
Reducing Chemotherapy-Induced DNA Damage via nAChR-Mediated Redox Reprograming—A New Mechanism for SCLC Chemoresistance Boosted by Nicotine

 , , ,
, , ,  ,
, 
Abstract
:Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture Conditions
2.2. Cell Viability
2.3. Cell Proliferation
2.4. Western Blotting
2.5. Apoptosis Analysis
2.6. Colony Formation Assay
2.7. Comet Assay
2.8. Tumor Digestion
2.9. Quantitative Real-Time PCR (RT-qPCR)
2.10. GSH, NADPH, and ROS Assays
2.11. A 60-Day Exposure of H841 Cells with Nicotine, Cotinine, Cisplatin or Their Combination
2.12. In Vivo Xenograft Experiments
2.13. Statistical Analysis
3. Results
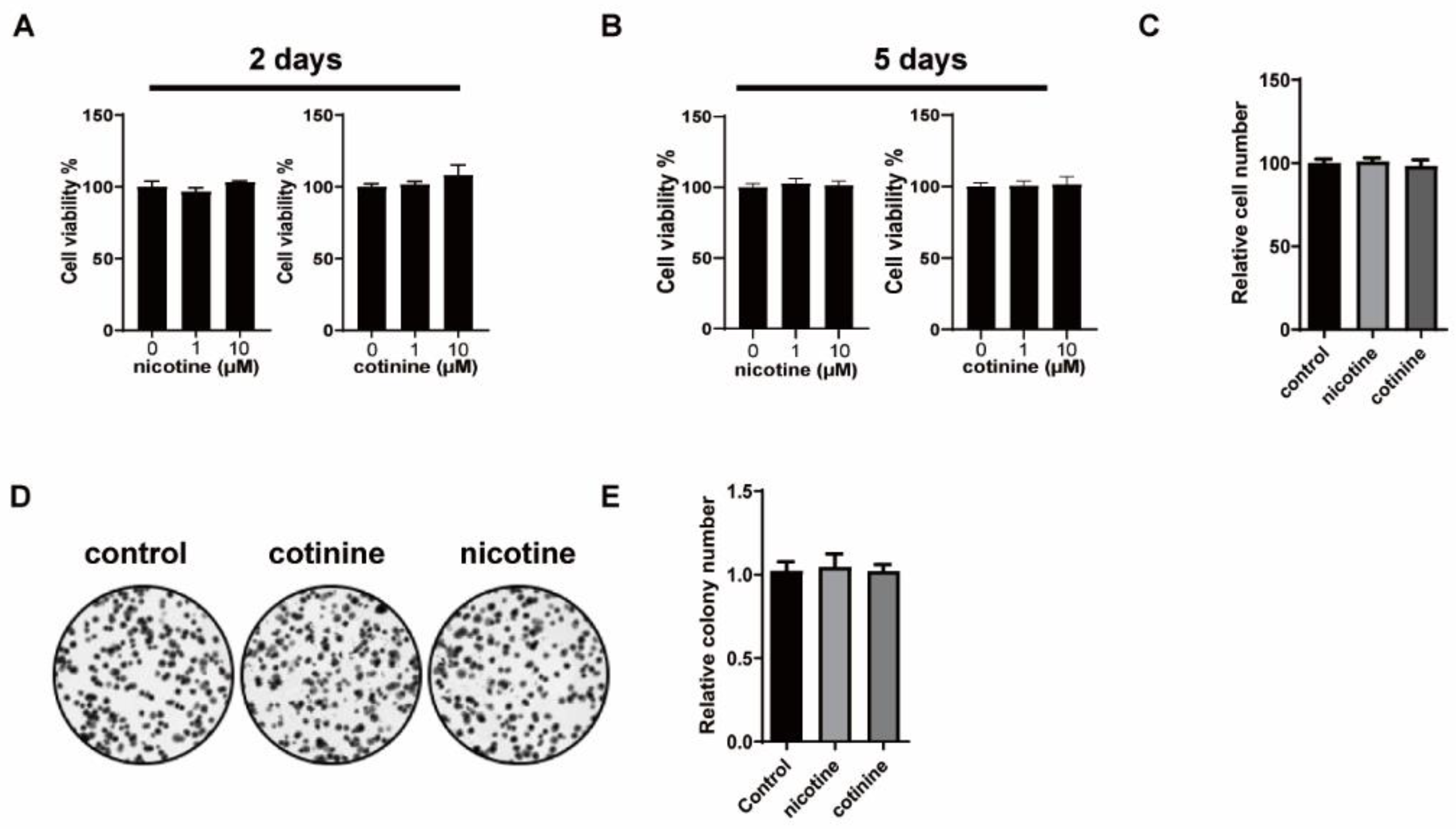
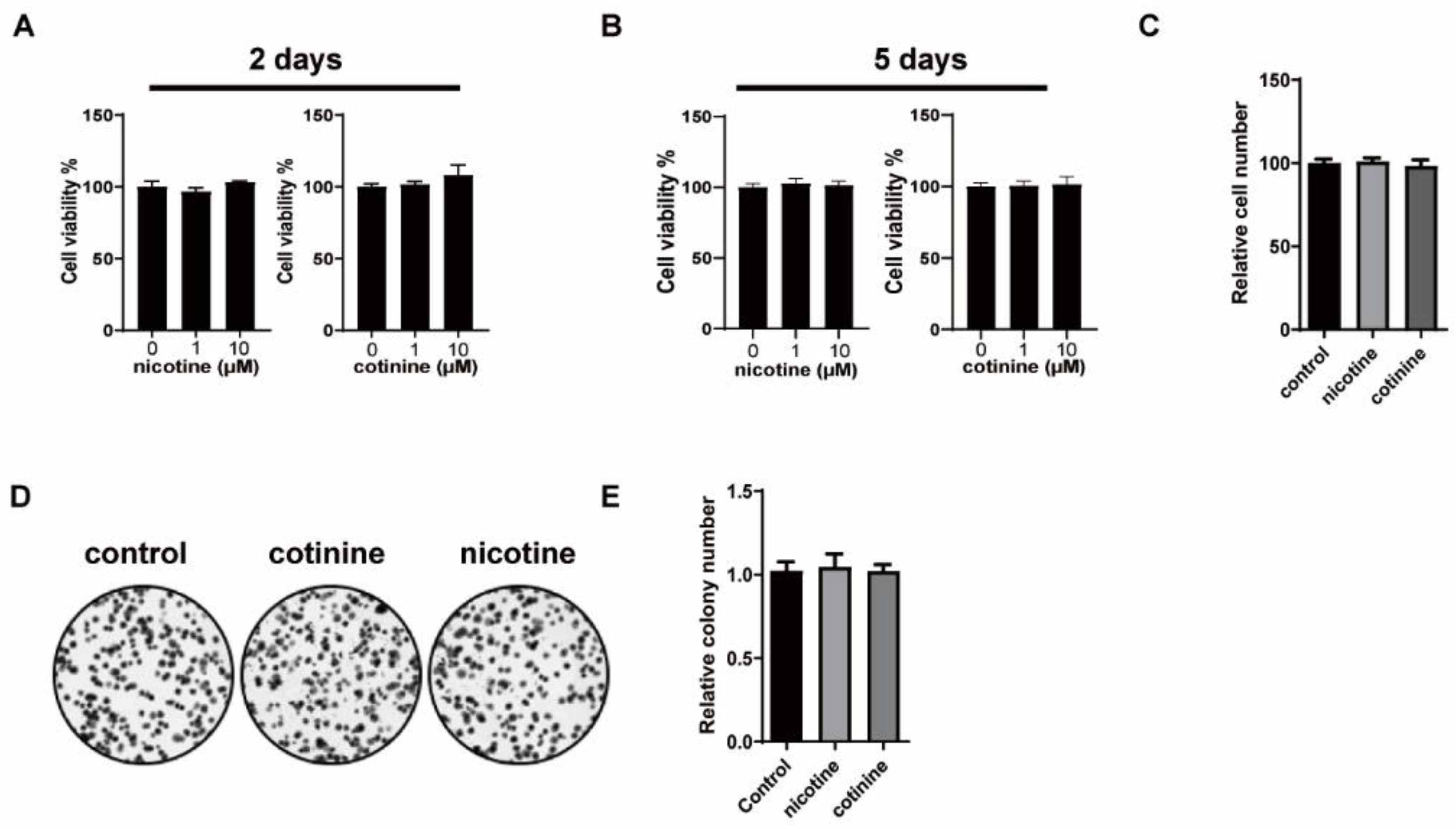
3.1. Nicotine or Cotinine Had No Effect on SCLC Cell Proliferation
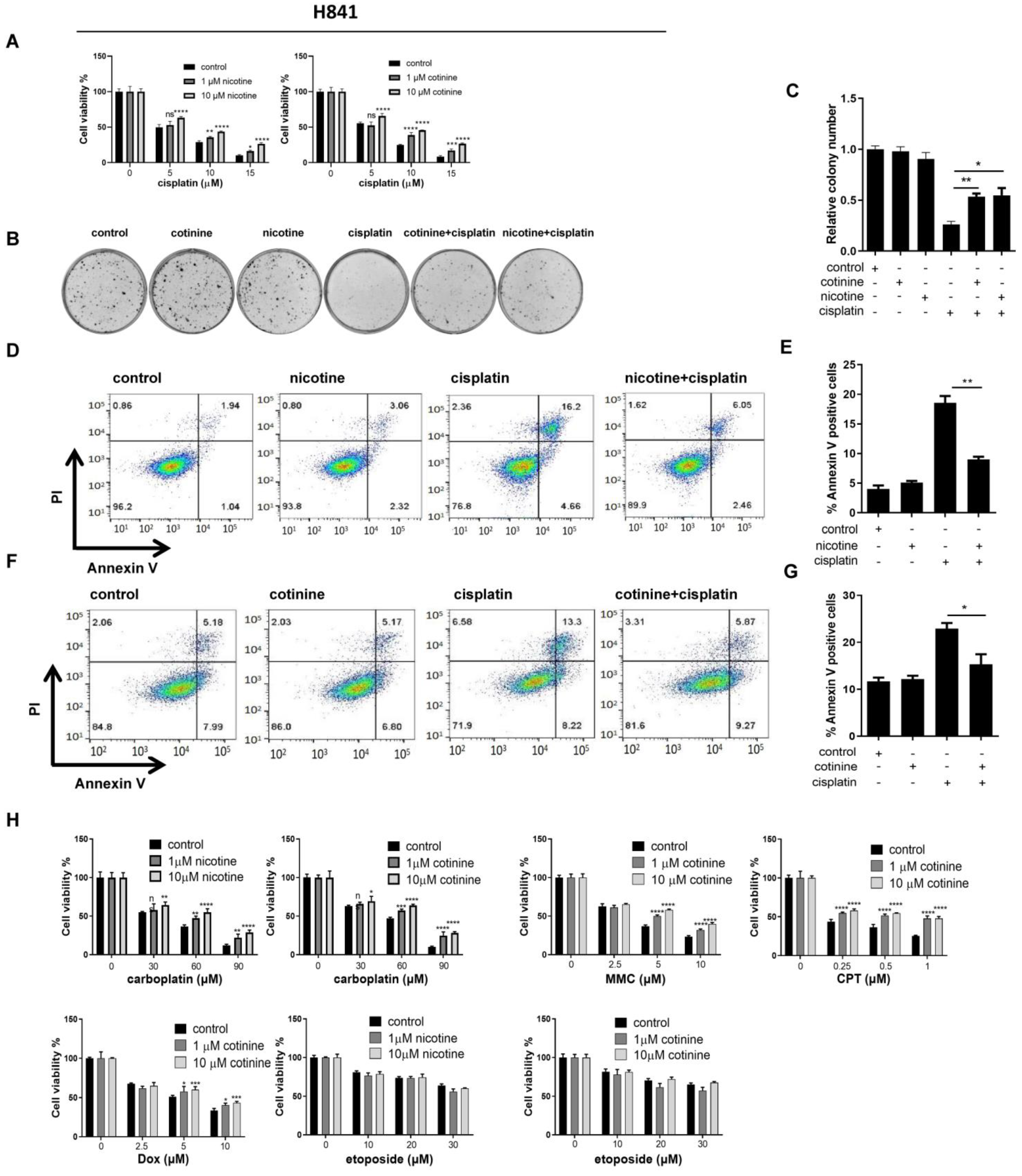
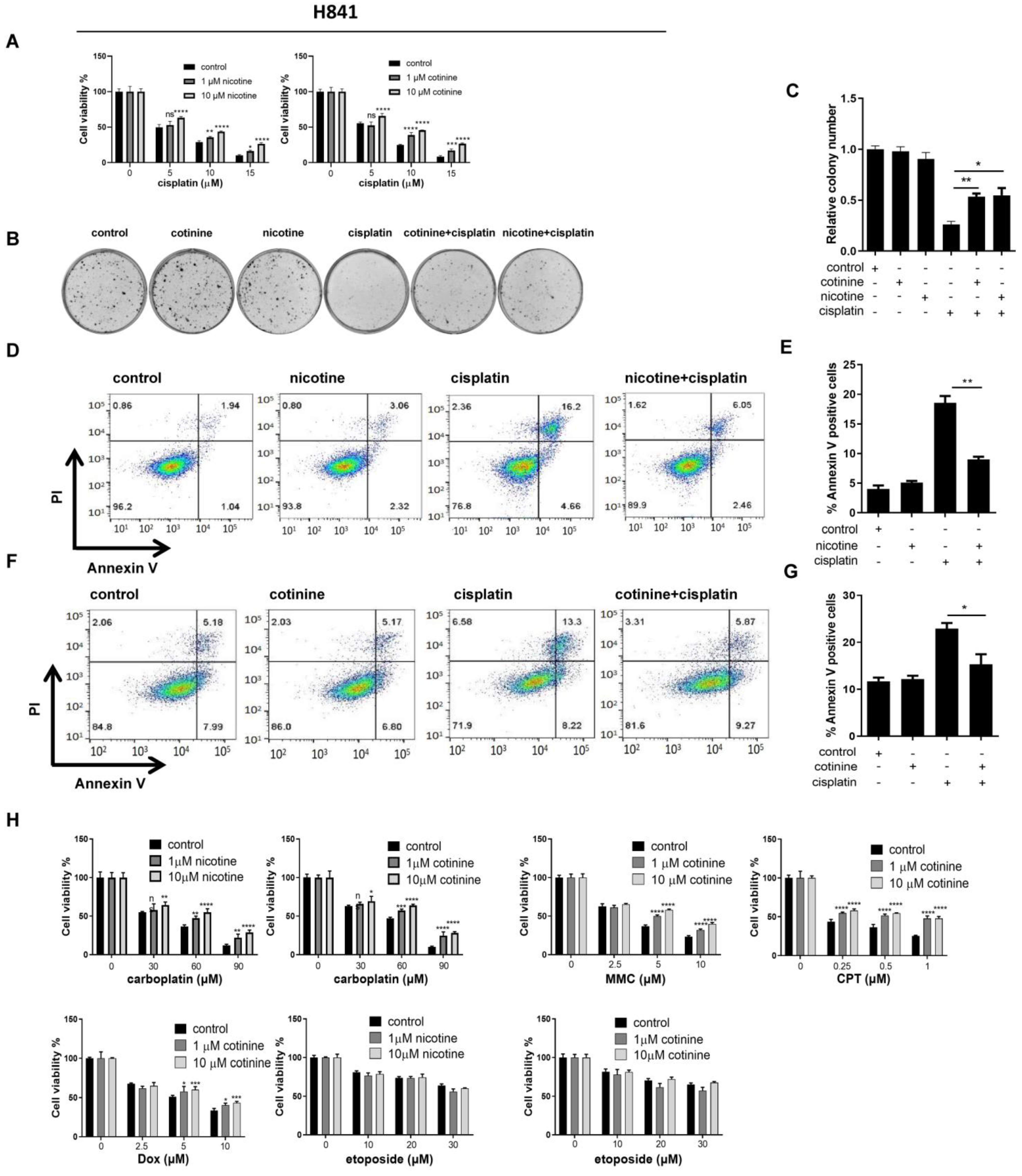
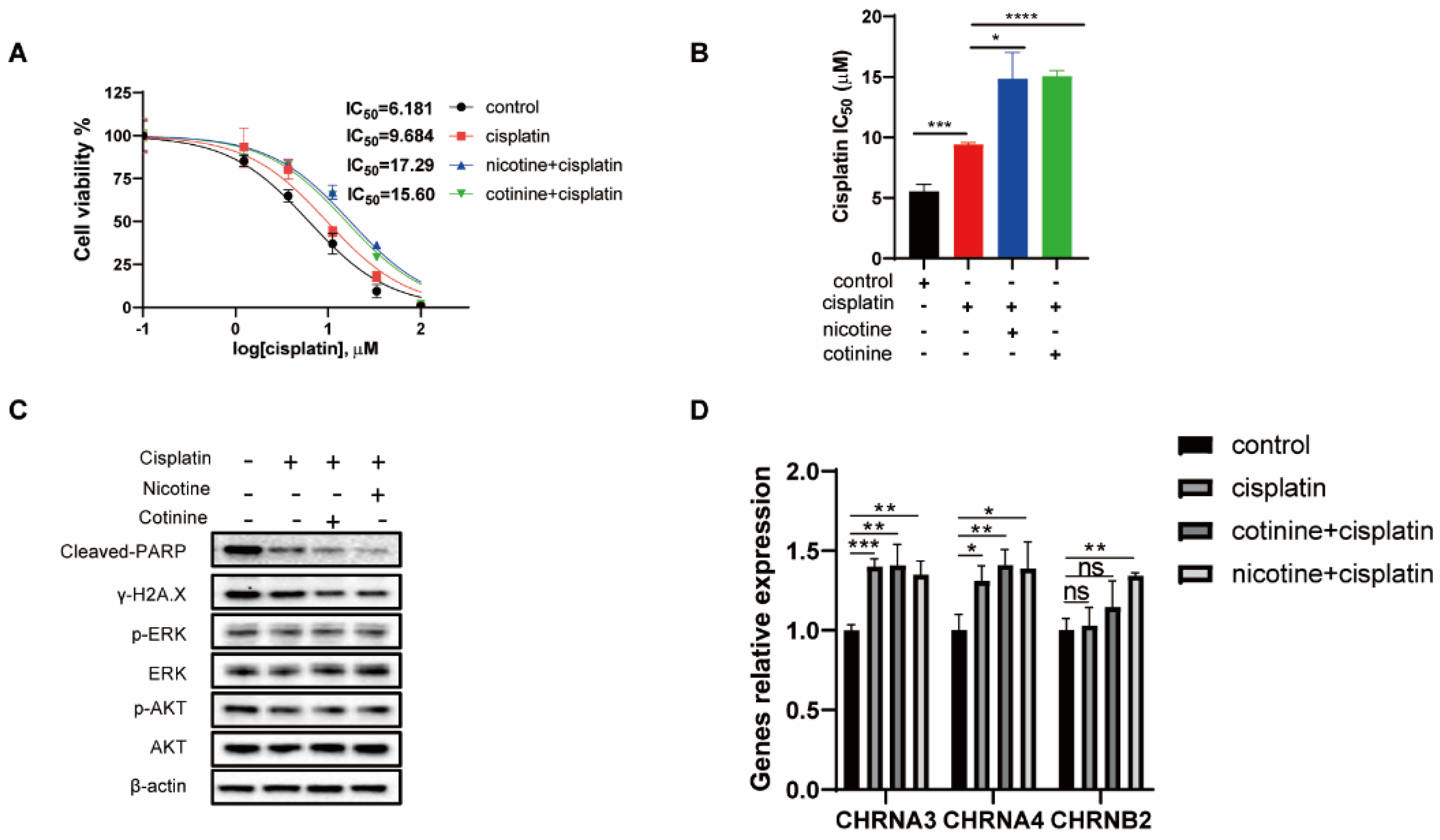
3.2. Nicotine or Cotinine Differentially Reduced the Sensitivity of SCLC Cells to Chemotherapies
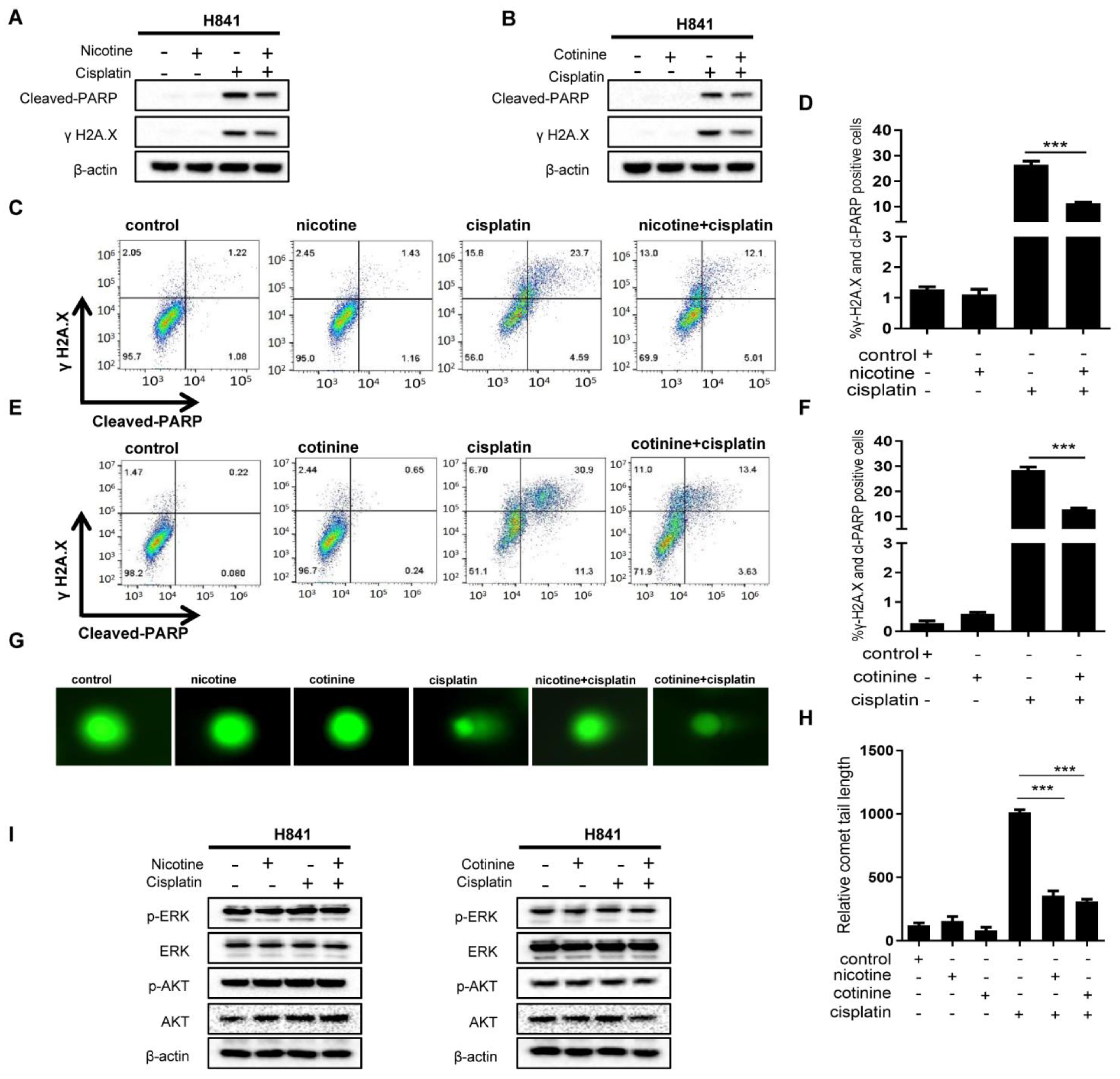
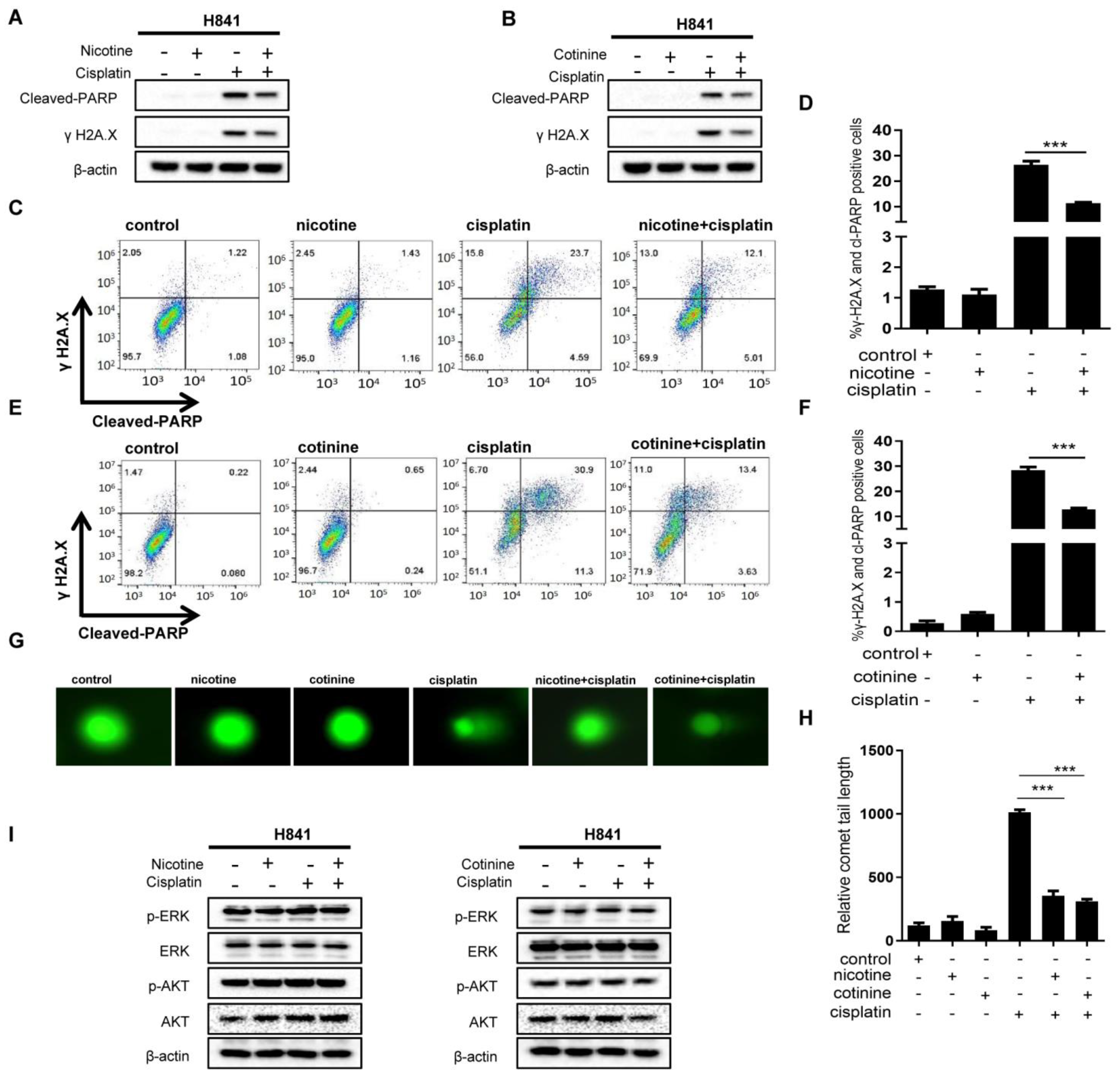
3.3. Nicotine or Cotinine Reduced Chemotherapy-Induced DNA Damage in SCLC Cells
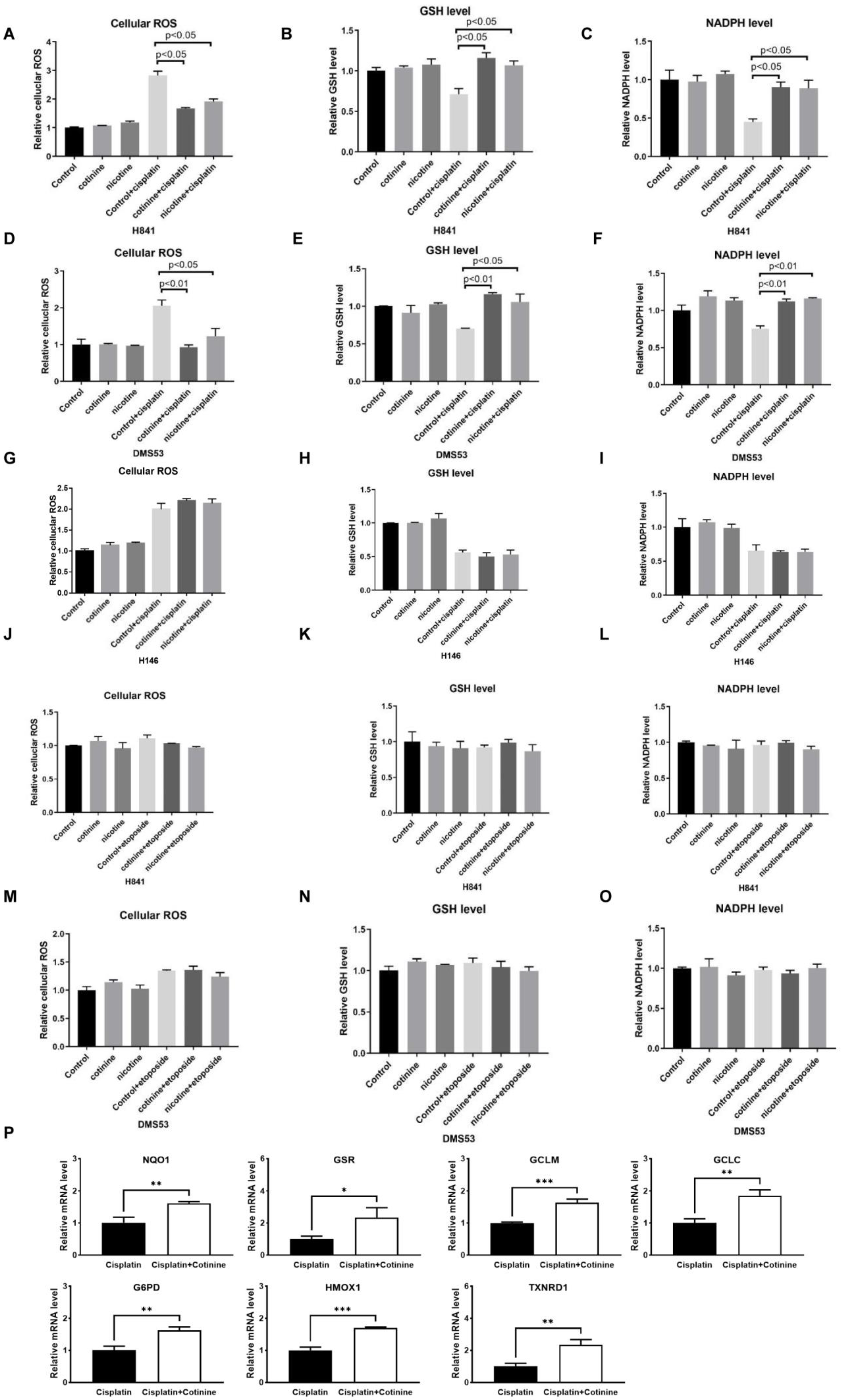
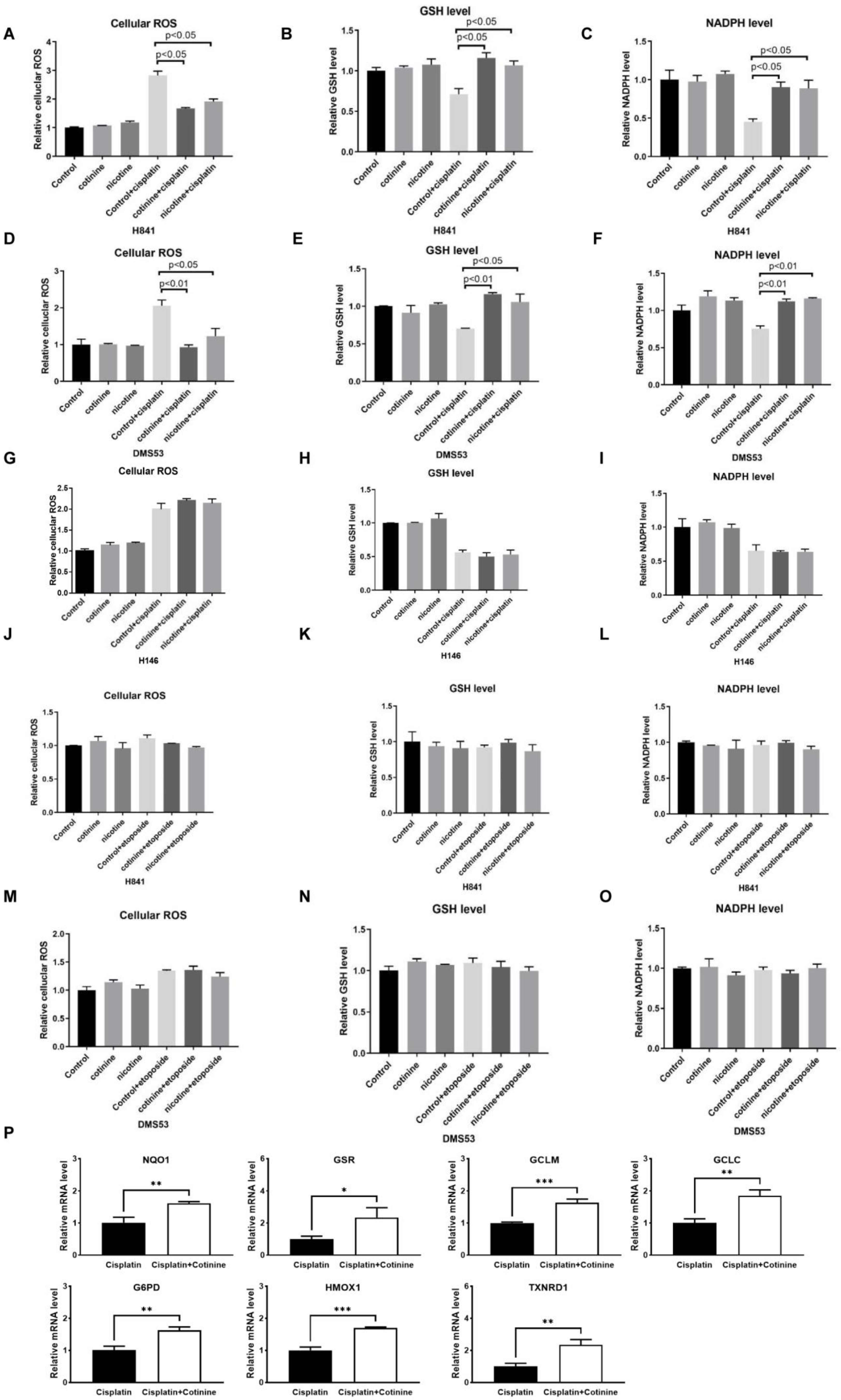
3.4. Nicotine or Cotinine Modulated Cellular Redox Processes, Contributing to the Reductions in Chemotherapy-Induced DNA Damage in SCLC Cells
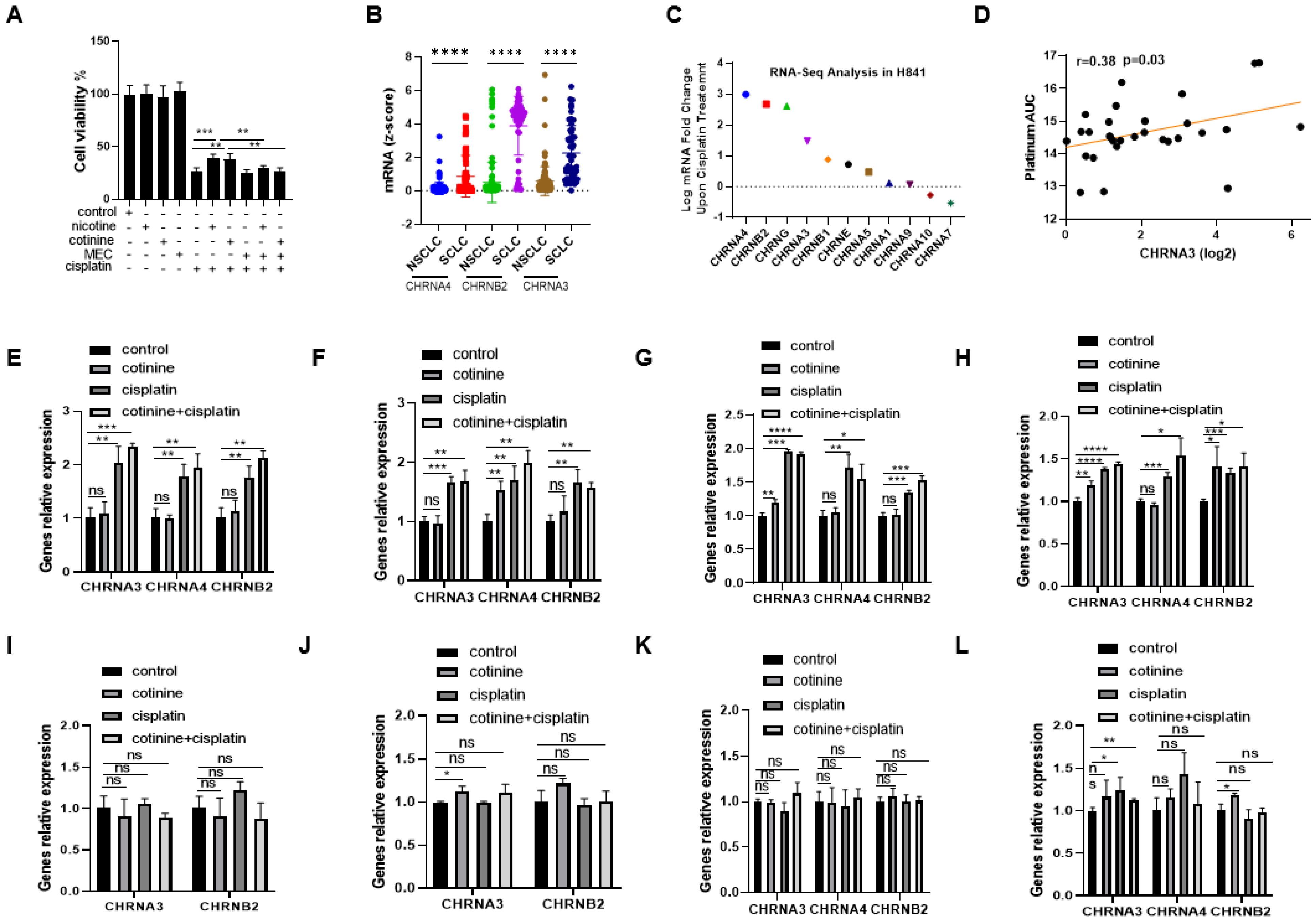
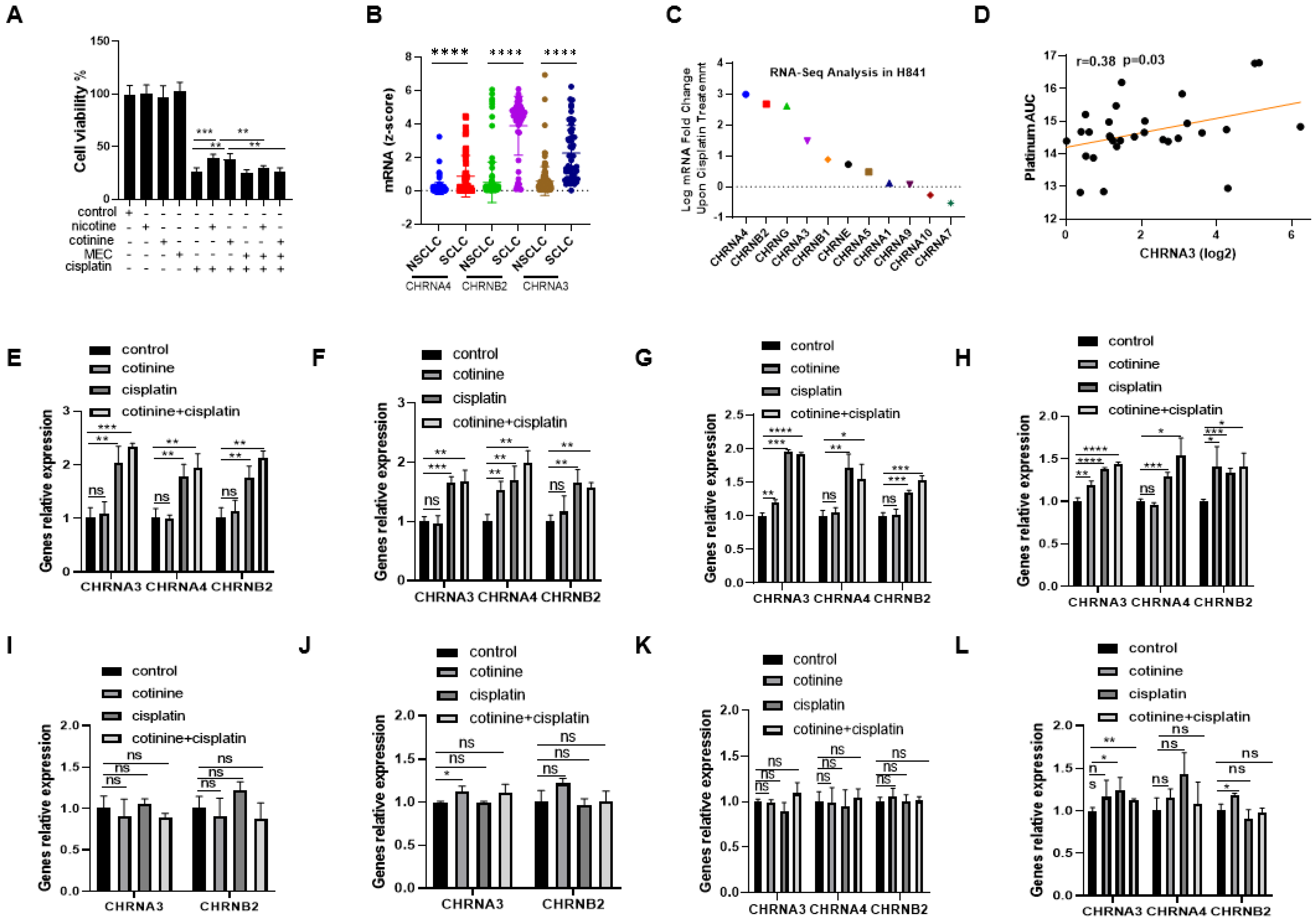
3.5. nAChRs as the UpStream Targets and Their UpRegulation upon Platinum-Based Chemotherapy Treatment as a Self-Defense Mechanism for SCLC to Become Less Sensitive to Chemotherapy While Nicotine or Cotinine Accelerate Resistance Acquisition
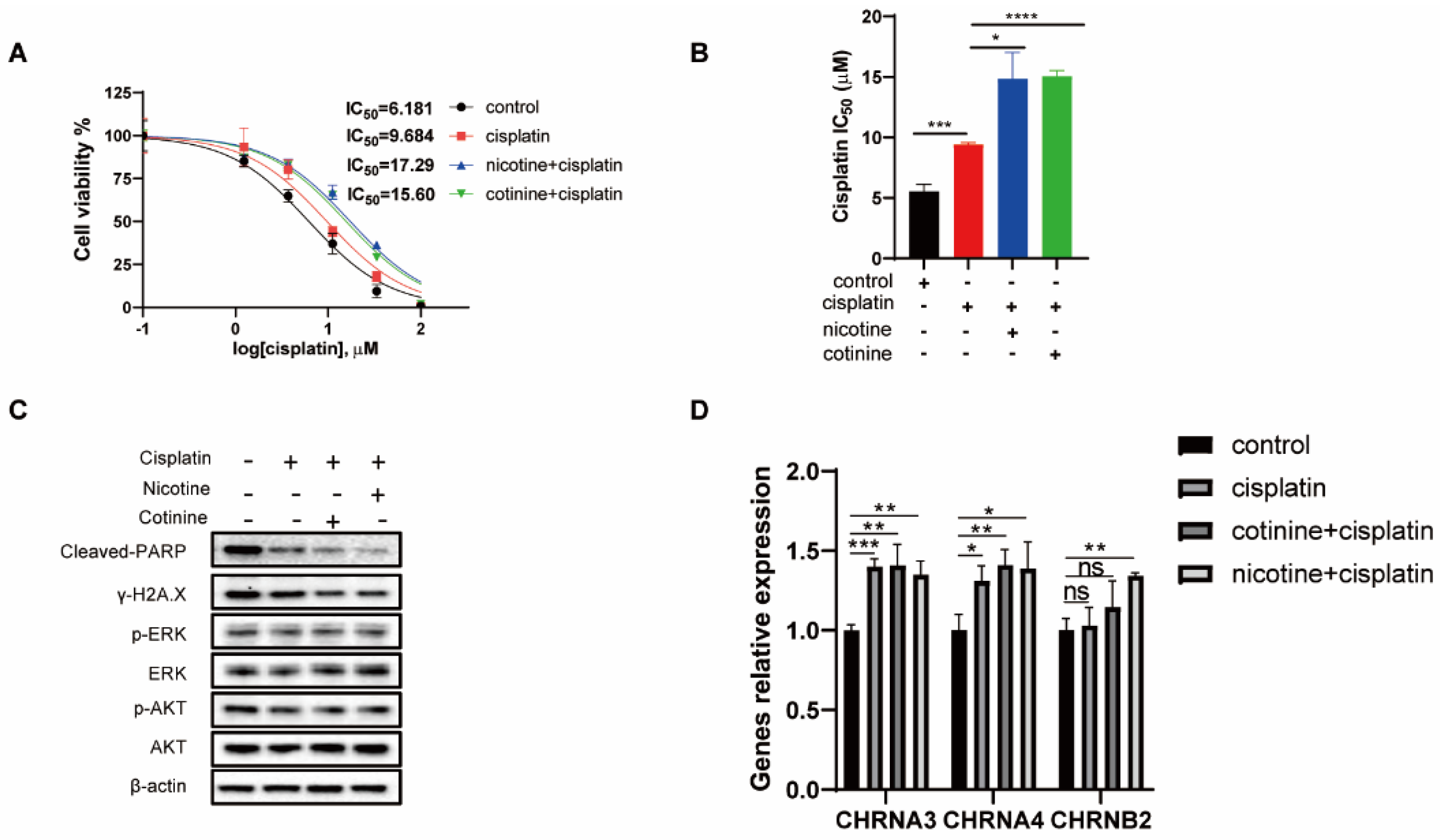
3.6. A 60-Day Nicotine or Cotinine Exposure on H841 Cells Boosted Cisplatin Resistance
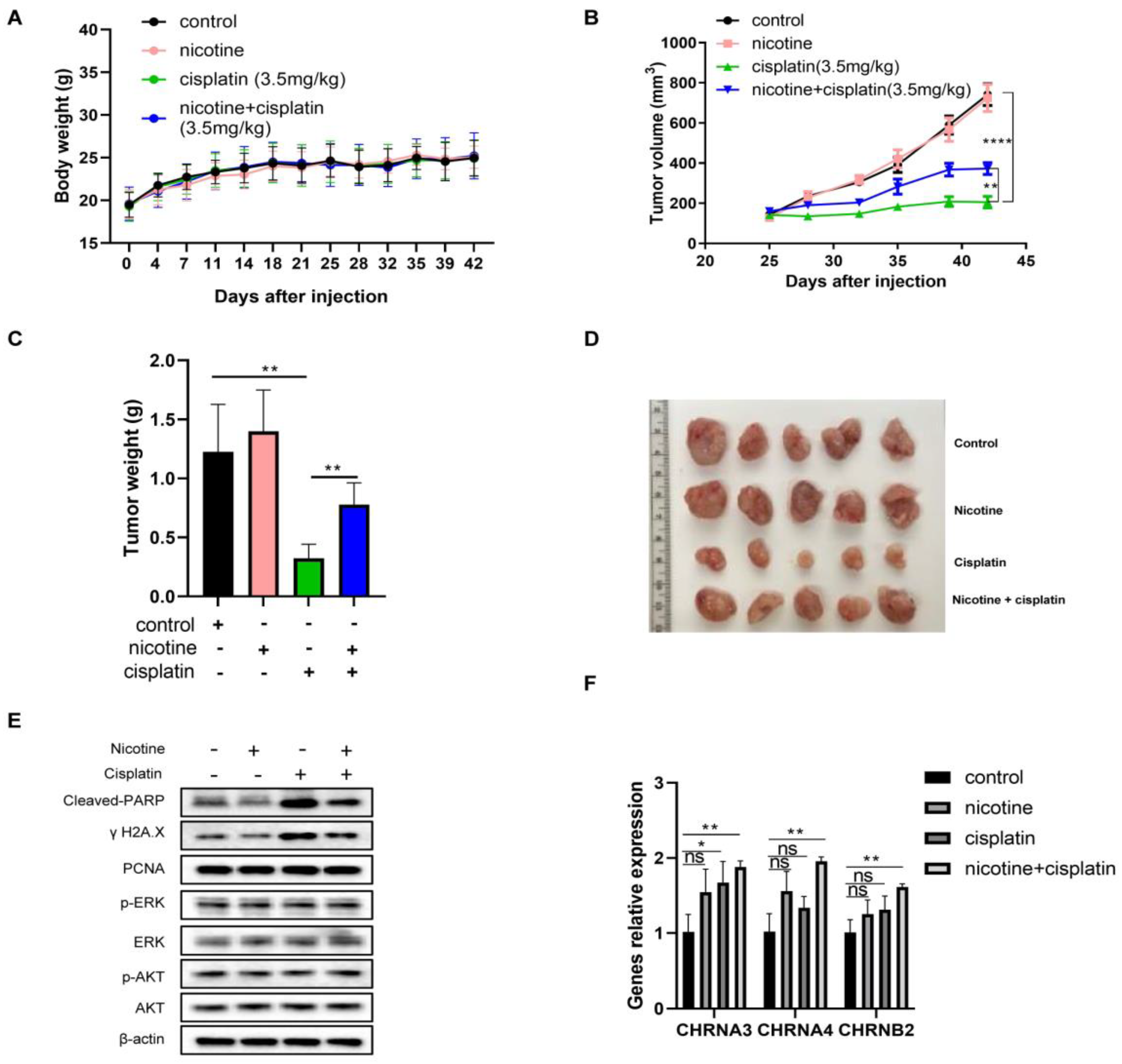
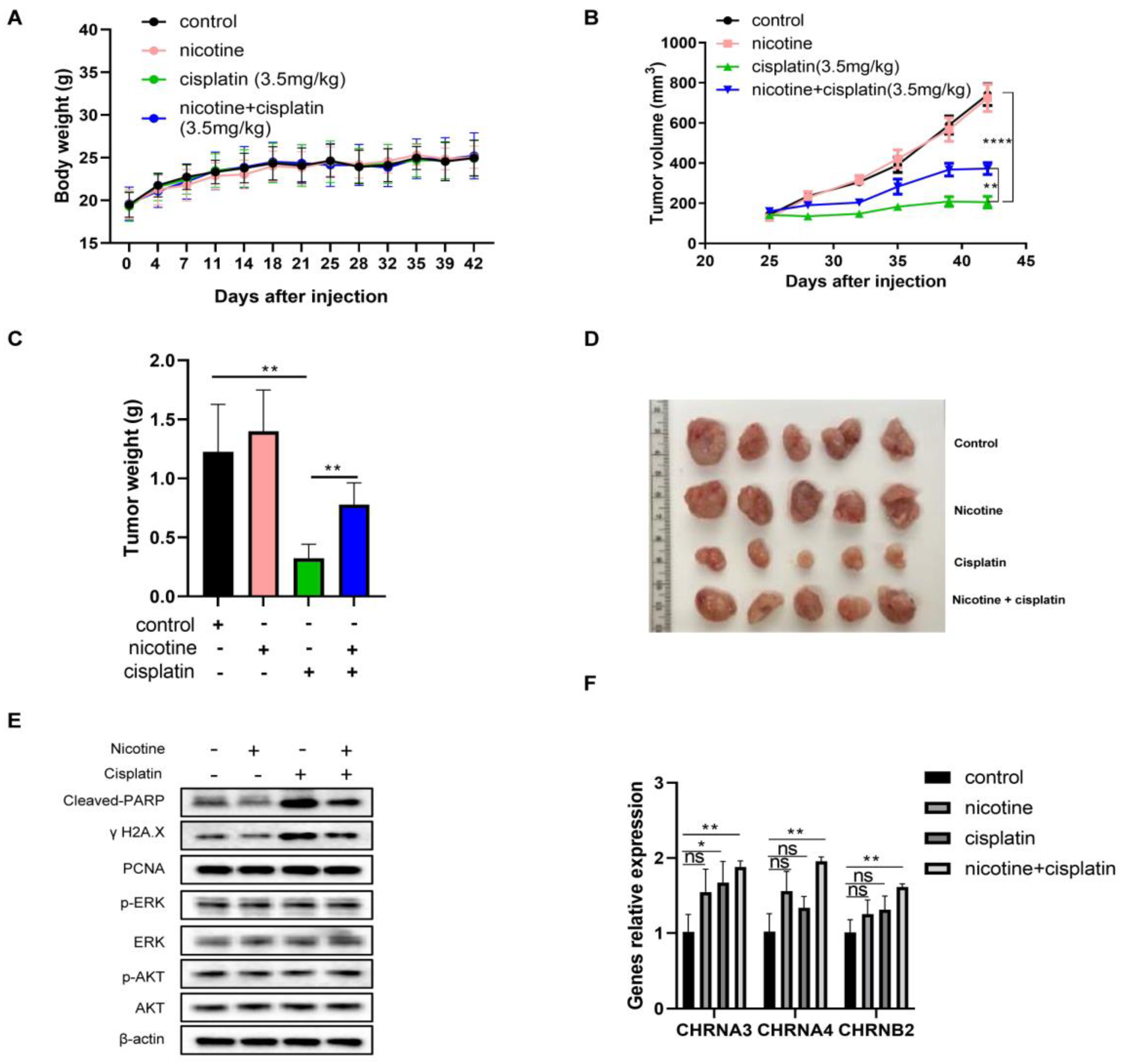
3.7. In Vivo Nicotine Exposure on H841-Derived SCLC Tumors Had No Effects on Tumor Growth but Significantly Compromised the Efficacy of Cisplatin Treatment, with Reductions in Cisplatin-Induced DNA Damage
