zhang, P.J.; A Gimotty, P.; Guerra, M.; Guo, W.; Xu, X. Acquired cancer stem cell phenotypes through Oct4-mediated dedifferentiation. Oncogene 2012, 31, 4898–4911. [Google Scholar] [CrossRef]
- Fleege, N.M.G.; Cobain, E.F. Breast Cancer Management in 2021: A Primer for the OB GYN. Best Pract. Res. Clin. Obstet. Gynaecol. 2022, 82, 30–45. [Google Scholar] [CrossRef] [PubMed]
- Giaquinto, A.N.; Sung, H.; Miller, K.D.; Kramer, J.L.; Newman, L.A.; Minihan, A.; Jemal, A.; Siegel, R.L. Breast cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 524–541. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Peng, C.; Tang, H.; Liu, X.; Peng, F. New Advances in the Research of Resistance to Neoadjuvant Chemotherapy in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 9644. [Google Scholar] [CrossRef]
- Romeo, V.; Accardo, G.; Perillo, T.; Basso, L.; Garbino, N.; Nicolai, E.; Maurea, S.; Salvatore, M. Assessment and Prediction of Response to Neoadjuvant Chemotherapy in Breast Cancer: A Comparison of Imaging Modalities and Future Perspectives. Cancers 2021, 13, 3521. [Google Scholar] [CrossRef] [PubMed]
- Mieog, J.S.D.; A van der Hage, J.; van de Velde, C.J.H. Neoadjuvant chemotherapy for operable breast cancer. Br. J. Surg. 2007, 94, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Litviakov, N.V.; Ibragimova, M.K.; Tsyganov, M.M.; Deriusheva, I.V.; Pevsner, A.M.; Garbukov, E.Y.; Doroshenko, A.V.; Slonimskaya, E.M. Association of the combination of stemness gene amplifications and copy number aberrations of wnt-signaling genes in breast tumors with metastasis. Sib. J. Oncol. 2020, 19, 78–88. [Google Scholar] [CrossRef]
- Yuan, Y.; Xie, G.; Yao, Q.; Liu, Y.; Du, S.; Liu, A.; Guo, Z.; Sun, A.; Ruan, J.; Chen, L.; et al. IL-6-induced epithelial-mesenchymal transition promotes the generation of breast cancer stem-like cells analogous to mammosphere cultures. Int. J. Oncol. 2011, 40, 1171–1179. [Google Scholar] [CrossRef] [PubMed]
- Poth, K.J.; Guminski, A.D.; Thomas, G.P.; Leo, P.J.; Jabbar, I.A.; Saunders, N.A. Cisplatin Treatment Induces a Transient Increase in Tumorigenic Potential Associated with High Interleukin-6 Expression in Head and Neck Squamous Cell CarcinomaCisplatin Induces Increased Tumor Initiation by HNSCC. Mol. Cancer Ther. 2010, 9, 2430–2439. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Kohli, J.; Demaria, M. Senescent cells in cancer therapy: Friends or foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Fang, J.; Chen, J. Tumor cell senescence response produces aggressive variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef]
- Wong, A.M.G.; Wong, A.M.G.; Can, L.K.; Kwong, J.; Chan, A.W.; Chen, J.; Kahn, M.; Wong, N. Abstract 2886: Investigation of tumor-intrinsic activated Wnt/B-catenin signaling in immune avoidance of HCC. Cancer Res. 2023, 83, 2886. [Google Scholar] [CrossRef]
- Nie, X.; Liu, H.; Liu, L.; Wang, Y.-D.; Chen, W.-D. Emerging Roles of Wnt Ligands in Human Colorectal Cancer. Front. Oncol. 2020, 10, 1341. [Google Scholar] [CrossRef]
- Ibragimova, M.K.; Tsyganov, M.M.; Pevzner, A.M.; Litviakov, N.V. Transcriptome of Breast Tumors With Different Amplification Status of the Long Arm of Chromosome 8. Anticancer Res. 2021, 41, 187–195. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, W.A.A.; Moens, S.; El Laithy, Y.; van der Veer, B.K.; Athanasouli, P.; Cortesi, E.E.; Baietti, M.F.; Koh, K.P.; Ventura, J.-J.; Amant, F.; et al. Wnt/β-Catenin Inhibition Disrupts Carboplatin Resistance in Isogenic Models of Triple-Negative Breast Cancer. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Hashemi, M.; Hasani, S.; Hajimazdarany, S.; Ghadyani, F.; Olyaee, Y.; Khodadadi, M.; Ziyarani, M.F.; Dehghanpour, A.; Salehi, H.; Kakavand, A.; et al. Biological functions and molecular interactions of Wnt/β-catenin in breast cancer: Revisiting signaling networks. Int. J. Biol. Macromol. 2023, 232, 123377. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Kinugasa, T.; Ohshima, K.; Yuge, K.; Ohchi, T.; Fujino, S.; Shiraiwa, S.; Katagiri, M.; Akagi, Y. Analysis of Wnt and β-catenin expression in advanced colorectal cancer. Anticancer Res. 2015, 35, 4403–4410. [Google Scholar] [PubMed]
