
Resveratrol Contrasts IL-6 Pro-Growth Effects and Promotes Autophagy-Mediated Cancer Cell Dormancy in 3D Ovarian Cancer: Role of miR-1305 and of Its Target ARH-I

 ,
,  ,
, 

Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Reagents
2.3. Antibodies
2.4. 3D Spheroids Proliferation Assay
2.5. Western Blotting
2.6. Clonogenic Assay
2.7. Cell Counting, Doubling Time, and Cell Cycle Analysis
2.8. Immunofluorescence
2.9. Co-Immunoprecipitation Assay
2.10. Cell Transfection
2.11. Assessment of Autophagy in Living Cells Expressing GFP-LC3
2.12. Bioinformatic Prediction of miR-1305/ARH-I Interaction
2.13. Bioinformatic Analysis of TCGA Clinical Data
2.14. Imaging Acquisition and Analysis
2.15. Statistical Analysis
3. Results
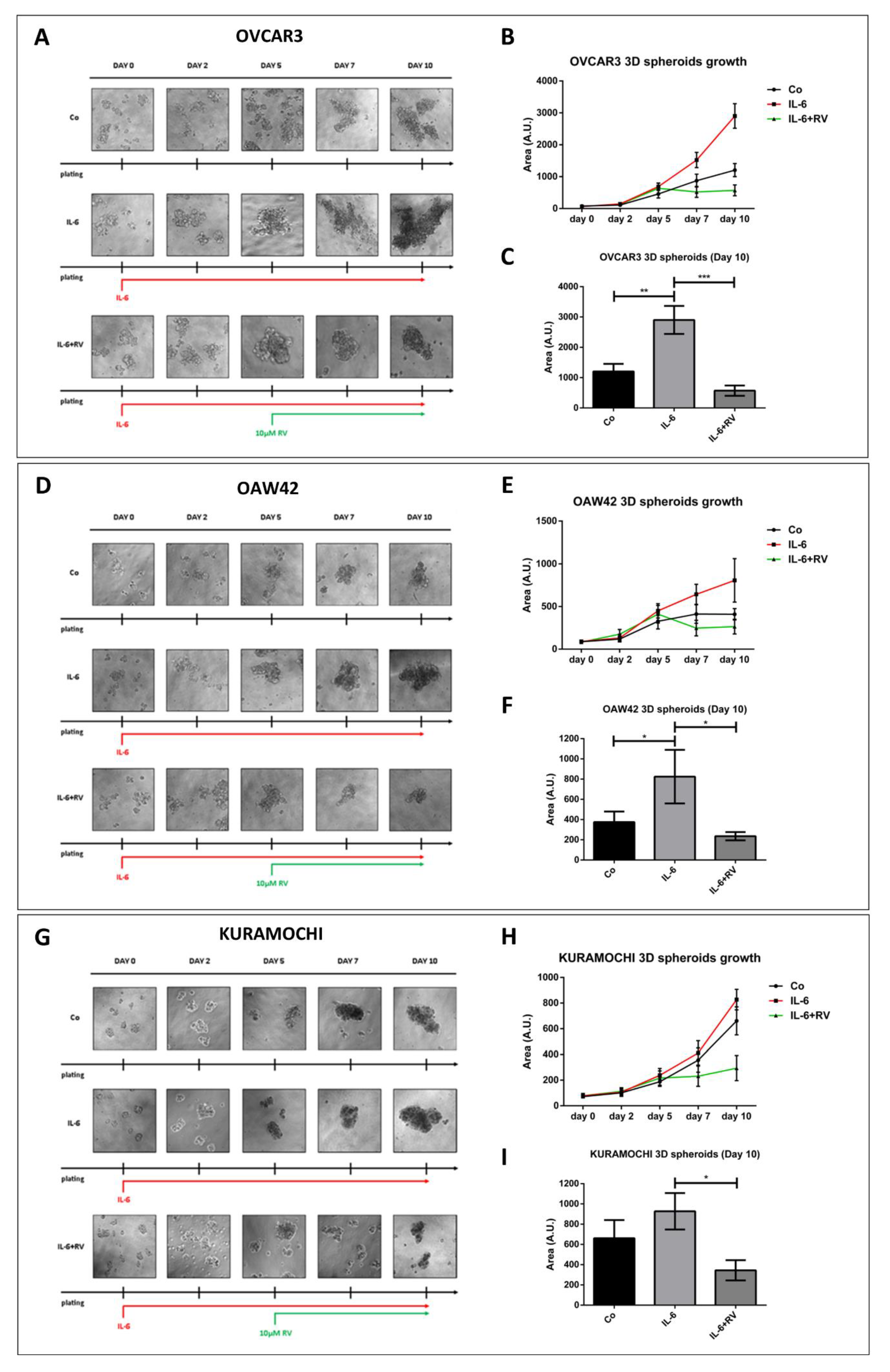
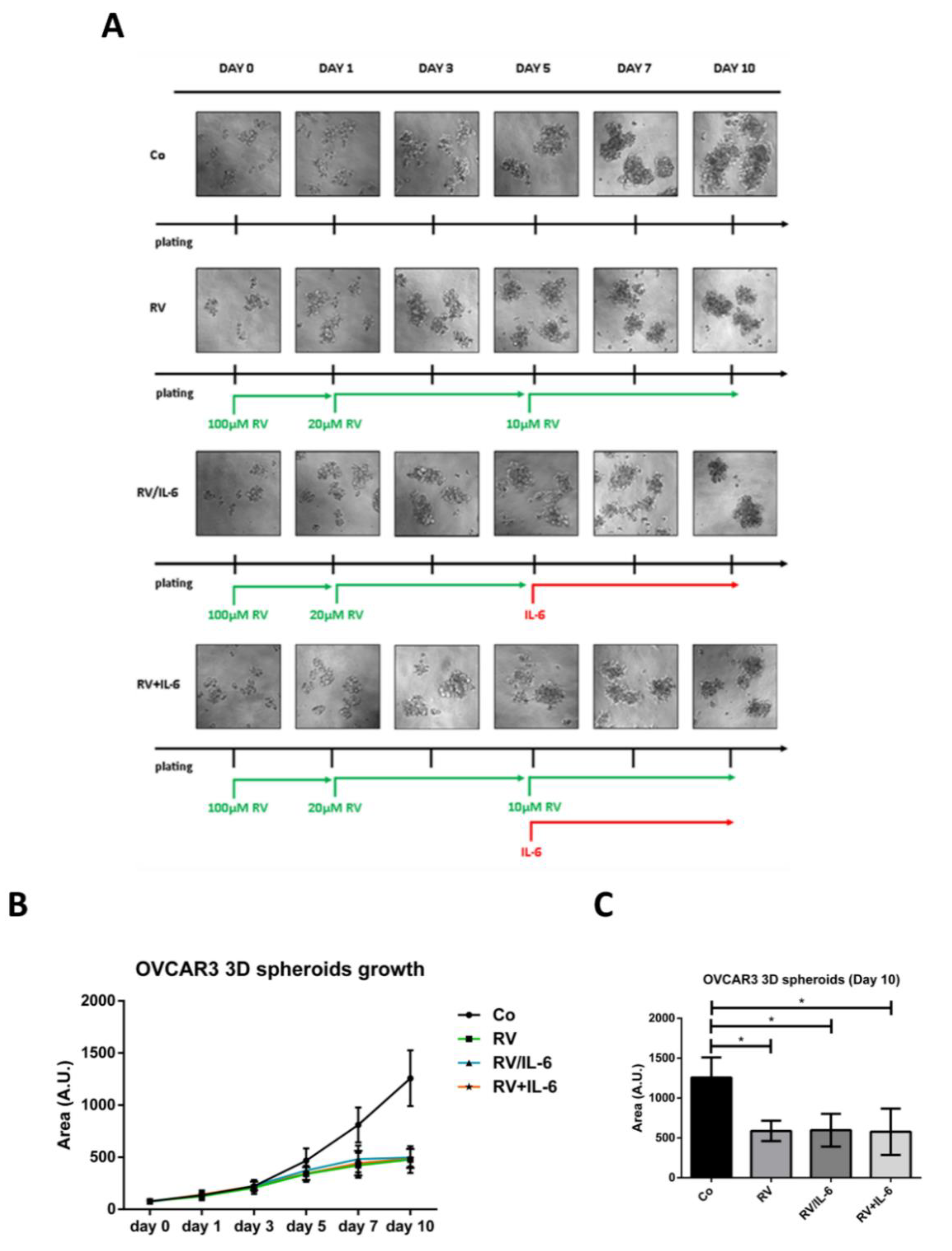
3.1. Resveratrol Contrasts IL-6 Stimulation of 3D Tumor Spheroid Growth
3.2. Resveratrol Induces a G0/G1 Cell Cycle Arrest in OVCAR3 Cells Exposed to IL-6
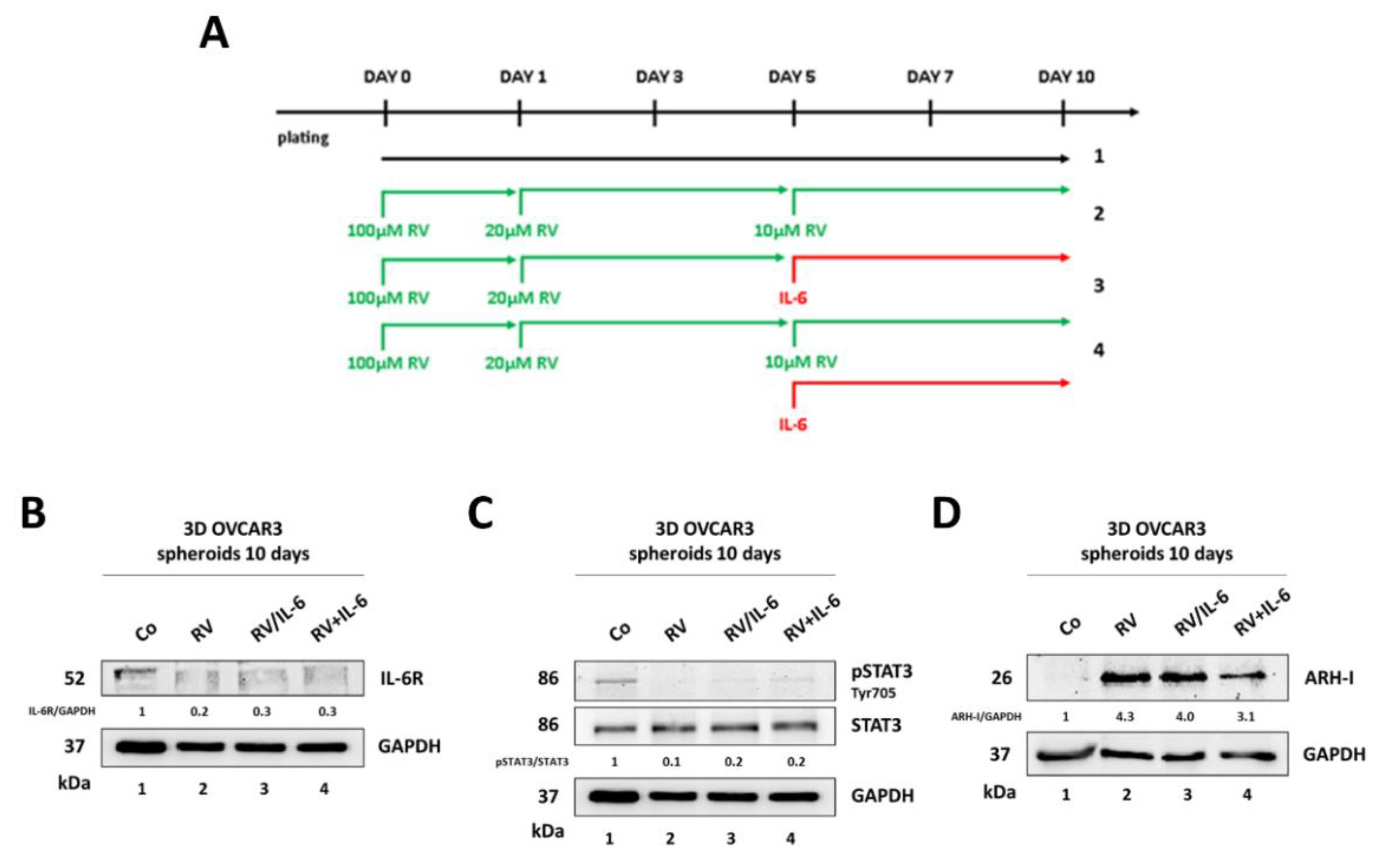
3.3. Ovarian Cancer Spheroids Pre-Treated with Resveratrol Do Not Rescue the Growth upon Challenge with IL-6
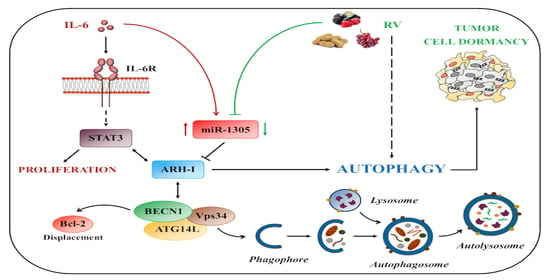
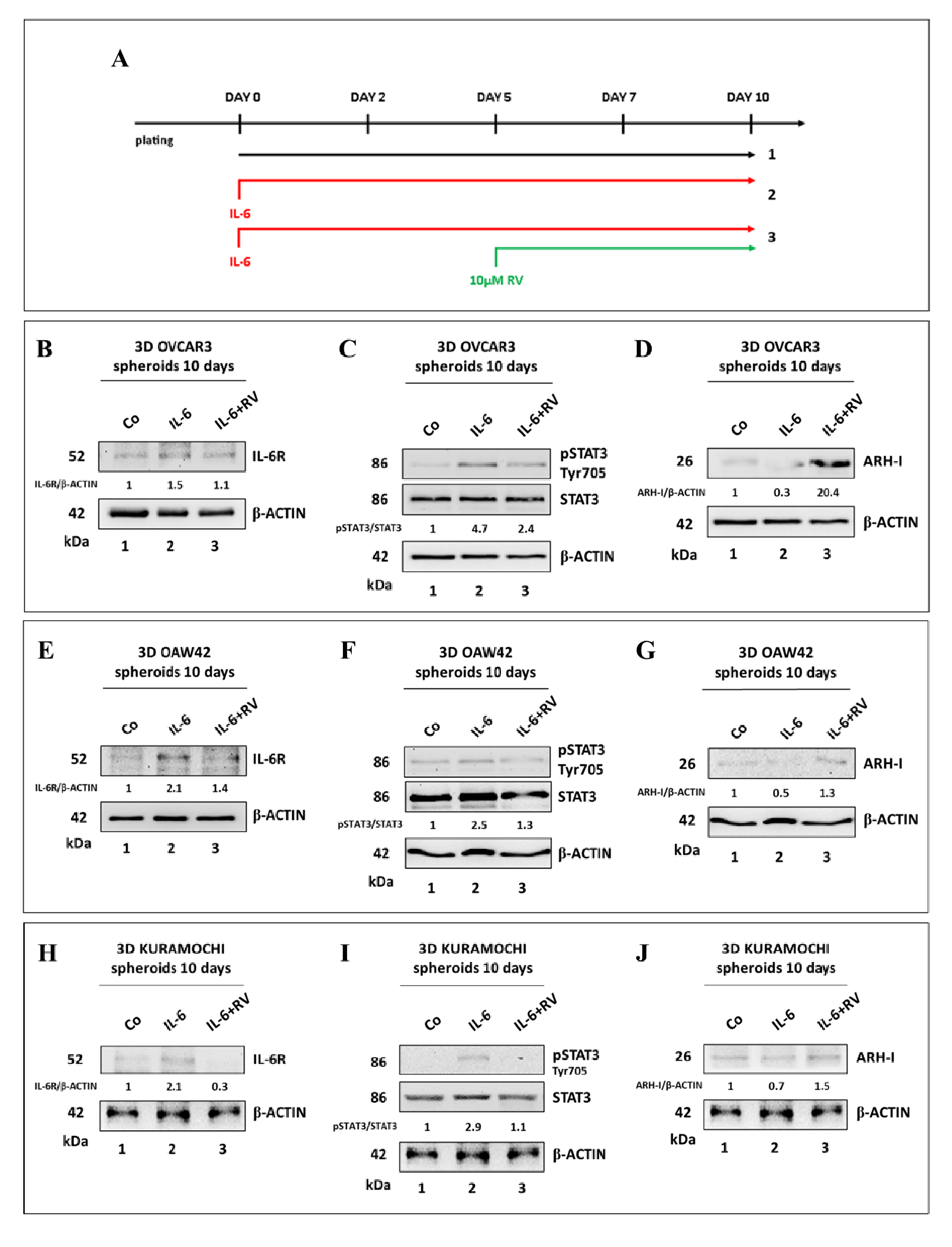
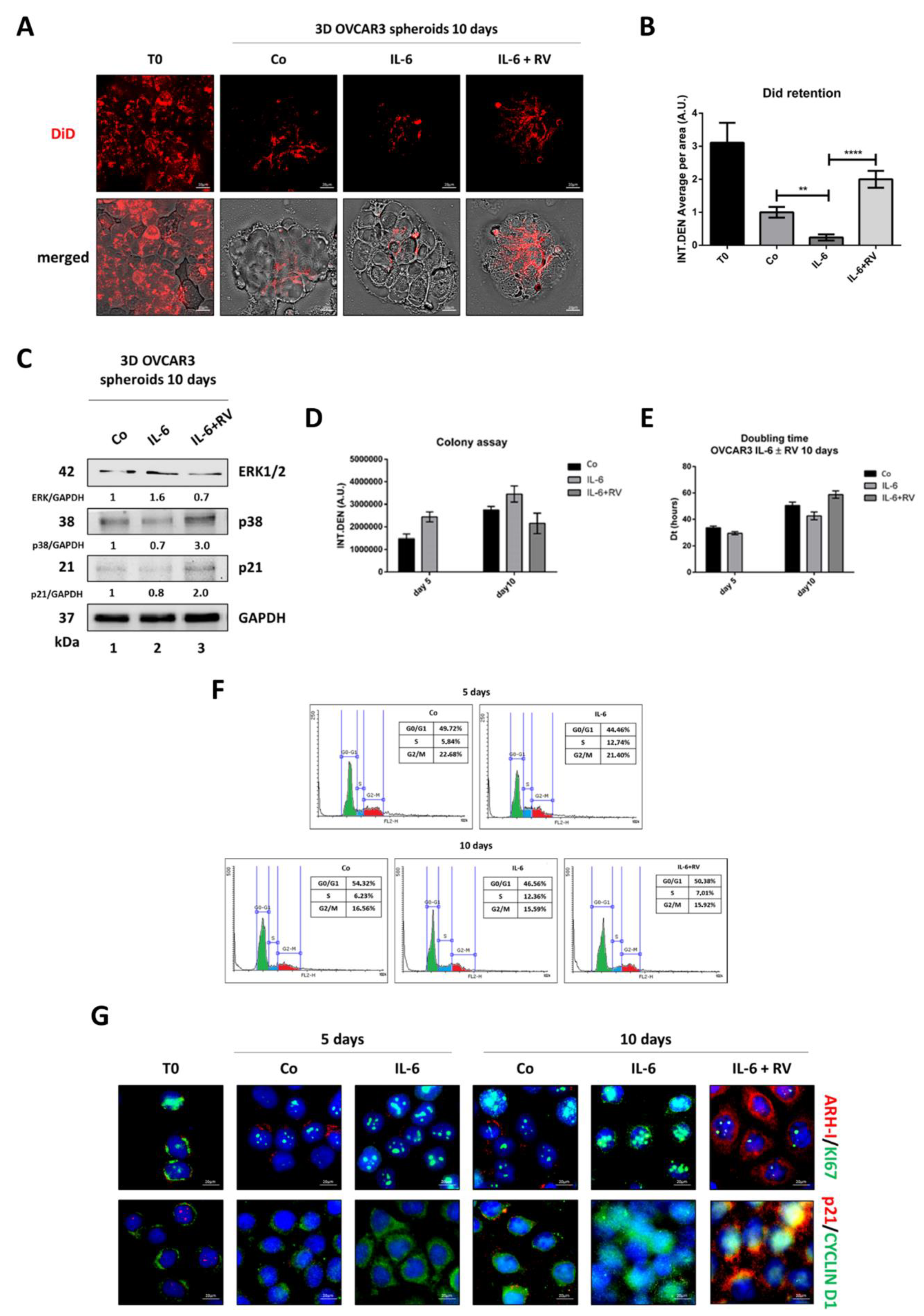
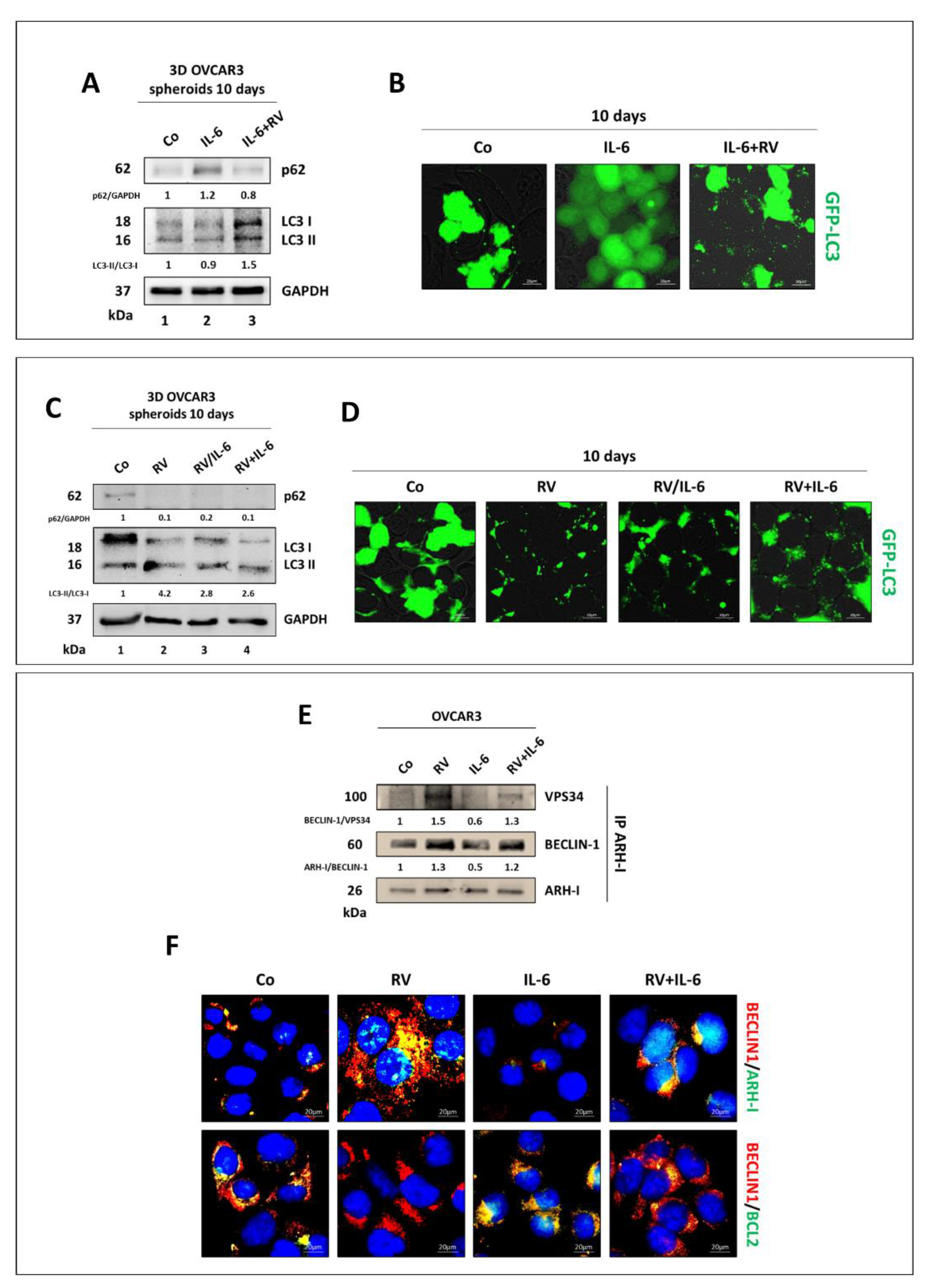
3.4. Resveratrol Promotes an ARH-I-Mediated Autophagy in Contrast to IL-6
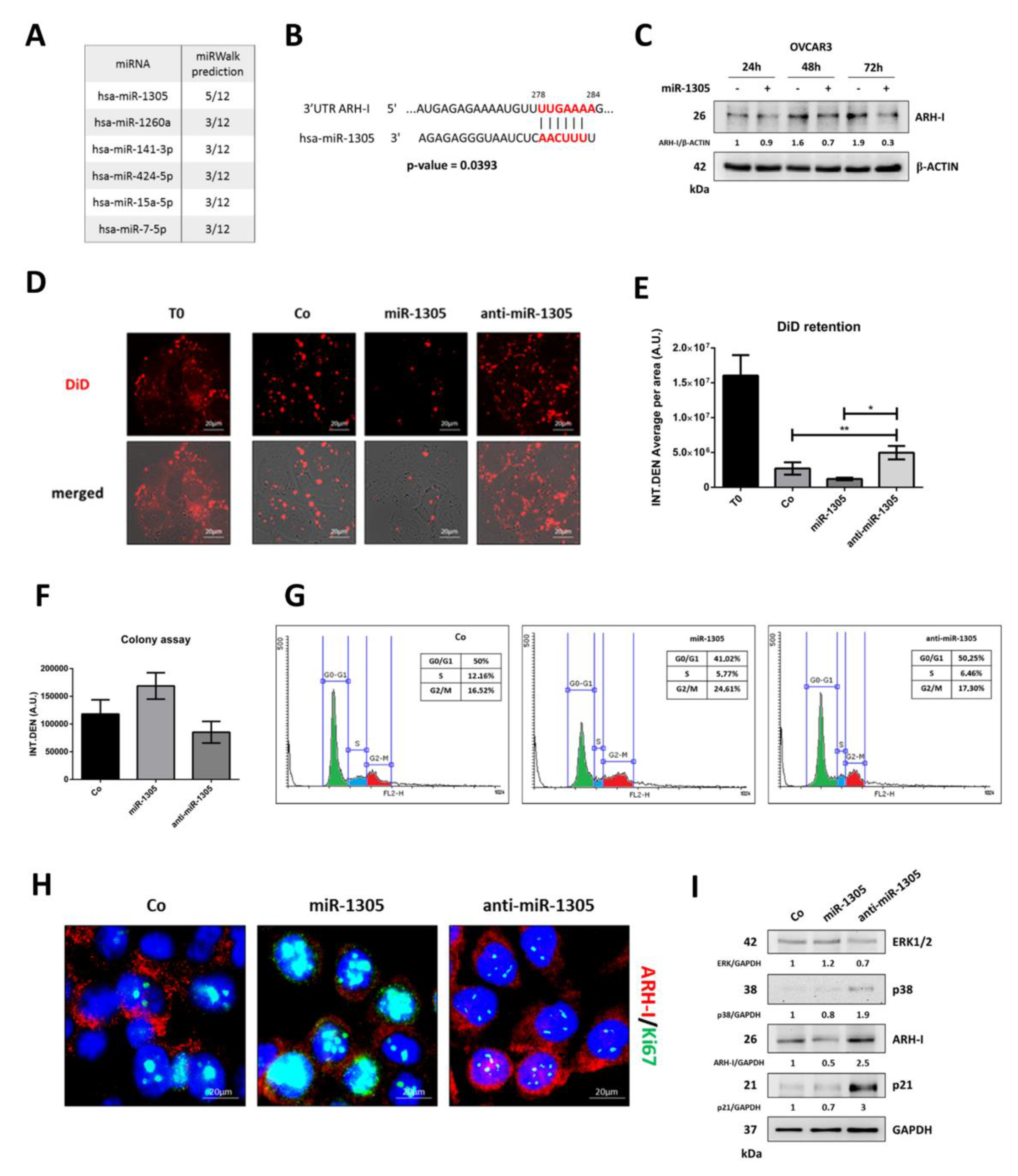
3.5. miR-1305 Downregulates ARH-I and Induces Ovarian Cancer Cell Proliferation Mimicking IL-6 Effects
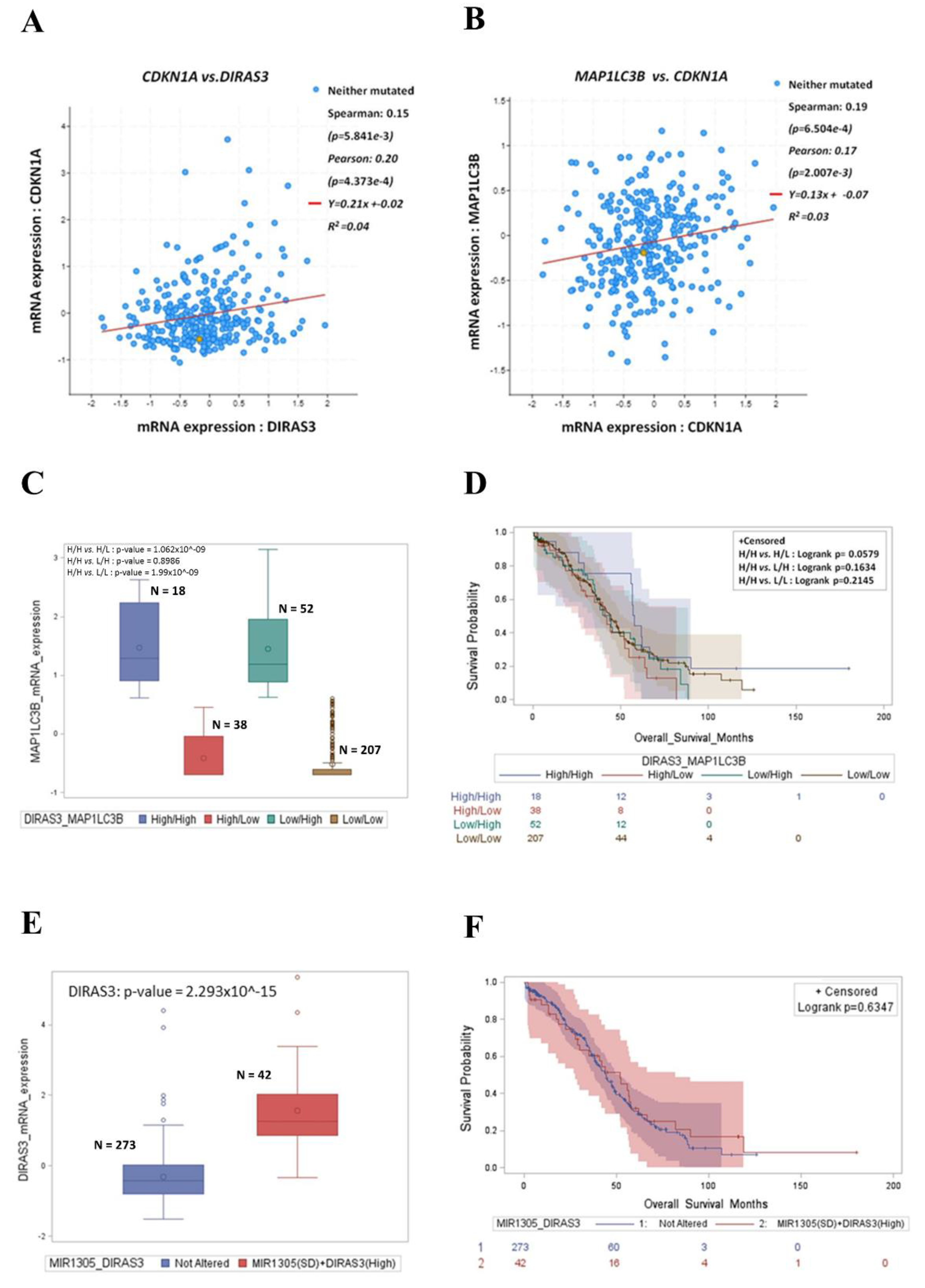
3.6. High Expression of DIRAS3 and MAP1LC3B Correlates with CDKN1A and Predicts Good Prognosis in Ovarian Cancer Patients
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gao, X.-L.; Zhang, M.; Tang, Y.-L.; Liang, X.-H. Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. OncoTargets Ther. 2017, 10, 5219–5228. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Inoue, M. Dormancy in cancer. Cancer Sci. 2019, 110, 474–480. [Google Scholar] [CrossRef]
