

Immunotherapy for Triple-Negative Breast Cancer: Combination Strategies to Improve Outcome

Abstract
Simple Summary
Abstract
1. Introduction
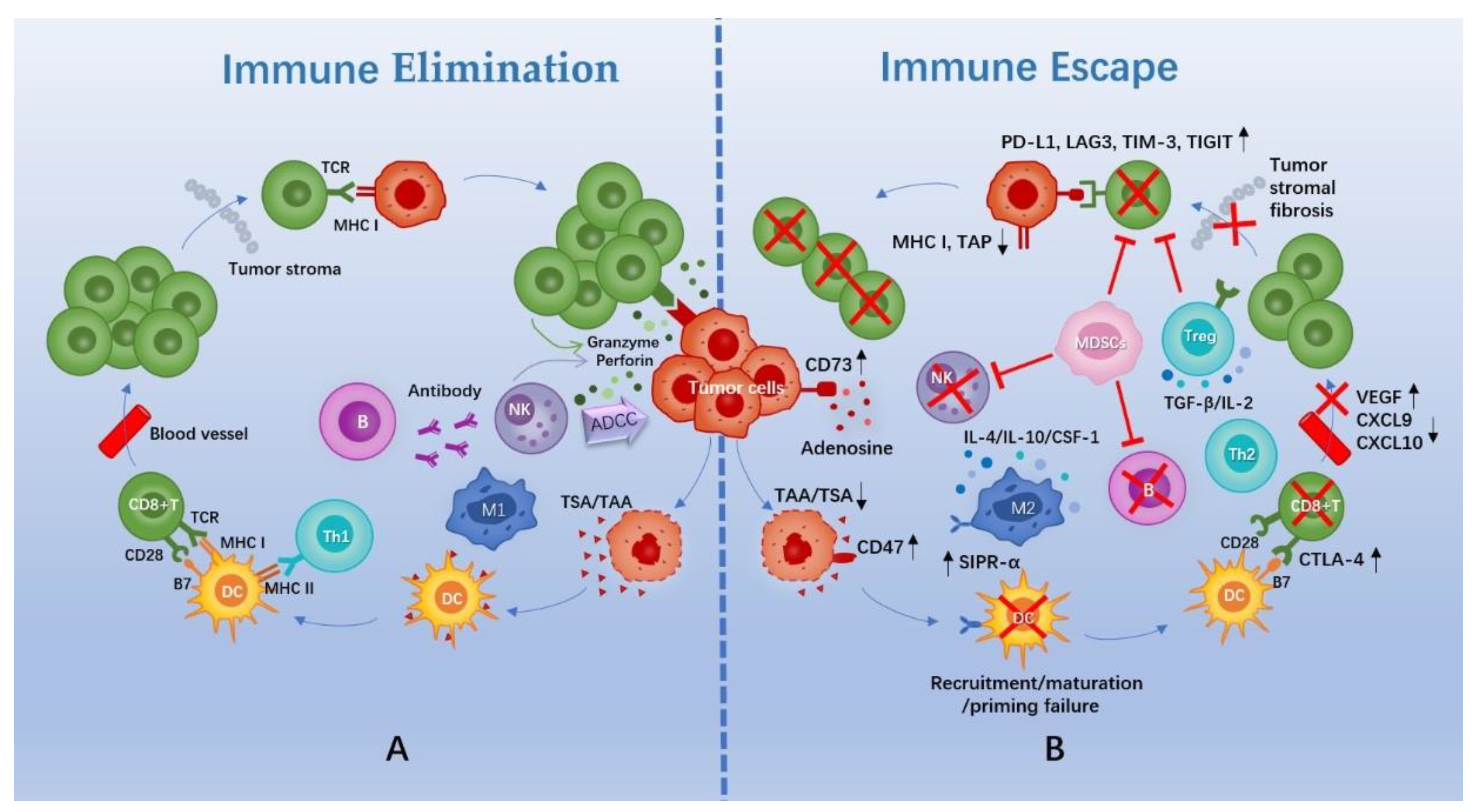
2. Rationale of Immunotherapy and the TIME of TNBC
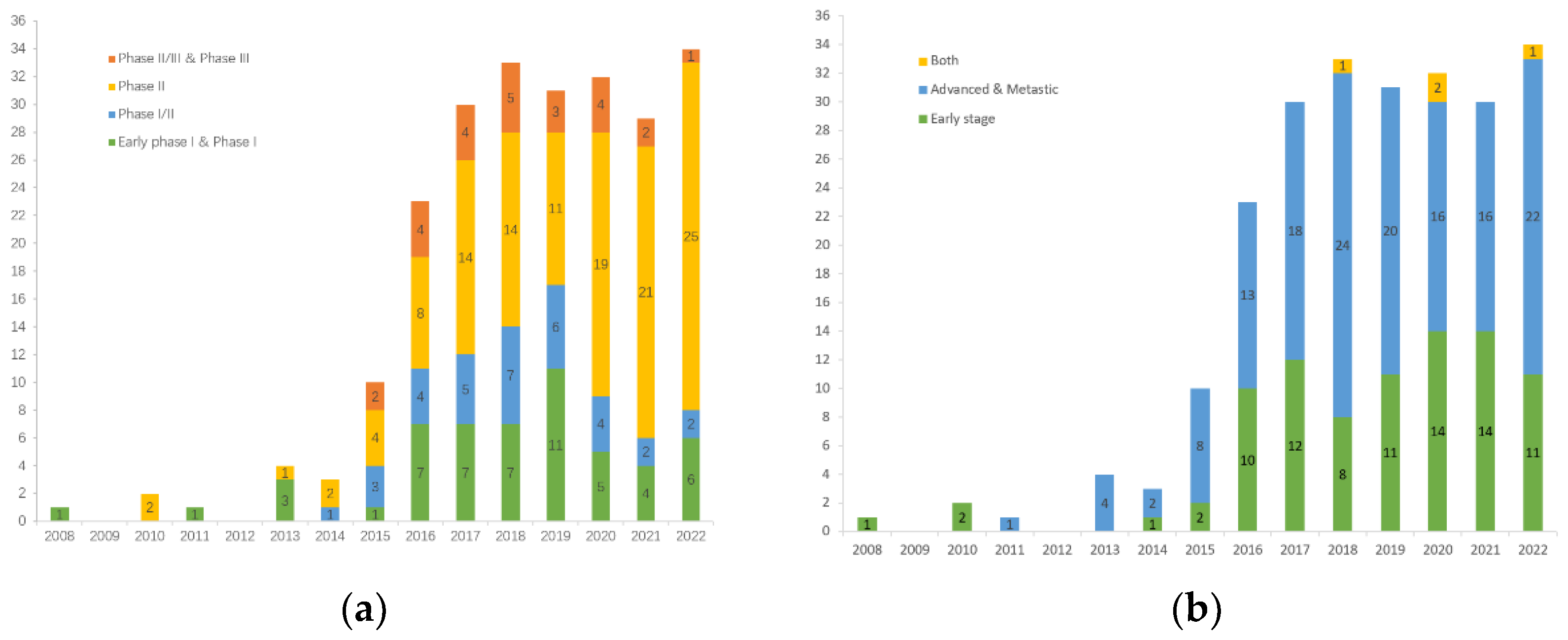
3. The Landscape of Clinical Trials on TNBC Immunotherapy
4. Performance of PD-(L)1 Inhibitors Monotherapy
4.1. In Advanced TNBC
4.2. In Early-Stage TNBC
5. Research Progress of PD-(L)1 Inhibitors in Combination with Chemotherapy
5.1. In Advanced TNBC
5.2. In Early-Stage TNBC
6. Research Progress of PD-(L)1 Inhibitors in Combination with Radiotherapy
7. Research Progress of PD-(L)1 Inhibitors in Combination with Targeted Therapy
7.1. Combination with PARPi
7.2. Combination with ADCs
7.3. Combination with Small Molecule Inhibitors
8. Exploration of PD-(L)1 Inhibitors in Combination with Other Immunotherapies
8.1. Combination with Other ICIs
8.2. TCVs and PD-(L)1 Inhibitors
8.3. Oncolytic Virus (OVs) and PD-(L)1 Inhibitors
8.4. ACT and PD-(L)1 Inhibitors
9. Potential Therapeutic Targets for Reversing Cold Tumors
10. Biomarkers for Predicting Immunological Response
10.1. PD-L1 Expression and TILs
10.2. TMB and Microsatellite Instability (MSI)/Mismatch Repair Deficiency (dMMR)
11. Pseudoprogression and Immune-Related Adverse Events
12. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lin, N.U.; Vanderplas, A.; Hughes, M.E.; Theriault, R.L.; Edge, S.B.; Wong, Y.N.; Blayney, D.W.; Niland, J.C.; Winer, E.P.; Weeks, J.C. Clinicopathologic features, patterns of recurrence, and survival among women with triple-negative breast cancer in the National Comprehensive Cancer Network. Cancer 2012, 118, 5463–5472. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS One 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef]
- Jiang, Y.Z.; Ma, D.; Suo, C.; Shi, J.; Xue, M.; Hu, X.; Xiao, Y.; Yu, K.D.; Liu, Y.R.; Yu, Y.; et al. Genomic and Transcriptomic Landscape of Triple-Negative Breast Cancers: Subtypes and Treatment Strategies. Cancer Cell 2019, 35, 428–440.e5. [Google Scholar] [CrossRef] [PubMed]
- De Melo Gagliato, D.; Cortes, J.; Curigliano, G.; Loi, S.; Denkert, C.; Perez-Garcia, J.; Holgado, E. Tumor-infiltrating lymphocytes in Breast Cancer and implications for clinical practice. Biochim. Biophys. Acta. Rev. Cancer 2017, 1868, 527–537. [Google Scholar] [CrossRef] [PubMed]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Kwa, M.J.; Adams, S. Checkpoint inhibitors in triple-negative breast cancer (TNBC): Where to go from here. Cancer 2018, 124, 2086–2103. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Campoli, M.; Ferrone, S. HLA antigen changes in malignant cells: Epigenetic mechanisms and biologic significance. Oncogene 2008, 27, 5869–5885. [Google Scholar] [CrossRef]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.M.; Chen, D.S. Immune escape to PD-L1/PD-1 blockade: Seven steps to success (or failure). Ann. Oncol. 2016, 27, 1492–1504. [Google Scholar] [CrossRef] [PubMed]
- Dou, A.; Fang, J. Heterogeneous Myeloid Cells in Tumors. Cancers 2021, 13, 3772. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S. Mechanisms of tumor escape in the context of the T-cell-inflamed and the non-T-cell-inflamed tumor microenvironment. Int. Immunol. 2016, 28, 383–391. [Google Scholar] [CrossRef]
- Dyck, L.; Mills, K.H.G. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur. J. Immunol. 2017, 47, 765–779. [Google Scholar] [CrossRef] [PubMed]
- Feng, M.; Jiang, W.; Kim, B.Y.S.; Zhang, C.C.; Fu, Y.X.; Weissman, I.L. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat. Rev. Cancer. 2019, 19, 568–586. [Google Scholar] [CrossRef]
- Sanmamed, M.F.; Chen, L. A Paradigm Shift in Cancer Immunotherapy: From Enhancement to Normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Xiao, Y.; Ma, D.; Zhao, S.; Suo, C.; Shi, J.; Xue, M.Z.; Ruan, M.; Wang, H.; Zhao, J.; Li, Q.; et al. Multi-Omics Profiling Reveals Distinct Microenvironment Characterization and Suggests Immune Escape Mechanisms of Triple-Negative Breast Cancer. Clin. Cancer Res. 2019, 25, 5002–5014. [Google Scholar] [CrossRef] [PubMed]
- Gruosso, T.; Gigoux, M.; Manem, V.S.K.; Bertos, N.; Zuo, D.; Perlitch, I.; Saleh, S.M.I.; Zhao, H.; Souleimanova, M.; Johnson, R.M.; et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J. Clin. Investig. 2019, 129, 1785–1800. [Google Scholar] [CrossRef] [PubMed]
- Bareche, Y.; Buisseret, L.; Gruosso, T.; Girard, E.; Venet, D.; Dupont, F.; Desmedt, C.; Larsimont, D.; Park, M.; Rothe, F.; et al. Unraveling Triple-Negative Breast Cancer Tumor Microenvironment Heterogeneity: Towards an Optimized Treatment Approach. J. Natl. Cancer Inst. 2020, 112, 708–719. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008, 26, 677–704. [Google Scholar] [CrossRef] [PubMed]
- Balar, A.V.; Weber, J.S. PD-1 and PD-L1 antibodies in cancer: Current status and future directions. Cancer Immunol. Immunother. 2017, 66, 551–564. [Google Scholar] [CrossRef]
- Solinas, C.; Aiello, M.; Rozali, E.; Lambertini, M.; Willard-Gallo, K.; Migliori, E. Programmed cell death-ligand 2: A neglected but important target in the immune response to cancer? Transl. Oncol. 2020, 13, 100811. [Google Scholar] [CrossRef]
- Nanda, R.; Chow, L.Q.; Dees, E.C.; Berger, R.; Gupta, S.; Geva, R.; Pusztai, L.; Pathiraja, K.; Aktan, G.; Cheng, J.D.; et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J. Clin. Oncol. 2016, 34, 2460–2467. [Google Scholar] [CrossRef]
- Adams, S.; Loi, S.; Toppmeyer, D.; Cescon, D.W.; De Laurentiis, M.; Nanda, R.; Winer, E.P.; Mukai, H.; Tamura, K.; Armstrong, A.; et al. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: Cohort B of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 405–411. [Google Scholar] [CrossRef]
- Adams, S.; Schmid, P.; Rugo, H.S.; Winer, E.P.; Loirat, D.; Awada, A.; Cescon, D.W.; Iwata, H.; Campone, M.; Nanda, R.; et al. Pembrolizumab monotherapy for previously treated metastatic triple-negative breast cancer: Cohort A of the phase II KEYNOTE-086 study. Ann. Oncol. 2019, 30, 397–404. [Google Scholar] [CrossRef]
- Winer, E.P.; Lipatov, O.; Im, S.A.; Goncalves, A.; Munoz-Couselo, E.; Lee, K.S.; Schmid, P.; Tamura, K.; Testa, L.; Witzel, I.; et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): A randomised, open-label, phase 3 trial. Lancet Oncol. 2021, 22, 499–511. [Google Scholar] [CrossRef]
