
The Genetic and Molecular Analyses of RAD51C and RAD51D Identifies Rare Variants Implicated in Hereditary Ovarian Cancer from a Genetically Unique Population

 , , , , , , , and
, , , , , , , and
Abstract
Simple Summary
Abstract
1. Introduction
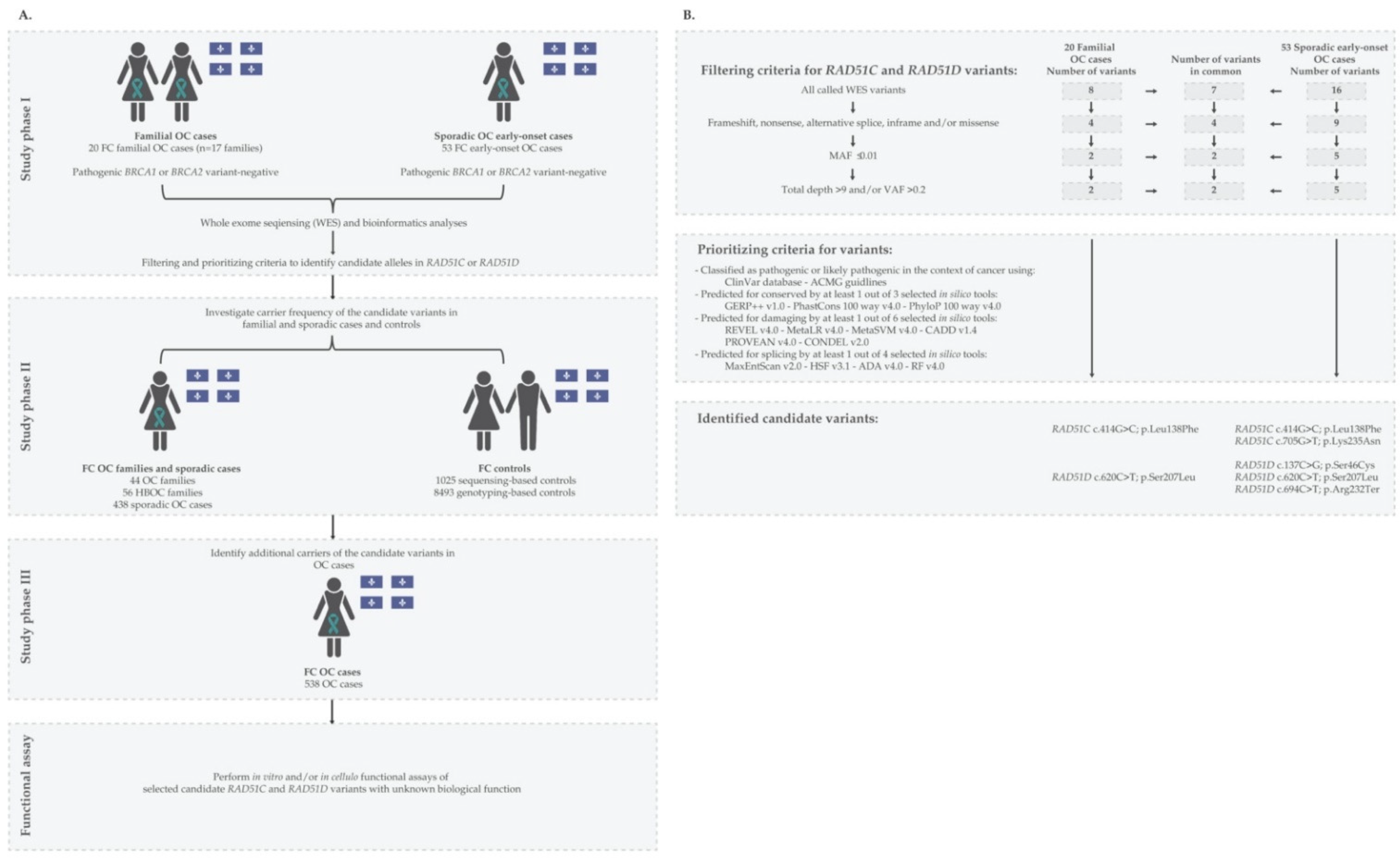
2. Materials and Methods
2.1. FC Study Participants
2.2. Identification and Verification of Candidate Variants
2.3. Investigating Carrier Frequencies of Candidate Variants in FC OC Cases and Controls
2.4. Surveying Allele and Carrier Frequencies of Candidate Variants in Genetic Databases of Non-FC Populations
2.5. LOH Analysis of RAD51C and RAD51D Loci in OC Tumour DNA from Candidate Variant Carriers
2.6. RNA Extraction and Reverse Transcription Analyses of RAD51C
2.7. Cell Lines
2.8. Complementation Assays and siRNA Transfections
2.9. Olaparib and Talazoparib Sensitivity Assays
2.10. Immunofluorescence Analysis
2.11. Protein Expression and Immunoblotting Analyses of RAD51D
3. Results
3.1. Identification and Characteristics of Candidate Variants
3.2. Carrier Frequency of Candidate Variants in OC Cases and Cancer-Free Controls of FC Ancestry
3.3. Clinico-Pathological Characteristics of OC Variant Carriers
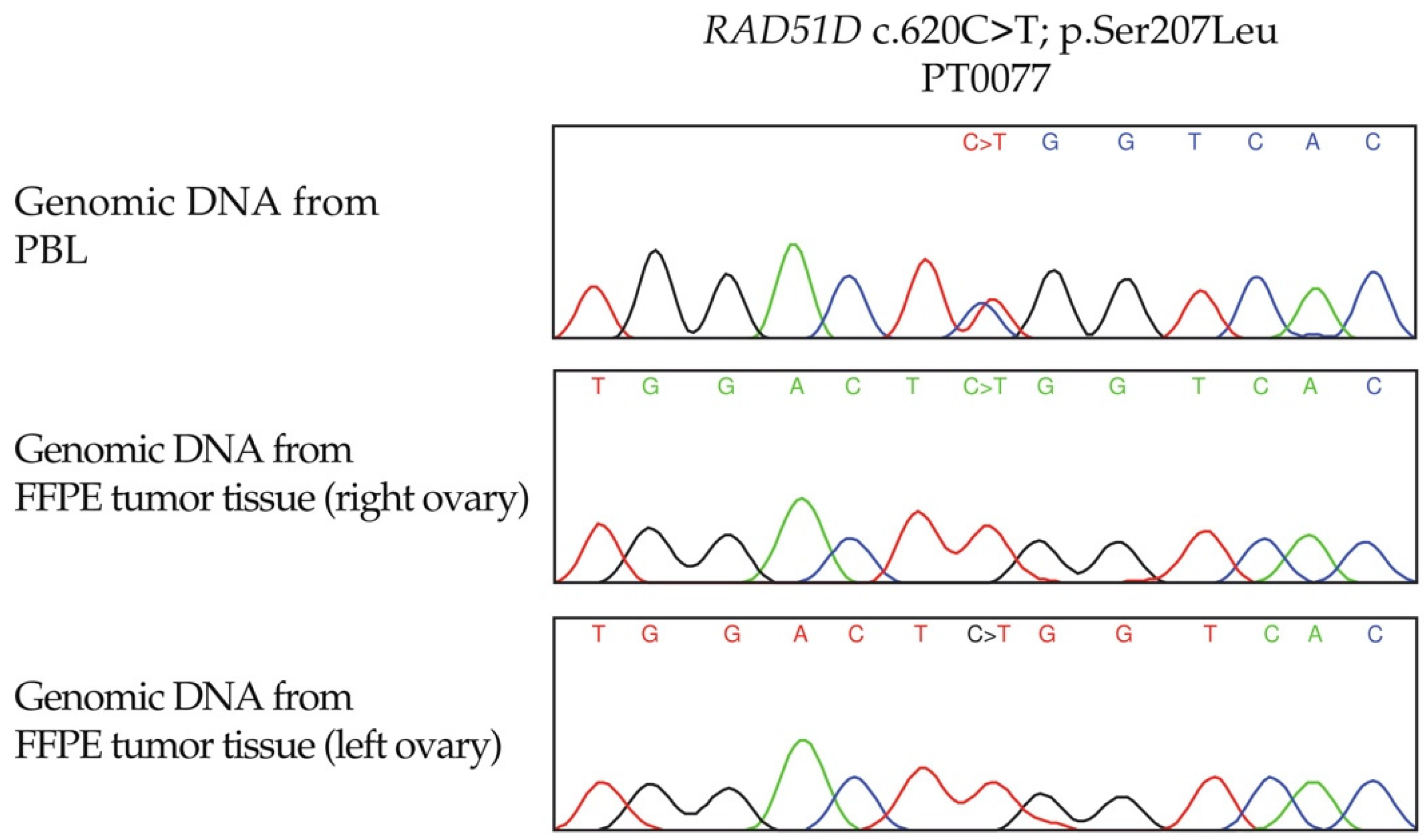
3.4. LOH Analyses of RAD51C and RAD51D Loci in OC Tumour DNA from Candidate Variant Carriers
3.5. In Vitro Investigation of Aberrant Splicing of RAD51C c.705G>T
3.6. In Cellulo Investigation of RAD51D p.Ser46Cys
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Möller, S.; Unger, R.H.; et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA J. Am. Med. Assoc. 2016, 315, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Parmigiani, G. Meta-Analysis of BRCA1 and BRCA2 Penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef]
- Meindl, A.; Hellebrand, H.; Wiek, C.; Erven, V.; Wappenschmidt, B.; Niederacher, D.; Freund, M.; Lichtner, P.; Hartmann, L.; Schaal, H.; et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010, 42, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Pavanello, M.; Chan, I.H.; Ariff, A.; Pharoah, P.D.; Gayther, S.A.; Ramus, S.J. Rare Germline Genetic Variants and the Risks of Epithelial Ovarian Cancer. Cancers 2020, 12, 3046. [Google Scholar] [CrossRef]
- Nielsen, F.C.; van Overeem Hansen, T.; Sørensen, C.S. Hereditary breast and ovarian cancer: New genes in confined pathways. Nat. Rev. Cancer 2016, 16, 599–612. [Google Scholar] [CrossRef]
- Min, A.; Im, S.-A.; Yoon, Y.-K.; Song, S.-H.; Nam, H.-J.; Hur, H.-S.; Kim, H.-P.; Lee, K.-H.; Han, S.-W.; Oh, D.-Y.; et al. RAD51C-Deficient Cancer Cells Are Highly Sensitive to the PARP Inhibitor Olaparib. Mol. Cancer Ther. 2013, 12, 865–877. [Google Scholar] [CrossRef]
- Gayarre, J.; Martin-Gimeno, P.; Osorio, A.; Paumard, B.; Barroso, A.; Fernández, V.; de la Hoya, M.; Rojo-Sebastián, A.; Caldés, T.; Palacios, J.; et al. Characterisation of the novel deleterious RAD51C p.Arg312Trp variant and prioritisation criteria for functional analysis of RAD51C missense changes. Br. J. Cancer 2017, 117, 1048–1062. [Google Scholar] [CrossRef]
