
Dissecting the Genetic and Non-Genetic Heterogeneity of Acute Myeloid Leukemia Using Next-Generation Sequencing and In Vivo Models



Abstract
:Simple Summary
Abstract
1. Introduction
2. The Genomic Landscape of AML
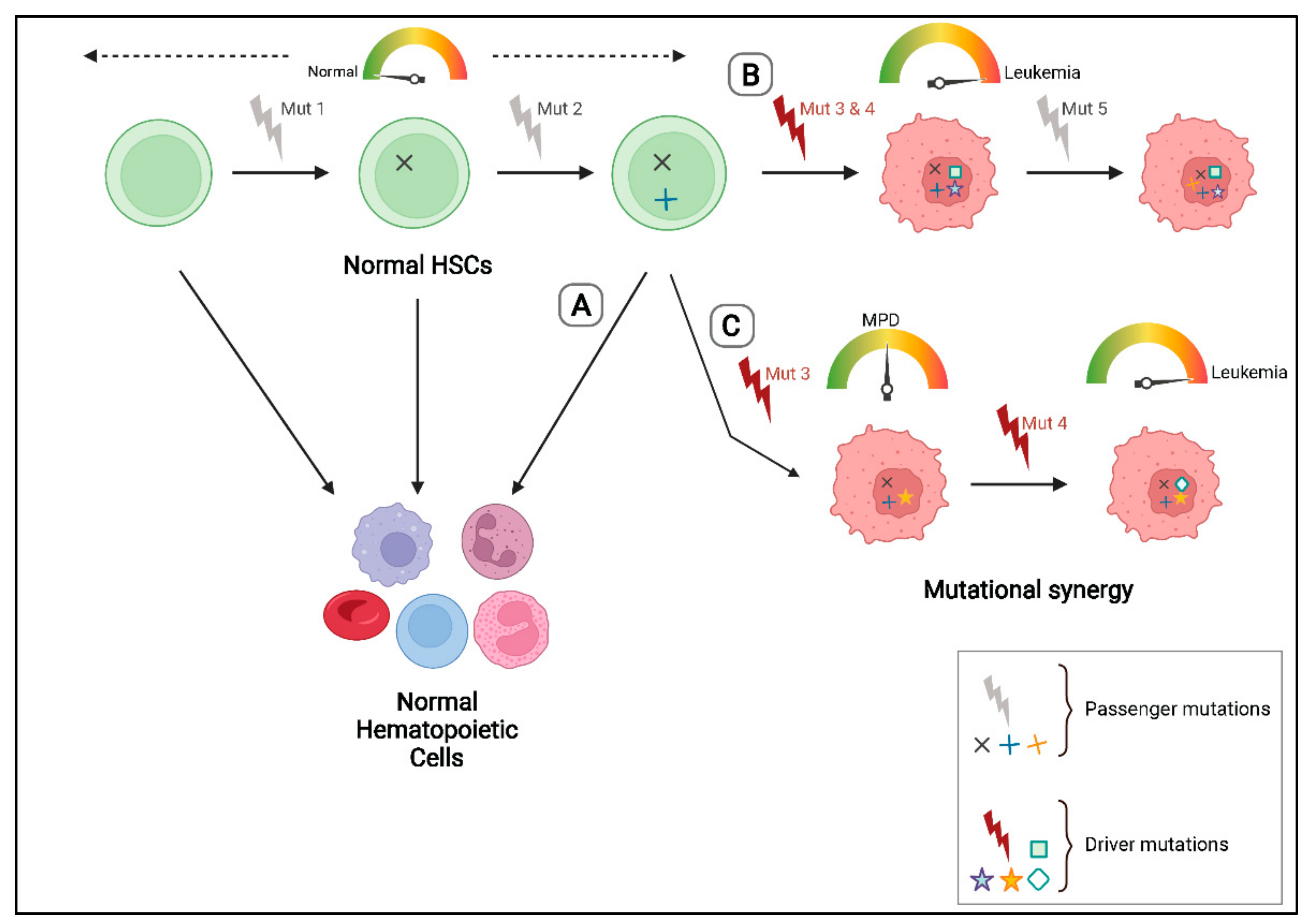
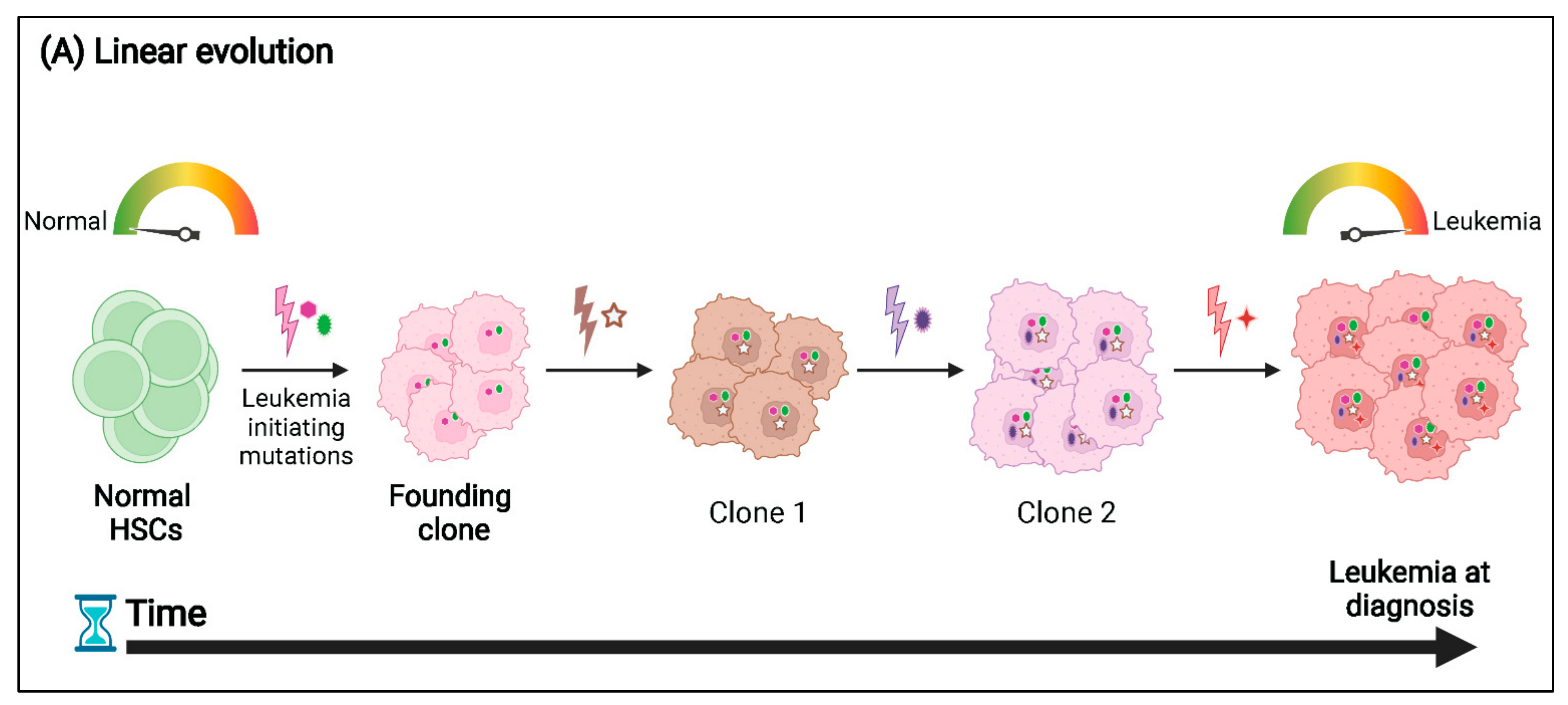
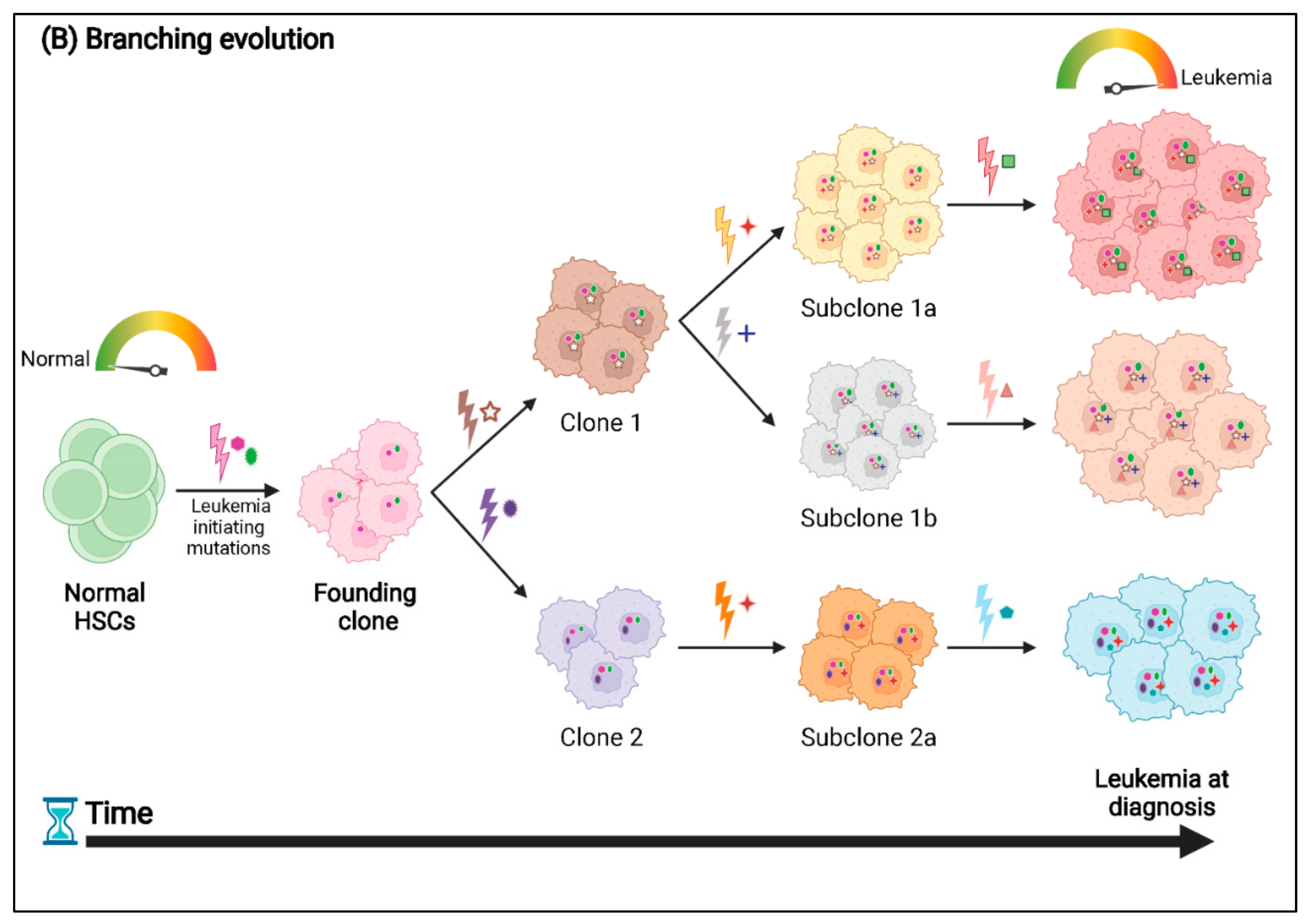
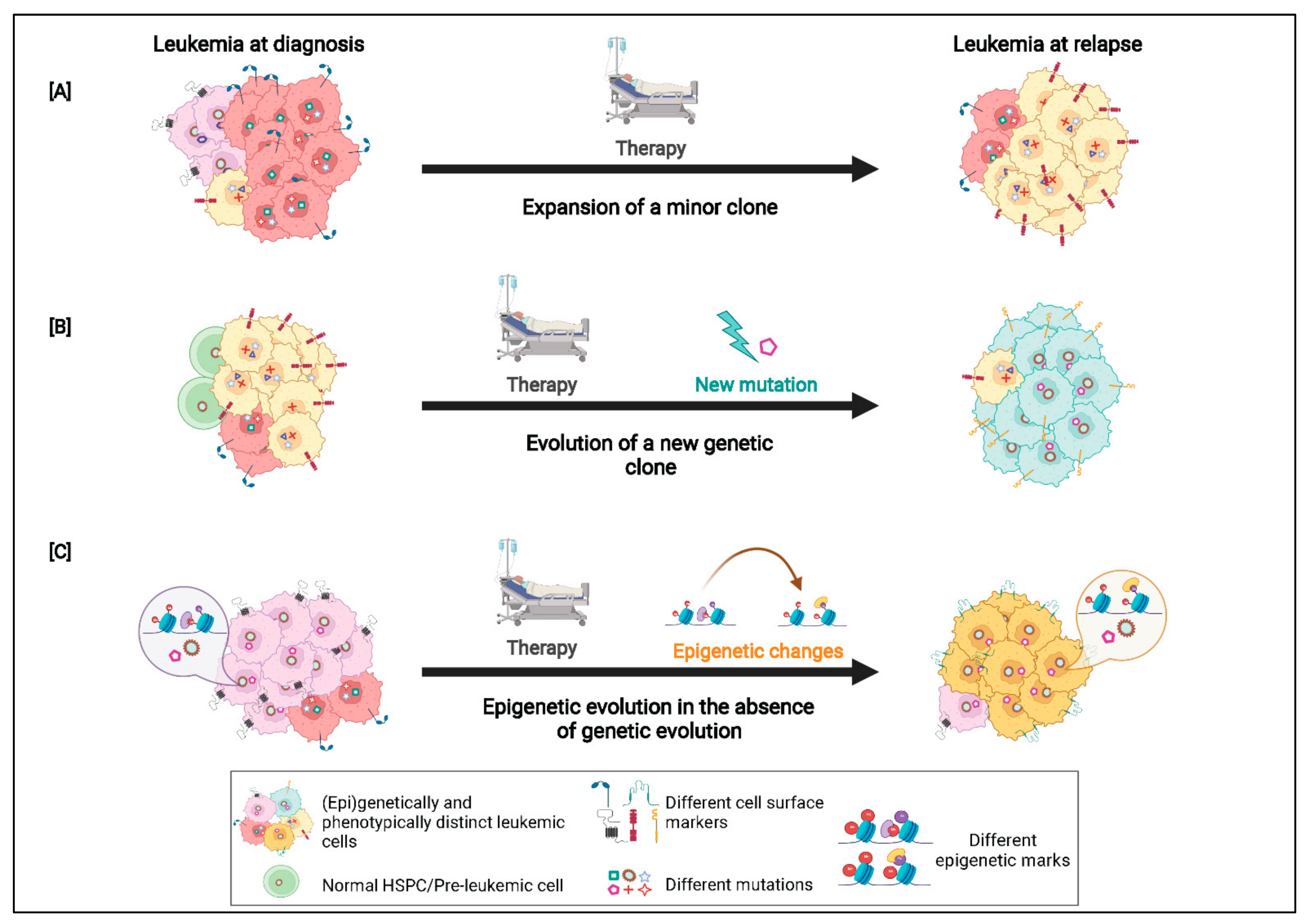
3. Patterns of Clonal Evolution
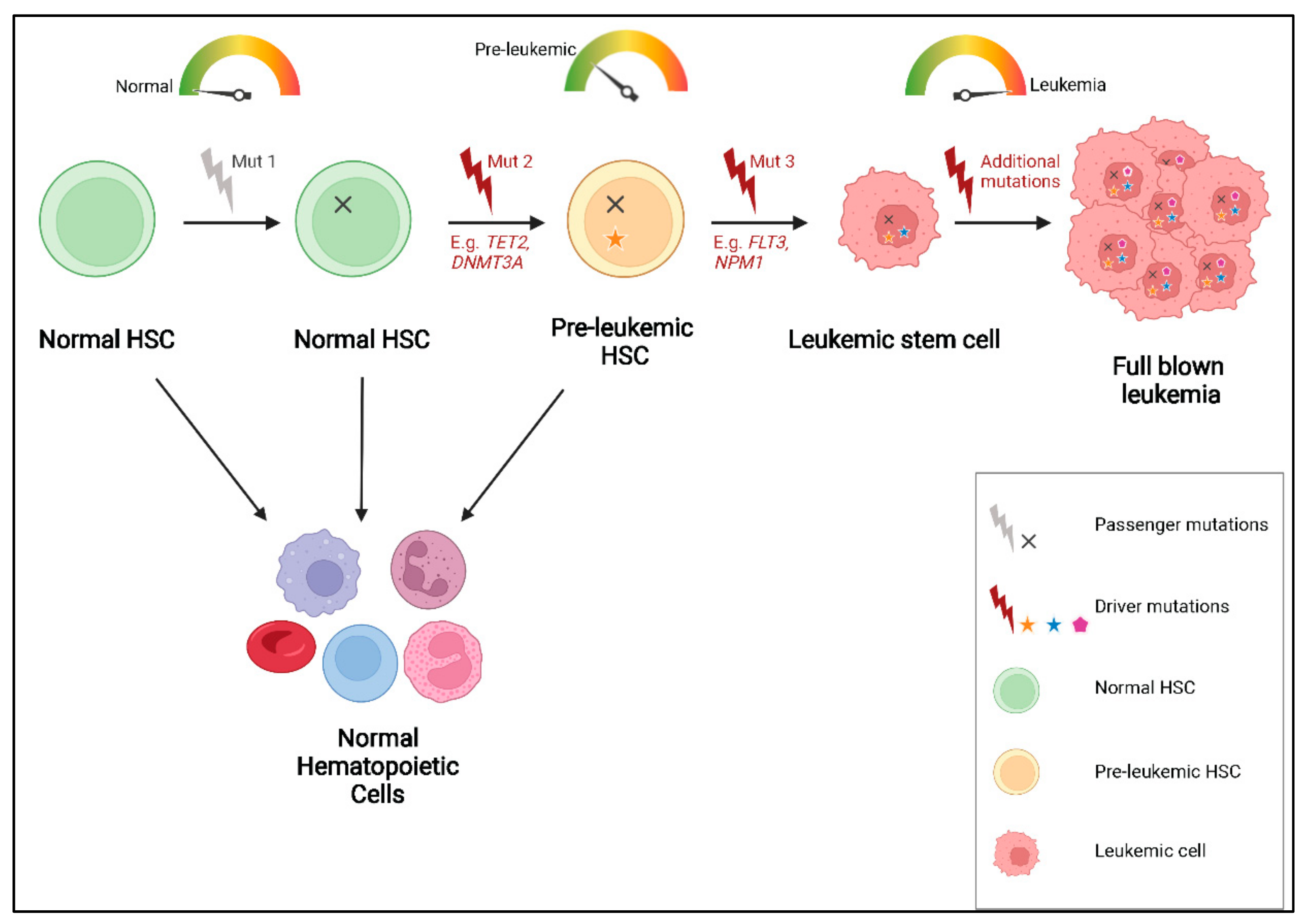
4. Mutational Order and Pre-Leukemic HSCs
5. Phenotypic-Genotypic Correlation in AML
6. The Epigenetic Landscape of AML
7. Conclusions and Future Implications for Research
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nowell, P.C. The Clonal Evolution of Tumour Cell Populations. Sci. New Ser. 1976, 194, 23–28. [Google Scholar]
- Vosberg, S.; Greif, P.A. Clonal Evolution of Acute Myeloid Leukemia from Diagnosis to Relapse. Genes Chromosom. Cancer 2019, 58, 839–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlo, L.M.F.; Pepper, J.W.; Reid, B.J.; Maley, C.C. Cancer as an Evolutionary and Ecological Process. Nat. Rev. Cancer 2006, 6, 924–935. [Google Scholar] [CrossRef]
- Greaves, M. Leukaemia “firsts” in Cancer Research and Treatment. Nat. Rev. Cancer 2016, 16, 163–172. [Google Scholar] [CrossRef]
- Landau, D.A.; Carter, S.L.; Getz, G.; Wu, C.J. Clonal Evolution in Hematological Malignancies and Therapeutic Implications. Leukemia 2014, 28, 34–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohner, K.; Dohner, H.; Döhner, K.; Döhner, H. Molecular Characterization of Acute Myeloid Leukemia. Haematologica 2008, 93, 976–982. [Google Scholar] [CrossRef]
- Network, T.C.G.A.R. Genomic and Epigenomic Landscapes of Adult De Novo Acute Myeloid Leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [Green Version]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Greaves, M.; Maley, C.C. Clonal Evolution in Cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef]
- Döhner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute Myeloid Leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [CrossRef] [Green Version]
- Löwenberg, B.; Ossenkoppele, G.J.; van Putten, W.; Schouten, H.C.; Graux, C.; Ferrant, A.; Sonneveld, P.; Maertens, J.; Jongen-Lavrencic, M.; von Lilienfeld-Toal, M.; et al. High-Dose Daunorubicin in Older Patients with Acute Myeloid Leukemia. N. Engl. J. Med. 2009, 361, 1235–1248. [Google Scholar] [CrossRef] [PubMed]
- Dombret, H.; Gardin, C. An Update of Current Treatments for Adult Acute Myeloid Leukemia. Blood 2016, 127, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Wei, A.H.; Pollyea, D.A.; Fathi, A.T.; Vyas, P.; DiNardo, C.D. New Directions for Emerging Therapies in Acute Myeloid Leukemia: The next Chapter. Blood Cancer J. 2020, 10, 107. [Google Scholar] [CrossRef]
