
Role of Patient-Derived Models of Cancer in Translational Oncology




Abstract
Simple Summary
Abstract
1. Introduction
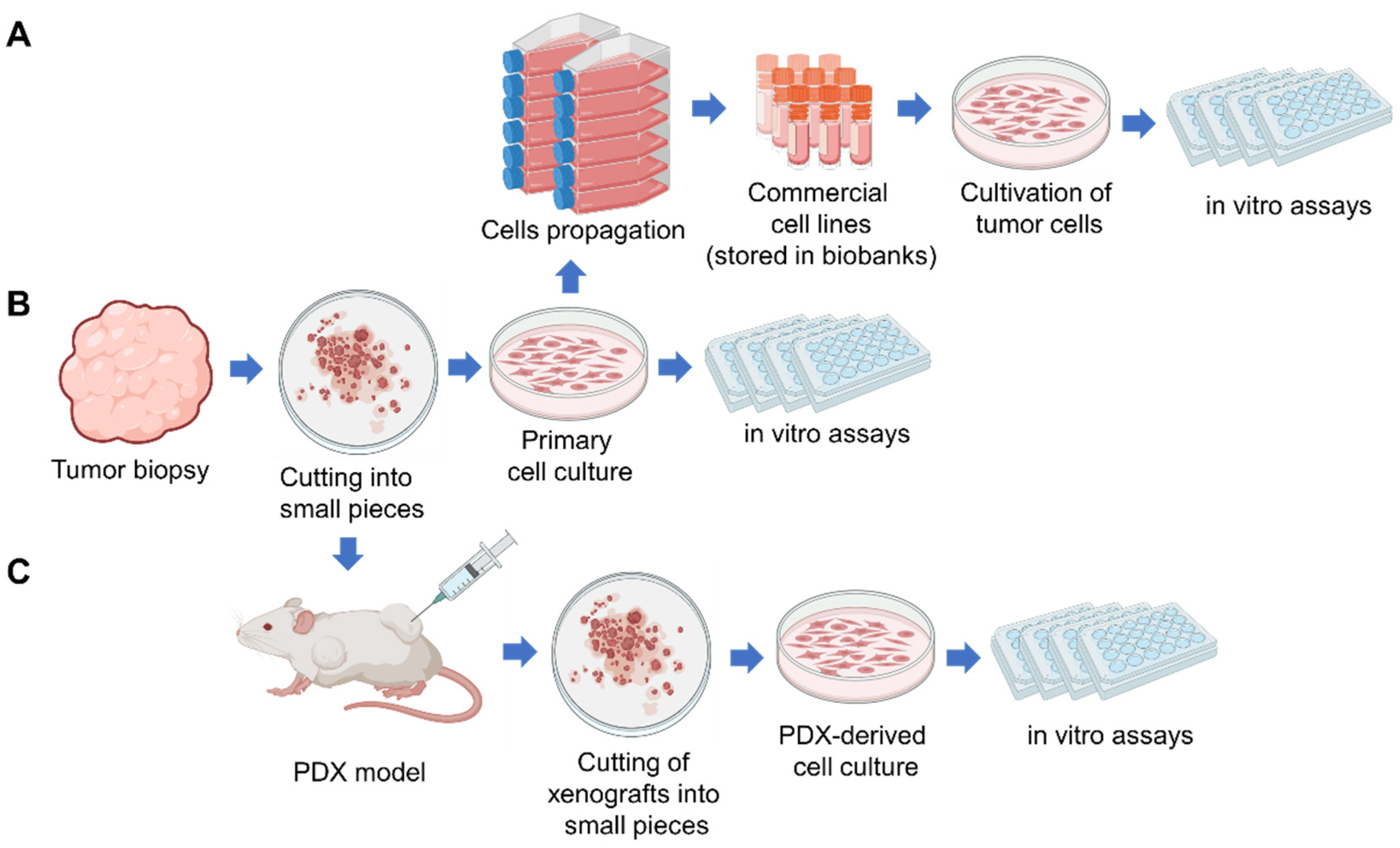
2. Patient-Derived Tumor Cell Cultures (PDC)
3. Patient-Derived Spheroids (PDS) and Organoids (PDO)
4. Complex PDO Model
5. Patient-Derived Tissue Slice Culture
6. Patient-Derived Xenografts
6.1. Mouse Patient-Derived Xenografts (mPDXs)
6.2. Zebrafish Patient-Derived Xenografts (zPDXs)
6.3. Chick Chorioallantoic Membrane Patient-Derived Xenografts (CAM-PDXs)
6.4. Humanized Mouse Patient-Derived Xenografts (Humanized mPDX)
6.5. PDX-Derived Organoids (PDXO) and PDX-Derived Cell Cultures (PDXC)
7. Databases and Computational Models of Patient-Derived Cancer Models
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J. Clin. 2022, 72, 1. [Google Scholar] [CrossRef] [PubMed]
- Boyd, M.R. The NCI Human Tumor Cell Line (60-Cell) Screen. In Anticancer Drug Development Guide; Humana Press Inc.: Totowa, NJ, USA, 2004; pp. 41–62. [Google Scholar] [CrossRef]
- Editorial Nature. Time to Tackle Cells’ Mistaken Identity. Nature 2015, 520, 264. [Google Scholar] [CrossRef]
- Horbach, S.P.J.M.; Halffman, W. The Ghosts of HeLa: How Cell Line Misidentification Contaminates the Scientific Literature. PLoS ONE 2017, 12, e0186281. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. US Cancer Institute to Overhaul Tumour Cell Lines. Nature 2016, 530, 391. [Google Scholar] [CrossRef]
- Mirabelli, P.; Coppola, L.; Salvatore, M. Cancer Cell Lines Are Useful Model Systems for Medical Research. Cancers 2019, 11, 1098. [Google Scholar] [CrossRef]
- Okada, S.; Vaeteewoottacharn, K.; Kariya, R. Application of Highly Immunocompromised Mice for the Establishment of Patient-Derived Xenograft (PDX) Models. Cells 2019, 8, 889. [Google Scholar] [CrossRef]
- Wang, J.; Chen, C.; Wang, L.; Xie, M.; Ge, X.; Wu, S.; He, Y.; Mou, X.; Ye, C.; Sun, Y. Patient-Derived Tumor Organoids: New Progress and Opportunities to Facilitate Precision Cancer Immunotherapy. Front. Oncol. 2022, 12, 872531. [Google Scholar] [CrossRef]
- Hou, X.; Du, C.; Lu, L.; Yuan, S.; Zhan, M.; You, P.; Du, H. Opportunities and Challenges of Patient-Derived Models in Cancer Research: Patient-Derived Xenografts, Patient-Derived Organoid and Patient-Derived Cells. World J. Surg. Oncol. 2022, 20, 37. [Google Scholar] [CrossRef]
- Lamb, J. The Connectivity Map: A New Tool for Biomedical Research. Nat. Rev. Cancer 2007, 7, 54–60. [Google Scholar] [CrossRef]
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A Resource for Therapeutic Biomarker Discovery in Cancer Cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia Enables Predictive Modelling of Anticancer Drug Sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Fekete, J.T.; Győrffy, B. A unified platform enabling biomarker ranking and validation for 1562 drugs using transcriptomic data of 1250 cancer cell lines. Comput. Struct. Biotechnol. J. 2022, 20, 2885–2894. [Google Scholar] [CrossRef]
- Zhong, C.H.; Tong, D.; Zhou, Z.Q.; Su, Z.Q.; Luo, Y.L.; Xing, J.; Bai, Y.L.; Guo, S.J.; Li, S.Y. Performance Evaluation of Detecting Circulating Tumor Cells and Tumor Cells in Bronchoalveolar Lavage Fluid in Diagnosis of Peripheral Lung Cancer. J. Thorac. Dis. 2018, 10, S830. [Google Scholar] [CrossRef]
- Giuntoli, R.L.; Webb, T.J.; Zoso, A.; Rogers, O.; Diaz-Montes, T.P.; Bristow, R.E.; Oelke, M. Ovarian Cancer-Associated Ascites Demonstrates Altered Immune Environment: Implications for Antitumor Immunity. Anticancer Res. 2009, 29, 2875–2884. [Google Scholar]
- Jones, J. Record of the First Physician to See Henrietta Lacks at the Johns Hopkins Hospital: History of the Beginning of the HeLa Cell Line. Am. J. Obstet. Gynecol. 1997, 176, s227–s228. [Google Scholar] [CrossRef]
- Rahbari, R.; Sheahan, T.; Modes, V.; Collier, P.; Macfarlane, C.; Badge, R.M. A Novel L1 Retrotransposon Marker for HeLa Cell Line Identification. Biotechniques 2009, 46, 277–284. [Google Scholar] [CrossRef]
- Salvadores, M.; Fuster-Tormo, F.; Supek, F. Matching Cell Lines with Cancer Type and Subtype of Origin via Mutational, Epigenomic, and Transcriptomic Patterns. Sci. Adv. 2020, 6, eaba1862. [Google Scholar] [CrossRef]
- Huo, K.G.; D’Arcangelo, E.; Tsao, M.S. Patient-Derived Cell Line, Xenograft and Organoid Models in Lung Cancer Therapy. Transl. Lung Cancer Res. 2020, 9, 2214. [Google Scholar] [CrossRef]
- Kim, S.Y.; Lee, J.Y.; Kim, D.H.; Joo, H.S.; Yun, M.R.; Jung, D.; Yun, J.; Heo, S.G.; Ahn, B.C.; Park, C.W.; et al. Patient-Derived Cells to Guide Targeted Therapy for Advanced Lung Adenocarcinoma. Sci. Rep. 2019, 9, 19909. [Google Scholar] [CrossRef]
- Wilding, J.L.; Bodmer, W.F. Cancer Cell Lines for Drug Discovery and Development. Cancer Res. 2014, 74, 2377–2384. [Google Scholar] [CrossRef]