- Curtin, N.J.; Szabo, C. Poly(ADP-ribose) polymerase inhibition: Past, present and future. Nat. Rev. Drug Discov. 2020, 19, 711–736. [Google Scholar] [CrossRef]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(ADP)-Ribose Polymerase Inhibition: Frequent Durable Responses in BRCA Carrier Ovarian Cancer Correlating With Platinum-Free Interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmaña, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib Monotherapy in Patients With Advanced Cancer and a Germline BRCA1/2 Mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Song, H.; Dicks, E.; Ramus, S.; Tyrer, J.P.; Intermaggio, M.P.; Hayward, J.; Edlund, C.K.; Conti, D.; Harrington, P.; Fraser, L.; et al. Contribution of Germline Mutations in the RAD51B, RAD51C, and RAD51D Genes to Ovarian Cancer in the Population. J. Clin. Oncol. 2015, 33, 2901–2907. [Google Scholar] [CrossRef]
- Suszynska, M.; Ratajska, M.; Kozlowski, P. BRIP1, RAD51C, and RAD51D mutations are associated with high susceptibility to ovarian cancer: Mutation prevalence and precise risk estimates based on a pooled analysis of ~30,000 cases. J. Ovarian Res. 2020, 13, 50. [Google Scholar] [CrossRef]
- Lilyquist, J.; LaDuca, H.; Polley, E.; Davis, B.T.; Shimelis, H.; Hu, C.; Hart, S.N.; Dolinsky, J.S.; Couch, F.J.; Goldgar, D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017, 147, 375–380. [Google Scholar] [CrossRef]
- Scriver, C.R. Human genetics: Lessons from Quebec populations. Annu. Rev. Genomics Hum. Genet. 2001, 2, 69–101. [Google Scholar] [CrossRef]
- Fierheller, C.; Alenezi, W.; Tonin, P. The Genetic Analyses of French Canadians of Quebec Facilitate the Characterization of New Cancer Predisposing Genes Implicated in Hereditary Breast and/or Ovarian Cancer Syndrome Families. Cancers 2021, 13, 3406. [Google Scholar] [CrossRef]
- Laberge, A.-M.; Michaud, J.; Richter, A.; Lemyre, E.; Lambert, M.; Brais, B.; Mitchell, G. Population history and its impact on medical genetics in Quebec. Clin. Genet. 2005, 68, 287–301. [Google Scholar] [CrossRef]
- Tonin, P.N.; Mes-Masson, A.-M.; Futreal, P.A.; Morgan, K.; Mahon, M.; Foulkes, W.; Cole, D.E.; Provencher, D.; Ghadirian, P.; Narod, S.A. Founder BRCA1 and BRCA2 Mutations in French Canadian Breast and Ovarian Cancer Families. Am. J. Hum. Genet. 1998, 63, 1341–1351. [Google Scholar] [CrossRef]
- Oros, K.K.; Ghadirian, P.; Greenwood, C.; Perret, C.; Shen, Z.; Paredes, Y.; Arcand, S.L.; Mes-Masson, A.-M.; Narod, S.A.; Foulkes, W.; et al. Significant proportion of breast and/or ovarian cancer families of French Canadian descent harbor 1 of 5BRCA1 andBRCA2 mutations. Int. J. Cancer 2004, 112, 411–419. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Ghadirian, P.; Akbari, M.R.; Hamel, N.; Giroux, S.; Sabbaghian, N.; Darnel, A.; Royer, R.; Poll, A.; Fafard, E.; et al. Identification of a novel truncating PALB2mutation and analysis of its contribution to early-onset breast cancer in French-Canadian women. Breast Cancer Res. 2007, 9, R83. [Google Scholar] [CrossRef]
- Tischkowitz, M.; Sabbaghian, N.; Hamel, N.; Pouchet, C.; Foulkes, W.D.; Mes-Masson, A.-M.; Provencher, D.M.; Tonin, P.N. Contribution of the PALB2 c.2323C>T [p.Q775X] Founder mutation in well-defined breast and/or ovarian cancer families and unselected ovarian cancer cases of French Canadian descent. BMC Med. Genet. 2013, 14, 5. [Google Scholar] [CrossRef]
