
To β or Not to β: How Important Is β-Catenin Dependent and Independent WNT Signaling in CLL?


Abstract
Simple Summary
Abstract
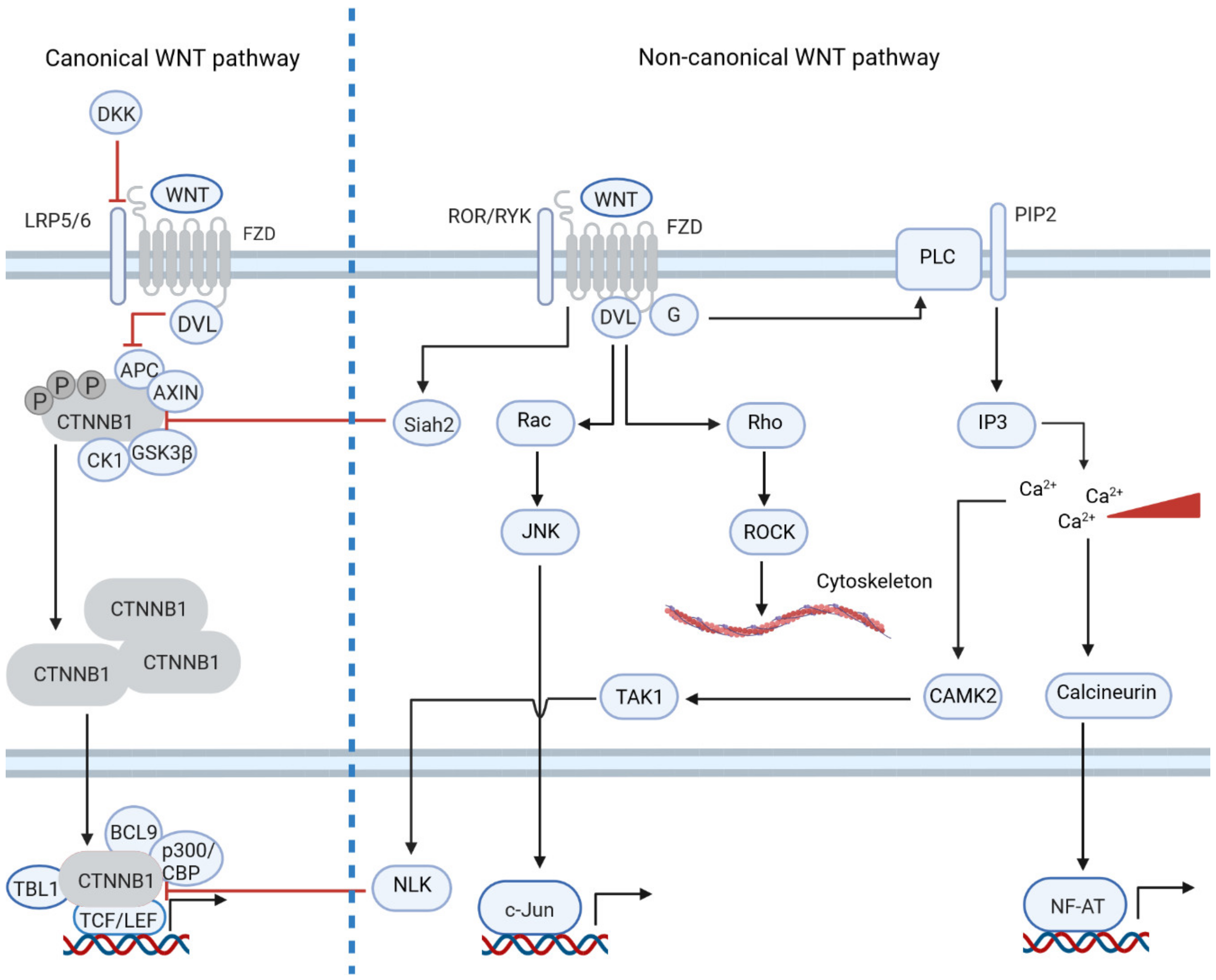
1. Introduction: The Physiological and Pathogenic Role of WNT Signaling
- Inhibition of PORCN to disrupt ligand secretion;
- Antibodies blocking FZD or ROR receptors;
- Inhibitors of positive regulators of the WNT pathway, such as RSPO3 or tankyrase;
- Compounds directly targeting β-catenin and β-catenin-mediated transcription;
- Activation of the WNT pathway by inhibition of negative regulators; this strategy was developed for tumors such as leukemia, prostate cancer or lymphoma.