- Dirix, L.Y.; Takacs, I.; Jerusalem, G.; Nikolinakos, P.; Arkenau, H.T.; Forero-Torres, A.; Boccia, R.; Lippman, M.E.; Somer, R.; Smakal, M.; et al. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase 1b JAVELIN Solid Tumor study. Breast. Cancer Res. Tr. 2018, 167, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Emens, L.A.; Cruz, C.; Eder, J.P.; Braiteh, F.; Chung, C.; Tolaney, S.M.; Kuter, I.; Nanda, R.; Cassier, P.A.; Delord, J.P.; et al. Long-term Clinical Outcomes and Biomarker Analyses of Atezolizumab Therapy for Patients With Metastatic Triple-Negative Breast Cancer A Phase 1 Study. Jama. Oncol. 2019, 5, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Bachelot, T.; Filleron, T.; Bieche, I.; Arnedos, M.; Campone, M.; Dalenc, F.; Coussy, F.; Sablin, M.P.; Debled, M.; Lefeuvre-Plesse, C.; et al. Durvalumab compared to maintenance chemotherapy in metastatic breast cancer: The randomized phase II SAFIR02-BREAST IMMUNO trial. Nat. Med. 2021, 27, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Pusztai, L.; Barlow, W.E.; Ganz, P.A.; Henry, N.L.; White, J.; Jagsi, R.; Mammen, J.M.V.; Lew, D.; Mejia, J.; Karantza, V.; et al. SWOG S1418/NRG-BR006: A randomized, phase III trial to evaluate the efficacy and safety of MK-3475 as adjuvant therapy for triple receptor-negative breast cancer with ≥1 cm residual invasive cancer or positive lymph nodes (> pN1mic) after neoadjuvant chemotherapy. Cancer Res. 2018, 78 (Suppl. 4), OT1-02-04. [Google Scholar] [CrossRef]
- Conte, P.F.; Dieci, M.V.; Bisagni, G.; De Laurentiis, M.; Tondini, C.A.; Schmid, P.; De Salvo, G.L.; Moratello, G.; Guarneri, V. Phase III randomized study of adjuvant treatment with the ANTI-PD-L1 antibody avelumab for high-risk triple negative breast cancer patients: The A-BRAVE trial. J. Clin. Oncol. 2020, 38, TPS598. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buque, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunological Effects of Conventional Chemotherapy and Targeted Anticancer Agents. Cancer Cell 2015, 28, 690–714. [Google Scholar] [CrossRef]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Wu, J.; Waxman, D.J. Immunogenic chemotherapy: Dose and schedule dependence and combination with immunotherapy. Cancer Lett. 2018, 419, 210–221. [Google Scholar] [CrossRef]
- Ahlmann, M.; Hempel, G. The effect of cyclophosphamide on the immune system: Implications for clinical cancer therapy. Cancer Chemoth. Pharm. 2016, 78, 661–671. [Google Scholar] [CrossRef]
- Cortes, J.; Cescon, D.W.; Rugo, H.S.; Nowecki, Z.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Holgado, E.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple-negative breast cancer (KEYNOTE-355): A randomised, placebo-controlled, double-blind, phase 3 clinical trial. Lancet 2020, 396, 1817–1828. [Google Scholar] [CrossRef]
- Cortes, J.; Rugo, H.S.; Cescon, D.W.; Im, S.A.; Yusof, M.M.; Gallardo, C.; Lipatov, O.; Barrios, C.H.; Perez-Garcia, J.; Iwata, H.; et al. Pembrolizumab plus Chemotherapy in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 387, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Tolaney, S.M.; Kalinsky, K.; Kaklamani, V.G.; D’Adamo, D.R.; Aktan, G.; Tsai, M.L.; O’Regan, R.M.; Kaufman, P.A.; Wilks, S.T.; Andreopoulou, E.; et al. Eribulin Plus Pembrolizumab in Patients with Metastatic Triple-Negative Breast Cancer (ENHANCE 1): A Phase Ib/II Study. Clin. Cancer Res. 2021, 27, 3061–3068. [Google Scholar] [CrossRef]
- Adams, S.; Diamond, J.R.; Hamilton, E.; Pohlmann, P.R.; Tolaney, S.M.; Chang, C.W.; Zhang, W.; Iizuka, K.; Foster, P.G.; Molinero, L.; et al. Atezolizumab Plus nab-Paclitaxel in the Treatment of Metastatic Triple-Negative Breast Cancer With 2-Year Survival Follow-up: A Phase 1b Clinical Trial. Jama. Oncol. 2019, 5, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): Updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2020, 21, 44–59. [Google Scholar] [CrossRef]
- Emens, L.A.; Adams, S.; Barrios, C.H.; Dieras, V.; Iwata, H.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Winer, E.P.; Patel, S.; et al. First-line atezolizumab plus nab-paclitaxel for unresectable, locally advanced, or metastatic triple-negative breast cancer: IMpassion130 final overall survival analysis. Ann. Oncol. 2021, 32, 983–993. [Google Scholar] [CrossRef]
- Miles, D.; Gligorov, J.; Andre, F.; Cameron, D.; Schneeweiss, A.; Barrios, C.; Xu, B.; Wardley, A.; Kaen, D.; Andrade, L.; et al. Primary results from IMpassion131, a double-blind, placebo-controlled, randomised phase III trial of first-line paclitaxel with or without atezolizumab for unresectable locally advanced/metastatic triple-negative breast cancer. Ann. Oncol. 2021, 32, 994–1004. [Google Scholar] [CrossRef]
- Franzoi, M.A.; de Azambuja, E. Atezolizumab in metastatic triple-negative breast cancer: IMpassion130 and 131 trials-how to explain different results? ESMO Open 2020, 5, e001112. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Chen, H.Y.; Mo, H.N.; Hu, X.D.; Gao, R.R.; Zhao, Y.H.; Liu, B.L.; Niu, L.J.; Sun, X.Y.; Yu, X.; et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 2021, 39, 1578–1593.e8. [Google Scholar] [CrossRef]
- Voorwerk, L.; Slagter, M.; Horlings, H.M.; Sikorska, K.; van de Vijver, K.K.; de Maaker, M.; Nederlof, I.; Kluin, R.J.C.; Warren, S.; Ong, S.; et al. Immune induction strategies in metastatic triple-negative breast cancer to enhance the sensitivity to PD-1 blockade: The TONIC trial. Nat. Med. 2019, 25, 920–928. [Google Scholar] [CrossRef]
- Hutchinson, K.E.; Yost, S.E.; Chang, C.W.; Johnson, R.M.; Carr, A.R.; McAdam, P.R.; Halligan, D.L.; Chang, C.C.; Schmolze, D.; Liang, J.; et al. Comprehensive Profiling of Poor-Risk Paired Primary and Recurrent Triple-Negative Breast Cancers Reveals Immune Phenotype Shifts. Clin. Cancer Res. 2020, 26, 657–668. [Google Scholar] [CrossRef]
- Szekely, B.; Bossuyt, V.; Li, X.; Wali, V.B.; Patwardhan, G.A.; Frederick, C.; Silber, A.; Park, T.; Harigopal, M.; Pelekanou, V.; et al. Immunological differences between primary and metastatic breast cancer. Ann. Oncol. 2018, 29, 2232–2239. [Google Scholar] [CrossRef] [PubMed]
- Nanda, R.; Liu, M.C.; Yau, C.; Shatsky, R.; Pusztai, L.; Wallace, A.; Chien, A.J.; Forero-Torres, A.; Ellis, E.; Han, H.; et al. Effect of Pembrolizumab Plus Neoadjuvant Chemotherapy on Pathologic Complete Response in Women With Early-Stage Breast Cancer: An Analysis of the Ongoing Phase 2 Adaptively Randomized I-SPY2 Trial. JAMA Oncol. 2020, 6, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Salgado, R.; Park, Y.H.; Munoz-Couselo, E.; Kim, S.B.; Sohn, J.; Im, S.A.; Foukakis, T.; Kuemmel, S.; Dent, R.; et al. Pembrolizumab plus chemotherapy as neoadjuvant treatment of high-risk, early-stage triple-negative breast cancer: Results from the phase 1b open-label, multicohort KEYNOTE-173 study. Ann. Oncol. 2020, 31, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Schmid, P.; Cortes, J.; Dent, R.; Pusztai, L.; McArthur, H.; Kummel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; et al. Event-free Survival with Pembrolizumab in Early Triple-Negative Breast Cancer. N. Engl. J. Med. 2022, 386, 556–567. [Google Scholar] [CrossRef]