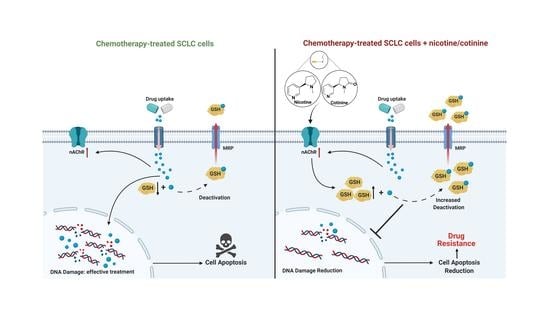
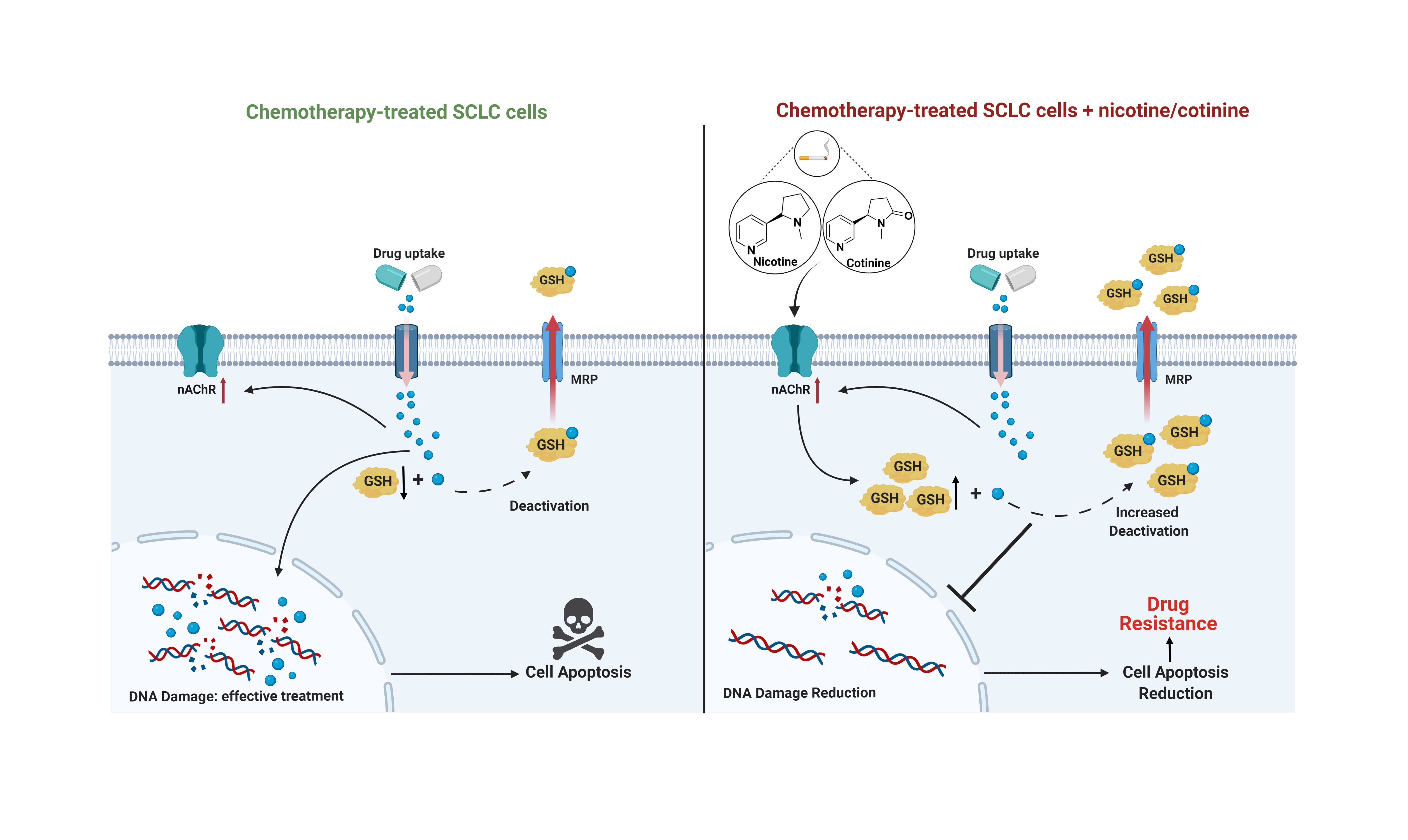
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SCLC | small cell lung cancer |
| NSCLC | non-small cell lung cancer |
| NRT | nicotine replacement therapy |
| nAChRs | nicotinic acetylcholine receptors |
| EMT | epithelial to mesenchymal transition |
| GSH | glutathione |
| NADPH | nicotinamide adenine dinucleotide phosphate |
| ROS | reactive oxygen species |
| MMC | mitomycin C |
| CPT | camptothecin |
| Dox | doxorubicin |
| FACS | fluorescence-activated cell sorting |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Rudin, C.M. Small cell lung cancer: Where do we go from here? Cancer 2015, 121, 664–672. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.N.; Owonikoko, T.K. Small cell lung cancer: Therapies and targets. Semin. Oncol. 2014, 41, 133–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, L.; Mansfield, A.S.; Szczesna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G.J.C.D. Systems biology of cisplatin resistance: Past, present and future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, G.; Rath, B. Smoking, inflammation and small cell lung cancer: Recent developments. Wien. Med. Wochenschr. 2015, 165, 379–386. [Google Scholar] [CrossRef]
- Raez, L.; Samuels, M.; Lilenbaum, R. Combined modality therapy for limited-disease small cell lung cancer. Curr. Treat. Options Oncol. 2005, 6, 69–74. [Google Scholar] [CrossRef]
- Cooper, S.; Spiro, S.G. Small cell lung cancer: Treatment review. Respirology 2006, 11, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Brambilla, E.; Faivre-Finn, C.; Sage, J. Small-cell lung cancer. Nat. Rev. Dis. Primers 2021, 7, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jiang, R.; Garces, Y.I.; Jatoi, A.; Stoddard, S.M.; Sun, Z.; Marks, R.S.; Liu, Y.; Yang, P. Prognostic factors for limited-stage small cell lung cancer: A study of 284 patients. Lung Cancer 2010, 67, 221–226. [Google Scholar] [CrossRef] [Green Version]