- Cucchi, D.G.J.; Polak, T.B.; Ossenkoppele, G.J.; Uyl–De Groot, C.A.; Cloos, J.; Zweegman, S.; Janssen, J.J.W.M. Two Decades of Targeted Therapies in Acute Myeloid Leukemia. Leukemia 2021, 35, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.E.O.; Magrini, V.; Demeter, R.; Miller, C.A.; Fulton, R.; Fulton, L.L.; Eades, W.C.; Elliott, K.; Heath, S.; Westervelt, P.; et al. Clonal Architecture of Secondary Acute Myeloid Leukemia Defined by Single-Cell Sequencing. PLoS Genet. 2014, 10, e1004462. [Google Scholar] [CrossRef]
- Paguirigan, A.L.; Smith, J.; Meshinchi, S.; Carroll, M.; Maley, C.; Radich, J.P. Single-Cell Genotyping Demonstrates Complex Clonal Diversity in Acute Myeloid Leukemia. Sci. Transl. Med. 2015, 7, 281re2. [Google Scholar] [CrossRef] [Green Version]
- Shouval, R.; Shlush, L.I.; Yehudai-Resheff, S.; Ali, S.; Pery, N.; Shapiro, E.; Tzukerman, M.; Rowe, J.M.; Zuckerman, T. Single Cell Analysis Exposes Intratumor Heterogeneity and Suggests That FLT3-ITD Is a Late Event in Leukemogenesis. Exp. Hematol. 2014, 42, 457–463. [Google Scholar] [CrossRef]
- Kohnken, R.; Porcu, P.; Mishra, A. Overview of the Use of Murine Models in Leukemia and Lymphoma Research. Front. Oncol. 2017, 7, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohanty, S.; Heuser, M. Mouse Models of Frequently Mutated Genes in Acute Myeloid Leukemia. Cancers 2021, 13, 6192. [Google Scholar] [CrossRef]
- Skayneh, H.; Jishi, B.; Hleihel, R.; Hamieh, M.; Darwiche, N.; Bazarbachi, A.; El Sabban, M.; El Hajj, H. A Critical Review of Animal Models Used in Acute Myeloid Leukemia Pathophysiology. Genes 2019, 10, 614. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, R.; Grisendi, S.; Pandolfi, P.P. Modelling Haematopoietic Malignancies in the Mouse and Therapeutical Implications. Oncogene 2002, 21, 3445–3458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopra, M.; Bohlander, S.K. The Cell of Origin and the Leukemia Stem Cell in Acute Myeloid Leukemia. Genes Chromosom. Cancer 2019, 58, 850–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowley, J.D. Chromosome Translocations Play a Key Role in Leukemogenesis. In Acute Leukemias VI; Büchner, T., Schellong, G., Ritter, J., Creutzig, U., Hiddemann, W., Wörmann, B., Eds.; Springer: Heidelberg, Germany, 1997; pp. 3–10. [Google Scholar]
- Mrózek, K.; Heerema, N.A.; Bloomfield, C.D. Cytogenetics in Acute Leukemia. Blood Rev. 2004, 18, 115–136. [Google Scholar] [CrossRef]
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA Sequencing of a Cytogenetically Normal Acute Myeloid Leukaemia Genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christen, F.; Hoyer, K.; Yoshida, K.; Hou, H.-A.; Waldhueter, N.; Heuser, M.; Hills, R.K.; Chan, W.; Hablesreiter, R.; Blau, O.; et al. Genomic Landscape and Clonal Evolution of Acute Myeloid Leukemia with t(8; 21): An International Study on 331 Patients. Blood 2019, 133, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Grove, C.S.; Vassiliou, G.S. Acute Myeloid Leukaemia: A Paradigm for the Clonal Evolution of Cancer? Dis. Model. Mech. 2014, 7, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.; Lutz, C.; van Delft, F.W.; Bateman, C.M.; Guo, Y.; Colman, S.M.; Kempski, H.; Moorman, A.V.; Titley, I.; Swansbury, J.; et al. Genetic Variegation of Clonal