- Charbonneau, H.; Desjardins, B.; Légaré, J.; Denis, H. The Population of the St. Lawrence Valley, 1608–1760 | Bibliographie sur l’histoire de Montréal. In A Population History of North America; Haines, M.R., Steckel, R.H., Eds.; Cambridge University Press: Cambridge, UK, 2000; pp. 99–142. [Google Scholar]
- Rivera, B.; Di Iorio, M.; Frankum, J.; Nadaf, J.; Fahiminiya, S.; Arcand, S.L.; Burk, D.L.; Grapton, D.; Tomiak, E.; Hastings, V.; et al. Functionally Null RAD51D Missense Mutation Associates Strongly with Ovarian Carcinoma. Cancer Res. 2017, 77, 4517–4529. [Google Scholar] [CrossRef]
- Akbari, M.R.; Tonin, P.; Foulkes, W.D.; Ghadirian, P.; Tischkowitz, M.; Narod, S.A. RAD51C germline mutations in breast and ovarian cancer patients. Breast Cancer Res. 2010, 12, 404. [Google Scholar] [CrossRef]
- Osher, D.J.; De Leeneer, K.; Michils, G.; Hamel, N.; Tomiak, E.; Poppe, B.; Leunen, K.; Legius, E.; Shuen, A.; Smith, E.; et al. Mutation analysis of RAD51D in non-BRCA1/2 ovarian and breast cancer families. Br. J. Cancer 2012, 106, 1460–1463. [Google Scholar] [CrossRef][Green Version]
- Awadalla, P.; Boileau, C.; Payette, Y.; Idaghdour, Y.; Goulet, J.-P.; Knoppers, B.; Hamet, P.; Laberge, C. Cohort profile of the CARTaGENE study: Quebec’s population-based biobank for public health and personalized genomics. Int. J. Epidemiol. 2012, 42, 1285–1299. [Google Scholar] [CrossRef]
- Gan-Or, Z.; Zhou, S.; Ambalavanan, A.; Leblond, C.S.; Xie, P.; Johnson, A.; Spiegelman, D.; Allen, R.P.; Earley, C.J.; Desautels, A.; et al. Analysis of functional GLO1 variants in the BTBD9 locus and restless legs syndrome. Sleep Med. 2015, 16, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Guillemette, L.; Allard, C.; Lacroix, M.; Patenaude, J.; Battista, M.-C.; Doyon, M.; Moreau, J.; Ménard, J.; Bouchard, L.; Ardilouze, J.-L.; et al. Genetics of Glucose regulation in Gestation and Growth (Gen3G): A prospective prebirth cohort of mother–child pairs in Sherbrooke, Canada. BMJ Open 2016, 6, e010031. [Google Scholar] [CrossRef] [PubMed]
- Belanger, M.H.; Dolman, L.; Arcand, S.L.; Shen, Z.; Chong, G.; Mes-Masson, A.-M.; Provencher, D.; Tonin, P.N. A targeted analysis identifies a high frequency of BRCA1 and BRCA2 mutation carriers in women with ovarian cancer from a founder population. J. Ovarian Res. 2015, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Koch, L. Exploring human genomic diversity with gnomAD. Nat. Rev. Genet. 2020, 21, 448. [Google Scholar] [CrossRef]
- Pedersen, B.S.; Brown, J.M.; Dashnow, H.; Wallace, A.D.; Velinder, M.; Tristani-Firouzi, M.; Schiffman, J.D.; Tvrdik, T.; Mao, R.; Best, D.H.; et al. Effective variant filtering and expected candidate variant yield in studies of rare human disease. NPJ Genom. Med. 2021, 6, 60. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W.; et al. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Ghosh, R.; Oak, N.; Plon, S.E. Evaluation of in silico algorithms for use with ACMG/AMP clinical variant interpretation guidelines. Genome Biol. 2017, 18, 225. [Google Scholar] [CrossRef]
- Jian, X.; Boerwinkle, E.; Liu, X. In silico prediction of splice-altering single nucleotide variants in the human genome. Nucleic Acids Res. 2014, 42, 13534–13544. [Google Scholar] [CrossRef]
- Gunning, A.C.; Fryer, V.; Fasham, J.; Crosby, A.H.; Ellard, S.; Baple, E.L.; Wright, C.F. Assessing performance of pathogenicity predictors using clinically relevant variant datasets. J. Med. Genet. 2021, 58, 547–555. [Google Scholar] [CrossRef]