2. Non-Canonical WNT Signaling Is Dysregulated in Tumors by Differential Gene Expression
3. Non-Canonical WNT Signaling Often Inhibits Canonical WNT Signaling
4. WNT Signaling Links Cancer Cells with Their Tumor Microenvironment
5. Chronic Lymphocytic Leukemia Is an Ideal Model for WNT Signaling and Its Role in the Interaction with the Microenvironment
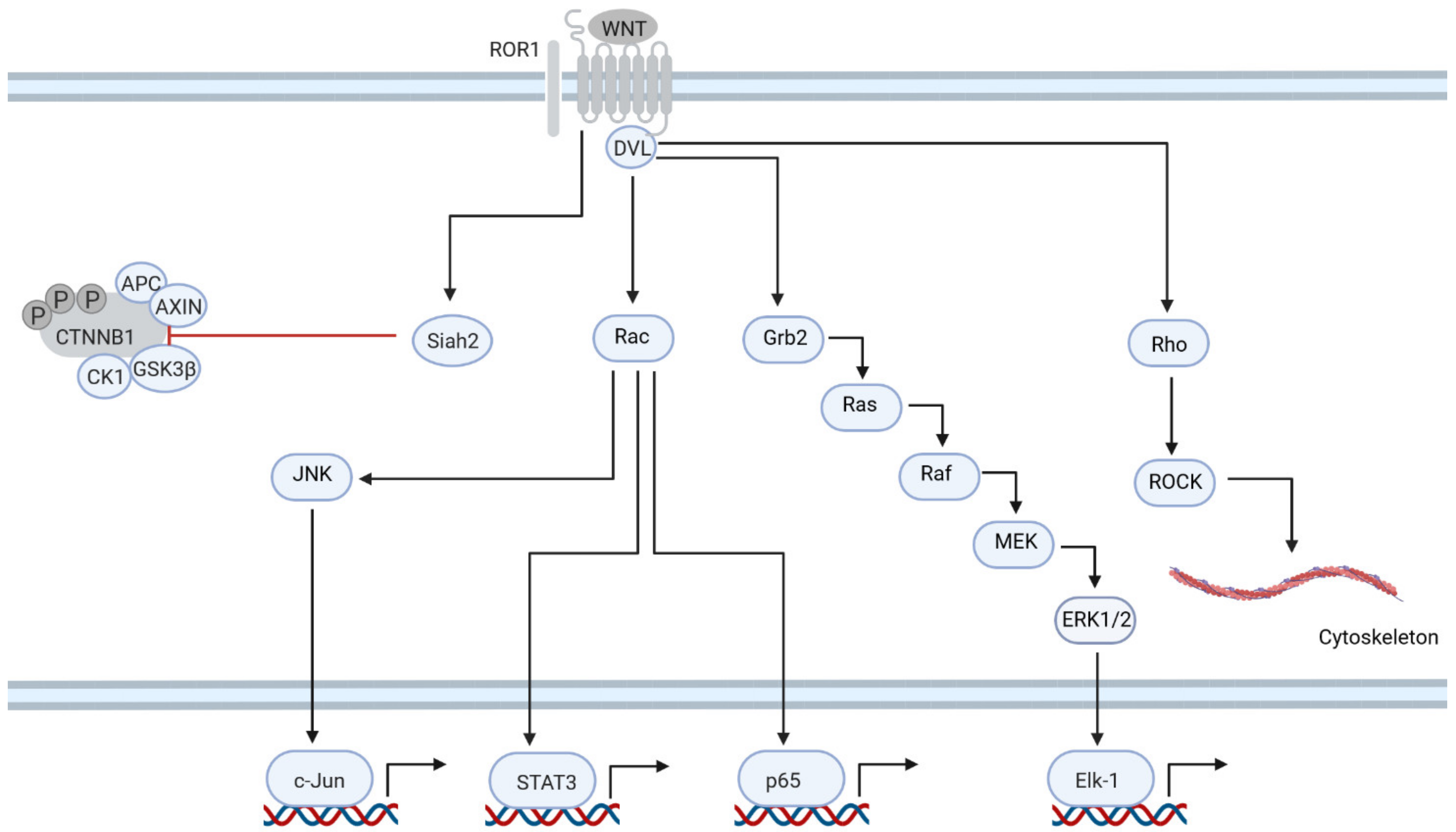
6. ROR1 Is Central to Non-Canonical Signaling in CLL
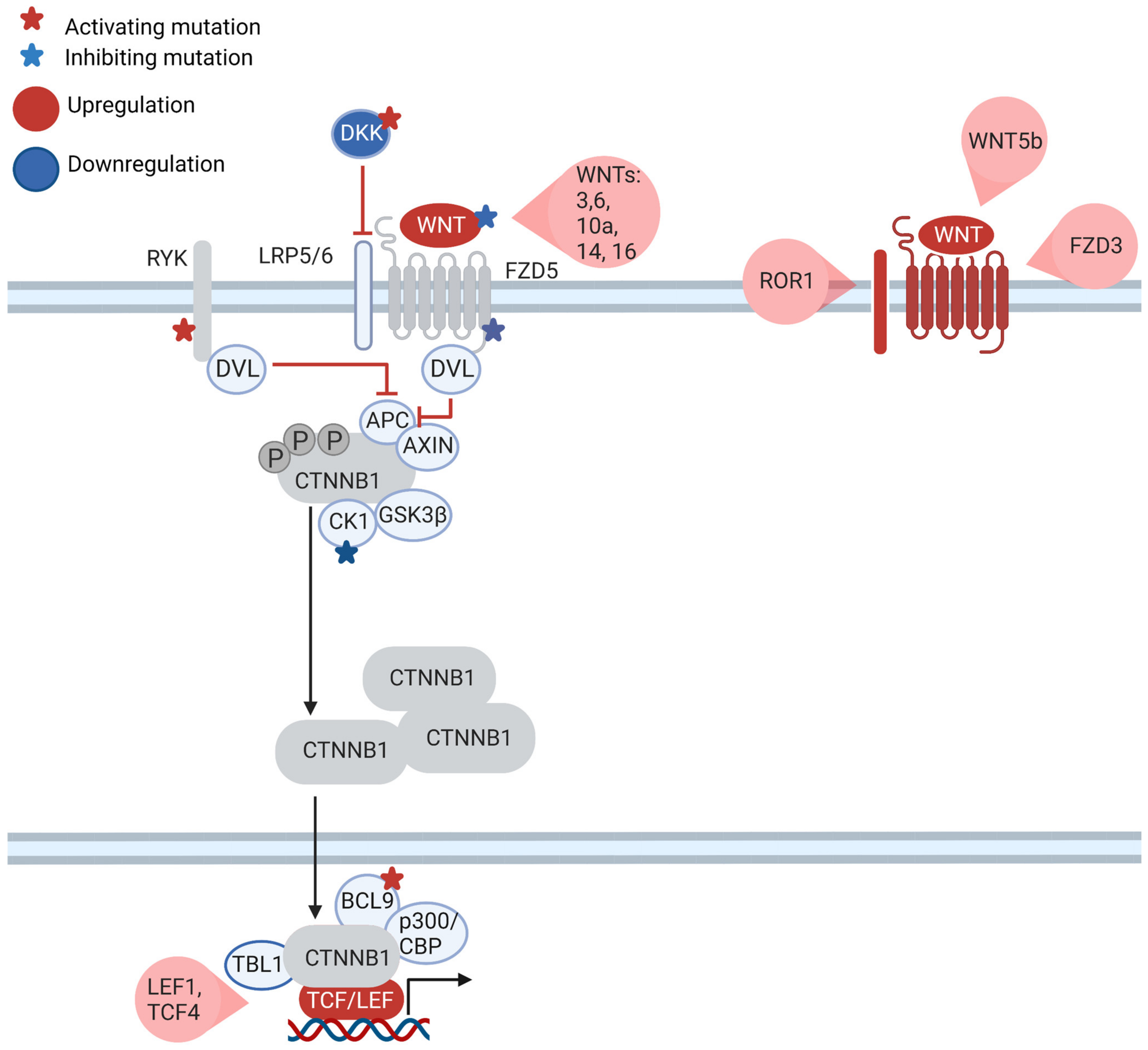
7. What Is the Role of the Canonical WNT Pathway in CLL?
8. WNT Is Part of the Communication of CLL Cells with the Microenvironment
9. The WNT Signaling Cascade Is a Therapeutic Target in CLL
10. Conclusions: Next Steps to Address Open Questions concerning WNT Signaling in CLL
Author Contributions
Funding
Conflicts of Interest
Acronyms
| APC | adenomatous polyposis coli |
| BCL2 | B-cell lymphoma protein 2 |
| BCL2L1 | gene encoding BCL2-xL protein |
| BCL2L11 | gene encoding BIM protein |
| BCL9 | B-cell lymphoma protein 9 |
| BCR | B-cell receptor |
| BMSC | bone marrow stromal cell |
| BRD7 | bromodomain-containing protein 7 |
| BTK | Bruton’s tyrosine kinase |
| CBP, CREBBP | CREB-binding protein |
| CCK4 | protein tyrosine kinase 7 |
| CCL | chemokine ligand |
| CHD8 | chromodomain-helicase-DNA-binding protein 8 |
| CLL | chronic lymphocytic leukemia |
| CRC | colorectal cancer |
| CSNK1E | gene encoding casein kinase 1ε |
| CTLA4 | cytotoxic T-lymphocyte-associated protein 4 |
| CTNNB1 | gene encoding β-catenin |
| CXCL | chemokine (C-X-C motif) ligand |
| DKK | dickkopf-related protein |
| DOCK2 | dedicator of cytokinesis 2 |
| DVL | dishevelled protein |
| ERK | mitogen-activated protein kinase |
| FAP | familial adenomatous polyposis |
| FCR | fludarabine, cyclophosphamide and rituximab therapeutic regime |
| FZD | frizzled family of receptors |
| GSK-3β | glycogen synthase kinase 3β |
| HCC | hepatocellular carcinoma |
| IGHV | immunoglobulin heavy-chain variable region gene |
| IL | interleukin |
| JNK | c-Jun N-terminal kinases |
| LEF1 | lymphoid enhancer-binding factor 1 |
| LRP | low-density lipoprotein receptor-related protein |
| LUAD | adenocarcinoma of the lung |
| MAPK | mitogen-activated protein kinase |
| MCL | mantle cell lymphoma |
| MMP9 | matrix metallopeptidase 9 |
| NFAT | nuclear factor of activated T-cells |
| NLCs | nurse-like cells |
| PD1 | programmed cell death protein 1 |
| PDGF | platelet-derived growth factor |
| PI3K | phosphoinositide 3-kinase |
| PLC | planar cell polarity pathway |
| PORCN | gene encoding porcupine |
| PRICKLE1 | planar cell polarity protein 1 |