- Loibl, S.; Untch, M.; Burchardi, N.; Huober, J.; Sinn, B.V.; Blohmer, J.U.; Grischke, E.M.; Furlanetto, J.; Tesch, H.; Hanusch, C.; et al. A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple-negative breast cancer: Clinical results and biomarker analysis of GeparNuevo study. Ann. Oncol. 2019, 30, 1279–1288. [Google Scholar] [CrossRef]
- Loibl, S.; Schneeweiss, A.; Huober, J.; Braun, M.; Rey, J.; Blohmer, J.U.; Furlanetto, J.; Zahm, D.M.; Hanusch, C.; Thomalla, J.; et al. Neoadjuvant durvalumab improves survival in early triple-negative breast cancer independent of pathological complete response. Ann. Oncol. 2022, 33, 1149–1158. [Google Scholar] [CrossRef]
- Gianni, L.; Huang, C.S.; Egle, D.; Bermejo, B.; Zamagni, C.; Thill, M.; Anton, A.; Zambelli, S.; Bianchini, G.; Russo, S.; et al. Pathologic complete response (pCR) to neoadjuvant treatment with or without atezolizumab in triple-negative, early high-risk and locally advanced breast cancer: NeoTRIP Michelangelo randomized study. Ann. Oncol. 2022, 33, 534–543. [Google Scholar] [CrossRef]
- Mittendorf, E.A.; Zhang, H.; Barrios, C.H.; Saji, S.; Jung, K.H.; Hegg, R.; Koehler, A.; Sohn, J.; Iwata, H.; Telli, M.L.; et al. Neoadjuvant atezolizumab in combination with sequential nab-paclitaxel and anthracycline-based chemotherapy versus placebo and chemotherapy in patients with early-stage triple-negative breast cancer (IMpassion031): A randomised, double-blind, phase 3 trial. Lancet 2020, 396, 1090–1100. [Google Scholar] [CrossRef]
- McLaughlin, M.; Patin, E.C.; Pedersen, M.; Wilkins, A.; Dillon, M.T.; Melcher, A.A.; Harrington, K.J. Inflammatory microenvironment remodelling by tumour cells after radiotherapy. Nat. Rev. Cancer 2020, 20, 203–217. [Google Scholar] [CrossRef]
- Ho, A.Y.; Barker, C.A.; Arnold, B.B.; Powell, S.N.; Hu, Z.I.; Gucalp, A.; Lebron-Zapata, L.; Wen, H.Y.; Kallman, C.; D’Agnolo, A.; et al. A phase 2 clinical trialassessing theefficacy and safety of pembrolizumab and radiotherapy in patients with metastatic triple-negative breast cancer. Cancer 2020, 126, 850–860. [Google Scholar] [CrossRef] [PubMed]
- David, S.; Savas, P.; Siva, S.; White, M.; Neeson, M.W.; White, S.; Marx, G.; Cheuk, R.; Grogan, M.; Farrell, M.; et al. A randomised phase II trial of single fraction or multi-fraction SABR (stereotactic ablative body radiotherapy) with atezolizumab in patients with advanced triple negative breast cancer (AZTEC trial). Cancer Res. 2022, 82, PD10–02. [Google Scholar] [CrossRef]
- Pantelidou, C.; Sonzogni, O.; De Oliveria Taveira, M.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. PARP Inhibitor Efficacy Depends on CD8(+) T-cell Recruitment via Intratumoral STING Pathway Activation in BRCA-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA Damage Response Promotes Antitumor Immunity through STING-Mediated T-cell Activation in Small Cell Lung Cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Reislander, T.; Lombardi, E.P.; Groelly, F.J.; Miar, A.; Porru, M.; Di Vito, S.; Wright, B.; Lockstone, H.; Biroccio, A.; Harris, A.; et al. BRCA2 abrogation triggers innate immune responses potentiated by treatment with PARP inhibitors. Nat. Commun. 2019, 10, 3143. [Google Scholar] [CrossRef]
- Vinayak, S.; Tolaney, S.M.; Schwartzberg, L.; Mita, M.; McCann, G.; Tan, A.R.; Wahner-Hendrickson, A.E.; Forero, A.; Anders, C.; Wulf, G.M.; et al. Open-label Clinical Trial of Niraparib Combined With Pembrolizumab for Treatment of Advanced or Metastatic Triple-Negative Breast Cancer. JAMA Oncol. 2019, 5, 1132–1140. [Google Scholar] [CrossRef]
- Senkus-Konefka, E.; Domchek, S.M.; Im, S.A.; Xu, B.; Armstrong, A.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Conte, P.; et al. Subgroup analysis of olaparib monotherapy versus chemotherapy by hormone receptor and BRCA mutation status in patients with HER2-negative metastatic breast cancer and a germline BRCA mutation: OlympiAD. Eur. J. Cancer 2018, 92, S19–S20. [Google Scholar] [CrossRef]
- Eiermann, W.; Rugo, H.S.; Diab, S.; Ettl, J.; Hurvitz, S.A.; Goncalves, A. Analysis of germline BRCA1/2 mutated (gBRCA(mut)) hormone receptor-positive (HR plus ) and triple negative breast cancer (TNBC) treated with talazoparib (TALA). J. Clin. Oncol. 2018, 36, 1070. [Google Scholar] [CrossRef]
- Pusztai, L.; Yau, C.; Wolf, D.M.; Han, H.S.; Du, L.; Wallace, A.M.; String-Reasor, E.; Boughey, J.C.; Chien, A.J.; Elias, A.D.; et al. Durvalumab with olaparib and paclitaxel for high-risk HER2-negative stage II/III breast cancer: Results from the adaptively randomized I-SPY2 trial. Cancer Cell. 2021, 39, 989–998 e985. [Google Scholar] [CrossRef]
- Yu, J.F.; Song, Y.P.; Tian, W.Z. How to select IgG subclasses in developing anti-tumor therapeutic antibodies. J. Hematol. Oncol. 2020, 13, 45. [Google Scholar] [CrossRef]
- Li, F.; Ulrich, M.; Jonas, M.; Stone, I.J.; Linares, G.; Zhang, X.Q.; Westendorf, L.; Benjamin, D.R.; Law, C.L. Tumor-Associated Macrophages Can Contribute to Antitumor Activity through Fc gamma R-Mediated Processing of Antibody-Drug Conjugates. Mol. Cancer Ther. 2017, 16, 1347–1354. [Google Scholar] [CrossRef]
- Cao, A.T.; Higgins, S.; Stevens, N.; Gardai, S.J.; Sussman, D. Additional mechanisms of action of ladiratuzumab vedotin contribute to increased immune cell activation within the tumor. Cancer Res. 2018, 78, 2742. [Google Scholar] [CrossRef]
- Bauzon, M.; Drake, P.M.; Barfield, R.M.; Cornali, B.M.; Rupniewski, I.; Rabuka, D. Maytansine-bearing antibody-drug conjugates induce in vitro hallmarks of immunogenic cell death selectively in antigen-positive target cells. Oncoimmunology 2019, 8, e1565859. [Google Scholar] [CrossRef]