Architecture and Propagating Cells in Leukaemia. Nature 2011, 469, 356–361. [Google Scholar] [CrossRef]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal Evolution in Relapsed Acute Myeloid Leukaemia Revealed by Whole-Genome Sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Li, L.; Piloto, O.; Nguyen, H.B.; Greenberg, K.; Takamiya, K.; Racke, F.; Huso, D.; Small, D. Knock-in of an Internal Tandem Duplication Mutation into Murine FLT3 Confers Myeloproliferative Disease in a Mouse Model. Blood J. Am. Soc. Hematol. 2008, 111, 3849–3858. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.; Sportoletti, P.; Ito, K.; Clohessy, J.G.; Teruya-Feldstein, J.; Kutok, J.L.; Pandolfi, P.P. The Cytoplasmic NPM Mutant Induces Myeloproliferation in a Transgenic Mouse Model. Blood J. Am. Soc. Hematol. 2010, 115, 3341–3345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallardo, M.; Caronno, A.; Pruneri, G.; Raviele, P.R.; Viale, A.; Pelicci, P.G.; Colombo, E. NPMc+ and FLT3_ITD Mutations Cooperate in Inducing Acute Leukaemia in a Novel Mouse Model. Leukemia 2013, 27, 2248–2251. [Google Scholar] [CrossRef] [PubMed]
- Milne, T.A. Mouse Models of MLL Leukemia: Recapitulating the Human Disease. Blood 2017, 129, 2217–2223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caudell, D.; Zhang, Z.; Yang, J.C.; Aplan, P.D. Expression of a CALM-AF10 Fusion Gene Leads to Hoxa Cluster Overexpression and Acute Leukemia in Transgenic Mice. Cancer Res. 2007, 67, 8022–8031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desai, R.; Taghavi, S.; Chase, J.C.; Chopra, M.; Chien, J.; Browett, P.J.; Kakadia, P.M.; Bohlander, S.K. Unexpected Variation in Leukemia Stem Cell Frequency and Genetic Heterogeneity in Two Murine Leukemia Models Initiated by AML1/ETO9a and CALM/AF10. Leukemia 2020, 34, 1706–1710. [Google Scholar] [CrossRef] [PubMed]
- Taghavi, S.; Desai, R.; Chien, J.; Super, H.; Browett, P.; Kakadia, P.M.; Bohlander, S.K. Characterization of Cooperating Mutations in Murine Acute Leukemias: Towards Establishing Leukemia Models with Genetic Lesions in Defined Cellular Pathways. Blood 2017, 130 (Suppl. S1), 3944. [Google Scholar]
- Rhoades, K.L.; Hetherington, C.J.; Harakawa, N.; Yergeau, D.A.; Zhou, L.; Liu, L.Q.; Little, M.T.; Tenen, D.G.; Zhang, D.E. Analysis of the Role of AML1-ETO in Leukemogenesis, Using an Inducible Transgenic Mouse Model. Blood 2000, 96, 2108–2115. [Google Scholar] [CrossRef]
- Miyamoto, T.; Weissman, I.L.; Akashi, K. AML1/ETO-Expressing Nonleukemic Stem Cells in Acute Myelogenous Leukemia with 8;21 Chromosomal Translocation. Proc. Natl. Acad. Sci. USA 2000, 97, 7521–7526. [Google Scholar] [CrossRef] [Green Version]
- Schessl, C.; Rawat, V.P.S.; Cusan, M.; Deshpande, A.; Kohl, T.M.; Rosten, P.M.; Spiekermann, K.; Humphries, R.K.; Schnittger, S.; Kern, W.; et al. The AML1-ETO Fusion Gene and the FLT3 Length Mutation Collaborate in Inducing Acute Leukemia in Mice. J. Clin. Investig. 2005, 115, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-Y.; Zhao, L.-J.; Wu, C.-F.; Liu, P.; Shi, L.; Liang, Y.; Xiong, S.-M.; Mi, J.-Q.; Chen, Z.; Ren, R.; et al. C-KIT Mutation Cooperates with Full-Length AML1-ETO to Induce Acute Myeloid Leukemia in Mice. Proc. Natl. Acad. Sci. USA 2011, 108, 2450–2455. [Google Scholar] [CrossRef] [Green Version]