- Davydov, E.V.; Goode, D.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef]
- Liu, X.; Wu, C.; Li, C.; Boerwinkle, E. dbNSFP v3.0: A One-Stop Database of Functional Predictions and Annotations for Human Nonsynonymous and Splice-Site SNVs. Hum. Mutat. 2016, 37, 235–241. [Google Scholar] [CrossRef]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D.; et al. REVEL: An Ensemble Method for Predicting the Pathogenicity of Rare Missense Variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef]
- Dong, C.; Wei, P.; Jian, X.; Gibbs, R.; Boerwinkle, E.; Wang, K.; Liu, X. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum. Mol. Genet. 2015, 24, 2125–2137. [Google Scholar] [CrossRef]
- González-Pérez, A.; López-Bigas, N. Improving the Assessment of the Outcome of Nonsynonymous SNVs with a Consensus Deleteriousness Score, Condel. Am. J. Hum. Genet. 2011, 88, 440–449. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Shamsani, J.; Kazakoff, S.H.; Armean, I.; McLaren, W.; Parsons, M.T.; A Thompson, B.; A O’Mara, T.; Hunt, S.; Waddell, N.; Spurdle, A.B. A plugin for the Ensembl Variant Effect Predictor that uses MaxEntScan to predict variant spliceogenicity. Bioinformatics 2019, 35, 2315–2317. [Google Scholar] [CrossRef]
- Desmet, F.-O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Béroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef]
- Fedick, A.; Su, J.; Jalas, C.; Northrop, L.; Devkota, B.; Ekstein, J.; Treff, N.R. High-Throughput Carrier Screening Using TaqMan Allelic Discrimination. PLoS ONE 2013, 8, e59722. [Google Scholar] [CrossRef]
- Loh, P.-R.; Danecek, P.; Palamara, P.F.; Fuchsberger, C.; Reshef, Y.A.; Finucane, H.K.; Schoenherr, S.; Forer, L.; McCarthy, S.; Abecasis, C.F.G.R.; et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 2016, 48, 1443–1448. [Google Scholar] [CrossRef]
- Durbin, R. Efficient haplotype matching and storage using the positional Burrows-Wheeler transform (PBWT). Bioinformatics 2014, 30, 1266–1272. [Google Scholar] [CrossRef]
- Gagnon, A.; Heyer, E. Fragmentation of the Québec population genetic pool (Canada): Evidence from the genetic contribution of founders per region in the 17th and 18th centuries. Am. J. Phys. Anthr. 2001, 114, 30–41. [Google Scholar] [CrossRef]
- Shuen, A.Y.; Lanni, S.; Panigrahi, G.B.; Edwards, M.; Yu, L.; Campbell, B.B.; Mandel, A.; Zhang, C.; Zhukova, N.; Alharbi, M.; et al. Functional Repair Assay for the Diagnosis of Constitutional Mismatch Repair Deficiency From Non-Neoplastic Tissue. J. Clin. Oncol. 2019, 37, 461–470. [Google Scholar] [CrossRef]
- Elkholi, I.E.; Di Iorio, M.; Fahiminiya, S.; Arcand, S.L.; Han, H.; Nogué, C.; Behl, S.; Hamel, N.; Giroux, S.; de Ladurantaye, M.; et al. Investigating the causal role of MRE11A p.E506* in breast and ovarian cancer. Sci. Rep. 2021, 11, 2409. [Google Scholar] [CrossRef]
- Garcin, E.B.; Gon, S.; Sullivan, M.R.; Brunette, G.J.; De Cian, A.; Concordet, J.-P.; Giovannangeli, C.; Dirks, W.G.; Eberth, S.; Bernstein, K.A.; et al. Differential Requirements for the RAD51 Paralogs in Genome Repair and Maintenance in Human Cells. PLoS Genet. 2019, 15, e1008355. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Soldner, F.; Beard, C.; Gao, Q.; Mitalipova, M.; DeKelver, R.C.; Katibah, G.E.; Amora, R.; Boydston, E.A.; Zeitler, B.; et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 2009, 27, 851–857. [Google Scholar] [CrossRef]