- Muller, P.; Martin, K.; Theurich, S.; Schreiner, J.; Savic, S.; Terszowski, G.; Lardinois, D.; Heinzelmann-Schwarz, V.A.; Schlaak, M.; Kvasnicka, H.M.; et al. Microtubule-Depolymerizing Agents Used in Antibody-Drug Conjugates Induce Antitumor Immunity by Stimulation of Dendritic Cells. Cancer Immunol. Res. 2014, 2, 741–755. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.A.; Mbofung, R.M.; Malu, S.; Zhang, M.; Ashkin, E.; Devi, S.; Williams, L.; Tieu, T.; Peng, W.Y.; Pradeep, S.; et al. The Effect of Topoisomerase I Inhibitors on the Efficacy of T-Cell-Based Cancer Immunotherapy. Jnci.-J. Natl. Cancer I 2018, 110, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Diab, S.; Alemany, C.; Basho, R.; Brown-Glaberman, U.; Meisel, J.; Pluard, T.; Cortes, J.; Dillon, P.; Ettl, J.; et al. Open label phase 1b/2 study of ladiratuzumab vedotin in combination with pembrolizumab for first-line treatment of patients with unresectable locally-advanced or metastatic triple-negative breast cancer. Cancer Res. 2020, 80, PD1-06. [Google Scholar] [CrossRef]
- Bergholz, J.S.; Zhao, J.J. How Compensatory Mechanisms and Adaptive Rewiring Have Shaped Our Understanding of Therapeutic Resistance in Cancer. Cancer Res. 2021, 81, 6074–6077. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Richmond, A.; Yan, C. Immunomodulatory Properties of PI3K/AKT/mTOR and MAPK/MEK/ERK Inhibition Augment Response to Immune Checkpoint Blockade in Melanoma and Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2022, 23, 7353. [Google Scholar] [CrossRef]
- Zhang, Z.; Richmond, A. The Role of PI3K Inhibition in the Treatment of Breast Cancer, Alone or Combined With Immune Checkpoint Inhibitors. Front Mol. Biosci. 2021, 8, 648663. [Google Scholar] [CrossRef]
- Ho, P.C.; Meeth, K.M.; Tsui, Y.C.; Srivastava, B.; Bosenberg, M.W.; Kaech, S.M. Immune-based antitumor effects of BRAF inhibitors rely on signaling by CD40L and IFNgamma. Cancer Res. 2014, 74, 3205–3217. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Savas, P.; Espinosa, E.; Boni, V.; Italiano, A.; White, S.; Cheng, K.; Lam, L.; Robert, L.; Laliman, V.; et al. Phase 1b study evaluating a triplet combination of ipatasertib (IPAT), atezolizumab, and a taxane as first-line therapy for locally advanced/metastatic triple-negative breast cancer (TNBC). Cancer Res. 2021, 81, PS12-28. [Google Scholar] [CrossRef]
- Brufsky, A.; Kim, S.B.; Zvirbule, Z.; Eniu, A.; Mebis, J.; Sohn, J.H.; Wongchenko, M.; Chohan, S.; Amin, R.; Yan, Y.; et al. A phase II randomized trial of cobimetinib plus chemotherapy, with or without atezolizumab, as first-line treatment for patients with locally advanced or metastatic triple-negative breast cancer (COLET): Primary analysis. Ann. Oncol. 2021, 32, 652–660. [Google Scholar] [CrossRef]
- Lanitis, E.; Irving, M.; Coukos, G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015, 33, 55–63. [Google Scholar] [CrossRef]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Y.; Jia, W.; Deng, H.; Li, G.; Deng, W.; Chen, J.; Kim, B.Y.S.; Jiang, W.; Liu, Q.; et al. Low-Dose Anti-Angiogenic Therapy Sensitizes Breast Cancer to PD-1 Blockade. Clin. Cancer Res. 2020, 26, 1712–1724. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Q.; Liu, Q.; Li, Y.; Li, Q.; Su, F.X.; Yao, H.R.; Su, S.C.; Wang, Q.R.; Jin, L.; Wang, Y.; et al. Efficacy and safety of camrelizumab combined with apatinib in advanced triple-negative breast cancer: An open-label phase II trial. J. Immunother. Cancer 2020, 8, e000696. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, M.; Jiang, Z.; Wang, X. A Comprehensive Immunologic Portrait of Triple-Negative Breast Cancer. Transl. Oncol. 2018, 11, 311–329. [Google Scholar] [CrossRef] [PubMed]
- Saleh, R.; Toor, S.M.; Khalaf, S.; Elkord, E. Breast Cancer Cells and PD-1/PD-L1 Blockade Upregulate the Expression of PD-1, CTLA-4, TIM-3 and LAG-3 Immune Checkpoints in CD4(+) T Cells. Vaccines 2019, 7, 149. [Google Scholar] [CrossRef] [PubMed]
- Rowshanravan, B.; Halliday, N.; Sansom, D.M. CTLA-4: A moving target in immunotherapy. Blood 2018, 131, 58–67. [Google Scholar] [CrossRef]
- Tang, F.; Du, X.; Liu, M.; Zheng, P.; Liu, Y. Anti-CTLA-4 antibodies in cancer immunotherapy: Selective depletion of intratumoral regulatory T cells or checkpoint blockade? Cell Biosci. 2018, 8, 30. [Google Scholar] [CrossRef]
- Santa-Maria, C.A.; Kato, T.; Park, J.H.; Flaum, L.E.; Jain, S.; Tellez, C.; Stein, R.M.; Shah, A.N.; Gross, L.; Uthe, R.; et al. Durvalumab and tremelimumab in metastatic breast cancer (MBC): Immunotherapy and immunopharmacogenomic dynamics. J. Clin. Oncol. 2017, 35, 3052. [Google Scholar] [CrossRef]
- Xu, B.H.; Li, Q.; Zhang, Q.Y.; Zhang, Y.; Ouyang, Q.C.; Zhang, Y.; Liu, Q.; Sun, T.; Xu, J.; Yang, J.; et al. Preliminary safety tolerability & efficacy results of KN046 (an anti-PD-L1/CTLA-4 bispecific antibody) in combination with Nab-paclitaxel in patients with metastatic triple-negative breast cancer (mTNBC). Cancer Res. 2021, 81, 1660. [Google Scholar]
- Ghalamfarsa, G.; Kazemi, M.H.; Mohseni, S.R.; Masjedi, A.; Hojjat-Farsangi, M.; Azizi, G.; Yousefi, M.; Jadidi-Niaragh, F. CD73 as a potential opportunity for cancer immunotherapy. Expert. Opin. Ther. Tar. 2019, 23, 127–142. [Google Scholar] [CrossRef] [PubMed]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef]
- Buisseret, L.; Loirat, D.; Aftimos, P.G.; Punie, K.; Maurer, C.; Debien, V.; Goncalves, A.; Ghiringhelli, F.; Taylor, D.; Clatot, F.; et al. Primary endpoint results of SYNERGY, a randomized phase II trial, first-line chemo-immunotherapy trial of durvalumab, paclitaxel, and carboplatin with or without the anti-CD73 antibody oleclumab in patients with advanced or metastatic triple-negative breast cancer (TNBC). Ann. Oncol. 2022, 33 (Suppl.7), S808–S869. [Google Scholar] [CrossRef]
- Shemesh, C.S.; Hsu, J.C.; Hosseini, I.; Shen, B.Q.; Rotte, A.; Twomey, P.; Girish, S.; Wu, B. Personalized Cancer Vaccines: Clinical Landscape, Challenges, and Opportunities. Mol. Ther. 2021, 29, 555–570. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer Neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef]
- Fritah, H.; Rovelli, R.; Chiang, C.L.; Kandalaft, L.E. The current clinical landscape of personalized cancer vaccines. Cancer Treat. Rev. 2022, 106, 102383. [Google Scholar] [CrossRef] [PubMed]
- Verma, V.; Shrimali, R.K.; Ahmad, S.; Dai, W.; Wang, H.; Lu, S.; Nandre, R.; Gaur, P.; Lopez, J.; Sade-Feldman, M.; et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat. Immunol. 2019, 20, 1231–1243. [Google Scholar] [CrossRef]
- Hemminki, O.; dos Santos, J.M.; Hemminki, A. Oncolytic viruses for cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 84. [Google Scholar] [CrossRef] [PubMed]
- Ylosmaki, E.; Cerullo, V. Design and application of oncolytic viruses for cancer immunotherapy. Curr. Opin. Biotech. 2020, 65, 25–36. [Google Scholar] [CrossRef]