- Gilliland, D.G.; Griffin, J.D. The Roles of FLT3 in Hematopoiesis and Leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, L.M.; Gilliland, D.G. Genetics of Myeloid Leukaemias. Annu. Rev. Genom. Hum. Genet. 2002, 3, 179–198. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, R.C.; Mar, B.G.; Mazzola, E.; Grauman, P.V.; Shareef, S.; Allen, S.L.; Pigneux, A.; Wetzler, M.; Stuart, R.K.; Erba, H.P.; et al. Acute Myeloid Leukemia Ontogeny Is Defined by Distinct Somatic Mutations. Blood 2015, 125, 1367–1376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Zhu, Y.-M.; Fan, X.; Shi, J.-Y.; Wang, Q.-R.; Yan, X.-J.; Gu, Z.-H.; Wang, Y.-Y.; Chen, B.; Jiang, C.-L.; et al. Gene Mutation Patterns and Their Prognostic Impact in a Cohort of 1185 Patients with Acute Myeloid Leukemia. Blood 2011, 118, 5593–5603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussaini, M.O.; Mirza, A.-S.; Komrokji, R.; Lancet, J.; Padron, E.; Song, J. Genetic Landscape of Acute Myeloid Leukemia Interrogated by Next-Generation Sequencing: A Large Cancer Center Experience. Cancer Genom.-Proteom. 2018, 15, 121–126. [Google Scholar]
- Klco, J.M.; Spencer, D.H.; Miller, C.A.; Griffith, M.; Lamprecht, T.L.; O’Laughlin, M.; Fronick, C.; Magrini, V.; Demeter, R.T.; Fulton, R.S.; et al. Functional Heterogeneity of Genetically Defined Subclones in Acute Myeloid Leukemia. Cancer Cell 2014, 25, 379–392. [Google Scholar] [CrossRef] [Green Version]
- Morita, K.; Wang, F.; Jahn, K.; Hu, T.; Tanaka, T.; Sasaki, Y.; Kuipers, J.; Loghavi, S.; Wang, S.A.; Yan, Y.; et al. Clonal Evolution of Acute Myeloid Leukemia Revealed by High-Throughput Single-Cell Genomics. Nat. Commun. 2020, 11, 5327. [Google Scholar] [CrossRef]
- Greif, P.A.; Hartmann, L.; Vosberg, S.; Stief, S.M.; Mattes, R.; Hellmann, I.; Metzeler, K.H.; Herold, T.; Bamopoulos, S.A.; Kerbs, P.; et al. Evolution of Cytogenetically Normal Acute Myeloid Leukemia During Therapy and Relapse: An Exome Sequencing Study of 50 Patients. Clin. Cancer Res. 2018, 24, 1716–1726. [Google Scholar] [CrossRef] [Green Version]
- Potter, N.; Miraki-Moud, F.; Ermini, L.; Titley, I.; Vijayaraghavan, G.; Papaemmanuil, E.; Campbell, P.; Gribben, J.; Taussig, D.; Greaves, M. Single Cell Analysis of Clonal Architecture in Acute Myeloid Leukaemia. Leukemia 2019, 33, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Sandén, C.; Lilljebjörn, H.; Orsmark Pietras, C.; Henningsson, R.; Saba, K.H.; Landberg, N.; Thorsson, H.; von Palffy, S.; Peña-Martinez, P.; Högberg, C.; et al. Clonal Competition within Complex Evolutionary Hierarchies Shapes AML over Time. Nat. Commun. 2020, 11, 579. [Google Scholar] [CrossRef] [Green Version]
- Desai, R.; Taghavi, S.; Lee, H.M.; Zandvakili, N.; Browett, P.; Kakadia, P.; Bohlander, S.K. Clonal Evolution in Murine Leukemia Models; European Hematology Association: Frankfurt, Germany, 2020. [Google Scholar]
