
Targeting mTORC1 Activity to Improve Efficacy of Radioligand Therapy in Cancer


Simple Summary
Abstract

1. Introduction to Radioligand Therapy
2. Overview of Regulation and Signaling of mTORC1 in Cancer
3. Inhibitors of mTOR for Cancer Therapy
4. Targeting mTORC1 for Improved RLT

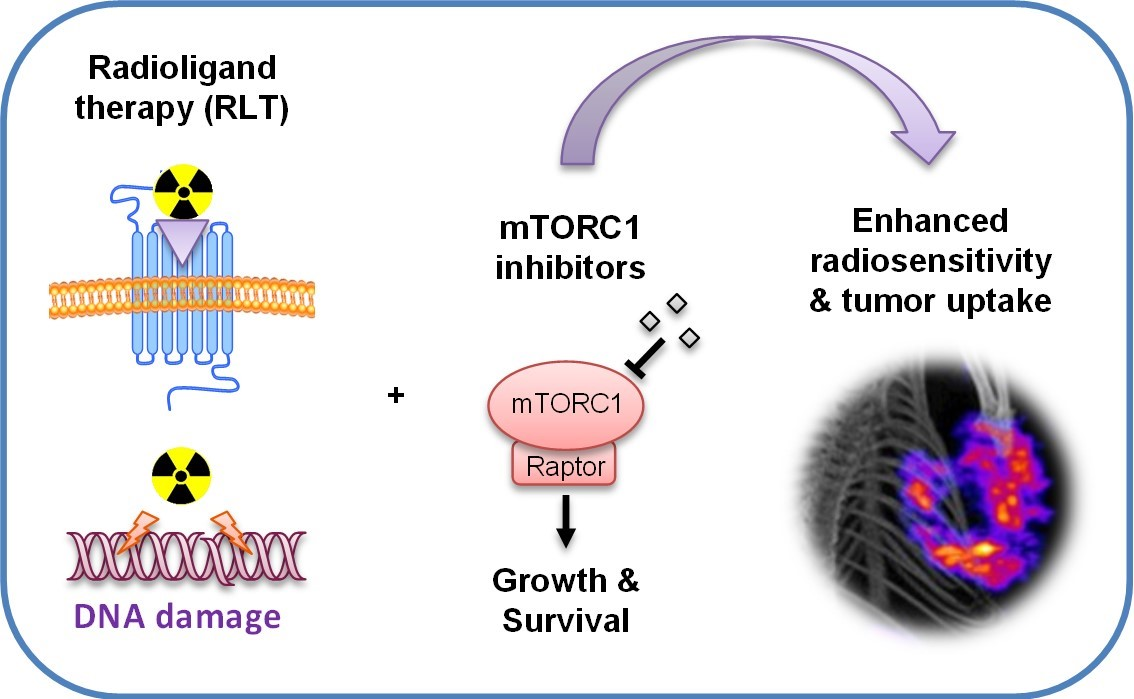{kind=link}
{kind=link}
{kind=link}
| Inhibitor | RLT | Cancer | Results | Ref. |
|---|---|---|---|---|
| Preclinical in vivo studies | ||||
| Rapamycin | [177Lu]Lu-RM2 | Human prostate cancer PC-3 xenograft mouse model | Significantly longer survival as compared with the monotherapies without treatment-related toxicity | [79] |
| Everolimus | [177Lu]Lu-DOTA-PP-F11N | Human epidermoid carcinoma A431/CCKBR xenograft mouse model | Everolimus significantly enhanced tumor-specific uptake of radiolabeled minigastrin without adverse effects | [81] |
| Metformin | [177Lu]Lu-DOTA-PP-F11N | Human epidermoid carcinoma A431/CCKBR xenograft mouse model | Metformin did not increase tumor uptake of radiolabeled minigastrin | [81] |
| Everolimus | [177Lu]Lu-DOTA-PP-F11N | Human epidermoid carcinoma A431/CCKBR xenograft mouse model | Increased tumor growth inhibition and extended survival as compared with the monotherapies without adverse effects | [82] |
| Everolimus | [177Lu]Lu-DOTA-TATE | Rat pancreatic cancer model with CA20948 tumors | Lack of therapeutic benefit. Metastases were found in combination and everolimus-treated groups | [83] |
| Everolimus | [177Lu]Lu-DOTA-TATE | Rat pancreatic cancer model with CA20948 tumors | Metastases were not dose-dependent or related to the duration of everolimus treatment | [84] |
| Everolimus | [177Lu]Lu-DOTA-TATE | Lewis rats: non-tumor bearing | Everolimus did not increase renal and hematological toxicity of the RLT | [85] |
| Clinical studies | ||||
| Everolimus | [177Lu]Lu-DOTA-TATE | Retrospective study; 24 patients with well- and moderately differentiated GEP-NETs (grades 1, 2) | The safety profile of everolimus was not influenced by the previous RLT. Partial responses (16.7%), stable disease (62.5%), and progressive disease (12.5%). | [86] |
| Everolimus | [177Lu]Lu-DOTA-TATE | Single-center retrospective study; 41 patients with NETs (80% with primary GEP-NETs) | No statistically significant differences in severe subacute hematotoxicity of RLT were seen in the everolimus pretreated group as compared with the untreated group | [87] |
| Everolimus | [177Lu]Lu-DOTA-TATE | Case study of a patient with grade II, well-differentiated rectal NET | Everolimus-induced SSTR overexpression, making the patient eligible for a second course of RLT | [88] |