- Butturini, E.; Carcereri De Prati, A.; Boriero, D.; Mariotto, S. Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 4305. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Bragado, P.; Aguirre-Ghiso, J.A. Mechanisms of disseminated cancer cell dormancy: An awakening field. Nat. Rev. Cancer 2014, 14, 611–622. [Google Scholar] [CrossRef]
- Greer, B.E.; Bundy, B.N.; Ozols, R.F.; Fowler, J.M.; Clarke-Pearson, D.; Burger, R.A.; Mannel, R.; DeGeest, K.; Hartenbach, E.M.; Baergen, R.N.; et al. Implications of second-look laparotomy in the context of optimally resected stage III ovarian cancer: A non-randomized comparison using an explanatory analysis: A Gynecologic Oncology Group study. Gynecol. Oncol. 2005, 99, 71–79. [Google Scholar] [CrossRef]
- Cabasag, C.J.; Arnold, M.; Butler, J.; Inoue, M.; Trabert, B.; Webb, P.M.; Bray, F.; Soerjomataram, I. The influence of birth cohort and calendar period on global trends in ovarian cancer incidence. Int. J. Cancer 2020, 146, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Jayson, G.C.; Kohn, E.C.; Kitchener, H.C.; Ledermann, J.A. Ovarian cancer. Lancet 2014, 384, 1376–1388. [Google Scholar] [CrossRef]
- Bast, R.C., Jr.; Matulonis, U.A.; Sood, A.K.; Ahmed, A.A.; Amobi, A.E.; Balkwill, F.R.; Wielgos-Bonvallet, M.; Bowtell, D.D.L.; Brenton, J.D.; Brugge, J.S.; et al. Critical questions in ovarian cancer research and treatment: Report of an American Association for Cancer Research Special Conference. Cancer 2019, 125, 1963–1972. [Google Scholar] [CrossRef] [PubMed]
- Peppicelli, S.; Andreucci, E.; Ruzzolini, J.; Laurenzana, A.; Margheri, F.; Fibbi, G.; Del Rosso, M.; Bianchini, F.; Calorini, L. The acidic microenvironment as a possible niche of dormant tumor cells. Cell. Mol. Life Sci. 2017, 74, 2761–2771. [Google Scholar] [CrossRef] [PubMed]