- Tucker, M.A.; Murray, N.; Shaw, E.G.; Ettinger, D.S.; Mabry, M.; Huber, M.H.; Feld, R.; Shepherd, F.A.; Johnson, D.H.; Grant, S.C.; et al. Second primary cancers related to smoking and treatment of small-cell lung cancer. J. Natl. Cancer Inst. 1997, 89, 1782–1788. [Google Scholar] [CrossRef] [Green Version]
- Rudin, C.M.; Awad, M.M.; Navarro, A.; Gottfried, M.; Peters, S.; Csőszi, T.; Cheema, P.K.; Rodriguez-Abreu, D.; Wollner, M.; Yang, J.C.; et al. Pembrolizumab or Placebo Plus Etoposide and Platinum as First-Line Therapy for Extensive-Stage Small-Cell Lung Cancer: Randomized, Double-Blind, Phase III KEYNOTE-604 Study. J. Clin. Oncol. 2020, 38, 2369. [Google Scholar] [CrossRef]
- Johnson, B.E.; Linnoila, R.I.; Williams, J.P.; Venzon, D.J.; Okunieff, P.; Anderson, G.B.; Richardson, G.E. Risk of second aerodigestive cancers increases in patients who survive free of small-cell lung cancer for more than 2 years. J. Clin. Oncol. 1995, 13, 101–111. [Google Scholar] [CrossRef]
- Johnston-Early, A.; Cohen, M.H.; Minna, J.D.; Paxton, L.M.; Fossieck, B.E.; Ihde, D.C.; Bunn, P.A.; Matthews, M.J.; Makuch, R. Smoking abstinence and small cell lung cancer survival: An association. Jama 1980, 244, 2175–2179. [Google Scholar] [CrossRef]
- Shields, P.G. Long-term nicotine replacement therapy: Cancer risk in context. Cancer Prev. Res. 2011, 4, 1719–1723. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.; Rizwani, W.; Banerjee, S.; Kovacs, M.; Haura, E.; Coppola, D.; Chellappan, S. Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS ONE 2009, 4, e7524. [Google Scholar] [CrossRef]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Kinkade, R.; Kovacs, M.; Rastogi, S.; Banerjee, S.; Carless, M.; Kim, E.; Coppola, D.; et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 2009, 124, 36–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernyavsky, A.I.; Shchepotin, I.B.; Galitovkiy, V.; Grando, S.A. Mechanisms of tumor-promoting activities of nicotine in lung cancer: Synergistic effects of cell membrane and mitochondrial nicotinic acetylcholine receptors. BMC Cancer 2015, 15, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, H.; Numakawa, T.; Ikeuchi, T. Nicotine-induced phosphorylation of Akt through epidermal growth factor receptor and Src in PC12h cells. J. Neurochem. 