| Everolimus | [177Lu]Lu-DOTA-TATE | Clinical phase I; 16 patients with advanced unresectable progressive well-differentiated GEP-NETs | The maximum tolerated dose of everolimus was 7.5 mg daily. Overall response (44%) and no progression during 6-month treatment. Partial response (80%) in pancreatic NET patients | [89] |
| Everolimus | [177Lu]Lu-DOTA-TATE | Clinical phase I/II; 11 patients with grade 1–2 NET of different origin | Everolimus at a dose of 10 mg daily with RLT was terminated due to toxicity and progression. One patient achieved partial response, and nine had stable disease. One patient developed disease progression. | [90] |
5. Conclusions and Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Grzmil, M.; Meisel, A.; Behé, M.; Schibli, R. An Overview of Targeted Radiotherapy. In Radiopharmaceutical Chemistry; Lewis, J.S., Windhorst, A.D., Zeglis, B.M., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 85–100. [Google Scholar]
- Naik, M.; Al-Nahhas, A.; Khan, S.R. Treatment of Neuroendocrine Neoplasms with Radiolabeled Peptides—Where Are We Now. Cancers 2022, 14, 761. [Google Scholar] [CrossRef]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Maggiolini, M. G protein-coupled receptors: Novel targets for drug discovery in cancer. Nat. Rev. Drug Discov. 2011, 10, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Artigas, C.; Mileva, M.; Flamen, P.; Karfis, I. Targeted radionuclide therapy: An emerging field in solid tumours. Curr. Opin. Oncol. 2021, 33, 493–499. [Google Scholar] [CrossRef]
- Kwekkeboom, D.J.; Kam, B.L.; van Essen, M.; Teunissen, J.J.; van Eijck, C.H.; Valkema, R.; de Jong, M.; de Herder, W.W.; Krenning, E.P. Somatostatin-receptor-based imaging and therapy of gastroenteropancreatic neuroendocrine tumors. Endocr. Relat. Cancer 2010, 17, R53–R73. [Google Scholar] [CrossRef] [PubMed]
- van Essen, M.; Krenning, E.P.; Kam, B.L.; de Jong, M.; Valkema, R.; Kwekkeboom, D.J. Peptide-receptor radionuclide therapy for endocrine tumors. Nat. Rev. Endocrinol. 2009, 5, 382–393. [Google Scholar] [CrossRef]
- Farolfi, A.; Mei, R.; Ali, S.; Castellucci, P. Theragnostics in prostate cancer. Q. J. Nucl. Med. Mol. Imaging 2021, 65, 333–341. [Google Scholar] [CrossRef]
- Hennrich, U.; Kopka, K. Lutathera®: The First FDA- and EMA-Approved Radiopharmaceutical for Peptide Receptor Radionuclide Therapy. Pharmaceuticals 2019, 12, 114. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef]
- FDA Approves Pluvicto/Locametz for Metastatic Castration-Resistant Prostate Cancer. J. Nucl. Med. 2022, 63, 13N.