- Vera-Ramirez, L.; Hunter, K.W. Tumor cell dormancy as an adaptive cell stress response mechanism. F1000Research 2017, 6, 2134. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gibson, S.B. Three dimensions of autophagy in regulating tumor growth: Cell survival/death, cell proliferation, and tumor dormancy. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 166265. [Google Scholar] [CrossRef] [PubMed]
- Ferraresi, A.; Girone, C.; Esposito, A.; Vidoni, C.; Vallino, L.; Secomandi, E.; Dhanasekaran, D.N.; Isidoro, C. How Autophagy Shapes the Tumor Microenvironment in Ovarian Cancer. Front. Oncol. 2020, 10, 599915. [Google Scholar] [CrossRef] [PubMed]
- Thuwajit, C.; Ferraresi, A.; Titone, R.; Thuwajit, P.; Isidoro, C. The metabolic cross-talk between epithelial cancer cells and stromal fibroblasts in ovarian cancer progression: Autophagy plays a role. Med. Res. Rev. 2018, 38, 1235–1254. [Google Scholar] [CrossRef] [PubMed]
- Bustos, S.O.; Antunes, F.; Rangel, M.C.; Chammas, R. Emerging Autophagy Functions Shape the Tumor Microenvironment and Play a Role in Cancer Progression—Implications for Cancer Therapy. Front. Oncol. 2020, 10, 606436. [Google Scholar] [CrossRef] [PubMed]
- Ferraresi, A.; Phadngam, S.; Morani, F.; Galetto, A.; Alabiso, O.; Chiorino, G.; Isidoro, C. Resveratrol inhibits IL-6-induced ovarian cancer cell migration through epigenetic up-regulation of autophagy. Mol. Carcinog. 2017, 56, 1164–1181. [Google Scholar] [CrossRef] [PubMed]
- Thongchot, S.; Ferraresi, A.; Vidoni, C.; Loilome, W.; Yongvanit, P.; Namwat, N.; Isidoro, C. Resveratrol interrupts the pro-invasive communication between cancer associated fibroblasts and cholangiocarcinoma cells. Cancer Lett. 2018, 430, 160–171. [Google Scholar] [CrossRef]