- Kent, D.G.; Green, A.R. Order Matters: The Order of Somatic Mutations Influences Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a027060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shlush, L.I.; Zandi, S.; Mitchell, A.; Chen, W.C.; Brandwein, J.M.; Gupta, V.; Kennedy, J.A.; Schimmer, A.D.; Schuh, A.C.; Yee, K.W.; et al. Identification of Pre-Leukaemic Haematopoietic Stem Cells in Acute Leukaemia. Nature 2014, 506, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shen, H.; Xu, T.; Yang, Z.; Qiu, H.; Sun, A.; Chen, S.; Wu, D.; Xu, Y. Persistent DNMT3A Mutation Burden in DNMT3A Mutated Adult Cytogenetically Normal Acute Myeloid Leukemia Patients in Long-Term Remission. Leuk. Res. 2016, 49, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.-Y.; Ding, L.-W.; Tan, K.-T.; Chien, W.; Mayakonda, A.; Lin, D.-C.; Loh, X.-Y.; Xiao, J.-F.; Meggendorfer, M.; Alpermann, T.; et al. Ordering of Mutations in Acute Myeloid Leukemia with Partial Tandem Duplication of MLL (MLL-PTD). Leukemia 2017, 31, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.; Snyder, T.M.; Corces-Zimmerman, M.R.; Vyas, P.; Weissman, I.L.; Quake, S.R.; Majeti, R. Clonal Evolution of Preleukemic Hematopoietic Stem Cells Precedes Human Acute Myeloid Leukemia. Sci. Transl. Med. 2012, 4, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-Related Clonal Hematopoiesis Associated with Adverse Outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steensma, D.P.; Bejar, R.; Jaiswal, S.; Lindsley, R.C.; Sekeres, M.A.; Hasserjian, R.P.; Ebert, B.L. Clonal Hematopoiesis of Indeterminate Potential and Its Distinction from Myelodysplastic Syndromes. Blood 2015, 126, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loberg, M.A.; Bell, R.K.; Goodwin, L.O.; Eudy, E.; Miles, L.A.; SanMiguel, J.M.; Young, K.; Bergstrom, D.E.; Levine, R.L.; Schneider, R.K.; et al. Sequentially Inducible Mouse Models Reveal That Npm1 Mutation Causes Malignant Transformation of Dnmt3a-Mutant Clonal Hematopoiesis. Leukemia 2019, 33, 1635–1649. [Google Scholar] [CrossRef]
- Todisco, G.; Creignou, M.; Gallì, A.; Guglielmelli, P.; Rumi, E.; Roncador, M.; Rizzo, E.; Nannya, Y.; Pietra, D.; Elena, C.; et al. Co-Mutation Pattern, Clonal Hierarchy, and Clone Size Concur to Determine Disease Phenotype of SRSF2P95-Mutated Neoplasms. Leukemia 2021, 35, 2371–2381. [Google Scholar] [CrossRef]
- Jiang, L.; Li, X.-P.; Dai, Y.-T.; Chen, B.; Weng, X.-Q.; Xiong, S.-M.; Zhang, M.; Huang, J.-Y.; Chen, Z.; Chen, S.-J. Multidimensional Study of the Heterogeneity of Leukemia Cells in t(8;21) Acute Myelogenous Leukemia Identifies the Subtype with Poor Outcome. Proc. Natl. Acad. Sci. USA 2020, 117, 20117–20126. [Google Scholar] [CrossRef]
- Cui, W.; Zhang, D.; Cunningham, M.T.; Tilzer, L. Leukemia-Associated Aberrant Immunophenotype in Patients with Acute Myeloid Leukemia: Changes at Refractory Disease or First Relapse and Clinicopathological Findings. Int. J. Lab. Hematol. 2014, 36, 636–649. [Google Scholar] [CrossRef] [PubMed]