| ROR | RAR-related orphan protein |
| RSPO | R-spondin |
| RYK | related to receptor tyrosine kinase |
| TFS | treatment-free survival |
| TNKS | gene encoding tankyrase |
| WIF1 | WNT inhibitory factor 1 |
| WNT | wingless family of ligands |
| YAP1 | yes-associated protein 1 |
References
- Anastas, J.N.; Moon, R.T. WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Canc. 2013, 13, 11–26. [Google Scholar]
- Parsons, M.J.; Tammela, T.; Dow, L.E. WNT as a Driver and Dependency in Cancer. Cancer Discov. 2021, 11, 2413–2429. [Google Scholar] [CrossRef]
- Corda, G.; Sala, A. Non-canonical WNT/PCP signalling in cancer: Fzd6 takes centre stage. Oncogenesis 2017, 6, e364. [Google Scholar] [CrossRef] [PubMed]
- Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Disc. 2014, 13, 513–532. [Google Scholar]
- Wu, Q.-L.; Zierold, C.; Ranheim, E.A. Dysregulation of Frizzled 6 is a critical component of B-cell leukemogenesis in a mouse model of chronic lymphocytic leukemia. Blood 2009, 113, 3031–3039. [Google Scholar] [CrossRef]
- Hasan, M.K.; Ghia, E.M.; Rassenti, L.Z.; Widhopf, G.F., 2nd; Kipps, T.J. Wnt5a enhances proliferation of chronic lymphocytic leukemia and ERK1/2 phosphorylation via a ROR1/DOCK2-dependent mechanism. Leukemia 2021, 35, 1621–1630. [Google Scholar] [CrossRef]
- Rosenbluh, J.; Nijhawan, D.; Cox, A.G.; Li, X.; Neal, J.T.; Schafer, E.J.; Zack, T.I.; Wang, X.; Tsherniak, A.; Schinzel, A.C.; et al. β-Catenin-Driven Cancers Require a YAP1 Transcriptional Complex for Survival and Tumorigenesis. Cell 2021, 151, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Neiheisel, A.; Kaur, M.; Ma, N.; Havard, P.; Shenoy, A.K. Wnt pathway modulators in cancer therapeutics: An update on completed and ongoing clinical trials. Int. J. Cancer. 2021, 150, 727–740. [Google Scholar] [CrossRef]
- Janovská, P.; Bryja, V. Wnt signalling pathways in chronic lymphocytic leukaemia and B-cell lymphomas. Br. J. Pharmacol. 2017, 174, 4701–4715. [Google Scholar] [CrossRef]
- Mikels, A.J.; Nusse, R. Purified Wnt5a Protein Activates or Inhibits β-Catenin–TCF Signaling Depending on Receptor Context. PLoS Biol. 2006, 4, e115. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Z.; Tang, Y.; Xiao, Q. The involvement of noncanonical Wnt signaling in cancers. Biomed. Pharm. 2021, 133, 110946. [Google Scholar] [CrossRef] [PubMed]