- Riffle, S.; Hegde, R.S. Modeling Tumor Cell Adaptations to Hypoxia in Multicellular Tumor Spheroids. J. Exp. Clin. Cancer Res. 2017, 36, 102. [Google Scholar] [CrossRef]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of Multicell Spheroids in Tissue Culture as a Model of Nodular Carcinomas. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef]
- Zhou, Z.; Cong, L.; Cong, X. Patient-Derived Organoids in Precision Medicine: Drug Screening, Organoid-on-a-Chip and Living Organoid Biobank. Front. Oncol. 2021, 11, 762184. [Google Scholar] [CrossRef]
- Gilazieva, Z.; Ponomarev, A.; Rutland, C.; Rizvanov, A.; Solovyeva, V. Promising Applications of Tumor Spheroids and Organoids for Personalized Medicine. Cancers 2020, 12, 2727. [Google Scholar] [CrossRef]
- Singh, S.K.; Abbas, S.; Saxena, A.K.; Tiwari, S.; Sharma, L.K.; Tiwari, M. Critical Role of Three-Dimensional Tumorsphere Size on Experimental Outcome. Biotechniques 2020, 69, 333–338. [Google Scholar] [CrossRef]
- Yang, H.; Wang, Y.; Wang, P.; Zhang, N.; Wang, P. Tumor Organoids for Cancer Research and Personalized Medicine. Cancer Biol. Med. 2022, 19, 319–333. [Google Scholar] [CrossRef]
- Verduin, M.; Hoeben, A.; De Ruysscher, D.; Vooijs, M. Patient-Derived Cancer Organoids as Predictors of Treatment Response. Front. Oncol. 2021, 820, 641980. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Generation of Cerebral Organoids from Human Pluripotent Stem Cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef]
- Kunz-Schughart, L.A.; Freyer, J.P.; Hofstaedter, F.; Ebner, R. The Use of 3-D Cultures for High-Throughput Screening: The Multicellular Spheroid Model. J. Biomol. Screen. 2004, 9, 273–285. [Google Scholar] [CrossRef]
- Yoon, W.H.; Lee, H.R.; Kim, S.; Kim, E.; Ku, J.H.; Shin, K.; Jung, S. Use of Inkjet-Printed Single Cells to Quantify Intratumoral Heterogeneity. Biofabrication 2020, 12, 035030. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 Stem Cells Build Crypt-Villus Structures In Vitro without a Mesenchymal Niche. Nature 2009, 459, 7244. [Google Scholar] [CrossRef]
- Vlachogiannis, G.; Hedayat, S.; Vatsiou, A.; Jamin, Y.; Fernández-Mateos, J.; Khan, K.; Lampis, A.; Eason, K.; Huntingford, I.; Burke, R.; et al. Patient-Derived Organoids Model Treatment Response of Metastatic Gastrointestinal Cancers. Science 2018, 359, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Shimokawa, M.; Date, S.; Takano, A.; Matano, M.; Nanki, K.; Ohta, Y.; Toshimitsu, K.; Nakazato, Y.; Kawasaki, K.; et al. A Colorectal Tumor Organoid Library Demonstrates Progressive Loss of Niche Factor Requirements during Tumorigenesis. Cell Stem Cell 2016, 18, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Broutier, L.; Mastrogiovanni, G.; Verstegen, M.M.A.; Francies, H.E.; Gavarró, L.M.; Bradshaw, C.R.; Allen, G.E.; Arnes-Benito, R.; Sidorova, O.; Gaspersz, M.P.; et al. Human Primary Liver Cancer-Derived Organoid Cultures for Disease Modeling and Drug Screening. Nat. Med. 2017, 23, 1424–1435. [Google Scholar] [CrossRef]
- Seino, T.; Kawasaki, S.; Shimokawa, M.; Tamagawa, H.; Toshimitsu, K.; Fujii, M.; Ohta, Y.; Matano, M.; Nanki, K.; Kawasaki, K.; et al. Human Pancreatic Tumor Organoids Reveal Loss of Stem Cell Niche Factor Dependence during Disease Progression. Cell Stem Cell 2018, 22, 454–467. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Vela, I.; Sboner, A.; Iaquinta, P.J.; Karthaus, W.R.; Gopalan, A.; Dowling, C.; Wanjala, J.N.; Undvall, E.A.; Arora, V.K.; et al. Organoid Cultures Derived from Patients with Advanced Prostate Cancer. Cell 2014, 159, 176–187. [Google Scholar] [CrossRef]
- Sachs, N.; de Ligt, J.; Kopper, O.; Gogola, E.; Bounova, G.; Weeber, F.; Balgobind, A.V.; Wind, K.; Gracanin, A.; Begthel, H.; et al. A Living Biobank of Breast Cancer Organoids Captures Disease Heterogeneity. Cell 2018, 172, 373–386. [Google Scholar] [CrossRef]
- Costa, E.C.; de Melo-Diogo, D.; Moreira, A.F.; Carvalho, M.P.; Correia, I.J. Spheroids Formation on Non-Adhesive Surfaces by Liquid Overlay Technique: Considerations and Practical Approaches. Biotechnol. J. 2018, 13, 1700417. [Google Scholar] [CrossRef]
- Timmins, N.E.; Nielsen, L.K. Generation of Multicellular Tumor Spheroids by the Hanging-Drop Method. Methods Mol. Med. 2007, 140, 141–151. [Google Scholar] [CrossRef]
- Kundu, S.C.; Subhas, R.R.; Kundu, C.; Reis, R.L. Biomaterials for 3D Tumor Modeling; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Lazzari, G.; Couvreur, P.; Mura, S. Multicellular Tumor Spheroids: A Relevant 3D Model for the: In Vitro Preclinical Investigation of Polymer Nanomedicines. Polym. Chem. 2017, 8, 4947–4969. [Google Scholar] [CrossRef]
- Valdoz, J.C.; Johnson, B.C.; Jacobs, D.J.; Franks, N.A.; Dodson, E.L.; Sanders, C.; Cribbs, C.G.; Van Ry, P.M. The ECM: To Scaffold, or Not to Scaffold, That Is the Question. Int. J. Mol. Sci. 2021, 22, 12690. [Google Scholar] [CrossRef] [PubMed]