- Rodrigue, A.; Margaillan, G.; Gomes, T.T.; Coulombe, Y.; Montalban, G.; Carvalho, S.D.C.E.S.; Milano, L.; Ducy, M.; De-Gregoriis, G.; Dellaire, G.; et al. A global functional analysis of missense mutations reveals two major hotspots in the PALB2 tumor suppressor. Nucleic Acids Res. 2019, 47, 10662–10677. [Google Scholar] [CrossRef]
- Castroviejo-Bermejo, M.; Cruz, C.; Llop-Guevara, A.; Gutiérrez-Enríquez, S.; Ducy, M.; Ibrahim, Y.H.; Oliver, A.G.; Pellegrino, B.; Bruna, A.; Guzmán, M.; et al. A RAD 51 assay feasible in routine tumor samples calls PARP inhibitor response beyond BRCA mutation. EMBO Mol. Med. 2018, 10, e9172. [Google Scholar] [CrossRef] [PubMed]
- Cavallone, L.; Arcand, S.L.; Maugard, C.M.; Nolet, S.; Gaboury, L.A.; Mes-Masson, A.-M.; Ghadirian, P.; Provencher, D.; Tonin, P.N. Comprehensive BRCA1 and BRCA2 mutation analyses and review of French Canadian families with at least three cases of breast cancer. Fam. Cancer 2010, 9, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Prim. 2016, 2, 16061. [Google Scholar] [CrossRef] [PubMed]
- Tonin, P.N.; Mes-Masson, A.-M.; A Narod, S.; Ghadirian, P.; Provencher, D. Founder BRCA1 and BRCA2 mutations in French Canadian ovarian cancer cases unselected for family history. Clin. Genet. 1999, 55, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Manderson, E.N.; Provencher, D.; Mes-Masson, A.-M.; Tonin, P.N. Comparative analysis of loss of heterozygosity of specific chromosome 3, 13, 17, and X loci andTP53 mutations in human epithelial ovarian cancer. Mol. Carcinog. 2002, 34, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Buechelmaier, E.S.; Powell, S.N. Rad51 Paralog Complexes BCDX2 and CX3 Act at Different Stages in the BRCA1-BRCA2-Dependent Homologous Recombination Pathway. Mol. Cell. Biol. 2013, 33, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.; Dumont, M.; Moisan, A.-M.; Gaborieau, V.; Vezina, H.; Durocher, F.; Chiquette, J.; Plante, M.; Avard, D.; Bessette, P.; et al. Evaluation of BRCA1 and BRCA2 mutation prevalence, risk prediction models and a multistep testing approach in French-Canadian families with high risk of breast and ovarian cancer. J. Med. Genet. 2007, 44, 107–121. [Google Scholar] [CrossRef]
- Ghadirian, P.; Robidoux, A.; Zhang, P.; Royer, R.; Akbari, M.; Zhang, S.; Fafard, E.; Costa, M.; Martin, G.; Potvin, C.; et al. The contribution of founder mutations to early-onset breast cancer in French-Canadian women. Clin. Genet. 2009, 76, 421–426. [Google Scholar] [CrossRef]
- Yang, X.; Song, H.; Leslie, G.; Engel, C.; Hahnen, E.; Auber, B.; Horváth, J.; Kast, K.; Niederacher, D.; Turnbull, C.; et al. Ovarian and Breast Cancer Risks Associated With Pathogenic Variants in RAD51C and RAD51D. JNCI J. Natl. Cancer Inst. 2020, 112, 1242–1250. [Google Scholar] [CrossRef]
- Cummings, S.; Roman, S.S.; Saam, J.; Bernhisel, R.; Brown, K.; Lancaster, J.M.; Usha, L. Age of ovarian cancer diagnosis among BRIP1, RAD51C, and RAD51D mutation carriers identified through multi-gene panel testing. J. Ovarian Res. 2021, 14, 61. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Yurgelun, M.B.; Berry, M.P.; Buys, S.S.; Dickson, P.; Domchek, S.M.; Elkhanany, A.; Friedman, S.; Garber, J.E.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020. J. Natl. Compr. Cancer Netw. 2020, 18, 380–391. [Google Scholar] [CrossRef]