- Thongchot, S.; Jamjuntra, P.; Therasakvichya, S.; Warnnissorn, M.; Ferraresi, A.; Thuwajit, P.; Isidoro, C.; Thuwajit, C. Interleukin-8 released by cancer-associated fibroblasts attenuates the autophagy and promotes the migration of ovarian cancer cells. Int. J. Oncol. 2021, 58, 14. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Mahapatra, K.K.; Praharaj, P.P.; Patil, S.; Bhutia, S.K. Recent progress of autophagy signaling in tumor microenvironment and its targeting for possible cancer therapeutics. Semin. Cancer Biol. 2021; in press. [Google Scholar] [CrossRef] [PubMed]
- Ngabire, D.; Kim, G.-D. Autophagy and Inflammatory Response in the Tumor Microenvironment. Int. J. Mol. Sci. 2017, 18, 2016. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Baquero, M.T.; Yang, H.; Yang, M.; Reger, A.S.; Kim, C.; Levine, D.A.; Clarke, C.H.; Liao, W.S.-L.; Bast, R.C., Jr. DIRAS3 regulates the autophagosome initiation complex in dormant ovarian cancer cells. Autophagy 2014, 10, 1071–1092, Erratum in Autophagy 2014, 10, 1482. [Google Scholar] [CrossRef]
- Bildik, G.; Liang, X.; Sutton, M.N.; Bast, R.C., Jr.; Lu, Z. DIRAS3: An Imprinted Tumor Suppressor Gene that Regulates RAS and PI3K-driven Cancer Growth, Motility, Autophagy, and Tumor Dormancy. Mol. Cancer Ther. 2022, 21, 25–37. [Google Scholar] [CrossRef]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Klionsky, D.J. Mammalian autophagy: Core molecular machinery and signaling regulation. Curr. Opin. Cell Biol. 2010, 22, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Ferraresi, A.; Titone, R.; Follo, C.; Castiglioni, A.; Chiorino, G.; Dhanasekaran, D.N.; Isidoro, C. The protein restriction mimetic Resveratrol is an autophagy inducer stronger than amino acid starvation in ovarian cancer cells. Mol. Carcinog. 2017, 56, 2681–2691. [Google Scholar] [CrossRef]
- Ferraresi, A.; Esposito, A.; Girone, C.; Vallino, L.; Salwa, A.; Ghezzi, I.; Thongchot, S.; Vidoni, C.; Dhanasekaran, D.N.; Isidoro, C. Resveratrol Contrasts LPA-Induced Ovarian Cancer Cell Migration and Platinum Resistance by Rescuing Hedgehog-Mediated Autophagy. Cells 2021, 10, 3213. [Google Scholar] [CrossRef] [PubMed]