- Nii, T.; Katayama, Y. Biomaterial-Assisted Regenerative Medicine. Int. J. Mol. Sci. 2021, 22, 8657. [Google Scholar] [CrossRef] [PubMed]
- Qu, H.; Fu, H.; Han, Z.; Sun, Y. Biomaterials for Bone Tissue Engineering Scaffolds: A Review. RSC Adv. 2019, 9, 26252–26262. [Google Scholar] [CrossRef] [PubMed]
- Groeber, F.; Holeiter, M.; Hampel, M.; Hinderer, S.; Schenke-Layland, K. Skin Tissue Engineering—In Vivo and In Vitro Applications. Adv. Drug Deliv. Rev. 2011, 63, 352–366. [Google Scholar] [CrossRef]
- Nii, T.; Makino, K.; Tabata, Y. Three-Dimensional Culture System of Cancer Cells Combined with Biomaterials for Drug Screening. Cancers 2020, 12, 2754. [Google Scholar] [CrossRef]
- Peña, B.; Laughter, M.; Jett, S.; Rowland, T.J.; Taylor, M.R.G.; Mestroni, L.; Park, D. Injectable Hydrogels for Cardiac Tissue Engineering. Macromol. Biosci. 2018, 16, 1800079. [Google Scholar] [CrossRef]
- Kwee, B.J.; Mooney, D.J. Biomaterials for Skeletal Muscle Tissue Engineering. Curr. Opin. Biotechnol. 2017, 47, 16–22. [Google Scholar] [CrossRef]
- Langhans, S.A. Three-Dimensional In Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front. Pharmacol. 2018, 9, 6. [Google Scholar] [CrossRef]
- Unnikrishnan, K.; Thomas, L.V.; Ram Kumar, R.M. Advancement of Scaffold-Based 3D Cellular Models in Cancer Tissue Engineering: An Update. Front. Oncol. 2021, 11, 733652. [Google Scholar] [CrossRef]
- Ramadan, Q.; Zourob, M. 3D Bioprinting at the Frontier of Regenerative Medicine, Pharmaceutical, and Food Industries. Front. Med. Technol. 2021, 2, 607648. [Google Scholar] [CrossRef]
- Maloney, E.; Clark, C.; Sivakumar, H.; Yoo, K.; Aleman, J.; Rajan, S.A.P.; Forsythe, S.; Mazzocchi, A.; Laxton, A.W.; Tatter, S.B.; et al. Immersion Bioprinting of Tumor Organoids in Multi-Well Plates for Increasing Chemotherapy Screening Throughput. Micromachines 2020, 11, 208. [Google Scholar] [CrossRef]
- Driehuis, E.; Kretzschmar, K.; Clevers, H. Establishment of Patient-Derived Cancer Organoids for Drug-Screening Applications. Nat. Protoc. 2020, 15, 3380–3409. [Google Scholar] [CrossRef]
- Van De Wetering, M.; Francies, H.E.; Francis, J.M.; Bounova, G.; Iorio, F.; Pronk, A.; Van Houdt, W.; Van Gorp, J.; Taylor-Weiner, A.; Kester, L.; et al. Prospective Derivation of a Living Organoid Biobank of Colorectal Cancer Patients. Cell 2015, 161, 933–945. [Google Scholar] [CrossRef]
- Nath, S.; Devi, G.R. Three-Dimensional Culture Systems in Cancer Research: Focus on Tumor Spheroid Model. Pharmacol. Ther. 2016, 163, 94–108. [Google Scholar] [CrossRef]
- Liu, L.; Yu, L.; Li, Z.; Li, W.; Huang, W.R. Patient-Derived Organoid (PDO) Platforms to Facilitate Clinical Decision Making. J. Transl. Med. 2021, 19, 40. [Google Scholar] [CrossRef]
- Baker, K. Organoids Provide an Important Window on Inflammation in Cancer. Cancers 2018, 10, 151. [Google Scholar] [CrossRef]
- Jabs, J.; Zickgraf, F.M.; Park, J.; Wagner, S.; Jiang, X.; Jechow, K.; Kleinheinz, K.; Toprak, U.H.; Schneider, M.A.; Meister, M.; et al. Screening Drug Effects in Patient-Derived Cancer Cells Links Organoid Responses to Genome Alterations. Mol. Syst. Biol. 2017, 13, 955. [Google Scholar] [CrossRef]
- Xu, H.; Lyu, X.; Yi, M.; Zhao, W.; Song, Y.; Wu, K. Organoid Technology and Applications in Cancer Research. J. Hematol. Oncol. 2018, 11, 116. [Google Scholar] [CrossRef]
- Zumwalde, N.A.; Haag, J.D.; Sharma, D.; Mirrielees, J.A.; Wilke, L.G.; Gould, M.N.; Gumperz, J.E. Analysis of Immune Cells from Human Mammary Ductal Epithelial Organoids Reveals Vδ2+ T Cells That Efficiently Target Breast Carcinoma Cells in the Presence of Bisphosphonate. Cancer Prev. Res. 2016, 9, 305–316. [Google Scholar] [CrossRef]
- Direkze, N.C.; Alison, M.R. Bone Marrow and Tumour Stroma: An Intimate Relationship. Hematol. Oncol. 2006, 24, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal Stem Cells within Tumour Stroma Promote Breast Cancer Metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Strong, A.L.; Pei, D.T.; Hurst, C.G.; Gimble, J.M.; Burow, M.E.; Bunnell, B.A. Obesity Enhances the Conversion of Adipose-Derived Stromal/Stem Cells into Carcinoma-Associated Fibroblast Leading to Cancer Cell Proliferation and Progression to an Invasive Phenotype. Stem Cells Int. 2017, 2017, 9216502. [Google Scholar] [CrossRef]
- Mishra, P.J.; Mishra, P.J.; Humeniuk, R.; Medina, D.J.; Alexe, G.; Mesirov, J.P.; Ganesan, S.; Glod, J.W.; Banerjee, D. Carcinoma-Associated Fibroblast-like Differentiation of Human Mesenchymal Stem Cells. Cancer Res. 2008, 68, 4331–4339. [Google Scholar] [CrossRef] [PubMed]
- Quante, M.; Tu, S.P.; Tomita, H.; Gonda, T.; Wang, S.S.W.; Takashi, S.; Baik, G.H.; Shibata, W.; DiPrete, B.; Betz, K.S.; et al. Bone Marrow-Derived Myofibroblasts Contribute to the Mesenchymal Stem Cell Niche and Promote Tumor Growth. Cancer Cell 2011, 19, 257–272. [Google Scholar] [CrossRef]