- Bernards, S.S.; Norquist, B.M.; Harrell, M.I.; Agnew, K.J.; Lee, M.K.; Walsh, T.; Swisher, E.M. Genetic characterization of early onset ovarian carcinoma. Gynecol. Oncol. 2016, 140, 221–225. [Google Scholar] [CrossRef]
- Wickramanyake, A.; Bernier, G.; Pennil, C.; Casadei, S.; Agnew, K.J.; Stray, S.M.; Mandell, J.; Garcia, R.L.; Walsh, T.; King, M.-C.; et al. Loss of function germline mutations in RAD51D in women with ovarian carcinoma. Gynecol. Oncol. 2012, 127, 552–555. [Google Scholar] [CrossRef]
- Kurosaki, T.; Maquat, L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar] [CrossRef]
- Masson, J.-Y.; Tarsounas, M.C.; Stasiak, A.Z.; Stasiak, A.; Shah, R.; McIlwraith, M.J.; Benson, F.E.; West, S.C. Identification and purification of two distinct complexes containing the five RAD51 paralogs. Genes Dev. 2001, 15, 3296–3307. [Google Scholar] [CrossRef]
- Sanoguera-Miralles, L.; Valenzuela-Palomo, A.; Bueno-Martínez, E.; Llovet, P.; Díez-Gómez, B.; Caloca, M.J.; Pérez-Segura, P.; Fraile-Bethencourt, E.; Colmena, M.; Carvalho, S.; et al. Comprehensive Functional Characterization and Clinical Interpretation of 20 Splice-Site Variants of the RAD51C Gene. Cancers 2020, 12, 3771. [Google Scholar] [CrossRef]
- Wiese, C.; Hinz, J.M.; Tebbs, R.S.; Nham, P.B.; Urbin, S.S.; Collins, D.W.; Thompson, L.H.; Schild, D. Disparate requirements for the Walker A and B ATPase motifs of human RAD51D in homologous recombination. Nucleic Acids Res. 2006, 34, 2833–2843. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | RAD51C | RAD51C | RAD51D | RAD51D | RAD51D |
|---|---|---|---|---|---|
| Genomic features (GRCh37/hg19) | |||||
| RefSeq transcript no. | NM_058216.3 | NM_058216.3 | NM_002878 | NM_002878 | NM_002878 |
| Genome change | g.56774063G>C | g.56780690G>T | g.33446137G>C | g.33430520G>A | g.33430317G>A |
| Coding change | c.414G>C | c.705G>T | c.137C>G | c.620C>T | c.694C>T |
| Protein change | p.Leu138Phe | p.Lys235Asn | p.Ser46Cys | p.Ser207Leu | p.Arg232Ter |
| Number of carriers discovered (Phase I) | |||||
| Familial OC cases (n = 20) | 1 | 0 | 0 | 2 | 0 |
| Sporadic OC early-onset cases (n = 53) | 1 | 1 | 1 | 2 | 1 |
| Allele frequencies in gnomAD 1 | |||||
| Non-Finish European | 0.00001 (1/102,736) | 0.00001 (1/102,610) | 0.0001 (16/118,138) | 0.0001 (6/118,136) | 0.00003 (4/126,578) |
| Carrier frequencies in FLOSSIES 2 | |||||
| European | 0 (0/7325) | 0 (0/7325) | 0.0002 (2/7325) | 0.0003 (3/7325) | 0.0001 (1/7325) |
| Clinical classification 3 | |||||
| ClinVar (number of submissions) | Pathogenic/Likely pathogenic (7) | Conflicting (7): Likely pathogenic (1); Uncertain significance (6) | Conflicting (8): Uncertain significance (7); Likely benign (1) | Conflicting (11): Pathogenic (2); Likely pathogenic (6); Uncertain significance (3) | Pathogenic (15) |
| ACMG guidelines (classification codes) | Likely pathogenic (PS1; PM2; PP3; PP5) | Pathogenic (PS3; PM2) | Uncertain significance (PM2; PP3) | Uncertain significance (PS3; M2; PP3) | Pathogenic (PVS1; PM2; PP3; PP5) |
| Predictions by in silico tools 4 | |||||
| GERP++ v1.0 | Conserved | Conserved | Conserved | Conserved | Conserved |