- Kast, V.; Loessner, D. 3D Models for Ovarian Cancer. In Ovarian Cancer: Molecular & Diagnostic Imaging and Treatment Strategies; Springer: Boston, MA, USA, 2021; pp. 139–149. [Google Scholar] [CrossRef]
- Beaufort, C.M.; Helmijr, J.C.A.; Piskorz, A.M.; Hoogstraat, M.; Ruigrok-Ritstier, K.; Besselink, N.; Murtaza, M.; van Ijcken, W.; Heine, A.; Smid, M.; et al. Ovarian Cancer Cell Line Panel (OCCP): Clinical Importance of In Vitro Morphological Subtypes. PLoS ONE 2014, 9, e103988, Erratum in PLoS ONE 2015, 10, e0122284. [Google Scholar] [CrossRef]
- Ha, J.H.; Jayaraman, M.; Nadhan, R.; Kashyap, S.; Mukherjee, P.; Isidoro, C.; Song, Y.S.; Dhanasekaran, D.N. Unraveling Autocrine Signaling Pathways through Metabolic Fingerprinting in Serous Ovarian Cancer Cells. Biomedicines 2021, 9, 1927. [Google Scholar] [CrossRef]
- Haley, J.; Tomar, S.; Pulliam, N.; Xiong, S.; Perkins, S.M.; Karpf, A.R.; Mitra, S.; Nephew, K.P.; Mitra, A.K. Functional characterization of a panel of high-grade serous ovarian cancer cell lines as representative experimental models of the disease. Oncotarget 2016, 7, 32810–32820. [Google Scholar] [CrossRef]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Investig. 2008, 118, 3917–3929. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Gomis, R.R.; Gawrzak, S. Tumor cell dormancy. Mol. Oncol. 2017, 11, 62–78. [Google Scholar] [CrossRef]
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84. [Google Scholar] [CrossRef]
- Pranzini, E.; Raugei, G.; Taddei, M.L. Metabolic Features of Tumor Dormancy: Possible Therapeutic Strategies. Cancers 2022, 14, 547. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef]
- Badgwell, D.B.; Lu, Z.; Le, K.; Gao, F.; Yang, M.; Suh, G.K.; Bao, J.-J.; Das, P.; Andreeff, M.; Chen, W.; et al. The tumor-suppressor gene ARHI (DIRAS3) suppresses ovarian cancer cell migration through inhibition of the Stat3 and FAK/Rho signaling pathways. Oncogene 2012, 31, 68–79. [Google Scholar] [CrossRef]