2002, 83, 1372–1379. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bao, C.; Mu, Q.; Chen, J.; Wang, J.; Mi, Y.; Sayari, A.; Chen, Y.; Guo, M. Reversal of cisplatin resistance by inhibiting PI3K/Akt signal pathway in human lung cancer cells. Neoplasma 2016, 63, 362–370. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Huang, H.; Pan, C.; Zhang, B.; Liu, X.; Zhang, L. Nicotine inhibits apoptosis induced by cisplatin in human oral cancer cells. Int. J. Oral Maxillofac. Surg. 2007, 36, 739–744. [Google Scholar] [CrossRef]
- Carlisle, D.L.; Liu, X.; Hopkins, T.M.; Swick, M.C.; Dhir, R.; Siegfried, J.M. Nicotine activates cell-signaling pathways through muscle-type and neuronal nicotinic acetylcholine receptors in non-small cell lung cancer cells. Pulm. Pharmacol. Ther. 2007, 20, 629–641. [Google Scholar] [CrossRef]
- Schaal, C.M.; Bora-Singhal, N.; Kumar, D.M.; Chellappan, S.P. Regulation of Sox2 and stemness by nicotine and electronic-cigarettes in non-small cell lung cancer. Mol. Cancer 2018, 17, 149. [Google Scholar] [CrossRef] [Green Version]
- Schaal, C.; Chellappan, S. Nicotine-Mediated Regulation of Nicotinic Acetylcholine Receptors in Non-Small Cell Lung Adenocarcinoma by E2F1 and STAT1 Transcription Factors. PLoS ONE 2016, 11, e0156451. [Google Scholar] [CrossRef]
- Improgo, M.R.; Tapper, A.R.; Gardner, P.D. Nicotinic acetylcholine receptor-mediated mechanisms in lung cancer. Biochem. Pharmacol. 2011, 82, 1015–1021. [Google Scholar] [CrossRef]
- Trevino, J.G.; Pillai, S.; Kunigal, S.; Singh, S.; Fulp, W.J.; Centeno, B.A.; Chellappan, S.P. Nicotine induces inhibitor of differentiation-1 in a Src-dependent pathway promoting metastasis and chemoresistance in pancreatic adenocarcinoma. Neoplasia 2012, 14, 1102–1114. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Sun, H.; Wu, H.; Zhang, H.; Zhang, X.; Xiao, D.; Ma, X.; Wang, Y. Nicotine Inhibits Cisplatin-Induced Apoptosis via Regulating alpha5-nAChR/AKT Signaling in Human Gastric Cancer Cells. PLoS ONE 2016, 11, e0149120. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Deng, X. Nicotine inactivation of the proapoptotic function of Bax through phosphorylation. J. Biol. Chem. 2005, 280, 10781–10789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, M.; Gao, F.; May, W.S.; Flagg, T.; Deng, X. Protein kinase Czeta abrogates the proapoptotic function of Bax through phosphorylation. J. Biol. Chem. 2007, 282, 21268–21277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishioka, T.; Luo, L.; Shen, L.; He, H.; Mariyannis, A.; Dai, W.; Chen, C. Nicotine increases the resistance of lung cancer cells to cisplatin through enhancing Bcl-2 stability. Br. J. Cancer 2014, 110, 1785–1792. [Google Scholar] [CrossRef] [Green Version]
- Mai, H.; May, W.S.; Gao, F.; Jin, Z.; Deng, X. A functional role for nicotine in Bcl2 phosphorylation and suppression of apoptosis. J. Biol. Chem. 2003, 278, 1886–1891. [Google Scholar] [CrossRef] [Green Version]
- Yalcin, E.; de la Monte, S. Tobacco nitrosamines as culprits in disease: Mechanisms reviewed. J. Physiol. Biochem. 2016, 72, 107–120. [Google Scholar] [CrossRef]
- Vainio, P.J.; Tuominen, R.K. Cotinine binding to nicotinic acetylcholine receptors in bovine chromaffin cell and rat brain membranes. Nicotine Tob. Res. 2001, 3, 177–182. [Google Scholar]
- Nakada, T.; Kiyotani, K.; Iwano, S.; Uno, T.; Yokohira, M.; Yamakawa, K.; Fujieda, M.; Saito, T.; Yamazaki, H.; Imaida, K.; et al. Lung tumorigenesis promoted by anti-apoptotic effects of cotinine, a nicotine metabolite through activation of PI3K/Akt pathway. J. Toxicol. Sci. 2012, 37, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef]
- Basu, A.; Krishnamurthy, S. Cellular responses to Cisplatin-induced DNA damage. J. Nucleic Acids 2010, 2010, 201367. [Google Scholar] [CrossRef] [Green Version]