- Flores-Hernández, E.; Velázquez, D.M.; Castañeda-Patlán, M.C.; Fuentes-García, G.; Fonseca-Camarillo, G.; Yamamoto-Furusho, J.K.; Romero-Avila, M.T.; García-Sáinz, J.A.; Robles-Flores, M. Canonical and non-canonical Wnt signaling are simultaneously activated by Wnts in colon cancer cells. Cell. Signal. 2020, 72, 109636. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, C.; Tong, J.; Su, Y.; Lin, Y.; Zhou, X.; Ye, L. WNT6 Promotes the Migration and Differentiation of Human Dental Pulp Cells Partly through c-Jun N-terminal Kinase Signaling Pathway. J. Endod. 2014, 40, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Krawetz, R. Wnt6 induces the specification and epithelialization of F9 embryonal carcinoma cells to primitive endoderm. Cell. Signal. 2008, 20, 506–517. [Google Scholar] [CrossRef]
- Ali, I.; Medegan, B.; Braun, D.P. Wnt9A Induction Linked to Suppression of Human Colorectal Cancer Cell Proliferation. Int. J. Mol. Sci. 2016, 17, 495. [Google Scholar] [CrossRef]
- Topol, L.; Jiang, X.; Choi, H.; Garrett-Beal, L.; Carolan, P.J.; Yang, Y. Wnt-5a inhibits the canonical Wnt pathway by promoting GSK-3–independent β-catenin degradation. J. Cell. Biol. 2003, 162, 899–908. [Google Scholar] [CrossRef]
- Nemeth, M.J.; Topol, L.; Anderson, S.M.; Yang, Y.; Bodine, D.M. Wnt5a inhibits canonical Wnt signaling in hematopoietic stem cells and enhances repopulation. Proc. Natl. Acad. Sci. USA 2007, 104, 15436–15441. [Google Scholar] [CrossRef]
- Atkinson, J.M.; Rank, K.B.; Zeng, Y.; Capen, A.; Yadav, V.; Manro, J.R.; Engler, T.A.; Chedid, M. Activating the Wnt/β-Catenin Pathway for the Treatment of Melanoma—Application of LY2090314, a Novel Selective Inhibitor of Glycogen Synthase Kinase-3. PLoS ONE 2015, 10, e0125028. [Google Scholar] [CrossRef] [PubMed]
- Kunnimalaiyaan, S.; Schwartz, V.K.; Jackson, I.A.; Clark Gamblin, T.; Kunnimalaiyaan, M. Antiproliferative and apoptotic effect of LY2090314, a GSK-3 inhibitor, in neuroblastoma in vitro. BMC Cancer 2018, 18, 506. [Google Scholar] [CrossRef]
- Zimmerman, Z.F.; Kulikauskas, R.M.; Bomsztyk, K.; Moon, R.T.; Chien, A.J. Activation of Wnt/β-catenin signaling increases apoptosis in melanoma cells treated with trail. PLoS ONE 2013, 8, e69593. [Google Scholar] [CrossRef]
- Patel, S. Wnt Signaling and Its Significance Within the Tumor Microenvironment: Novel Therapeutic Insights. Front. Immunol. 2019, 10, 2872. [Google Scholar] [CrossRef] [PubMed]
- Bosch, F.; Dalla-Favera, R. Chronic lymphocytic leukaemia: From genetics to treatment. Nat. Rev. Clin. Oncol. 2019, 16, 681–704. [Google Scholar]
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: Atlanta, GA, USA, 2022. [Google Scholar]
- Yosifov, D.Y.; Wolf, C.; Stilgenbauer, S.; Mertens, D. From Biology to Therapy: The CLL Success Story. HemaSphere 2019, 3, e175. [Google Scholar] [CrossRef] [PubMed]