- Al Habyan, S.; Kalos, C.; Szymborski, J.; McCaffrey, L. Multicellular detachment generates metastatic spheroids during intra-abdominal dissemination in epithelial ovarian cancer. Oncogene 2018, 37, 5127–5135. [Google Scholar] [CrossRef] [PubMed]
- Kenny, H.A.; Dogan, S.; Zillhardt, M.; Mitra, A.K.; Yamada, S.D.; Krausz, T.; Lengyel, E. Organotypic Models of Metastasis: A Three-dimensional Culture Mimicking the Human Peritoneum and Omentum for the Study of the Early Steps of Ovarian Cancer Metastasis. In Ovarian Cancer, Cancer Treatment and Research; Springer: Boston, MA, USA, 2009; Volume 149, pp. 335–351. [Google Scholar] [CrossRef]
- Chen, Y.; Zaman, M.S.; Deng, G.; Majid, S.; Saini, S.; Liu, J.; Tanaka, Y.; Dahiya, R. MicroRNAs 221/222 and Genistein-Mediated Regulation of ARHI Tumor Suppressor Gene in Prostate Cancer. Cancer Prev. Res. 2011, 4, 76–86. [Google Scholar] [CrossRef]
- Liu, W.; Zhuang, R.; Feng, S.; Bai, X.; Jia, Z.; Kapora, E.; Tan, W. Long non-coding RNA ASB16-AS1 enhances cell proliferation, migration and invasion via functioning as a ceRNA through miR-1305/Wnt/β-catenin axis in cervical cancer. Biomed. Pharmacother. 2020, 125, 109965. [Google Scholar] [CrossRef]
- Cai, Y.; Hao, Y.; Ren, H.; Dang, Z.; Xu, H.; Xue, X.; Gao, Y. miR-1305 Inhibits The Progression of Non-Small Cell Lung Cancer by Regulating MDM2. Cancer Manag. Res. 2019, 11, 9529–9540. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, L.; Wang, B.; Tao, C.; Shi, L.; Xu, M. Circ0120816 acts as an oncogene of esophageal squamous cell carcinoma by inhibiting miR-1305 and releasing TXNRD1. Cancer Cell Int. 2020, 20, 526. [Google Scholar] [CrossRef]
- Lee, J.Y.; Ryu, D.; Lim, S.W.; Ryu, K.J.; Choi, M.E.; Yoon, S.E.; Kim, K.; Park, C.; Kim, S.J. Exosomal miR-1305 in the oncogenic activity of hypoxic multiple myeloma cells: A biomarker for predicting prognosis. J. Cancer 2021, 12, 2825–2834. [Google Scholar] [CrossRef]