- Baer, M.R. High Frequency of Immunophenotype Changes in Acute Myeloid Leukemia at Relapse: Implications for Residual Disease Detection (Cancer and Leukemia Group B Study 8361). Blood 2001, 97, 3574–3580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Du, W.; Liu, W.; Li, X.; Li, H.; Huang, S.-A. Comprehensive Flow Cytometry Phenotype in Acute Leukemia at Diagnosis and at Relapse. APMIS 2010, 118, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Langebrake, C.; Brinkmann, I.; Teigler-Schlegel, A.; Creutzig, U.; Griesinger, F.; Puhlmann, U.; Reinhardt, D. Immunophenotypic Differences between Diagnosis and Relapse in Childhood AML: Implications for MRD Monitoring. Cytom. Part B Clin. Cytom. 2005, 63, 1–9. [Google Scholar] [CrossRef]
- Arnone, M.; Konantz, M.; Hanns, P.; Paczulla Stanger, A.M.; Bertels, S.; Godavarthy, P.S.; Christopeit, M.; Lengerke, C. Acute Myeloid Leukemia Stem Cells: The Challenges of Phenotypic Heterogeneity. Cancers 2020, 12, 3742. [Google Scholar] [CrossRef]
- De Boer, B.; Prick, J.; Pruis, M.G.; Keane, P.; Imperato, M.R.; Jaques, J.; Brouwers-Vos, A.Z.; Hogeling, S.M.; Woolthuis, C.M.; Nijk, M.T.; et al. Prospective Isolation and Characterization of Genetically and Functionally Distinct AML Subclones. Cancer Cell 2018, 34, 674–689. [Google Scholar] [CrossRef] [Green Version]
- Angelini, D.F.; Ottone, T.; Guerrera, G.; Lavorgna, S.; Cittadini, M.; Buccisano, F.; De Bardi, M.; Gargano, F.; Maurillo, L.; Divona, M.; et al. A Leukemia-Associated CD34/CD123/CD25/CD99 + Immunophenotype Identifies FLT3 -Mutated Clones in Acute Myeloid Leukemia. Clin. Cancer Res. 2015, 21, 3977–3985. [Google Scholar] [CrossRef] [Green Version]
- Zandvakili, N.; Lee, H.M.; Desai, R.; Oryshchuk, A.; Browett, P.J.; Kakadia, P.M.; Bohlander, S.K. Clonal Evolution in a Murine CALM-AF10 Leukemia: Evidence of Functional Heterogeneity of Leukemia Stem Cells. Blood 2021, 138 (Suppl. S1), 1158. [Google Scholar] [CrossRef]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational Landscape and Significance across 12 Major Cancer Types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Garrett-Bakelman, F.E.; Chung, S.S.; Sanders, M.A.; Hricik, T.; Rapaport, F.; Patel, J.; Dillon, R.; Vijay, P.; Brown, A.L.; et al. Distinct Evolution and Dynamics of Epigenetic and Genetic Heterogeneity in Acute Myeloid Leukemia. Nat. Med. 2016, 22, 792–799. [Google Scholar] [CrossRef] [Green Version]
- Nuno, K.; Azizi, A.; Koehnke, T.; Corces, M.R.; Majeti, R. Multi-Omic Analysis Identifies Epigenetic Evolution in Relapsed Acute Myeloid Leukemia. Blood 2020, 136, 13–14. [Google Scholar] [CrossRef]