- Xu, X.; Sun, P.-L.; Li, J.-Z.; Jheon, S.; Lee, C.-T.; Chung, J.-H. Aberrant Wnt1/β-catenin expression is an independent poor prognostic marker of non-small cell lung cancer after surgery. J. Thorac. Oncol. 2011, 6, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Blagodatski, A.; Poteryaev, D.; Katanaev, V.L. Targeting the Wnt pathways for therapies. Mol. Cell. Ther. 2014, 2, 28. [Google Scholar] [CrossRef] [PubMed]
- Shaw, H.V.; Koval, A.; Katanaev, V.L. Targeting the Wnt signalling pathway in cancer: Prospects and perils. Swiss Med. Wkly. 2019, 149, w20129. [Google Scholar] [CrossRef] [PubMed]
- Jun, S.; Jung, Y.-S.; Na Suh, H.; Wang, W.; Kim, M.J.; Oh, Y.S.; Lien, E.M.; Shen, X.; Matsumoto, Y.; McCrea, P.D.; et al. LIG4 mediates Wnt signalling-induced radioresistance. Nat. Commun. 2016, 7, 10994. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line Name | CNA List of WNT Signalling Pathway Genes | Total Score on CNA Genes WNT Signalling | Prediction of Capacity |
|---|---|---|---|
| BT-474 | GSK3B-Gain -1, FZD9-Gain +1, TSF7L2-Gain -1, CCND1-Gain +1 | 0 | Dedifferentiation and no recovery from postchemotherapy shock |
| BT-549 | WNT2B-Gain +1, TCF7-Loss -1, SKP1-Loss -1, MAPK9-Gain +1 | 0 | No dedifferentiation and no recovery from postchemotherapy shock |
| MDA- MB-231 | GSK3B-Loss +1, CCND3-Gain +1, FZD9-Loss -1, CSNK2A2-Loss +1 | 2 | Dedifferentiation and recovery from postchemotherapy shock |
| MDA- MD-468 | WNT2B-Gain +1, MAPK9-Gain +1, CSNK2B-Gain -1, CCND3-Gain +1, FZD9-Loss -1, WNT8B-Loss -1, BTRC-Loss +1, TSF7L2-Loss +1, CSNK2A2-Gain -1, FZD2-Loss -1, WNT3-Loss -1, WNT9B-Loss -1, | −2 | Dedifferentiation and no recovery from postchemotherapy shock |
| MCF7 | SCRP5-Loss +1, WNT8B-Loss -1, BTRC-Loss +1, TSF7L2-Loss +1, CCND1-Gain +1, CSNK2A2-Loss +1 | 4 | Dedifferentiation and recovery from postchemotherapy shock |
| SK-BR-3 | GSK3B-Loss +1, APC-Loss+1, TCF7-Loss -1, SKP1-Loss -1, MAPK9-Loss -1, CCND3-Loss -1, PPP2RD5-Loss -1, FZD9-Gain +1, WNT8B-Loss -1, BTRC-Loss +1, CSNK2A2-Gain -1, FZD2-Loss -1, WNT3-Loss -1, WNT9B-Loss -1 | −6 | Dedifferentiation and no recovery from postchemotherapy shock |
| T47D | FZD9-Gain +1, TSF7L2-Loss +1, CCND1-Gain +1, CSNK2A2-Loss +1, FZD2-Gain +1, WNT3-Gain +1, WNT9B-Gain +1 | 7 | Dedifferentiation and recovery from postchemotherapy shock |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsydenova, I.A.; Dolgasheva, D.S.; Gaptulbarova, K.A.; Ibragimova, M.K.; Tsyganov, M.M.; Kravtsova, E.A.; Nushtaeva, A.A.; Litviakov, N.V. WNT-Conditioned Mechanism of Exit from Postchemotherapy Shock of Differentiated Tumour Cells. Cancers 2023, 15, 2765. https://doi.org/10.3390/cancers15102765
Tsydenova IA, Dolgasheva DS, Gaptulbarova KA, Ibragimova MK, Tsyganov MM, Kravtsova EA, Nushtaeva AA, Litviakov NV. WNT-Conditioned Mechanism of Exit from Postchemotherapy Shock of Differentiated Tumour Cells. Cancers. 2023; 15(10):2765. https://doi.org/10.3390/cancers15102765
Chicago/Turabian StyleTsydenova, Irina A., Daria S. Dolgasheva, Ksenia A. Gaptulbarova, Marina K. Ibragimova, Matvei M. Tsyganov, Ekaterina A. Kravtsova, Anna A. Nushtaeva, and Nikolai V. Litviakov. 2023. "WNT-Conditioned Mechanism of Exit from Postchemotherapy Shock of Differentiated Tumour Cells" Cancers 15, no. 10: 2765. https://doi.org/10.3390/cancers15102765
APA StyleTsydenova, I. A., Dolgasheva, D. S., Gaptulbarova, K. A., Ibragimova, M. K., Tsyganov, M. M., Kravtsova, E. A., Nushtaeva, A. A., & Litviakov, N. V. (2023). WNT-Conditioned Mechanism of Exit from Postchemotherapy Shock of Differentiated Tumour Cells. Cancers, 15(10), 2765. https://doi.org/10.3390/cancers15102765