- Jin, S.; Collin, J.; Zhu, L.; Montaner, D.; Armstrong, L.; Neganova, I.; Lako, M. A Novel Role for miR-1305 in Regulation of Pluripotency-Differentiation Balance, Cell Cycle, and Apoptosis in Human Pluripotent Stem Cells. Stem Cells 2016, 34, 2306–2317. [Google Scholar] [CrossRef]
- Rosen, D.G.; Wang, L.; Jain, A.N.; Lu, K.H.; Luo, R.Z.; Yu, Y.; Liu, J.; Bast, R.C., Jr. Expression of the Tumor Suppressor Gene ARHI in Epithelial Ovarian Cancer Is Associated with Increased Expression of p21WAF1/CIP1 and Prolonged Progression-Free Survival. Clin. Cancer Res. 2004, 10, 6559–6566. [Google Scholar] [CrossRef][Green Version]
- Vallino, L.; Ferraresi, A.; Vidoni, C.; Secomandi, E.; Esposito, A.; Dhanasekaran, D.N.; Isidoro, C. Modulation of non-coding RNAs by resveratrol in ovarian cancer cells: In silico analysis and literature review of the anti-cancer pathways involved. J. Tradit. Complement. Med. 2020, 10, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Berretta, M.; Bignucolo, A.; Di Francia, R.; Comello, F.; Facchini, G.; Ceccarelli, M.; Iaffaioli, R.V.; Quagliariello, V.; Maurea, N. Resveratrol in Cancer Patients: From Bench to Bedside. Int. J. Mol. Sci. 2020, 21, 2945. [Google Scholar] [CrossRef] [PubMed]
- Musial, C.; Siedlecka-Kroplewska, K.; Kmiec, Z.; Gorska-Ponikowska, M. Modulation of Autophagy in Cancer Cells by Dietary Polyphenols. Antioxidants 2021, 10, 123. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, E.M.; Welters, M.J.; Nortier, J.W.; Van Der Burg, S.H.; Kroep, J.R. Interleukin-6/interleukin-6 Receptor Pathway as a New Therapy Target in Epithelial Ovarian Cancer. Curr. Pharm. Des. 2012, 18, 3816–3827. [Google Scholar] [CrossRef] [PubMed]
- Isobe, A.; Sawada, K.; Kinose, Y.; Ohyagi-Hara, C.; Nakatsuka, E.; Makino, H.; Ogura, T.; Mizuno, T.; Suzuki, N.; Morii, E.; et al. Interleukin 6 Receptor Is an Independent Prognostic Factor and a Potential Therapeutic Target of Ovarian Cancer. PLoS ONE 2015, 10, e0118080. [Google Scholar] [CrossRef]