- Karpinich, N.O.; Tafani, M.; Rothman, R.J.; Russo, M.A.; Farber, J.L. The course of etoposide-induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochromec. J. Biol. Chem. 2002, 277, 16547–16552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olive, P.L.; Banáth, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Kuo, M.T. Role of glutathione in the regulation of Cisplatin resistance in cancer chemotherapy. Met. Based Drugs 2010, 2010, 430939. [Google Scholar] [CrossRef]
- Vascellari, S.; Valletta, E.; Perra, D.; Pinna, E.; Serra, A.; Isaia, F.; Pani, A.; Pivetta, T. Cisplatin, glutathione and the third wheel: A copper-(1,10-phenanthroline) complex modulates cisplatin–GSH interactions from antagonism to synergism in cancer cells resistant to cisplatin. RSC Adv. 2019, 9, 5362–5376. [Google Scholar] [CrossRef] [Green Version]
- Conklin, K.A. Chemotherapy-associated oxidative stress: Impact on chemotherapeutic effectiveness. Integr. Cancer Ther. 2004, 3, 294–300. [Google Scholar] [CrossRef]
- Marullo, R.; Werner, E.; Degtyareva, N.; Moore, B.; Altavilla, G.; Ramalingam, S.S.; Doetsch, P.W. Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions. PLoS ONE 2013, 8, e81162. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-M.; Kim, H.-K.; Shim, W.; Anwar, M.A.; Kwon, J.-W.; Kwon, H.-K.; Kim, H.J.; Jeong, H.; Kim, H.M.; Hwang, D.; et al. Mechanism of cisplatin-induced cytotoxicity is correlated to impaired metabolism due to mitochondrial ROS generation. PLoS ONE 2015, 10, e0135083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brozovic, A.; Ambriović-Ristov, A.; Osmak, M. The relationship between cisplatin-induced reactive oxygen species, glutathione, and BCL-2 and resistance to cisplatin. Crit. Rev. Toxicol. 2010, 40, 347–359. [Google Scholar] [CrossRef]
- He, P.J.; Ge, R.F.; Mao, W.J.; Chung, P.S.; Ahn, J.C.; Wu, H.T. Oxidative stress induced by carboplatin promotes apoptosis and inhibits migration of HN-3 cells. Oncol. Lett. 2018, 16, 7131–7138. [Google Scholar] [CrossRef]
- Husain, K.; Whitworth, C.; Hazelrigg, S.; Rybak, L. Carboplatin-induced oxidative injury in rat inferior colliculus. Int. J. Toxicol. 2003, 22, 335–342. [Google Scholar] [CrossRef]