- Waghray, M.; Yalamanchili, M.; Dziubinski, M.; Zeinali, M.; Erkkinen, M.; Yang, H.; Schradle, K.A.; Urs, S.; Di Magliano, M.P.; Welling, T.H.; et al. GM-CSF Mediates Mesenchymal–Epithelial Cross-Talk in Pancreatic Cancer. Cancer Discov. 2016, 6, 886–899. [Google Scholar] [CrossRef]
- Pereira, B.A.; Vennin, C.; Papanicolaou, M.; Chambers, C.R.; Herrmann, D.; Morton, J.P.; Cox, T.R.; Timpson, P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer 2019, 5, 724–741. [Google Scholar] [CrossRef]
- Ping, Q.; Yan, R.; Cheng, X.; Wang, W.; Zhong, Y.; Hou, Z.; Shi, Y.; Wang, C.; Li, R. Cancer-Associated Fibroblasts: Overview, Progress, Challenges, and Directions. Cancer Gene Ther. 2021, 28, 984–999. [Google Scholar] [CrossRef]
- Kalluri, R. The Biology and Function of Fibroblasts in Cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A Framework for Advancing Our Understanding of Cancer-Associated Fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Östman, A.; Augsten, M. Cancer-Associated Fibroblasts and Tumor Growth—Bystanders Turning into Key Players. Curr. Opin. Genet. Dev. 2009, 19, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct Populations of Inflammatory Fibroblasts and Myofibroblasts in Pancreatic Cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Liu, J.; Li, P.; Wang, L.; Li, M.; Ge, Z.; Noordam, L.; Lieshout, R.; Verstegen, M.M.A.; Ma, B.; Su, J.; et al. Cancer-Associated Fibroblasts Provide a Stromal Niche for Liver Cancer Organoids That Confers Trophic Effects and Therapy Resistance. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 407–431. [Google Scholar] [CrossRef]
- Tsai, S.; McOlash, L.; Palen, K.; Johnson, B.; Duris, C.; Yang, Q.; Dwinell, M.B.; Hunt, B.; Evans, D.B.; Gershan, J.; et al. Development of Primary Human Pancreatic Cancer Organoids, Matched Stromal and Immune Cells and 3D Tumor Microenvironment Models. BMC Cancer 2018, 18, 335. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, J.; Holokai, L.; Syu, L.J.; Steele, N.G.; Chang, J.; Wang, J.; Ahmed, S.; Dlugosz, A.; Zavros, Y. Hedgehog Signaling Induces PD-L1 Expression and Tumor Cell Proliferation in Gastric Cancer. Oncotarget 2018, 9, 37439. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, K.K.; Cattaneo, C.M.; Weeber, F.; Chalabi, M.; van de Haar, J.; Fanchi, L.F.; Slagter, M.; van der Velden, D.L.; Kaing, S.; Kelderman, S.; et al. Generation of Tumor-Reactive T Cells by Co-Culture of Peripheral Blood Lymphocytes and Tumor Organoids. Cell 2018, 174, 1586–1598. [Google Scholar] [CrossRef] [PubMed]
- Padmanaban, V.; Grasset, E.M.; Neumann, N.M.; Fraser, A.K.; Henriet, E.; Matsui, W.; Tran, P.T.; Cheung, K.J.; Georgess, D.; Ewald, A.J. Organotypic Culture Assays for Murine and Human Primary and Metastatic-Site Tumors. Nat. Protoc. 2020, 15, 2413–2442. [Google Scholar] [CrossRef] [PubMed]
- Finnberg, N.K.; Gokare, P.; Lev, A.; Grivennikov, S.I.; MacFarlane, A.W.; Campbell, K.S.; Winters, R.M.; Kaputa, K.; Farma, J.M.; Abbas, A.E.-S.; et al. Application of 3D Tumoroid Systems to Define Immune and Cytotoxic Therapeutic Responses Based on Tumoroid and Tissue Slice Culture Molecular Signatures. Oncotarget 2017, 8, 66747. [Google Scholar] [CrossRef]
- Lingampally, A.; Jones, M.R.; Bagari, S.; Chen, C.; Rivetti, S.; Bellusci, S. Use of the Reversible Myogenic to Lipogenic Transdifferentiation Switch for the Design of Pre-Clinical Drug Screening in Idiopathic Pulmonary Fibrosis. Front. Bioeng. Biotechnol. 2020, 8, 569865. [Google Scholar] [CrossRef]
- Angeles, M.; Torrejon, M.; Gangoso, E.; Pollard, S.M. Modelling Glioblastoma Tumour-Host Cell Interactions Using Adult Brain Organotypic Slice Co-Culture. DMM Dis. Model. Mech. 2018, 11, dmm031435. [Google Scholar] [CrossRef]
- Kenerson, H.L.; Sullivan, K.M.; Seo, Y.D.; Stadeli, K.M.; Ussakli, C.; Yan, X.; Lausted, C.; Pillarisetty, V.G.; Park, J.O.; Riehle, K.J.; et al. Tumor Slice Culture as a Biologic Surrogate of Human Cancer. Ann. Transl. Med. 2020, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Kenerson, H.L.; Sullivan, K.M.; Labadie, K.P.; Pillarisetty, V.G.; Yeung, R.S. Protocol for Tissue Slice Cultures from Human Solid Tumors to Study Therapeutic Response. STAR Protoc. 2021, 2, 100574. [Google Scholar] [CrossRef] [PubMed]
- Carranza-Torres, I.E.; Guzmán-Delgado, N.E.; Coronado-Martínez, C.; Bañuelos-García, J.I.; Viveros-Valdez, E.; Morán-Martínez, J.; Carranza-Rosales, P. Organotypic Culture of Breast Tumor Explants as a Multicellular System for the Screening of Natural Compounds with Antineoplastic Potential. BioMed Res. Int. 2015, 2015, 618021. [Google Scholar] [CrossRef] [PubMed]
- Zech, H.B.; Berger, J.; Mansour, W.Y.; Nordquist, L.; von Bargen, C.M.; Bußmann, L.; Oetting, A.; Christiansen, S.; Möckelmann, N.; Böttcher, A.; et al. Patient Derived Ex Vivo Tissue Slice Cultures Demonstrate a Profound DNA Double-Strand Break Repair Defect in HPV-Positive Oropharyngeal Head and Neck Cancer. Radiother. Oncol. 2022, 168, 138–146. [Google Scholar] [CrossRef]