- Reubi, J.C.; Schaer, J.C.; Waser, B. Cholecystokinin(CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res. 1997, 57, 1377–1386. [Google Scholar] [PubMed]
- Goetze, J.P.; Nielsen, F.C.; Burcharth, F.; Rehfeld, J.F. Closing the gastrin loop in pancreatic carcinoma: Coexpression of gastrin and its receptor in solid human pancreatic adenocarcinoma. Cancer 2000, 88, 2487–2494. [Google Scholar] [CrossRef] [PubMed]
- Hellmich, M.R.; Rui, X.L.; Hellmich, H.L.; Fleming, R.Y.D.; Evers, B.M.; Townsend, C.M., Jr. Human colorectal cancers express a constitutively active cholecystokinin-B/gastrin receptor that stimulates cell growth. J. Biol. Chem. 2000, 275, 32122–32128. [Google Scholar] [CrossRef] [PubMed]
- Sauter, A.W.; Mansi, R.; Hassiepen, U.; Muller, L.; Panigada, T.; Wiehr, S.; Wild, A.M.; Geistlich, S.; Béhé, M.; Rottenburger, C.; et al. Targeting of the Cholecystokinin-2 Receptor with the Minigastrin Analog (177)Lu-DOTA-PP-F11N: Does the Use of Protease Inhibitors Further Improve In Vivo Distribution? J. Nucl. Med. 2019, 60, 393–399. [Google Scholar] [CrossRef]
- Rottenburger, C.; Nicolas, G.P.; McDougall, L.; Kaul, F.; Cachovan, M.; Vija, A.H.; Schibli, R.; Geistlich, S.; Schumann, A.; Rau, T.; et al. Cholecystokinin 2 Receptor Agonist 177Lu-PP-F11N for Radionuclide Therapy of Medullary Thyroid Carcinoma: Results of the Lumed Phase 0a Study. J. Nucl. Med. 2020, 61, 520–526. [Google Scholar] [CrossRef]
- Fani, M.; Peitl, P.K.; Velikyan, I. Current Status of Radiopharmaceuticals for the Theranostics of Neuroendocrine Neoplasms. Pharmaceuticals 2017, 10, 30. [Google Scholar] [CrossRef]
- Müller, C.; Béhé, M.; Geistlich, S.; van der Meulen, N.P.; Schibli, R. Targeted Radiotherapeutics from ‘Bench-to-Bedside’. Chimia 2020, 74, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, D.; Lang, L.; Zhu, Z.; Wang, L.; Wu, P.; Niu, G.; Li, F.; Chen, X. 68Ga-NOTA-Aca-BBN(7–14) PET/CT in Healthy Volunteers and Glioma Patients. J. Nucl. Med. 2016, 57, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, J.; Chi, C.; Xiao, X.; Wang, J.; Lang, L.; Ali, I.; Niu, G.; Zhang, L.; Tian, J.; et al. First-in-human study of PET and optical dual-modality image-guided surgery in glioblastoma using 68Ga-IRDye800CW-BBN. Theranostics 2018, 8, 2508–2520. [Google Scholar] [CrossRef]
- Zaccagna, F.; Grist, J.T.; Quartuccio, N.; Riemer, F.; Fraioli, F.; Caracò, C.; Halsey, R.; Aldalilah, Y.; Cunningham, C.H.; Massoud, T.F.; et al. Imaging and treatment of brain tumors through molecular targeting: Recent clinical advances. Eur. J. Radiol. 2021, 142, 109842. [Google Scholar] [CrossRef]
- Piwowarska-Bilska, H.; Kurkowska, S.; Birkenfeld, B. Individualization of Radionuclide Therapies: Challenges and Prospects. Cancers 2022, 14, 3418. [Google Scholar] [CrossRef] [PubMed]
- Puliafito, I.; Esposito, F.; Prestifilippo, A.; Marchisotta, S.; Sciacca, D.; Vitale, M.P.; Giuffrida, D. Target Therapy in Thyroid Cancer: Current Challenge in Clinical Use of Tyrosine Kinase Inhibitors and Management of Side Effects. Front. Endocrinol. 2022, 13, 860671. [Google Scholar] [CrossRef] [PubMed]