- Jin, S.; Wang, Q.; Wu, H.; Pang, D.; Xu, S. Oncolytic viruses for triple negative breast cancer and beyond. Biomark Res. 2021, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Hecht, J.R.; Chan, A.; Baurain, J.F.; Martin, M.; Longo-Munoz, F.; Kalinsky, K.; Raman, S.; Liu, C.X.; Cha, E.; Chan, E. Preliminary safety data of intrahepatic talimogene laherparepvec and intravenous atezolizumab in patients with triple negative breast cancer. Cancer Res. 2020, 80, P3-09. [Google Scholar] [CrossRef]
- Kistler, M.; Nangia, C.; To, C.; Sender, L.; Lee, J.; Jones, F.; Jafari, O.; Seery, T.; Rabizadeh, S.; Niazi, K.; et al. Safety and efficacy from first-in-human immunotherapy combining NK and T cell activation with off-the-shelf high-affinity CD16 NK cell line (haNK) in patients with 2nd-line or greater metastatic triple-negative breast cancer (TNBC). Cancer Res. 2020, 80, P5-04-02. [Google Scholar] [CrossRef]
- Han, D.; Liu, J.; Chen, C.; Dong, L.; Liu, Y.; Chang, R.; Huang, X.; Liu, Y.; Wang, J.; Dougherty, U.; et al. Anti-tumour immunity controlled through mRNA m(6)A methylation and YTHDF1 in dendritic cells. Nature 2019, 566, 270–274. [Google Scholar] [CrossRef]
- Sun, X.; Wu, B.; Chiang, H.C.; Deng, H.; Zhang, X.; Xiong, W.; Liu, J.; Rozeboom, A.M.; Harris, B.T.; Blommaert, E.; et al. Tumour DDR1 promotes collagen fibre alignment to instigate immune exclusion. Nature 2021, 599, 673–678. [Google Scholar] [CrossRef]
- Mohammadpour, H.; MacDonald, C.R.; Qiao, G.; Chen, M.; Dong, B.; Hylander, B.L.; McCarthy, P.L.; Abrams, S.I.; Repasky, E.A. β2 adrenergic receptor-mediated signaling regulates the immunosuppressive potential of myeloid-derived suppressor cells. J. Clin. Investig. 2019, 129, 5537–5552. [Google Scholar] [CrossRef]
- Bucsek, M.J.; Qiao, G.; MacDonald, C.R.; Giridharan, T.; Evans, L.; Niedzwecki, B.; Liu, H.; Kokolus, K.M.; Eng, J.W.; Messmer, M.N.; et al. β-Adrenergic Signaling in Mice Housed at Standard Temperatures Suppresses an Effector Phenotype in CD8(+) T Cells and Undermines Checkpoint Inhibitor Therapy. Cancer Res. 2017, 77, 5639–5651. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Rong, X.; Zhao, G.; Zhou, Y.; Xiao, Y.; Ma, D.; Jin, X.; Wu, Y.; Yan, Y.; Yang, H.; et al. The microbial metabolite trimethylamine N-oxide promotes antitumor immunity in triple-negative breast cancer. Cell Metab. 2022, 34, 581–594 e588. [Google Scholar] [CrossRef]
- Emens, L.A.; Molinero, L.; Loi, S.; Rugo, H.S.; Schneeweiss, A.; Dieras, V.; Iwata, H.; Barrios, C.H.; Nechaeva, M.; Nguyen-Duc, A.; et al. Atezolizumab and nab-Paclitaxel in Advanced Triple-Negative Breast Cancer: Biomarker Evaluation of the IMpassion130 Study. J. Natl. Cancer Inst. 2021, 113, 1005–1016. [Google Scholar] [CrossRef]
- Badve, S.S.; Penault-Llorca, F.; Reis-Filho, J.S.; Deurloo, R.; Siziopikou, K.P.; D’Arrigo, C.; Viale, G. Determining PD-L1 Status in Patients With Triple-Negative Breast Cancer: Lessons Learned From IMpassion130. J. Natl. Cancer Inst. 2022, 114, 664–675. [Google Scholar] [CrossRef]
- Chebib, I.; Mino-Kenudson, M. PD-L1 immunohistochemistry: Clones, cutoffs, and controversies. APMIS 2022, 130, 295–313. [Google Scholar] [CrossRef]
- Rugo, H.S.; Loi, S.; Adams, S.; Schmid, P.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Winer, E.P.; Kockx, M.M.; et al. PD-L1 Immunohistochemistry Assay Comparison in Atezolizumab plus nab-Paclitaxel-Treated Advanced Triple-Negative Breast Cancer. J. Natl. Cancer Inst. 2021, 113, 1733–1743. [Google Scholar] [CrossRef] [PubMed]
- Ghebeh, H.; Mansour, F.A.; Colak, D.; Alfuraydi, A.A.; Al-Thubiti, A.A.; Monies, D.; Al-Alwan, M.; Al-Tweigeri, T.; Tulbah, A. Higher PD-L1 Immunohistochemical Detection Signal in Frozen Compared to Matched Paraffin-Embedded Formalin-Fixed Tissues. Antibodies 2021, 10, 24. [Google Scholar] [CrossRef] [PubMed]
- Reisenbichler, E.S.; Han, G.; Bellizzi, A.; Bossuyt, V.; Brock, J.; Cole, K.; Fadare, O.; Hameed, O.; Hanley, K.; Harrison, B.T.; et al. Prospective multi-institutional evaluation of pathologist assessment of PD-L1 assays for patient selection in triple negative breast cancer. Mod. Pathol. 2020, 33, 1746–1752. [Google Scholar] [CrossRef] [PubMed]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef]
- Loi, S.; Adams, S.; Schmid, P.; Cortes, J.; Cescon, D.W.; Winer, E.P.; Toppmeyer, D.L.; Rugo, H.S.; De Laurentiis, M.; Nanda, R.; et al. Relationship between tumor infiltrating lymphocyte (TIL) levels and response to pembrolizumab (pembro) in metastatic triple-negative breast cancer (mTNBC): Results from KEYNOTE-086. Ann. Oncol. 2017, 28, v608. [Google Scholar] [CrossRef]
- Loi, S.; Winer, E.; Lipatov, O.; Im, S.A.; Goncalves, A.; Cortes, J.; Lee, K.S.; Schmid, P.; Testa, L.; Witzel, I.; et al. Relationship between tumor-infiltrating lymphocytes (TILs) and outcomes in the KEYNOTE-119 study of pembrolizumab vs chemotherapy for previously treated metastatic triple-negative breast cancer (mTNBC). Cancer Res. 2020, 80, PD5-03. [Google Scholar] [CrossRef]
- Loi, S.; Michiels, S.; Adams, S.; Loibl, S.; Budczies, J.; Denkert, C.; Salgado, R. The journey of tumor-infiltrating lymphocytes as a biomarker in breast cancer: Clinical utility in an era of checkpoint inhibition. Ann. Oncol. 2021, 32, 1236–1244. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Ott, P.A.; Bang, Y.J.; Piha-Paul, S.A.; Razak, A.R.A.; Bennouna, J.; Soria, J.C.; Rugo, H.S.; Cohen, R.B.; O’Neil, B.H.; Mehnert, J.M.; et al. T-Cell-Inflamed Gene-Expression Profile, Programmed Death Ligand 1 Expression, and Tumor Mutational Burden Predict Efficacy in Patients Treated With Pembrolizumab Across 20 Cancers: KEYNOTE-028. J. Clin. Oncol. 2019, 37, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Barroso-Sousa, R.; Jain, E.; Cohen, O.; Kim, D.; Buendia-Buendia, J.; Winer, E.; Lin, N.; Tolaney, S.M.; Wagle, N. Prevalence and mutational determinants of high tumor mutation burden in breast cancer. Ann. Oncol. 2020, 31, 387–394. [Google Scholar] [CrossRef]
- Karn, T.; Denkert, C.; Weber, K.E.; Holtrich, U.; Hanusch, C.; Sinn, B.V.; Higgs, B.W.; Jank, P.; Sinn, H.P.; Huober, J.; et al. Tumor mutational burden and immune infiltration as independent predictors of response to neoadjuvant immune checkpoint inhibition in early TNBC in GeparNuevo. Ann. Oncol. 2020, 31, 1216–1222. [Google Scholar] [CrossRef] [PubMed]