- Ghia, E.M.; Rassenti, L.Z.; Chen, L.; Cui, B.; Deboever, C.; Frazer, K.A.; Kipps, T.J. ROR1 Negative Chronic Lymphocytic Leukemia (CLL) Have a Distinctive Gene Expression Signature and May Represent an Indolent-Disease Subtype. Blood 2015, 126, 2932. [Google Scholar] [CrossRef]
- Poppova, L.; Janovska, P.; Plevova, K.; Radova, L.; Plesingerova, H.; Borsky, M.; Kotaskova, J.; Kantorova, B.; Hlozkova, M.; Figulova, J.; et al. Decreased WNT3 expression in chronic lymphocytic leukaemia is a hallmark of disease progression and identifies patients with worse prognosis in the subgroup with mutated IGHV. Br. J. Haematol. 2016, 175, 851–859. [Google Scholar] [CrossRef]
- Wu, W.; Zhu, H.; Fu, Y.; Shen, W.; Miao, K.; Hong, M.; Xu, W.; Fan, L.; Young, K.H.; Liu, P.; et al. High LEF1 expression predicts adverse prognosis in chronic lymphocytic leukemia and may be targeted by ethacrynic acid. Oncotarget 2016, 7, 21631–21643. [Google Scholar] [CrossRef]
- Baskar, S.; Kwong, K.Y.; Hofer, T.; Levy, J.M.; Kennedy, M.G.; Lee, E.; Staudt, L.M.; Wilson, W.H.; Wiestner, A.; Rader, C. Unique cell surface expression of receptor tyrosine kinase ROR1 in human B-cell chronic lymphocytic leukemia. Clin. Cancer Res. 2008, 14, 396–404. [Google Scholar] [CrossRef]
- Daneshmanesh, A.H.; Mikaelsson, E.; Jeddi-Tehrani, M.; Bayat, A.A.; Ghods, R.; Ostadkarampour, M.; Akhondi, M.; Lagercrantz, S.; Larsson, C.; Osterborg, A.; et al. Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. Int. J. Cancer 2008, 123, 1190–1195. [Google Scholar] [CrossRef]
- Li, P. Stat3 Activates the Receptor Tyrosine Kinase Like Orphan Receptor-1 Gene in Chronic Lymphocytic Leukemia Cells. PLoS ONE 2010, 5, e11859. [Google Scholar] [CrossRef]
- Lu, D.; Zhao, Y.; Tawatao, R.; Cottam, H.B.; Sen, M.; Leoni, L.M.; Kipps, T.J.; Corr, M.; Carson, D.A. Activation of the Wnt signaling pathway in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2004, 101, 3118–3123. [Google Scholar] [CrossRef]
- Kaucká, M.; Plevová, K.; Pavlová, S.; Janovská, P.; Mishra, A.; Verner, J.; Procházková, J.; Krejcí, P.; Kotasková, J.; Ovesná, P.; et al. The Planar Cell Polarity Pathway Drives Pathogenesis of Chronic Lymphocytic Leukemia by the Regulation of B-Lymphocyte Migration. Cancer Res. 2013, 73, 1491–1501. [Google Scholar] [CrossRef]
- Hasan, M.K.; Rassenti, L.; Widhopf, G.F., 2nd; Yu, J.; Kipps, T.J. Wnt5a causes ROR1 to complex and activate cortactin to enhance migration of chronic lymphocytic leukemia cells. Leukemia 2019, 33, 653–661. [Google Scholar] [CrossRef]