- Vaira, V.; Fedele, G.; Pyne, S.; Fasoli, E.; Zadra, G.; Bailey, D.; Snyder, E.; Faversani, A.; Coggi, G.; Flavin, R.; et al. Preclinical Model of Organotypic Culture for Pharmacodynamic Profiling of Human Tumors. Proc. Natl. Acad. Sci. USA 2010, 107, 8352–8356. [Google Scholar] [CrossRef]
- Jiang, X.; Seo, Y.D.; Chang, J.H.; Coveler, A.; Nigjeh, E.N.; Pan, S.; Jalikis, F.; Yeung, R.S.; Crispe, I.N.; Pillarisetty, V.G. Long-Lived Pancreatic Ductal Adenocarcinoma Slice Cultures Enable Precise Study of the Immune Microenvironment. Oncoimmunology 2017, 6, e1333210. [Google Scholar] [CrossRef]
- Van De Merbel, A.F.; Van Der Horst, G.; Van Der Mark, M.H.; Van Uhm, J.I.M.; Van Gennep, E.J.; Kloen, P.; Beimers, L.; Pelger, R.C.M.; Van Der Pluijm, G. An Ex Vivo Tissue Culture Model for the Assessment of Individualized Drug Responses in Prostate and Bladder Cancer. Front. Oncol. 2018, 8, 400. [Google Scholar] [CrossRef]
- Roelants, C.; Pillet, C.; Franquet, Q.; Sarrazin, C.; Peilleron, N.; Giacosa, S.; Guyon, L.; Fontanell, A.; Fiard, G.; Long, J.A.; et al. Ex-Vivo Treatment of Tumor Tissue Slices as a Predictive Preclinical Method to Evaluate Targeted Therapies for Patients with Renal Carcinoma. Cancers 2020, 12, 232. [Google Scholar] [CrossRef]
- Stenzel, P.J.; Hörner, N.; Foersch, S.; Wagner, D.C.; Tsaur, I.; Thomas, A.; Haferkamp, A.; Macher-Goeppinger, S.; Roth, W.; Porubsky, S.; et al. Nivolumab Reduces PD1 Expression and Alters Density and Proliferation of Tumor Infiltrating Immune Cells in a Tissue Slice Culture Model of Renal Cell Carcinoma. Cancers 2021, 13, 4511. [Google Scholar] [CrossRef]
- Sönnichsen, R.; Hennig, L.; Blaschke, V.; Winter, K.; Körfer, J.; Hähnel, S.; Monecke, A.; Wittekind, C.; Jansen-Winkeln, B.; Thieme, R.; et al. Individual Susceptibility Analysis Using Patient-Derived Slice Cultures of Colorectal Carcinoma. Clin. Color. Cancer 2018, 17, 4511. [Google Scholar] [CrossRef]
- Wulf-Goldenberg, A.; Hoffmann, J.; Becker, M.; Brzezicha, B.; Walther, W. Patient-Derived Xenografts from Solid Tumors (PDX) for Models of Metastasis. In Methods in Molecular Biology; Humana: New York, NY, USA, 2021; pp. 43–58. [Google Scholar] [CrossRef]
- Ivanics, T.; Bergquist, J.R.; Liu, G.; Kim, M.P.; Kang, Y.; Katz, M.H.; Perez, M.V.R.; Thomas, R.M.; Fleming, J.B.; Truty, M.J. Patient-Derived Xenograft Cryopreservation and Reanimation Outcomes Are Dependent on Cryoprotectant Type. Lab. Investig. 2018, 98, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, T.G.; Thériault, B.L.; Campbell, E.J.; Nachtigal, M.W. Primary Culture of Ovarian Surface Epithelial Cells and Ascites-Derived Ovarian Cancer Cells from Patients. Nat. Protoc. 2006, 1, 2643–2649. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Li, J.; Cadilha, B.L.; Markota, A.; Voigt, C.; Huang, Z.; Lin, P.P.; Wang, D.D.; Dai, J.; Kranz, G.; et al. Epithelial-Type Systemic Breast Carcinoma Cells with a Restricted Mesenchymal Transition Are a Major Source of Metastasis. Sci. Adv. 2019, 5, eaav4275. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Pi, C.; Li, K.; Sheng, L.; Zuo, Y.; Yuan, J.; Zou, Y.; Zhang, X.; Zhao, W.; Lee, R.J.; et al. Patient-Derived Xenograft: A More Standard “Avatar” Model in Preclinical Studies of Gastric Cancer. Front. Oncol. 2022, 12, 898563. [Google Scholar] [CrossRef] [PubMed]
- McCauley, H.A.; Wells, J.M. Pluripotent Stem Cell-Derived Organoids: Using Principles of Developmental Biology to Grow Human Tissues in a Dish. Development 2017, 144, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Abdolahi, S.; Ghazvinian, Z.; Muhammadnejad, S.; Saleh, M.; Asadzadeh Aghdaei, H.; Baghaei, K. Patient-Derived Xenograft (PDX) Models, Applications and Challenges in Cancer Research. J. Transl. Med. 2022, 20, 206. [Google Scholar] [CrossRef]
- Kamili, A.; Gifford, A.J.; Li, N.; Mayoh, C.; Chow, S.O.; Failes, T.W.; Eden, G.L.; Cadiz, R.; Xie, J.; Lukeis, R.E.; et al. Accelerating Development of High-Risk Neuroblastoma Patient-Derived Xenograft Models for Preclinical Testing and Personalised Therapy. Br. J. Cancer 2020, 122, 680–691. [Google Scholar] [CrossRef]
- DeRose, Y.S.; Wang, G.; Lin, Y.C.; Bernard, P.S.; Buys, S.S.; Ebbert, M.T.; Factor, R.; Matsen, C.; Milash, B.A.; Nelson, E.; et al. Tumor grafts derived from women with breast cancer authentically reflect tumor pathology, growth, metastasis and disease outcomes. Nat. Med. 2011, 17, 1514–1520. [Google Scholar] [CrossRef]
- Pearson, A.T.; Finkel, K.A.; Warner, K.A.; Nör, F.; Tice, D.; Martins, M.D.; Jackson, T.L.; Nör, J.E. Patient-Derived Xenograft (PDX) Tumors Increase Growth Rate with Time. Oncotarget 2016, 7, 7993. [Google Scholar] [CrossRef]