- Winer, E.P.; Lipatov, O.; Im, S.A.; Goncalves, A.; Munoz-Couselo, E.; Lee, K.S.; Schmid, P.; Testa, L.; Witzel, I.; Ohtani, S.; et al. Association of tumor mutational burden (TMB) and clinical outcomes with pembrolizumab (pembro) versus chemotherapy (chemo) in patients with metastatic triple-negative breast cancer (mTNBC) from KEYNOTE-119. J. Clin. Oncol. 2020, 38, 1013. [Google Scholar] [CrossRef]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell 2021, 39, 154–173. [Google Scholar] [CrossRef]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Prasad, V.; Kaestner, V.; Mailankody, S. Cancer Drugs Approved Based on Biomarkers and Not Tumor Type-FDA Approval of Pembrolizumab for Mismatch Repair-Deficient Solid Cancers. JAMA Oncol. 2018, 4, 157–158. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef]
- Ren, X.Y.; Song, Y.; Wang, J.; Chen, L.Y.; Pang, J.Y.; Zhou, L.R.; Shen, S.J.; Cao, X.; Wang, Y.X.; Shao, M.M.; et al. Mismatch Repair Deficiency and Microsatellite Instability in Triple-Negative Breast Cancer: A Retrospective Study of 440 Patients. Front Oncol. 2021, 11, 570623. [Google Scholar] [CrossRef]
- Horimoto, Y.; Hlaing, M.T.; Saeki, H.; Kitano, S.; Nakai, K.; Sasaki, R.; Kurisaki-Arakawa, A.; Arakawa, A.; Otsuji, N.; Matsuoka, S.; et al. Microsatellite instability and mismatch repair protein expressions in lymphocyte-predominant breast cancer. Cancer Sci. 2020, 111, 2647–2654. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Hamid, O.; Ribas, A.; Robert, C.; Kefford, R.; Hwu, W.J.; Weber, J.S.; Joshua, A.M.; Gangadhar, T.C.; Dronca, R.S.; et al. Atypical patterns of response in patients (pts) with metastatic melanoma treated with pembrolizumab (MK-3475) in KEYNOTE-001. J. Clin. Oncol. 2015, 33, 3000. [Google Scholar] [CrossRef]
- Champiat, S.; Dercle, L.; Ammari, S.; Massard, C.; Hollebecque, A.; Postel-Vinay, S.; Chaput, N.; Eggermont, A.; Marabelle, A.; Soria, J.C.; et al. Hyperprogressive Disease Is a New Pattern of Progression in Cancer Patients Treated by Anti-PD-1/PD-L1. Clinical. Cancer Res. 2017, 23, 1920–1928. [Google Scholar] [CrossRef]
- Tazdait, M.; Mezquita, L.; Lahmar, J.; Ferrara, R.; Bidault, F.; Ammari, S.; Balleyguier, C.; Planchard, D.; Gazzah, A.; Soria, J.C.; et al. Patterns of responses in metastatic NSCLC during PD-1 or PDL-1 inhibitor therapy: Comparison of RECIST 1.1, irRECIST and iRECIST criteria. Eur. J. Cancer 2018, 88, 38–47. [Google Scholar] [CrossRef]
- Chiou, V.L.; Burotto, M. Pseudoprogression and Immune-Related Response in Solid Tumors. J. Clin. Oncol. 2015, 33, 3541–3543. [Google Scholar] [CrossRef]
- Schmid, P.; Cruz, C.; Braiteh, F.S.; Eder, J.P.; Tolaney, S.; Kuter, I.; Nanda, R.; Chung, C.; Cassier, P.; Delord, J.P.; et al. Atezolizumab in metastatic TNBC (mTNBC): Long-term clinical outcomes and biomarker analyses. Cancer Res. 2017, 77, 2986. [Google Scholar] [CrossRef]
- Borcoman, E.; Kanjanapan, Y.; Champiat, S.; Kato, S.; Servois, V.; Kurzrock, R.; Goel, S.; Bedard, P.; Le Tourneau, C. Novel patterns of response under immunotherapy. Ann. Oncol. 2019, 30, 385–396. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Hoos, A.; O’Day, S.; Weber, J.S.; Hamid, O.; Lebbe, C.; Maio, M.; Binder, M.; Bohnsack, O.; Nichol, G.; et al. Guidelines for the evaluation of immune therapy activity in solid tumors: Immune-related response criteria. Clin. Cancer Res. 2009, 15, 7412–7420. [Google Scholar] [CrossRef] [PubMed]
- Seymour, L.; Bogaerts, J.; Perrone, A.; Ford, R.; Schwartz, L.H.; Mandrekar, S.; Lin, N.U.; Litiere, S.; Dancey, J.; Chen, A.; et al. iRECIST: Guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 2017, 18, E143–E152. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Giobbie-Hurder, A.; Gargano, M.; Suda, M.; Ramaiya, N.H.; Hodi, F.S. Developing a Common Language for Tumor Response to Immunotherapy: Immune-Related Response Criteria Using Unidimensional Measurements. Clin. Cancer Res. 2013, 19, 3936–3943. [Google Scholar] [CrossRef]
- Majd, N.; de Groot, J. Challenges and strategies for successful clinical development of immune checkpoint inhibitors in glioblastoma. Expert Opin Pharm. 2019, 20, 1609–1624. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, Q.; Chen, L.; Qian, K.; Xu, X. Severe immune-related hepatitis and myocarditis caused by PD-1 inhibitors in the treatment of triple-negative breast cancer: A case report. Ann. Transl. Med. 2022, 10, 424. [Google Scholar] [CrossRef]
- Wang, P.F.; Chen, Y.; Song, S.Y.; Wang, T.J.; Ji, W.J.; Li, S.W.; Liu, N.; Yan, C.X. Immune-Related Adverse Events Associated with Anti-PD-1/PD-L1 Treatment for Malignancies: A Meta-Analysis. Front Pharm. 2017, 8, 730. [Google Scholar] [CrossRef]
- Balibegloo, M.; Nejadghaderi, S.A.; Sadeghalvad, M.; Soleymanitabar, A.; Nezamabadi, S.S.; Saghazadeh, A.; Rezaei, N. Adverse events associated with immune checkpoint inhibitors in patients with breast cancer: A systematic review and meta-analysis. Int. Immunopharmacol. 2021, 96, 107796. [Google Scholar] [CrossRef] [PubMed]
- Xin, Y.; Shen, G.; Zheng, Y.; Guan, Y.; Huo, X.; Li, J.; Ren, D.; Zhao, F.; Liu, Z.; Li, Z.; et al. Immune checkpoint inhibitors plus neoadjuvant chemotherapy in early triple-negative breast cancer: A systematic review and meta-analysis. BMC Cancer 2021, 21, 1261. [Google Scholar] [CrossRef] [PubMed]
- Villacampa, G.; Tolosa, P.; Salvador, F.; Sanchez-Bayona, R.; Villanueva, L.; Dienstmann, R.; Ciruelos, E.; Pascual, T. Addition of immune checkpoint inhibitors to chemotherapy versus chemotherapy alone in first-line metastatic triple-negative breast cancer: A systematic review and meta-analysis. Cancer Treat. Rev. 2022, 104, 102352. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef]
- Ceschi, A.; Noseda, R.; Palin, K.; Verhamme, K. Immune Checkpoint Inhibitor-Related Cytokine Release Syndrome: Analysis of WHO Global Pharmacovigilance Database. Front Pharmacol. 2020, 11, 557. [Google Scholar] [CrossRef] [PubMed]
- Ciner, A.T.; Hochster, H.S.; August, D.A.; Carpizo, D.R.; Spencer, K.R. Delayed cytokine release syndrome after neoadjuvant nivolumab: A case report and literature review. Immunotherapy 2021, 13, 1071–1078. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Clinical Trial | Phase | Status | Arms (n) | Population (n) | PD-L1 Status | Major Outcomes |
|---|---|---|---|---|---|---|
| Trials in advanced TNBC | ||||||
| KEYNOTE-012 (NCT01848834) | Ib | Completed | Pemb | Pre-treated: PD-L1 (+) (32) | +(stroma/ ≥1% TC a) | ORR: 18.5% |
| mPFS: 1.9 months | ||||||
| mOS: 11.2 months | ||||||
| KEYNOTE-086 (NCT02447003) | II | Completed | Pemb | Cohort A (170): pre-treated | Overall | ORR: 5.3% |
| mPFS: 2.0 months | ||||||
| mOS: 9.0 months | ||||||
| +(CPS b ≥ 1) | ORR: 5.7% | |||||
| mPFS: 2.0 months | ||||||