- Hasan, M.K.; Rassenti, L.; Widhopf, G.F., 2nd; Kipps, T.J. Wnt5a Induces ROR1-Dependent Upregulation of MMP9 and Enhanced Invasiveness in Chronic Lymphocytic Leukemia. Blood 2019, 134, 4274. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, L.; Yu, J.; Ghia, E.M.; Choi, M.Y.; Zhang, L.; Zhang, S.; Sanchez-Lopez, E.; Widhopf, G.F., 2nd; Messer, K.; et al. Cirmtuzumab blocks Wnt5a/ROR1 stimulation of NF-kB to repress autocrine STAT3 activation in chronic lymphocytic leukemia. Blood 2019, 134, 1084–1094. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, S.; Ghia, E.M.; Choi, M.Y.; Zhang, J.; Chen, L.; Widhopf, G.F., 2nd; Rassenti, L.Z.; Kipps, T.J. Cirmtuzumab Inhibits Non-Canonical Wnt Signaling without Enhancing Canonical Wnt/β-Catenin Signaling in Chronic Lymphocytic Leukemia. Blood 2018, 132, 2652. [Google Scholar] [CrossRef]
- Ghia, E.M.; Rassenti, L.Z.; Choi, M.Y.; Quijada-Álamo, M.; Chu, E.; Widhopf, G.F., 2nd; Kipps, T.J. High expression level of ROR1 and ROR1-signaling associates with venetoclax resistance in chronic lymphocytic leukemia. Leukemia 2022, 36, 1609–1618. [Google Scholar] [CrossRef]
- Guo, Y. Aberrantly expressed Wnt5a in nurse-like cells drives resistance to Venetoclax in chronic lymphocytic leukemia. Cell Death Discov. 2022, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Wang, L. Somatic mutation as a mechanism of Wnt/β-catenin pathway activation in CLL. Blood 2014, 124, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A., Jr.; Tschumper, R.C.; Wu, X.; Shanafelt, T.D.; Eckel-Passow, J.; Huddleston, P.M., 3rd; Slager, S.L.; Kay, N.E.; Jelinek, D.F. LEF-1 is a prosurvival factor in chronic lymphocytic leukemia and is expressed in the preleukemic state of monoclonal B-cell lymphocytosis. Blood 2010, 116, 2975–2983. [Google Scholar] [CrossRef] [PubMed]
- Moskalev, E.A.; Luckert, K.; Vorobjev, I.A.; Mastitsky, S.E.; Gladkikh, A.A.; Stephan, A.; Schrenk, M.; Kaplanov, K.D.; Kalashnikova, O.B.; Pötz, O.; et al. Concurrent epigenetic silencing of Wnt/β-catenin pathway inhibitor genes in B cell chronic lymphocytic leukaemia. BMC Cancer 2012, 12, 213. [Google Scholar] [CrossRef]
- Jiang, M.; Kang, Y.; Sewastianik, T.; Wang, J.; Tanton, H.; Alder, K.; Dennis, P.; Xin, Y.; Wang, Z.; Liu, R.; et al. BCL9 provides multi-cellular communication properties in colorectal cancer by interacting with paraspeckle proteins. Nat. Commun. 2012, 11, 19. [Google Scholar] [CrossRef]
- Lu, W.; Yamamoto, V.; Ortega, B.; Baltimore, D. Mammalian Ryk Is a Wnt Coreceptor Required for Stimulation of Neurite Outgrowth. Cell 2004, 119, 97–108. [Google Scholar] [CrossRef]