- Recasens, A.; Munoz, L. Targeting Cancer Cell Dormancy. Trends Pharmacol. Sci. 2019, 40, 128–141. [Google Scholar] [CrossRef]
- Ruksha, T.G. MicroRNAs’ control of cancer cell dormancy. Cell Div. 2019, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Irshad, R.; Husain, M. Natural products in the reprogramming of cancer epigenetics. Toxicol. Appl. Pharmacol. 2021, 417, 115467. [Google Scholar] [CrossRef]
- Vidoni, C.; Ferraresi, A.; Secomandi, E.; Vallino, L.; Dhanasekaran, D.N.; Isidoro, C. Epigenetic targeting of autophagy for cancer prevention and treatment by natural compounds. Semin. Cancer Biol. 2020, 66, 34–44. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esposito, A.; Ferraresi, A.; Salwa, A.; Vidoni, C.; Dhanasekaran, D.N.; Isidoro, C. Resveratrol Contrasts IL-6 Pro-Growth Effects and Promotes Autophagy-Mediated Cancer Cell Dormancy in 3D Ovarian Cancer: Role of miR-1305 and of Its Target ARH-I. Cancers 2022, 14, 2142. https://doi.org/10.3390/cancers14092142
Esposito A, Ferraresi A, Salwa A, Vidoni C, Dhanasekaran DN, Isidoro C. Resveratrol Contrasts IL-6 Pro-Growth Effects and Promotes Autophagy-Mediated Cancer Cell Dormancy in 3D Ovarian Cancer: Role of miR-1305 and of Its Target ARH-I. Cancers. 2022; 14(9):2142. https://doi.org/10.3390/cancers14092142
Chicago/Turabian StyleEsposito, Andrea, Alessandra Ferraresi, Amreen Salwa, Chiara Vidoni, Danny N. Dhanasekaran, and Ciro Isidoro. 2022. "Resveratrol Contrasts IL-6 Pro-Growth Effects and Promotes Autophagy-Mediated Cancer Cell Dormancy in 3D Ovarian Cancer: Role of miR-1305 and of Its Target ARH-I" Cancers 14, no. 9: 2142. https://doi.org/10.3390/cancers14092142
APA StyleEsposito, A., Ferraresi, A., Salwa, A., Vidoni, C., Dhanasekaran, D. N., & Isidoro, C. (2022). Resveratrol Contrasts IL-6 Pro-Growth Effects and Promotes Autophagy-Mediated Cancer Cell Dormancy in 3D Ovarian Cancer: Role of miR-1305 and of Its Target ARH-I. Cancers, 14(9), 2142. https://doi.org/10.3390/cancers14092142