| PhyloP 100 way v4.0 | Conserved | Conserved | Conserved | Conserved | Conserved |
| PhastCons 100 way v4.0 | Conserved | Conserved | Conserved | Conserved | Conserved |
| REVEL v4.0 | Pathogenic | Benign | Pathogenic | Pathogenic | - |
| MetaLR v4.0 | Tolerated | Tolerated | Tolerated | Damaging | - |
| MetaSVM v4.0 | Tolerated | Tolerated | Tolerated | Damaging | - |
| CONDEL v2.0 | Damaging | Tolerated | Damaging | Damaging | - |
| PROVEAN v4.0 | Damaging | Tolerated | Damaging | Damaging | - |
| CADD v1.4 | Damaging | Damaging | Damaging | Damaging | Damaging |
| ADA v1.1 | - | Affecting splicing | - | - | - |
| RF v1.1 | - | Affecting splicing | - | - | - |
| HSF v3.1 | - | Affecting splicing | - | - | - |
| MaxEntScan v2.0 | - | Affecting splicing | - | - | - |
| Variant | Study Groups | Cancer Case Tested | Number of Participants or (Families) per Group | Number of Carriers (%) | p Value 1 |
|---|---|---|---|---|---|
| RAD51C c.414G>C | OC families | OC | 49 (44) | 1/44 (2.3) | 0.081 |
| HBOC families | OC or BC | 56 (56) | 0 | - | |
| Sporadic OC cases | OC | 438 | 0 | - | |
| Sequencing-based controls | - | 1025 | 1/1025 (0.1) | - | |
| RAD51C c.705G>T | OC families | OC | 49 (44) | 0 | - |
| HBOC families | OC or BC | 56 (56) | 0 | - | |
| Sporadic OC cases | OC | 438 | 1/438 (0.2) | 0.299 | |
| Sequencing-based controls | - | 1025 | 0 | - | |
| RAD51D c.137C>G | OC families | OC | 49 (44) | 0 | - |
| HBOC families | OC or BC | 56 (56) | 0 | - | |
| Sporadic OC cases | OC | 438 | 1/438 (0.2) 2 | 0.299 | |
| Sequencing-based controls | - | 1025 | 0 | - | |
| RAD51D c.694C>T | OC families | OC | 49 (44) | 0 | - |
| HBOC families | OC or BC | 56 (56) | 0 | - | |
| Sporadic OC cases | OC | 438 | 1/438 (0.2) 2 | 0.299 | |
| Sequencing-based controls | - | 1025 | 0 | - | |
| RAD51D c.620C>T | OC families | OC | 49 (44) | 1/44 (2.3) | 0.081 |
| HBOC families | OC or BC | 56 (56) | 0 | - | |
| Sporadic OC cases | OC | 438 | 15/438 (3.4) 3 | <0.0001 | |
| Sequencing-based controls | - | 1025 | 1/1025 (0.1) | - |
| Carrier ID 1 | Gene | Coding Change 2 | Protein Change | Germline Status | Laterality of Disease | LOH Analyses of Available DNA from Fresh Frozen Tumour | LOH Analyses of Available DNA from Formalin-Fixed Paraffin-Embedded Tumour | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Right ovary | Left ovary | Laterality unknown or alternative tissue | Right ovary | Left ovary | ||||||
| PT0095 | RAD51C | c.414G>C | p.Leu138Phe | Heterozygous | Unilateral (Left) | - | - | - | - | - |
| PT0094 | RAD51C | c.414G>C | p.Leu138Phe | Heterozygous | Bilateral | - | - | Partial loss in ascites | - | - |
| PT0124 | RAD51C | c.705G>T | p.Lys235Asn | Heterozygous | Bilateral | Partial loss | - | - | - | - |
| PT0125 | RAD51C | c.705G>T | p.Lys235Asn | Heterozygous | Bilateral | - | Complete loss | - | - | - |
| PT0126 3 | RAD51C | c.705G>T | p.Lys235Asn | Heterozygous | Bilateral | Heterozygous | - | - | - | - |
| PT0127 | RAD51C | c.705G>T | p.Lys235Asn | Heterozygous | Unknown | - | - | - | - | - |
| PT0143 | RAD51D | c.694C>T | p.Arg232Ter | Heterozygous | Bilateral | - | - | - | - | - |
| PT0058 | RAD51D | c.137C>G | p.Ser46Cys | Heterozygous | Bilateral | - | - | Heterozygous | Partial loss | - |