| mOS: 8.8 months | ||||||
| − | ORR: 4.7% | |||||
| mPFS: 1.9 months | ||||||
| mOS: 9.7 months | ||||||
| Cohort B (84): pre-untreated, PD-L1 (+) | +(CPS ≥ 1) | ORR: 21.4% | ||||
| mPFS: 2.1 months | ||||||
| mOS: 18.0 months | ||||||
| KEYNOTE-119 (NCT02555657) | III | Completed | Pemb (312) vs. CT c (310) | Pre-treated: 1–2 prior therapy (622) | Overall | ORR: 9.6 vs. 10.6% |
| mPFS: 2.1 vs. 3.3 months | ||||||
| mOS: 9.9 vs. 10.8 months | ||||||
| +(CPS ≥ 1) | ORR: 12.3 vs. 9.4% | |||||
| mPFS: 2.1 vs. 3.1 months | ||||||
| mOS: 10.7 vs. 10.2 months | ||||||
| +(CPS ≥ 10) | ORR: 17.7 vs. 9.2% | |||||
| mPFS: 2.1 vs. 3.4 months | ||||||
| mOS: 12.7 vs. 11.6 months | ||||||
| +(CPS ≥ 20) | ORR: 26.3 vs. 11.5% | |||||
| mPFS: 3.4 vs. 2.4 months | ||||||
| mOS: 14.9 vs. 12.5 months | ||||||
| JAVELIN (NCT01772004) | Ib | Completed | Avel | Received a median of 2 prior therapies (58) | Overall | ORR: 5.2% |
| mPFS: 5.9 months | ||||||
| mOS: 9.2 months | ||||||
| +(≥ 10% IC d) | ORR: 22.2% | |||||
| − | ORR: 2.6% | |||||
| NCT01375842 | Ia | Completed | Atez | mTNBC: 58% ≥ 2 prior therapies (116) | Overall | ORR: 10% |
| mPFS: 1.4 months | ||||||
| mOS: 8.9 months | ||||||
| +(≥ 1% IC) | ORR: 12% | |||||
| mOS: 10.1 months | ||||||
| − | ORR: 0% | |||||
| mOS: 6.0 months | ||||||
| SAFIR02-BREAST IMMUNO (NCT02299999) | II | Completed | Durv (47) vs. CT (35) | Maintenance setting (82) | Overall | mOS: 21.2 vs. 14 months |
| +(≥ 1% IC) | mOS: 27.3 vs. 12.1 months | |||||
| − | mOS: 19.5 vs. 14 months | |||||
| Trials in early-stage TNBC as adjuvant therapy | ||||||
| SWOG 1418 (NCT02954874) | III | Ongoing | Pemb vs. observation | TNBC with ≥ 1 cm RIC or LN (+) after NACT | NA | |
| A-Brave (NCT02926196) | III | Ongoing | Avel vs. observation | High-risk TNBC | NA |
| Clinical Trial | Phase | Status | Arms (n) | Population (n) | PD-L1 Status | Major Outcomes |
|---|---|---|---|---|---|---|
| Trials in advanced TNBC | ||||||
| KEYNOTE-355 (NCT02819518) | III | Ongoing | Pemb + CT a (566) vs. placebo + CT (281) | First-line treatment in mTNBC (847) | ITT population | mPFS: 7.5 vs. 5.6 months |
| mOS: 17.2 vs. 15.5 months | ||||||
| +(CPS b ≥ 1) | mPFS: 7.6 vs. 5.6 months | |||||
| mOS: 17.6 vs. 16.0 months | ||||||
| +(CPS ≥ 10) | mPFS: 9.7 vs. 5.6 months | |||||
| mOS: 23.0 vs. 16.1 months | ||||||
| KEYNOTE-150/ ENHANCE 1 (NCT02513472) | Ib/II | Completed | Pemb + eribulin mesylate | ≤2 prior lines therapies in the metastatic setting (167) | Overall | ORR in total: 23.4% |
| stratum 1: 25.8% | ||||||
| stratum 2: 21.8% | ||||||
| mPFS in total: 4.1 months | ||||||
| stratum 1: 4.2 months | ||||||
| stratum 2: 4.1 months | ||||||
| mOS in total: 16.1 months | ||||||
| stratum 1: 17.4 months | ||||||
| stratum 2: 15.5 months | ||||||
| +(CPS ≥ 1) | ORR in stratum 1: 34.5% | |||||
| ORR in stratum 2: 24.4% | ||||||
| mPFS in stratum 1: 6.1 months | ||||||
| mPFS in stratum 2: 4.1 months | ||||||
| mOS in stratum 1: 21.0 months | ||||||
| mOS in stratum 2: 14.0 months | ||||||
| − | ORR in stratum 1: 16.1% | |||||
| ORR in stratum 2: 18.2% | ||||||
| mPFS in stratum 1: 3.5 months | ||||||
| mPFS in stratum 2: 3.9 months | ||||||
| mOS in stratum 1: 15.2 months | ||||||
| mOS in stratum 2: 15.5 months | ||||||
| TORCHLIGHT (NCT04085276) | III | Recruiting | Tori + nab-P vs. placebo + nab-P | ≤1 line of CT in the metastatic setting | NA | |
| NCT04537286 | II | Recruiting | Cari + nab-P + Cp | First-line treatment in mTNBC | NA | |
| NCT02755272 | II | Recruiting | Pemb + Cb + gemcitabine vs. Cb + gemcitabine | >2 prior lines therapies in the metastatic setting | NA | |
| TONIC (NCT02499367) | II | Ongoing | A/C/Cp/ RT/no induction + Nivo (70) | mTNBC (70) | ORR in total: 20% | |
| Cp induction ORR: 23% | ||||||
| A induction ORR: 35% | ||||||
| mPFS in total: 1.9 months | ||||||
| TONIC-2 (NCT04159818) | II | Recruiting | Cp/ low dose A/no induction + Nivo | Metastatic or incurable locally advanced TNBC | NA | |
| NCT01633970 | Ib | Completed | Atez + nab-P (33) | ≤2 lines prior CT in the metastatic setting (33) | ORR: 39.4% | |
| mPFS: 5.5 months | ||||||
| mOS: 14.7 months | ||||||
| IMpassion130 (NCT02425891) | III | Completed | Atez + nab-P (451) vs. placebo + nab-P (451) | First-line treatment in mTNBC (902) | ITT population | mPFS: 7.2 vs. 5.5 months |
| mOS: 21.0 vs. 18.7 months | ||||||
| +(≥1% IC c) | mPFS: 7.5 vs. 5.0 months | |||||
| mOS: 25.4 vs. 17.9 months | ||||||
| IMpassion131 (NCT03125902) | III | Ongoing | Atez + P (431) vs. placebo + P (220) | First-line treatment in mTNBC (651) | ITT population | mPFS: 5.7 vs. 5.6 months |
| mOS: 19.2 vs. 22.8 months | ||||||
| +(≥1% IC) | mPFS: 6.0 vs. 5.7 months | |||||
| mOS: 22.1 vs. 28.3 months | ||||||
| IMpassion132 (NCT03371017) | III | Recruiting | Atez + CT d vs. placebo + CT | First-line treatment for locally advanced inoperable or mTNBC | NA | |
| ALICE (NCT03164993) | II | Ongoing | Atez + PLD + C vs. placebo + PLD + C | ≤ 1 line previous CT in the metastatic setting | NA | |
| GIM25-CAPT (NCT05266937) | II | Recruiting | Atez + nab-P + Cb | First-line therapy in PD-L1-positive mTNBC | NA | |
| EL1SSAR (NCT04148911) | III | Ongoing | Atez + nab-P | First-line therapy in PD-L1-positive mTNBC | NA | |
| Trials in early-stage TNBC as neoadjuvant therapy | ||||||
| I-SPY2 (NCT01042379) | II | Recruiting | Pemb + P→AC (29) vs. P→AC (85) | HER-2 negative, stage II or III at high risk (250, including 114 TNBC) | pCR rates in TNBC: 60% vs. 22% | |
| KEYNOTE-173 (NCT02622074) | Ib | Completed | Pemb + (nab-P ± Cb→AC) (60) | High-risk, early-stage TNBC (60) | Overall pCR rate: 60% | |
| KEYNOTE-522 (NCT03036488) | III | Ongoing | Pemb + (PCb→AC/EC) (784) vs. placebo + (PCb→AC/EC) (390) (→surgery→Pemb/placebo for up to 9 cycles) | Stage II-III TNBC (1174) | Overall | pCR rates e: 64.8% vs. 51.2% |
| 3-year EFS: 84.5% vs. 76.8% | ||||||
| +(CPS ≥ 1) | pCR rates: 68.9% vs. 54.9% | |||||
| − | pCR rates: 45.3% vs. 30.3% | |||||
| NeoPACT (NCT03639948) | II | Ongoing | Pemb + Cb + docetaxel | Early-stage TNBC | NA | |
| NCT04613674 | III | Recruiting | Camr + CT vs. placebo + CT | Early or Locally Advanced TNBC | NA | |
| GeparNuevo (NCT02685059) | II | Completed | Durv×2w f→durv + (nab-P →EC) (88) vs. placebo + (nab-P →EC) (86) (→surgery→physician’s choice) | Primary, cT1b-cT4a-d disease, centrally confirmed TNBC (174) | Overall pCR rates: 53.4% vs. 44.2% | |
| pCR rates in the window cohort: 61.0% vs. 41.4% | ||||||
| 3-year iDFS: 85.6% vs. 77.2% | ||||||