- Lazarian, G.; Friedrich, C.; Quinquenel, A.; Tran, J.; Ouriemmi, S.; Dondi, E.; Martin, A.; Mihoub, I.; Chiron, D.; Bellanger, C.; et al. Stabilization of β-catenin upon B-cell receptor signaling promotes NF-kB target genes transcription in mantle cell lymphoma. Oncogene 2020, 39, 2934–2947. [Google Scholar] [CrossRef] [PubMed]
- Mangolini, M.; Götte, F.; Moore, A.; Ammon, T.; Oelsner, M.; Lutzny-Geier, G.; Klein-Hitpass, L.; Williamson, J.C.; Lehner, P.J.; Dürig, J.; et al. Notch2 controls non-autonomous Wnt-signalling in chronic lymphocytic leukaemia. Nat. Commun. 2018, 9, 3839. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.Y.; Widhopf, G.F., 2nd; Ghia, E.M.; Kidwell, R.L.; Hasan, M.K.; Yu, J.; Rassenti, L.Z.; Chen, L.; Chen, Y.; Pittman, E.; et al. Phase I Trial: Cirmtuzumab Inhibits ROR1 Signaling and Stemness Signatures in Patients with Chronic Lymphocytic Leukemia. Cell Stem Cell 2018, 22, 951–959. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Choi, M.Y.; Siddiqi, T.; Barrientos, J.C.; Wierda, W.G.; Isufi, I.; Tuscano, J.M.; Lamanna, N.; Subbiah, S.; Koff, J.L.; et al. Phase 1/2 study of cirmtuzumab and ibrutinib in mantle cell lymphoma (MCL) or chronic lymphocytic leukemia (CLL). J. Clin. Oncol. 2021, 39, 7556. [Google Scholar] [CrossRef]
- Zenz, T.; Luetge, A.; Lu, J.; Jennifer, H.; Dietrich, S.; Sellner, L.; Huber, W. Transcriptional Profiling Reveals Strong Impact of Major Molecular Disease Subgroups and Mixed Epistasis in Chronic Lymphocytic Leukemia. Blood 2019, 134, 1742. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Molecular Target | Function | Drug | Function | Strategy Type | Cancer Type |
|---|---|---|---|---|---|
| PORCN | Posttranslational processing of WNT proteins | WNT-C59 WNT974 RXC004 CGX1321 XNW7201 ETC-1922159 IWP compounds | competitive inhibitor competitive inhibitor competitive inhibitor competitive inhibitor competitive inhibitor competitive inhibitor competitive inhibitor | 1 | Breast Pancreas Colorectal Melanoma Head and neck Cervix Esophageal cancer Lung Gastric Liver |
| RSPO3 | Enhancer of WNT signaling | Rosmantuzumab OMP-131R10 | humanized antibody | 3 | Advanced relapsed tumors and refractory solid tumors |
| FZD8 | Receptor | Ipafricept OMP-54F28 | truncated decoy receptor | 2 | Liver Ovary Pancreas |
| FZD1/2/5/7/8 | Receptors | Vantictumab OMP-18R5 | human IgG2 monoclonal antibody | 2 | Solid tumors |
| FZD10 | Receptors | OTSA101-DTPA- 111ln | radiolabeled monoclonal antibody | 2 | Sarcoma |
| TNKS | Positive regulator of the canonical pathway | AZ1366 G007-LK NVP-TNKS656 XAV939 MSC2504877 | competitive inhibitor competitive inhibitor competitive inhibitor competitive inhibitor competitive inhibitor | 3 | Cancer |