- Das, A.B.; Smith-Díaz, C.C.; Vissers, M.C.M. Emerging Epigenetic Therapeutics for Myeloid Leukemia: Modulating Demethylase Activity with Ascorbate. Haematologica 2020, 106, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Goldman, S.L.; Hassan, C.; Khunte, M.; Soldatenko, A.; Jong, Y.; Afshinnekoo, E.; Mason, C.E. Epigenetic Modifications in Acute Myeloid Leukemia: Prognosis, Treatment, and Heterogeneity. Front. Genet. 2019, 10, 133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Chen, X.; Wang, J.; Meydan, C.; Glass, J.L.; Shih, A.H.; Delwel, R.; Levine, R.L.; Mason, C.E.; Melnick, A.M. Somatic Mutations Drive Specific, but Reversible, Epigenetic Heterogeneity States in AML. Cancer Discov. 2020, 10, 1934–1949. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, M.E.; Abdel-Wahab, O.; Lu, C.; Ward, P.S.; Patel, J.; Shih, A.; Li, Y.; Bhagwat, N.; Vasanthakumar, A.; Fernandez, H.F. Leukemic IDH1 and IDH2 Mutations Result in a Hypermethylation Phenotype, Disrupt TET2 Function, and Impair Hematopoietic Differentiation. Cancer Cell 2010, 18, 553–567. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Mason, C.E.; Melnick, A. Genetic and Epigenetic Heterogeneity in Acute Myeloid Leukemia. Curr. Opin. Genet. Dev. 2016, 36, 100–106. [Google Scholar] [CrossRef] [Green Version]
- Yun, H.; Narayan, N.; Vohra, S.; Giotopoulos, G.; Mupo, A.; Madrigal, P.; Sasca, D.; Lara-Astiaso, D.; Horton, S.J.; Agrawal-Singh, S.; et al. Mutational Synergy during Leukemia Induction Remodels Chromatin Accessibility, Histone Modifications and Three-Dimensional DNA Topology to Alter Gene Expression. Nat. Genet. 2021, 53, 1443–1455. [Google Scholar] [CrossRef]
- Nuno, K.A.; Azizi, A.; Koehnke, T.; Ediriwickrema, A.; Corces, M.R.; Majeti, R. Chromatin Accessibility Analysis Reveals Epigenetic Evolution Is a Common Mechanism of Relapse in Acute Myeloid Leukemia. Blood 2021, 138 (Suppl. S1), 677. [Google Scholar] [CrossRef]
- Sonnet, M.; Claus, R.; Becker, N.; Zucknick, M.; Petersen, J.; Lipka, D.B.; Oakes, C.C.; Andrulis, M.; Lier, A.; Milsom, M.D.; et al. Early Aberrant DNA Methylation Events in a Mouse Model of Acute Myeloid Leukemia. Genome Med. 2014, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Lin, T.-C.; Hou, H.-A.; Chou, W.-C.; Ou, D.-L.; Yu, S.-L.; Tien, H.-F.; Lin, L.-I. CEBPA Methylation as a Prognostic Biomarker in Patients with de Novo Acute Myeloid Leukemia. Leukemia 2011, 25, 32–40. [Google Scholar] [CrossRef]
- Schlenk, R.F.; Döhner, H. Genomic Applications in the Clinic: Use in Treatment Paradigm of Acute Myeloid Leukemia. In Hematology/The Education Program of the American Society of Hematology; American Society of Hematology: New Orleans, LA, USA, 2013; pp. 324–330. [Google Scholar] [CrossRef] [Green Version]
- Simonetti, G.; Mengucci, C.; Padella, A.; Fonzi, E.; Picone, G.; Delpino, C.; Nanni, J.; De Tommaso, R.; Franchini, E.; Papayannidis, C.; et al. Integrated Genomic-Metabolic Classification of Acute Myeloid Leukemia Defines a Subgroup with NPM1 and Cohesin/DNA Damage Mutations. Leukemia 2021, 35, 2813–2826. [Google Scholar] [CrossRef] [PubMed]
- Dowling, P.; Tierney, C.; Dunphy, K.; Miettinen, J.J.; Heckman, C.A.; Bazou, D.; O’Gorman, P. Identification of Protein Biomarker Signatures for Acute Myeloid Leukemia (AML) Using Both Nontargeted and Targeted Approaches. Proteomes 2021, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Grønningsæter, I.S.; Reikvam, H.; Aasebø, E.; Bartaula-Brevik, S.; Tvedt, T.H.; Bruserud, Ø.; Hatfield, K.J. Targeting Cellular Metabolism in Acute Myeloid Leukemia and The Role of Patient Heterogeneity. Cells 2020, 9, 1155. [Google Scholar] [CrossRef]