- Navarro, E.; Gonzalez-Lafuente, L.; Perez-Liebana, I.; Buendia, I.; Lopez-Bernardo, E.; Sanchez-Ramos, C.; Prieto, I.; Cuadrado, A.; Satrustegui, J.; Cadenas, S.; et al. Heme-Oxygenase I and PCG-1alpha Regulate Mitochondrial Biogenesis via Microglial Activation of Alpha7 Nicotinic Acetylcholine Receptors Using PNU282987. Antioxid. Redox Signal. 2017, 27, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro, E.; Buendia, I.; Parada, E.; Leon, R.; Jansen-Duerr, P.; Pircher, H.; Egea, J.; Lopez, M.G. Alpha7 nicotinic receptor activation protects against oxidative stress via heme-oxygenase I induction. Biochem. Pharmacol. 2015, 97, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Parada, E.; Egea, J.; Buendia, I.; Negredo, P.; Cunha, A.C.; Cardoso, S.; Soares, M.P.; Lopez, M.G. The microglial alpha7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid. Redox Signal. 2013, 19, 1135–1148. [Google Scholar] [CrossRef] [Green Version]
- Parada, E.; Egea, J.; Romero, A.; del Barrio, L.; Garcia, A.G.; Lopez, M.G. Poststress treatment with PNU282987 can rescue SH-SY5Y cells undergoing apoptosis via alpha7 nicotinic receptors linked to a Jak2/Akt/HO-1 signaling pathway. Free. Radic. Biol. Med. 2010, 49, 1815–1821. [Google Scholar] [CrossRef]
- Qian, J.; Mummalaneni, S.K.; Alkahtani, R.M.; Mahavadi, S.; Murthy, K.S.; Grider, J.R.; Lyall, V. Nicotine-induced effects on nicotinic acetylcholine receptors (nAChRs), Ca2+ and brain-derived neurotrophic factor (BDNF) in STC-1 cells. PLoS ONE 2016, 11, e0166565. [Google Scholar] [CrossRef] [PubMed]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R.; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 69, 503–508. [Google Scholar] [CrossRef] [PubMed]
- DepMap, Broad (2020): DepMap 20Q4 Public.Figshare. Available online: https://figshare.com/articles/dataset/DepMap_20Q4_Public/13237076/4 (accessed on 12 October 2021). [CrossRef]
- Benowitz, N.L.; Jacob, P. Daily intake of nicotine during cigarette smoking. Clin. Pharmacol. Ther. 1984, 35, 499–504. [Google Scholar] [CrossRef]