- Nyati, M.K.; Morgan, M.A.; Feng, F.Y.; Lawrence, T.S. Integration of EGFR inhibitors with radiochemotherapy. Nat. Rev. Cancer 2006, 6, 876–885. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.H. Perspective: Multimodality radionuclide therapy of progressive disseminated lymphoma and neuroendocrine tumors as a paradigm for cancer control. Cancer Biother. Radiopharm. 2012, 27, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Lundsten, S.; Spiegelberg, D.; Raval, N.R.; Nestor, M. The radiosensitizer Onalespib increases complete remission in (177)Lu-DOTATATE-treated mice bearing neuroendocrine tumor xenografts. Eur. J. Nucl. Med. Mol. Imaging 2020, 47, 980–990. [Google Scholar] [CrossRef]
- Chan, T.G.; O’Neill, E.; Habjan, C.; Cornelissen, B. Combination Strategies to Improve Targeted Radionuclide Therapy. J. Nucl. Med. 2020, 61, 1544–1552. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Tomasoni, R.; Mondino, A. The tuberous sclerosis complex: Balancing proliferation and survival. Biochem. Soc. Trans. 2011, 39, 466–471. [Google Scholar] [CrossRef][Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef]
- Avruch, J.; Hara, K.; Lin, Y.; Liu, M.; Long, X.; Ortiz-Vega, S.; Yonezawa, K. Insulin and amino-acid regulation of mTOR signaling and kinase activity through the Rheb GTPase. Oncogene 2006, 25, 6361–6372. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Corradetti, M.N.; Inoki, K.; Bardeesy, N.; DePinho, R.A.; Guan, K.L. Regulation of the TSC pathway by LKB1: Evidence of a molecular link between tuberous sclerosis complex and Peutz-Jeghers syndrome. Genes Dev. 2004, 18, 1533–1538. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Fayard, E.; Tintignac, L.A.; Baudry, A.; Hemmings, B.A. Protein kinase B/Akt at a glance. J. Cell Sci. 2005, 118, 5675–5678. [Google Scholar] [CrossRef]
- Hollander, M.C.; Blumenthal, G.M.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef] [PubMed]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; de Stanchina, E.; Teresky, A.K.; Jin, S.; Lowe, S.; Levine, A.J. The regulation of AMPK β1, TSC2, and PTEN expression by p53: Stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 2007, 67, 3043–3053. [Google Scholar] [CrossRef]
- Grzmil, M.; Hemmings, B.A. Translation regulation as a therapeutic target in cancer. Cancer Res. 2012, 72, 3891–3900. [Google Scholar] [CrossRef]
- Mayer, C.; Grummt, I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene 2006, 25, 6384–6391. [Google Scholar] [CrossRef] [PubMed]
- Proud, C.G. Signalling to translation: How signal transduction pathways control the protein synthetic machinery. Biochem. J. 2007, 403, 217–234. [Google Scholar] [CrossRef] [PubMed]
- Dorrello, N.V.; Peschiaroli, A.; Guardavaccaro, D.; Colburn, N.H.; Sherman, N.E.; Pagano, M. S6K1- and βTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 2006, 314, 467–471. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.S.; Jansen, A.P.; Komar, A.A.; Zheng, X.; Merrick, W.C.; Costes, S.; Lockett, S.J.; Sonenberg, N.; Colburn, N.H. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol. Cell. Biol. 2003, 23, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem. Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Bounds, P.L.; Dang, C.V. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Marín-Hernández, Á.; Gallardo-Perez, J.C.; Ralph, S.J.; Rodriguez-Enriquez, S.; Moreno-Sanchez, R. HIF-1α modulates energy metabolism in cancer cells by inducing over-expression of specific glycolytic isoforms. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.L.; Ragel, B.T.; Whang, K.; Gillespie, D. Inhibition of hypoxia inducible factor-1α (HIF-1α) decreases vascular endothelial growth factor (VEGF) secretion and tumor growth in malignant gliomas. J. Neuro-Oncol. 2006, 78, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Deleyto-Seldas, N.; Efeyan, A. The mTOR—Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 2021, 9, 655731. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Park, J.M.; Grunwald, D.; Kim, D.H. An expanded role for mTORC1 in autophagy. Mol. Cell. Oncol. 2016, 3, e1010958. [Google Scholar] [CrossRef]
- Sehgal, S.N. Sirolimus: Its discovery, biological properties, and mechanism of action. Transplant. Proc. 2003, 35, S7–S14. [Google Scholar] [CrossRef]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Huang, Z.; Wu, Y.; Zhou, X.; Qian, J.; Zhu, W.; Shu, Y.; Liu, P. Clinical efficacy of mTOR inhibitors in solid tumors: A systematic review. Future Oncol. 2015, 11, 1687–1699. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wullschleger, S.; Shpiro, N.; McGuire, V.A.; Sakamoto, K.; Woods, Y.L.; McBurnie, W.; Fleming, S.; Alessi, D.R. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem. J. 2008, 412, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Pafundi, P.C.; Morgillo, F.; Di Liello, R.; Galiero, R.; Nevola, R.; Marfella, R.; Monaco, L.; Rinaldi, L.; Adinolfi, L.E.; et al. Metformin: An old drug against old age and associated morbidities. Diabetes Res. Clin. Pract. 2020, 160, 108025. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Yan, H.; Frost, P.; Gera, J.; Lichtenstein, A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol. Cancer Ther. 2005, 4, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Wang, X.; Proud, C.G. mTOR inhibitors in cancer therapy. F1000Research 2016, 5. [Google Scholar] [CrossRef]
- Chiarini, F.; Evangelisti, C.; McCubrey, J.A.; Martelli, A.M. Current treatment strategies for inhibiting mTOR in cancer. Trends Pharmacol. Sci. 2015, 36, 124–135. [Google Scholar] [CrossRef]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, M.; de Stanchina, E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature 2016, 534, 272–276. [Google Scholar] [CrossRef]
- Su, Y.C.; Yu, C.C.; Hsu, F.T.; Fu, S.L.; Hwang, J.J.; Hung, L.C.; Lee, M.S.; Chiou, W.Y.; Lin, H.Y.; Hung, S.K. Everolimus sensitizes Ras-transformed cells to radiation in vitro through the autophagy pathway. Int. J. Mol. Med. 2014, 34, 1417–1422. [Google Scholar] [CrossRef][Green Version]
- Kirova, Y.M.; Servois, V.; Chargari, C.; Amessis, M.; Zerbib, M.; Beuzeboc, P. Further developments for improving response and tolerance to irradiation for advanced renal cancer: Concurrent (mTOR) inhibitor RAD001 and helical tomotherapy. Investig. New Drugs 2012, 30, 1241–1243. [Google Scholar] [CrossRef]
- Albert, J.M.; Kim, K.W.; Cao, C.; Lu, B. Targeting the Akt/mammalian target of rapamycin pathway for radiosensitization of breast cancer. Mol. Cancer Ther. 2006, 5, 1183–1189. [Google Scholar] [CrossRef] [PubMed]