- Bohlander, S.K. ABCs of Genomics. Hematol. Am. Soc. Hematol. Educ. Program. 2013, 2013, 316–323. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequencing Technology | Description |
|---|---|
| Genetic Analysis | |
| Whole genome sequencing | Identifies the complete range of genomic alterations, including point mutations, indels, copy number changes as well as structural chromosomal rearrangements |
| Whole exome sequencing | Reveals changes in the coding region of the genome |
| Targeted or gene panel sequencing | Deep sequencing of a panel of genes that are recurringly mutated or have a prognostic significance in AML |
| Gene expression analysis | |
| mRNA sequencing | Identifies alternative transcripts, gene fusions and allele-specific expression patterns in the coding transcriptome |
| Whole transcriptome analysis | Detects changes in expression of both coding and noncoding RNA |
| Epigenetic analysis | |
| Methyl sequencing | For studying changes in methylation patterns at a single nucleotide level, mainly using bisulfite treatment. Several methyl-seq. strategies have been developed including whole genome bisulfite sequencing and targeted approaches as well as reduced representation bisulfite sequencing, which enriches for CpG islands |
| ChIP-seq analysis | Combines chromatin immunoprecipitation with NGS for the identification of binding sites of DNA-associated proteins across the genome. Used to map histone modifications and transcription factor binding sites. |
| ATAC-seq analysis | Used for determining regions of chromatin accessibility and to map DNA binding proteins for the identification of active promoters, enhancers and cis-regulatory elements. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desai, R.H.; Zandvakili, N.; Bohlander, S.K. Dissecting the Genetic and Non-Genetic Heterogeneity of Acute Myeloid Leukemia Using Next-Generation Sequencing and In Vivo Models. Cancers 2022, 14, 2182. https://doi.org/10.3390/cancers14092182
Desai RH, Zandvakili N, Bohlander SK. Dissecting the Genetic and Non-Genetic Heterogeneity of Acute Myeloid Leukemia Using Next-Generation Sequencing and In Vivo Models. Cancers. 2022; 14(9):2182. https://doi.org/10.3390/cancers14092182
Chicago/Turabian StyleDesai, Rhea H., Niloofar Zandvakili, and Stefan K. Bohlander. 2022. "Dissecting the Genetic and Non-Genetic Heterogeneity of Acute Myeloid Leukemia Using Next-Generation Sequencing and In Vivo Models" Cancers 14, no. 9: 2182. https://doi.org/10.3390/cancers14092182
APA StyleDesai, R. H., Zandvakili, N., & Bohlander, S. K. (2022). Dissecting the Genetic and Non-Genetic Heterogeneity of Acute Myeloid Leukemia Using Next-Generation Sequencing and In Vivo Models. Cancers, 14(9), 2182. https://doi.org/10.3390/cancers14092182