- Ben-David, U.; Ha, G.; Tseng, Y.Y.; Greenwald, N.F.; Oh, C.; Shih, J.; McFarland, J.M.; Wong, B.; Boehm, J.S.; Beroukhim, R.; et al. Patient-Derived Xenografts Undergo Mouse-Specific Tumor Evolution. Nat. Genet. 2017, 49, 1567–1575. [Google Scholar] [CrossRef]
- Woo, X.Y.; Giordano, J.; Srivastava, A.; Zhao, Z.M.; Lloyd, M.W.; de Bruijn, R.; Suh, Y.S.; Patidar, R.; Chen, L.; Scherer, S.; et al. Conservation of Copy Number Profiles during Engraftment and Passaging of Patient-Derived Cancer Xenografts. Nat. Genet. 2021, 53, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Trusolino, L.; Martino, C.; Bencardino, K.; Lonardi, S.; Bergamo, F.; Zagonel, V.; Leone, F.; Depetris, I.; Martinelli, E.; et al. Dual-Targeted Therapy with Trastuzumab and Lapatinib in Treatment-Refractory, KRAS Codon 12/13 Wild-Type, HER2-Positive Metastatic Colorectal Cancer (HERACLES): A Proof-of-Concept, Multicentre, Open-Label, Phase 2 Trial. Lancet Oncol. 2016, 17, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Bruckheimer, E.; Rajeshkumar, N.V.; Garrido-Laguna, I.; De Oliveira, E.; Rubio-Viqueira, B.; Strawn, S.; Wick, M.J.; Martell, J.; Sidransky, D. A Pilot Clinical Study of Treatment Guided by Personalized Tumorgrafts in Patients with Advanced Cancer. Mol. Cancer Ther. 2011, 10, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Laguna, I.; Uson, M.; Rajeshkumar, N.V.; Tan, A.C.; De Oliveira, E.; Karikari, C.; Villaroel, M.C.; Salomon, A.; Taylor, G.; Sharma, R.; et al. Tumor Engraftment in Nude Mice and Enrichment in Stroma-Related Gene Pathways Predict Poor Survival and Resistance to Gemcitabine in Patients with Pancreatic Cancer. Clin. Cancer Res. 2011, 17, 5793–5800. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, G.J. Applications of Patient-Derived Tumor Xenograft Models and Tumor Organoids. J. Hematol. Oncol. 2020, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, Y.; Yao, T.; Jia, R. Benefits of Zebrafish Xenograft Models in Cancer Research. Front. Cell Dev. Biol. 2021, 7, 616551. [Google Scholar] [CrossRef]
- Roth, S.M.; Berens, E.B.; Sharif, G.M.; Glasgow, E.; Wellstein, A. Cancer Cell Invasion and Metastasis in Zebrafish Models (Danio rerio). In Methods in Molecular Biology; Humana: New York, NY, USA, 2021; pp. 3–16. [Google Scholar] [CrossRef]
- Ali, Z.; Vildevall, M.; Rodriguez, G.V.; Tandiono, D.; Vamvakaris, I.; Evangelou, G.; Lolas, G.; Syrigos, K.N.; Villanueva, A.; Wick, M.; et al. Zebrafish Patient-Derived Xenograft Models Predict Lymph Node Involvement and Treatment Outcome in Non-Small Cell Lung Cancer. J. Exp. Clin. Cancer Res. 2022, 41, 58. [Google Scholar] [CrossRef]
- Lawson, N.D.; Weinstein, B.M. In Vivo Imaging of Embryonic Vascular Development Using Transgenic Zebrafish. Dev. Biol. 2002, 248, 307–318. [Google Scholar] [CrossRef]
- Cross, L.M.; Cook, M.A.; Lin, S.; Chen, J.N.; Rubinstein, A.L. Rapid Analysis of Angiogenesis Drugs in a Live Fluorescent Zebrafish Assay. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 911–912. [Google Scholar] [CrossRef]
- Gamble, J.T.; Elson, D.J.; Greenwood, J.A.; Tanguay, R.L.; Kolluri, S.K. The Zebrafish Xenograft Models for Investigating Cancer and Cancer Therapeutics. Biology 2021, 10, 252. [Google Scholar] [CrossRef]
- Rebelo de Almeida, C.; Mendes, R.V.; Pezzarossa, A.; Gago, J.; Carvalho, C.; Alves, A.; Nunes, V.; Brito, M.J.; Cardoso, M.J.; Ribeiro, J.; et al. Zebrafish Xenografts as a Fast Screening Platform for Bevacizumab Cancer Therapy. Commun. Biol. 2020, 3, 299. [Google Scholar] [CrossRef] [PubMed]
- Di Franco, G.; Usai, A.; Piccardi, M.; Cateni, P.; Palmeri, M.; Pollina, L.E.; Gaeta, R.; Marmorino, F.; Cremolini, C.; Dente, L.; et al. Zebrafish Patient-Derived Xenograft Model to Predict Treatment Outcomes of Colorectal Cancer Patients. Biomedicines 2022, 10, 1474. [Google Scholar] [CrossRef] [PubMed]
- Fior, R.; Póvoa, V.; Mendes, R.V.; Carvalho, T.; Gomes, A.; Figueiredo, N.; Ferreira, F.R. Single-Cell Functional and Chemosensitive Profiling of Combinatorial Colorectal Therapy in Zebrafish Xenografts. Proc. Natl. Acad. Sci. USA 2017, 114, E8234–E8243. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.G.; Young, L.E.A.; Moore, L.H.; Chernyavskaya, Y.; Wei, M.; Markussen, K.H.; Ptacek, A.; Dockins, S.; Sanders, W.C.; Sun, R.C.; et al. Optimization of Human Cancer Cell Xenograft into Zebrafish Larvae for Anti-Cancer Drug Screening. Mol. Biosyst. 2021, preprint. [Google Scholar]
- Chu, P.Y.; Koh, A.P.F.; Antony, J.; Huang, R.Y.J. Applications of the Chick Chorioallantoic Membrane as an Alternative Model for Cancer Studies. Cells Tissues Organs 2022, 211, 222–237. [Google Scholar] [CrossRef]
- Van Weerden, W.M. Patient-Derived Xenograft Models in Cancer Research. Cancers 2021, 13, 815. [Google Scholar] [CrossRef]