- Cao, C.; Subhawong, T.; Albert, J.M.; Kim, K.W.; Geng, L.; Sekhar, K.R.; Gi, Y.J.; Lu, B. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006, 66, 10040–10047. [Google Scholar] [CrossRef] [PubMed]
- Exner, S.; Arrey, G.; Prasad, V.; Grötzinger, C. mTOR Inhibitors as Radiosensitizers in Neuroendocrine Neoplasms. Front. Oncol. 2020, 10, 578380. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.C.; Hung, S.K.; Liao, H.F.; Lee, C.C.; Lin, H.Y.; Lai, H.C.; Li, S.C.; Ho, H.C.; Huang, H.B.; Su, Y.C. RAD001 enhances the radiosensitivity of SCC4 oral cancer cells by inducing cell cycle arrest at the G2/M checkpoint. Anticancer Res. 2014, 34, 2927–2935. [Google Scholar]
- Guba, M.; von Breitenbuch, P.; Steinbauer, M.; Koehl, G.; Flegel, S.; Hornung, M.; Bruns, C.J.; Zuelke, C.; Farkas, S.; Anthuber, M.; et al. Rapamycin inhibits primary and metastatic tumor growth by antiangiogenesis: Involvement of vascular endothelial growth factor. Nat. Med. 2002, 8, 128–135. [Google Scholar] [CrossRef]
- Shinohara, E.T.; Cao, C.; Niermann, K.; Mu, Y.; Zeng, F.; Hallahan, D.E.; Lu, B. Enhanced radiation damage of tumor vasculature by mTOR inhibitors. Oncogene 2005, 24, 5414–5422. [Google Scholar] [CrossRef]
- Zhang, Q.; Bindokas, V.; Shen, J.; Fan, H.; Hoffman, R.M.; Xing, H.R. Time-course imaging of therapeutic functional tumor vascular normalization by antiangiogenic agents. Mol. Cancer Ther. 2011, 10, 1173–1184. [Google Scholar] [CrossRef]
- Dumont, R.A.; Tamma, M.; Braun, F.; Borkowski, S.; Reubi, J.C.; Maecke, H.; Weber, W.A.; Mansi, R. Targeted radiotherapy of prostate cancer with a gastrin-releasing peptide receptor antagonist is effective as monotherapy and in combination with rapamycin. J. Nucl. Med. 2013, 54, 762–769. [Google Scholar] [CrossRef]
- Kranzbühler, B.; Salemi, S.; Umbricht, C.A.; Müller, C.; Burger, I.A.; Sulser, T.; Eberli, D. Pharmacological upregulation of prostate-specific membrane antigen (PSMA) expression in prostate cancer cells. Prostate 2018, 78, 758–765. [Google Scholar] [CrossRef]
- Grzmil, M.; Qin, Y.; Schleuniger, C.; Frank, S.; Imobersteg, S.; Blanc, A.; Spillmann, M.; Berger, P.; Schibli, R.; Behe, M. Pharmacological inhibition of mTORC1 increases CCKBR-specific tumor uptake of radiolabeled minigastrin analogue [177Lu]Lu-PP-F11N. Theranostics 2020, 10, 10861–10873. [Google Scholar] [CrossRef]
- Grzmil, M.; Imobersteg, S.; Blanc, A.; Frank, S.; Schibli, R.; Behe, M.P. Therapeutic Response of CCKBR-Positive Tumors to Combinatory Treatment with Everolimus and the Radiolabeled Minigastrin Analogue [177Lu]Lu-PP-F11N. Pharmaceutics 2021, 13, 2156. [Google Scholar] [CrossRef] [PubMed]
- Pool, S.E.; Bison, S.; Koelewijn, S.J.; van der Graaf, L.M.; Melis, M.; Krenning, E.P.; de Jong, M. mTOR inhibitor RAD001 promotes metastasis in a rat model of pancreatic neuroendocrine cancer. Cancer Res. 2013, 73, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Bison, S.M.; Pool, S.E.; Koelewijn, S.J.; van der Graaf, L.M.; Groen, H.C.; Melis, M.; de Jong, M. Peptide receptor radionuclide therapy (PRRT) with [177Lu-DOTA0,Tyr3]octreotate in combination with RAD001 treatment: Further investigations on tumor metastasis and response in the rat pancreatic CA20948 tumor model. EJNMMI Res. 2014, 4, 21. [Google Scholar] [CrossRef] [PubMed]