| β-Catenin | Transcription activator | BC2059 | protein–protein interaction inhibitor | Desmoid tumor Osteosarcoma AML CML Myelodysplastic syndrome Multiple myeloma Colon Head and neck Liver Melanoma B cell lymphoma Gastric Pancreas | |
| CGP049090 | protein–protein interaction inhibitor | ||||
| CWP232291 | β-Catenin degrader | ||||
| MSAB | β-Catenin degrader | ||||
| E7386 | protein–protein interaction inhibitor | ||||
| PKF115-584 | protein–protein interaction inhibitor | ||||
| PKF118-310 | protein–protein interaction inhibitor | ||||
| SAH-BLC9 | protein–protein interaction inhibitor | 4 | |||
| ICG-001 | protein–protein interaction inhibitor | ||||
| PRI-724 | protein–protein interaction inhibitor | ||||
| SM08502 | competitive inhibitor | ||||
| LF3 | protein–protein interaction inhibitor | ||||
| DKK-1 | Antagonist of WNT ligands | BHQ880 DKN-01 | monoclonal Ab monoclonal Ab | 5 | Liver Biliary tract cancer Gastric Prostate Ovary Multiple myeloma Lung |
| GSK-3β | Negative regulator of the canonical pathway | LY2090314 9-ING-41 | competitive inhibitor competitive inhibitor | 5 | Leukemia Pancreas Lymphoma Sarcoma Glioblastoma Breast Bladder Kidney Ovary Bone |
| ROR1 | Co-receptor of the non-canonical pathway | Cirmtuzumab ROR1R-CAR-T NBE-002 VLS-101 KAN 0439834 ES425 | monoclonal Ab CAR-T therapy Ab–drug conjugate Ab–drug conjugate competitive inhibitor bispecific Ab | 2 | CLL SLL MCL Breast Lung |
| ROR2 | Co-receptor of the non-canonical pathway | CAB-ROR2-ADC | Ab–drug conjugate | 2 | Lung Breast Sarcoma |
| WNT5 | Ligand of non-canonical pathway | Foxy-5 | WNT5a mimetic | 3 | Colon Breast Prostate |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urbanek, K.D.; Stilgenbauer, S.; Mertens, D. To β or Not to β: How Important Is β-Catenin Dependent and Independent WNT Signaling in CLL? Cancers 2023, 15, 194. https://doi.org/10.3390/cancers15010194
Urbanek KD, Stilgenbauer S, Mertens D. To β or Not to β: How Important Is β-Catenin Dependent and Independent WNT Signaling in CLL? Cancers. 2023; 15(1):194. https://doi.org/10.3390/cancers15010194
Chicago/Turabian StyleUrbanek, Karol D., Stephan Stilgenbauer, and Daniel Mertens. 2023. "To β or Not to β: How Important Is β-Catenin Dependent and Independent WNT Signaling in CLL?" Cancers 15, no. 1: 194. https://doi.org/10.3390/cancers15010194
APA StyleUrbanek, K. D., Stilgenbauer, S., & Mertens, D. (2023). To β or Not to β: How Important Is β-Catenin Dependent and Independent WNT Signaling in CLL? Cancers, 15(1), 194. https://doi.org/10.3390/cancers15010194