- Reagan-Shaw, S.; Nihal, M.; Ahmad, N. Dose translation from animal to human studies revisited. FASEB J. 2008, 22, 659–661. [Google Scholar] [CrossRef] [Green Version]
- Fiore, M.C.; D’Angelo, H.; Baker, T. Effective cessation treatment for patients with cancer who smoke—The fourth pillar of cancer care. JAMA Netw. Open 2019, 2, e1912264. [Google Scholar] [CrossRef] [Green Version]
- Warren, G.W.; Evans, W.K.; Dresler, C. Critical determinants of cancer treatment outcomes: Smoking must be addressed at the highest levels in cancer care. J. Thorac. Oncol. 2021, 16, 891–893. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Bian, T.; Song, L.; Jiang, Y.; Huo, Z.; Salloum, R.G.; Warren, G.W.; Kaye, F.J.; Fujioka, N.; Jin, L.; et al. Reducing Chemotherapy-Induced DNA Damage via nAChR-Mediated Redox Reprograming—A New Mechanism for SCLC Chemoresistance Boosted by Nicotine. Cancers 2022, 14, 2272. https://doi.org/10.3390/cancers14092272
Wang Y, Bian T, Song L, Jiang Y, Huo Z, Salloum RG, Warren GW, Kaye FJ, Fujioka N, Jin L, et al. Reducing Chemotherapy-Induced DNA Damage via nAChR-Mediated Redox Reprograming—A New Mechanism for SCLC Chemoresistance Boosted by Nicotine. Cancers. 2022; 14(9):2272. https://doi.org/10.3390/cancers14092272
Chicago/Turabian StyleWang, Yuzhi, Tengfei Bian, Lina Song, Yunhan Jiang, Zhiguang Huo, Ramzi G. Salloum, Graham W. Warren, Frederic J. Kaye, Naomi Fujioka, Lingtao Jin, and et al. 2022. "Reducing Chemotherapy-Induced DNA Damage via nAChR-Mediated Redox Reprograming—A New Mechanism for SCLC Chemoresistance Boosted by Nicotine" Cancers 14, no. 9: 2272. https://doi.org/10.3390/cancers14092272
APA StyleWang, Y., Bian, T., Song, L., Jiang, Y., Huo, Z., Salloum, R. G., Warren, G. W., Kaye, F. J., Fujioka, N., Jin, L., & Xing, C. (2022). Reducing Chemotherapy-Induced DNA Damage via nAChR-Mediated Redox Reprograming—A New Mechanism for SCLC Chemoresistance Boosted by Nicotine. Cancers, 14(9), 2272. https://doi.org/10.3390/cancers14092272