| 3-year DDFS: 91.7% vs. 78.4% | ||||||
| 3-year OS: 95.2% vs. 83.5% | ||||||
| NeoTRIPaPDL1 (NCT02620280) | III | Ongoing | Atez + nab-P + Cb (138) vs. nab-P + Cb (142) (→surgery→adjuvant anthracycline regimen as per investigator’s choice) | Early high-risk and locally advanced TNBC (280) | ITT population | pCR rates: 48.6% vs. 44.4% |
| +(≥1% IC) | pCR rates: 59.5% vs. 51.9% | |||||
| IMpassion031 (NCT03197935) | III | Ongoing | Atez + (nab-P →AC) (165) vs. placebo + (nab-P →AC) (168) (→surgery→ adjuvant Atez/placebo for up to 11 cycles) | Stage II–III TNBC (333) | Overall | pCR rates: 58% vs. 41% |
| +(≥1% IC) | pCR rates: 69% vs. 49% | |||||
| − | pCR rates: 48% vs. 34% | |||||
| NSABP B-59 (NCT03281954) | III | Ongoing | Atez + (PCb→AC) vs. placebo + (PCb →AC) (→surgery→adjuvant Atez/placebo until 1 year after the first dose) | Stage II–III TNBC | NA | |
| NCT02530489 | II | Ongoing | Atez + nab-P (→surgery→adjuvant Atez for 4 cycles) | TNBC that were non-responders to initial AC chemotherapy | NA | |
| Trials in early-stage TNBC as adjuvant therapy | ||||||
| NCT03487666 | II | Ongoing | Nivo vs. capecitabine vs. Nivo + capecitabine | TNBC with ≥ 1 cm RIC or LN (+) after NACT | NA | |
| IMpassion030 (NCT03498716) | III | Recruiting | Atez + A/P-based CT vs. CT | Operable-stage II-III TNBC | NA | |
| NCT03756298 | II | Recruiting | Atez + capecitabine vs. capecitabine | TNBC with RIC after NACT | NA |
| Clinical Trial | Phase | Status | Arms | Population |
|---|---|---|---|---|
| NCT02730130 | II | Ongoing | Pemb + RT | mTNBC: a median of 3 lines prior systemic therapy |
| AZTEC (NCT03464942) | II | Ongoing | Atez + RT | Advanced TNBC: <2 lines of prior systemic therapy |
| NCT03483012 | II | Ongoing | Atez + RT | mTNBC with brain metastases |
| KEYNOTE-162 (NCT02657889) | I/II | Completed | Pemb + niraparib | Advanced TNBC: a median of 1 prior line of therapy (range, 0–3) in the metastatic setting |
| I-SPY2 (NCT01042379) | II | Recruiting | Durv + olaparib + paclitaxel vs. paclitaxel | Stage II-III TNBC: preoperative treatment |
| DORA (NCT03167619) | II | Ongoing | Durv + olaparib | Platinum-treated mTNBC |
| KEYLYNK-009 (NCT04191135) | II/III | Ongoing | Pemb + olaparib vs. Pemb + Cb + gemcitabine | Locally recurrent inoperable or metastatic TNBC: after induction with first-line CT + Pemb |
| NCT03594396 | I/II | Ongoing | Olaparib + Durv | Stage II/III TNBC or low ER breast cancer: preoperative treatment |
| NCT03310957 | Ib/II | Recruiting | Pemb + ladiratuzumab vedotin | Unresectable locally advanced or metastatic TNBC: first-line treatment |
| ASCENT-04 (NCT05382286) | III | Recruiting | Pemb + SG vs. pemb + TPC | Previously untreated, locally advanced inoperable, or metastatic PD-L1-positive TNBC |
| NCT04468061 | II | Recruiting | Pemb + SG vs. SG | PD-L1-negative mTNBC |
| ASPRIA (NCT04434040) | II | Recruiting | Atez + SG | Early-stage TNBC with RIC after NACT |
| NCT03394287 | II | Completed | Camr + apatinib | Advanced TNBC: <3 lines of systemic therapy |
| NCT05447702 | II | Not yet recruiting | Camr + apatinib + CT | Neoadjuvant therapy for stage II-III TNBC |
| NCT04303741 | II | Ongoing | Camr + apatinib + eribulin | Unresectable recurrent or mTNBC; pre-treated with anthracycline and taxane |
| NCT04427293 | I | Recruiting | Pemb + Lenvatinib | Early-stage TNBC: preoperative treatment |
| NCT04335006 | III | Recruiting | Care + nab-P + apatinib vs. Care + nab-P vs. nab-P | Locally advanced or metastatic TNBC: first-line treatment |
| NCT03800836 | Ib | Completed | Atez + ipatasertib + P/nab-P | mTNBC: first-line treatment |
| BARBICAN (NCT05498896) | II | Ongoing | Atez + PAC + ipatasertib vs. Atez + PAC | Early-stage TNBC: preoperative treatment |
| NCT04177108 | III | Ongoing | Atez/placebo + ipatasertib/placebo + P | Locally advanced unresectable or metastatic TNBC |
| COLET (NCT02322814) | II | Completed | Atez + cobimetinib + P (cohorts II)/Atez + cobimetinib + nab-P (cohort III) | First-line treatment for mTNBC |
| NCT02536794 | II | Completed | Durv + tremelimumab | Pre-treated mTNBC |
| NCT03872791 | Ib/II | Ongoing | KN046 vs. KN046 + nab-P | mTNBC |
| SYNERGY (NCT03616886) | Ib/II | Ongoing | Durv + oleclumab +PCb vs. Durv + PCb | First-line treatment for mTNBC |
| NCT04584112 | Ib | Ongoing | Atez + tiragolumab + CT | First-line treatment for PD-L1 (+) mTNBC |
| NCT05227664 | II | Recruiting | AK117 + P/nab-P vs. AK112 + P/nab-P vs. AK117+AK112 + P/nab-P | First-line treatment for mTNBC |
| NCT03362060 | I | Ongoing | Pemb + PVX-410 vaccine | Pre-treated HLA-A2 (+) mTNBC |
| NCT02826434 | I | Ongoing | Durv + PVX-410 | HLA-A2 (+) stage II or III TNBC |
| NCT03606967 | II | Recruiting | CT →Durv + tremelimumab + Vaccine vs. CT →Durv + tremelimumab | First-line treatment for PD-L1-negative mTNBC |
| NCT03199040 | I | Ongoing | Durv + DNA vaccine vs. DNA vaccine | Early-stage TNBC |
| NSABP FB-14 (NCT04024800) | II | Ongoing | AE37 peptide vaccine + Pemb | Advanced TNBC: ≤ 1 line of systemic therapy |
| NCT03387085 | Ib/II | Ongoing | Combination of multiple treatments | mTNBC: ≥ 2 lines of prior therapy |
| NCT04445844 | II | Recruiting | Retifanlimab + pelareorep | mTNBC: received 1–2 prior lines of systemic therapy |
| NCT03004183 | II | Ongoing | ADV/HSV-tk + RT + Pemb + | Pre-treated mTNBC |
| NCT03256344 | I | Completed | Atez + talimogene laherparepvec | mTNBC with liver metastases |
| NCT05081492 | I | Recruiting | CF33-hNIS-antiPDL1 | mTNBC: ≥ 2 prior lines of therapy for metastatic disease |
| NCT04185311 | I | Ongoing | Talimogene laherparepvec + nivolumab + ipilimumab | Localized, palpable HER-2 negative breast cancer |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Zhang, F.; Liu, Z.; Fan, Z. Immunotherapy for Triple-Negative Breast Cancer: Combination Strategies to Improve Outcome. Cancers 2023, 15, 321. https://doi.org/10.3390/cancers15010321
Li L, Zhang F, Liu Z, Fan Z. Immunotherapy for Triple-Negative Breast Cancer: Combination Strategies to Improve Outcome. Cancers. 2023; 15(1):321. https://doi.org/10.3390/cancers15010321
Chicago/Turabian StyleLi, Liying, Fan Zhang, Zhenyu Liu, and Zhimin Fan. 2023. "Immunotherapy for Triple-Negative Breast Cancer: Combination Strategies to Improve Outcome" Cancers 15, no. 1: 321. https://doi.org/10.3390/cancers15010321
APA StyleLi, L., Zhang, F., Liu, Z., & Fan, Z. (2023). Immunotherapy for Triple-Negative Breast Cancer: Combination Strategies to Improve Outcome. Cancers, 15(1), 321. https://doi.org/10.3390/cancers15010321