- Zellmer, J.; Yen, H.Y.; Kaiser, L.; Mille, E.; Gildehaus, F.J.; Böning, G.; Steiger, K.; Hacker, M.; Bartenstein, P.; Todica, A.; et al. Toxicity of a combined therapy using the mTOR-inhibitor everolimus and PRRT with [177Lu]Lu-DOTA-TATE in Lewis rats. EJNMMI Res. 2020, 10, 41. [Google Scholar] [CrossRef]
- Kamp, K.; Gumz, B.; Feelders, R.A.; Kwekkeboom, D.J.; Kaltsas, G.; Costa, F.P.; de Herder, W.W. Safety and efficacy of everolimus in gastrointestinal and pancreatic neuroendocrine tumors after 177Lu-octreotate. Endocr. Relat. Cancer 2013, 20, 825–831. [Google Scholar] [CrossRef]
- Medaer, E.; Verslype, C.; Van Cutsem, E.; Dekervel, J.; Clement, P.M.; Nackaerts, K.; Laenen, A.; Gheysens, O.; Goffin, K.; Jentjens, S.; et al. Influence of pretreatment with everolimus or sunitinib on the subacute hematotoxicity of 177Lu-DOTATATE PRRT. Acta Oncol. 2020, 59, 644–651. [Google Scholar] [CrossRef]
- Mileva, M.; Wimana, Z.; Flamen, P.; Karfis, I. Everolimus-induced somatostatin receptor overexpression in a rectal neuroendocrine tumor patient may promote somatostatin receptor-guided radionuclide therapy (peptide receptor radiotherapy) as an additional treatment option. World J. Nucl. Med. 2021, 20, 316–318. [Google Scholar] [CrossRef]
- Claringbold, P.G.; Turner, J.H. NeuroEndocrine Tumor Therapy with Lutetium-177-octreotate and Everolimus (NETTLE): A Phase I Study. Cancer Biother. Radiopharm. 2015, 30, 261–269. [Google Scholar] [CrossRef]
- Aljubran, A.; Badran, A.; Alrowaily, M.; Raef, H.; Alzahrani, A.M.; Almuhaideb, A.; Almanea, H.; El-Dali, A.; Tuli, M.; Bazarbashi, S. Efficacy of Everolimus Combined with 177Lu-Dotatate in the Treatment of Neuroendocrine Tumors. Cancer Biother. Radiopharm. 2022; in press. [Google Scholar] [CrossRef]
- Noh, W.C.; Mondesire, W.H.; Peng, J.; Jian, W.; Zhang, H.; Dong, J.; Mills, G.B.; Hung, M.C.; Meric-Bernstam, F. Determinants of rapamycin sensitivity in breast cancer cells. Clin. Cancer Res. 2004, 10, 1013–1023. [Google Scholar] [CrossRef]
- Hsu, H.S.; Lin, M.H.; Jang, Y.H.; Kuo, T.T.; Liu, C.C.; Cheng, T.H. The 4E-BP1/eIF4E ratio is a determinant for rapamycin response in esophageal cancer cells. J. Thorac. Cardiovasc. Surg. 2015, 149, 378–385. [Google Scholar] [CrossRef]
- Meng, L.H.; Zheng, X.F. Toward rapamycin analog (rapalog)-based precision cancer therapy. Acta Pharmacol. Sin. 2015, 36, 1163–1169. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grzmil, M.; Wiesmann, F.; Schibli, R.; Behe, M. Targeting mTORC1 Activity to Improve Efficacy of Radioligand Therapy in Cancer. Cancers 2023, 15, 17. https://doi.org/10.3390/cancers15010017
Grzmil M, Wiesmann F, Schibli R, Behe M. Targeting mTORC1 Activity to Improve Efficacy of Radioligand Therapy in Cancer. Cancers. 2023; 15(1):17. https://doi.org/10.3390/cancers15010017
Chicago/Turabian StyleGrzmil, Michal, Fabius Wiesmann, Roger Schibli, and Martin Behe. 2023. "Targeting mTORC1 Activity to Improve Efficacy of Radioligand Therapy in Cancer" Cancers 15, no. 1: 17. https://doi.org/10.3390/cancers15010017
APA StyleGrzmil, M., Wiesmann, F., Schibli, R., & Behe, M. (2023). Targeting mTORC1 Activity to Improve Efficacy of Radioligand Therapy in Cancer. Cancers, 15(1), 17. https://doi.org/10.3390/cancers15010017