- Sharrow, A.C.; Ishihara, M.; Hu, J.; Kim, I.H.; Wu, L. Using the Chicken Chorioallantoic Membrane In Vivo Model to Study Gynecological and Urological Cancers. J. Vis. Exp. 2020, 2020, e60651. [Google Scholar] [CrossRef]
- Komatsu, A.; Higashi, Y.; Matsumoto, K. Various CAM Tumor Models. Enzymes 2019, 46, 37–57. [Google Scholar] [CrossRef]
- Uloza, V.; Kuzminienė, A.; Palubinskienė, J.; Balnytė, I.; Ulozienė, I.; Valančiūtė, A. Model of Human Recurrent Respiratory Papilloma on Chicken Embryo Chorioallantoic Membrane for Tumor Angiogenesis Research. Histol. Histopathol. 2017, 32, 7. [Google Scholar] [CrossRef]
- Uloza, V.; Kuzminiene, A.; Šalomskaite-Davalgiene, S.; Palubinskiene, J.; Balnyte, I.; Uloziene, I.; Šaferis, V.; Valančiute, A. Effect of Laryngeal Squamous Cell Carcinoma Tissue Implantation on the Chick Embryo Chorioallantoic Membrane: Morphometric Measurements and Vascularity. BioMed Res. Int. 2015, 2015, 629754. [Google Scholar] [CrossRef] [PubMed]
- DeBord, L.C.; Pathak, R.R.; Villaneuva, M.; Liu, H.-C.; Harrington, D.A.; Yu, W.; Lewis, M.T.; Sikora, A.G. The Chick Chorioallantoic Membrane (CAM) as a Versatile Patient-Derived Xenograft (PDX) Platform for Precision Medicine and Preclinical Research. Am. J. Cancer Res. 2018, 8, 8. [Google Scholar]
- Pawlikowska, P.; Tayoun, T.; Oulhen, M.; Faugeroux, V.; Rouffiac, V.; Aberlenc, A.; Pommier, A.L.; Honore, A.; Marty, V.; Bawa, O.; et al. Exploitation of the Chick Embryo Chorioallantoic Membrane (CAM) as a Platform for Anti-Metastatic Drug Testing. Sci. Rep. 2020, 10, 16876. [Google Scholar] [CrossRef] [PubMed]
- Sys, G.; Van Bockstal, M.; Forsyth, R.; Balke, M.; Poffyn, B.; Uyttendaele, D.; Bracke, M.; De Wever, O. Tumor Grafts Derived from Sarcoma Patients Retain Tumor Morphology, Viability, and Invasion Potential and Indicate Disease Outcomes in the Chick Chorioallantoic Membrane Model. Cancer Lett. 2012, 326, 69–78. [Google Scholar] [CrossRef]
- Pizon, M.; Schott, D.; Pachmann, U.; Schobert, R.; Pizon, M.; Wozniak, M.; Bobinski, R.; Pachmann, K. Chick Chorioallantoic Membrane (CAM) Assays as a Model of Patient-Derived Xenografts from Circulating Cancer Stem Cells (CCSCs) in Breast Cancer Patients. Cancers 2022, 14, 1476. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Lee, S.; Kim, K.; Kim, S.H.; Chung, Y.J.; Lee, C. Studying Cancer Immunotherapy Using Patient-Derived Xenografts (PDXs) in Humanized Mice. Exp. Mol. Med. 2018, 50, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, M.; Chou, F.S.; Link, K.A.; Mizukawa, B.; Perry, R.L.; Carroll, M.; Mulloy, J.C. AML Xenograft Efficiency Is Significantly Improved in NOD/SCID-IL2RG Mice Constitutively Expressing Human SCF, GM-CSF and IL-3. Leukemia 2010, 24, 1785–1788. [Google Scholar] [CrossRef]
- Morton, J.J.; Bird, G.; Keysar, S.B.; Astling, D.P.; Lyons, T.R.; Anderson, R.T.; Glogowska, M.J.; Estes, P.; Eagles, J.R.; Le, P.N.; et al. XactMice: Humanizing Mouse Bone Marrow Enables Microenvironment Reconstitution in a Patient-Derived Xenograft Model of Head and Neck Cancer. Oncogene 2016, 35, 3. [Google Scholar] [CrossRef]
- Guillen, K.P.; Fujita, M.; Butterfield, A.J.; Scherer, S.D.; Bailey, M.H.; Chu, Z.; DeRose, Y.S.; Zhao, L.; Cortes-Sanchez, E.; Yang, C.H.; et al. A Human Breast Cancer-Derived Xenograft and Organoid Platform for Drug Discovery and Precision Oncology. Nat. Cancer 2022, 3, 232–250. [Google Scholar] [CrossRef]
- Huang, L.; Bockorny, B.; Paul, I.; Akshinthala, D.; Frappart, P.O.; Gandarilla, O.; Bose, A.; Sanchez-Gonzalez, V.; Rouse, E.E.; Lehoux, S.D.; et al. PDX-Derived Organoids Model In Vivo Drug Response and Secrete Biomarkers. JCI Insight 2020, 5, e135544. [Google Scholar] [CrossRef]
- Xu, X.; Qian, W.; Guo, S.; Li, H.Q. Abstract 180: Systematic Genomic Analysis of Matched PDX, PDX-Derived Organoids (PDXO), and PDX-Derived Cell Lines (PDXC). Cancer Res. 2020, 80, 180–181. [Google Scholar] [CrossRef]
- Xu, X.; Shang, L.; Wang, P.; Zhou, J.; Ouyang, X.; Zheng, M.; Mao, B.; Zhang, L.; Chen, B.; Wang, J.; et al. Creating Matched In Vivo/In Vitro Patient-Derived Model Pairs of PDX and PDX-Derived Organoids for Cancer Pharmacology Research. J. Vis. Exp. 2021, 2021, e61382. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinská, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Mælandsmo, G.M.; et al. Patient-Derived Xenograft Models: An Emerging Platform for Translational Cancer Research. Cancer Discov. 2014, 4, 9981013. [Google Scholar] [CrossRef] [PubMed]
- Tentler, J.J.; Tan, A.C.; Weekes, C.D.; Jimeno, A.; Leong, S.; Pitts, T.M.; Arcaroli, J.J.; Messersmith, W.A.; Eckhardt, S.G. Patient-Derived Tumour Xenografts as Models for Oncology Drug Development. Nat. Rev. Clin. Oncol. 2012, 9, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Den Breems, N.Y.; Eftimie, R. The Re-Polarisation of M2 and M1 Macrophages and Its Role on Cancer Outcomes. J. Theor. Biol. 2016, 390, 23–39. [Google Scholar] [CrossRef]