| PT0145 | RAD51D | c.137C>G | p.Ser46Cys | Heterozygous | Bilateral | Partial loss | - | - | - | - |
| PT0080 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Bilateral | - | - | Partial loss in omentum | - | - |
| PT0071 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Bilateral | Partial loss | - | - | Partial loss | - |
| PT0073 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Unilateral (Left) | - | - | - | - | - |
| PT0090 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Bilateral | - | - | - | - | - |
| PT0078 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Bilateral | - | - | - | - | - |
| PT0079 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Unilateral (Left) | - | - | - | - | - |
| PT0089 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Bilateral | - | - | - | - | - |
| PT0059 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Bilateral | - | - | Complete loss in ovary | - | - |
| PT0065 3 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Bilateral | - | - | Heterozygous in ovary | - | - |
| PT0075 3 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Unilateral (Right) | Partial loss | - | - | Complete loss | - |
| PT0076 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Bilateral | - | - | - | Complete loss | Partial loss |
| PT0077 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Bilateral | - | - | - | Complete loss | Complete loss |
| PT0074 3 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Bilateral | Partial loss | - | - | - | - |
| PT0144 | RAD51D | c.620C>T | p.Ser207Leu | Heterozygous | Bilateral | - | - | Heterozygous | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alenezi, W.M.; Milano, L.; Fierheller, C.T.; Serruya, C.; Revil, T.; Oros, K.K.; Behl, S.; Arcand, S.L.; Nayar, P.; Spiegelman, D.; et al. The Genetic and Molecular Analyses of RAD51C and RAD51D Identifies Rare Variants Implicated in Hereditary Ovarian Cancer from a Genetically Unique Population. Cancers 2022, 14, 2251. https://doi.org/10.3390/cancers14092251
Alenezi WM, Milano L, Fierheller CT, Serruya C, Revil T, Oros KK, Behl S, Arcand SL, Nayar P, Spiegelman D, et al. The Genetic and Molecular Analyses of RAD51C and RAD51D Identifies Rare Variants Implicated in Hereditary Ovarian Cancer from a Genetically Unique Population. Cancers. 2022; 14(9):2251. https://doi.org/10.3390/cancers14092251
Chicago/Turabian StyleAlenezi, Wejdan M., Larissa Milano, Caitlin T. Fierheller, Corinne Serruya, Timothée Revil, Kathleen K. Oros, Supriya Behl, Suzanna L. Arcand, Porangana Nayar, Dan Spiegelman, and et al. 2022. "The Genetic and Molecular Analyses of RAD51C and RAD51D Identifies Rare Variants Implicated in Hereditary Ovarian Cancer from a Genetically Unique Population" Cancers 14, no. 9: 2251. https://doi.org/10.3390/cancers14092251
APA StyleAlenezi, W. M., Milano, L., Fierheller, C. T., Serruya, C., Revil, T., Oros, K. K., Behl, S., Arcand, S. L., Nayar, P., Spiegelman, D., Gravel, S., Mes-Masson, A.-M., Provencher, D., Foulkes, W. D., El Haffaf, Z., Rouleau, G., Bouchard, L., Greenwood, C. M. T., Masson, J.-Y., ... Tonin, P. N. (2022). The Genetic and Molecular Analyses of RAD51C and RAD51D Identifies Rare Variants Implicated in Hereditary Ovarian Cancer from a Genetically Unique Population. Cancers, 14(9), 2251. https://doi.org/10.3390/cancers14092251