- Eftimie, R.; Hamam, H. Modelling and Investigation of the CD4+ T Cells—Macrophages Paradox in Melanoma Immunotherapies. J. Theor. Biol. 2017, 420, 82–104. [Google Scholar] [CrossRef]
- Shin, S.Y.; Müller, A.K.; Verma, N.; Lev, S.; Nguyen, L.K. Systems Modelling of the EGFR-PYK2-c-Met Interaction Network Predicts and Prioritizes Synergistic Drug Combinations for Triple-Negative Breast Cancer. PLoS Comput. Biol. 2018, 14, e1006192. [Google Scholar] [CrossRef]
- Li, X.; Jolly, M.K.; George, J.T.; Pienta, K.J.; Levine, H. Computational Modeling of the Crosstalk between Macrophage Polarization and Tumor Cell Plasticity in the Tumor Microenvironment. Front. Oncol. 2019, 9, 10. [Google Scholar] [CrossRef]
- Portman, N.; Lim, E. A New Sophistication for Breast Cancer PDXs. Nat. Cancer 2022, 3, 138–140. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Method | Description | Reference |
|---|---|---|
| Liquid overlay technique (LOT) | Cell suspension is seeded on a non-adhesive substrate, such as low adhesion or agar-coated plates preferably with round bottom, to prevent cells from attaching to surfaces. Advantages of LOT technique are relative ease of implementation, low price and ability to track spheroids in real time. The disadvantages are inconsistent size and shape of spheroids. | [40] |
| Hanging drop | Cell suspension (20–40 μL) is dropped onto a cap, which is then turned over, and cell aggregation occurs at the top of the drop due to surface tension and gravity. Advantages of this method are ease of handling and low price. The disadvantages are that it is labor intensive, limited in cultivation time and there are difficulties in observing the spheroids formation. | [41] |
| Suspension culture based on agitation or magnetic levitation | Cell suspension is cultivated in a rotating flask or bioreactor with agitation or magnetic levitation. Constant stirring prevents cells from setting and attaching to the surfaces of a device, and the use of a medium with increased viscosity stimulates intercellular adhesion. Advantage of this method is large yield of spheroids (~300). The disadvantage is large variation of obtained spheroids in size and not uniformity. | [42] |
| Micromolding microwells | A recently developed method for obtaining spheroids using arrays of micropits made by microforming or photolithography. Low adhesion surfaces are obtained using non-adhesive materials such as polydimethylsiloxane or by coating with agarose. Advantages of this method are spheroids formation with specific size and composition, small amount of cells, media, and reagents are required. The disadvantages are complexity and high cost of the equipment, as well as the inability to extract and characterize in detail the formed spheroids. | [43] |
| Scaffold-based | Cells are embedded into the matrix resembling extracellular matrix (ECM) of biological origin (collagen, fibrin or Matrigel) or synthetic (hyaluronic acid (HA), polyethylene glycol (PEG), polylactic acid (PA), polyglycolic acid (PGA)). The advantage of this method is replicating cell–ECM interactions. The disadvantages are difficult visualization of 3D structures with automated imaging systems and variations of biological scaffolds from batch-to-batch. | [44,45,46,47,48,49,50,51,52] |
| Immersion Bioprinting | Cells are mixed with hydrogel and bioprinted (20 μL) into a viscous gelatine bath, layered in 96-well plate. The gelatine bath prevents adhesion of cells to the plate and supports a spherical form of a spheroid. The gelatine bath later on aspirated and substituted with culture medium. The advantage of this method is consistency of spheroids with maintaining a high throughput format. | [53,54] |
| Model | Advantages | Disadvantages |
|---|---|---|
| Primary PDC—patient-derived cell culture |
|
|
| PDS—patient-derived spheroids |
|
|
| PDO—patient-derived organoids |
|
|
| PDTSC—patient-derived tissue slice culture |
|
|
| Mouse PDX—patient-derived xenografts |
|
|
| Zebrafish PDX |
|
|
| CAM-PDX |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Idrisova, K.F.; Simon, H.-U.; Gomzikova, M.O. Role of Patient-Derived Models of Cancer in Translational Oncology. Cancers 2023, 15, 139. https://doi.org/10.3390/cancers15010139
Idrisova KF, Simon H-U, Gomzikova MO. Role of Patient-Derived Models of Cancer in Translational Oncology. Cancers. 2023; 15(1):139. https://doi.org/10.3390/cancers15010139
Chicago/Turabian StyleIdrisova, K. F., H.-U. Simon, and M. O. Gomzikova. 2023. "Role of Patient-Derived Models of Cancer in Translational Oncology" Cancers 15, no. 1: 139. https://doi.org/10.3390/cancers15010139
APA StyleIdrisova, K. F., Simon, H.-U., & Gomzikova, M. O. (2023). Role of Patient-Derived Models of Cancer in Translational Oncology. Cancers, 15(1), 139. https://doi.org/10.3390/cancers15010139