
Molecular Mechanisms and Current Treatment Options for Cancer Cachexia

 ,
,  , , , , and
, , , , and
Abstract
:Simple Summary
Abstract
1. Introduction
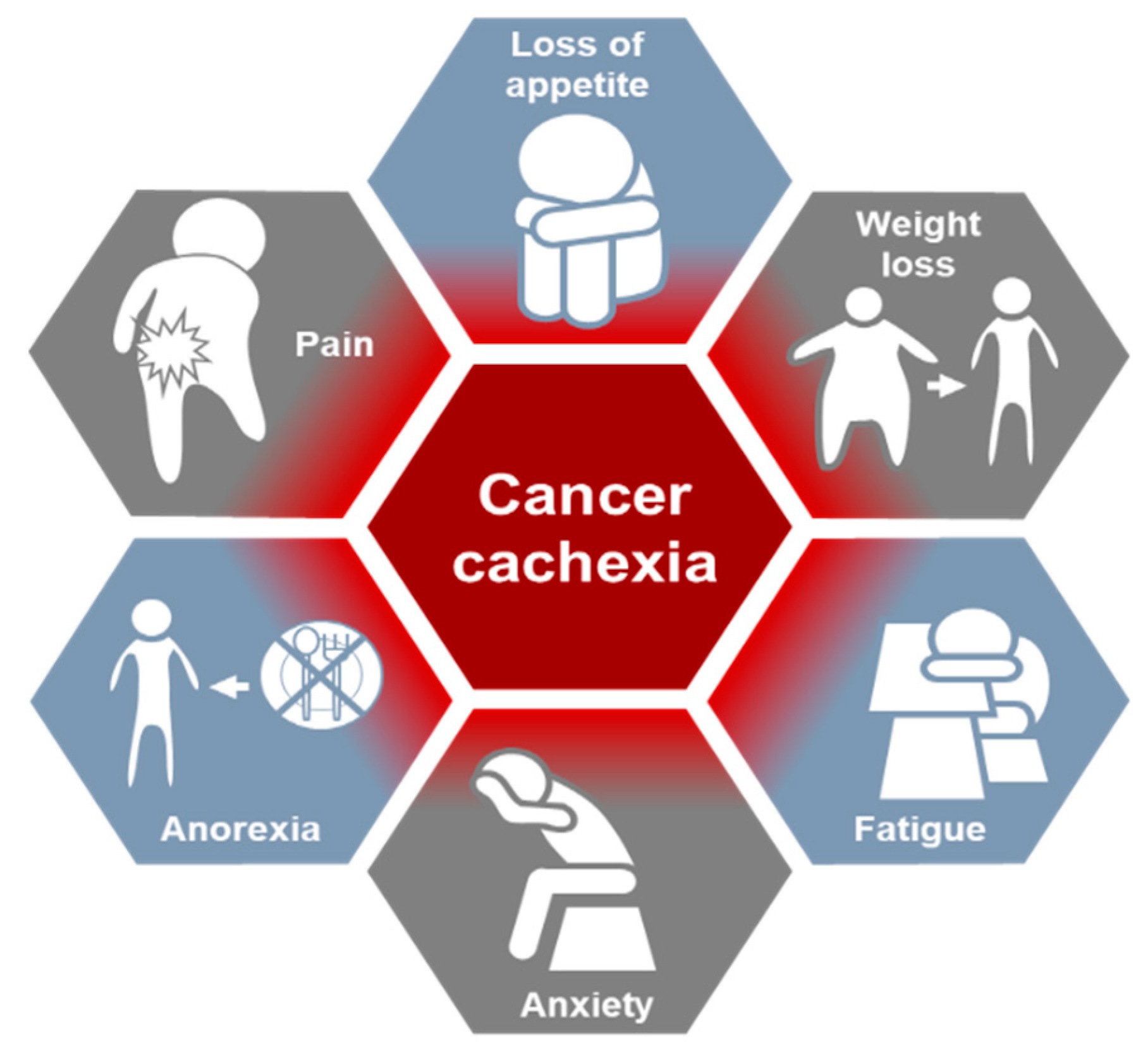
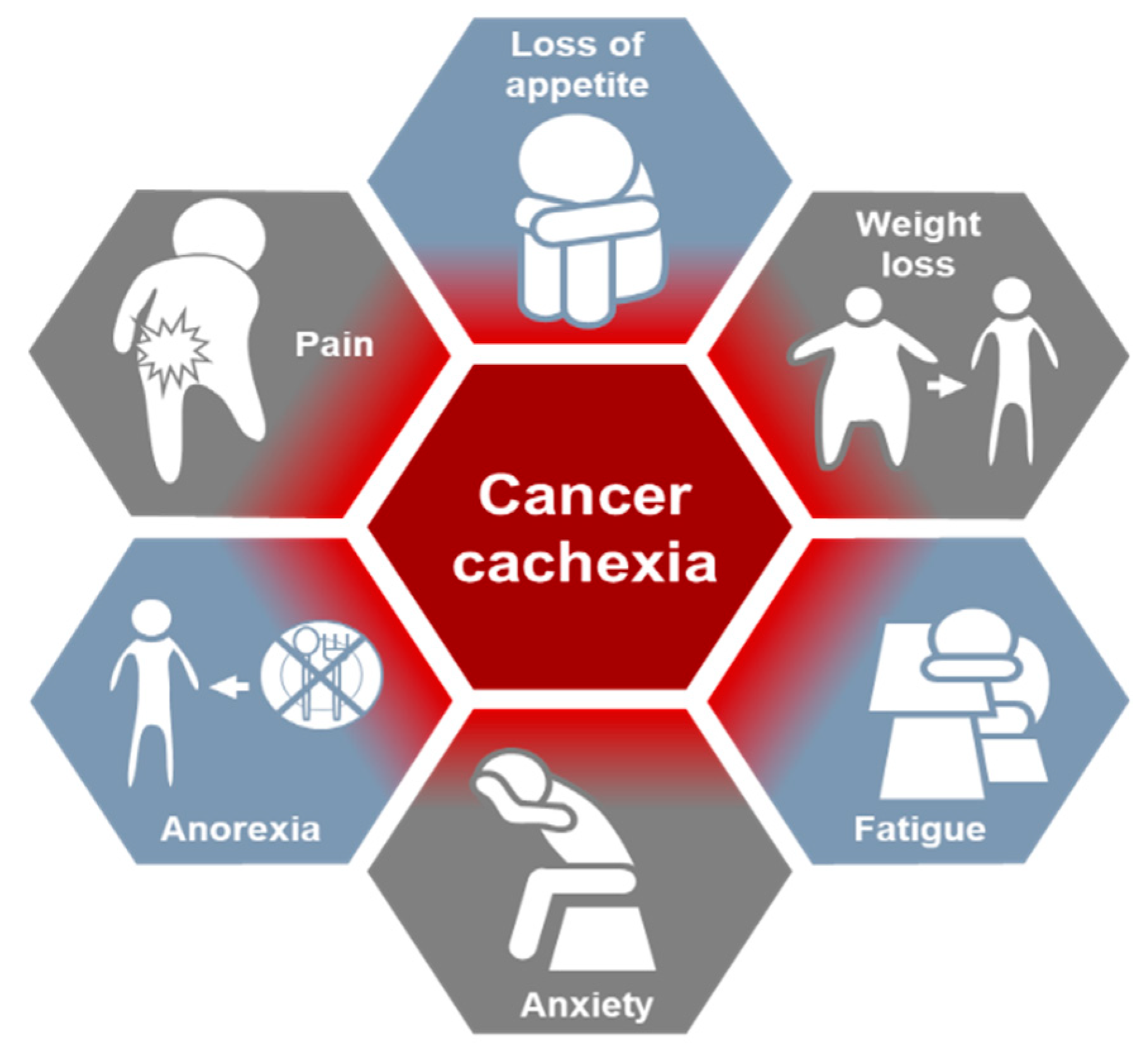
2. Cancer Cachexia
3. The Molecular Mechanism Underlying Cancer Cachexia
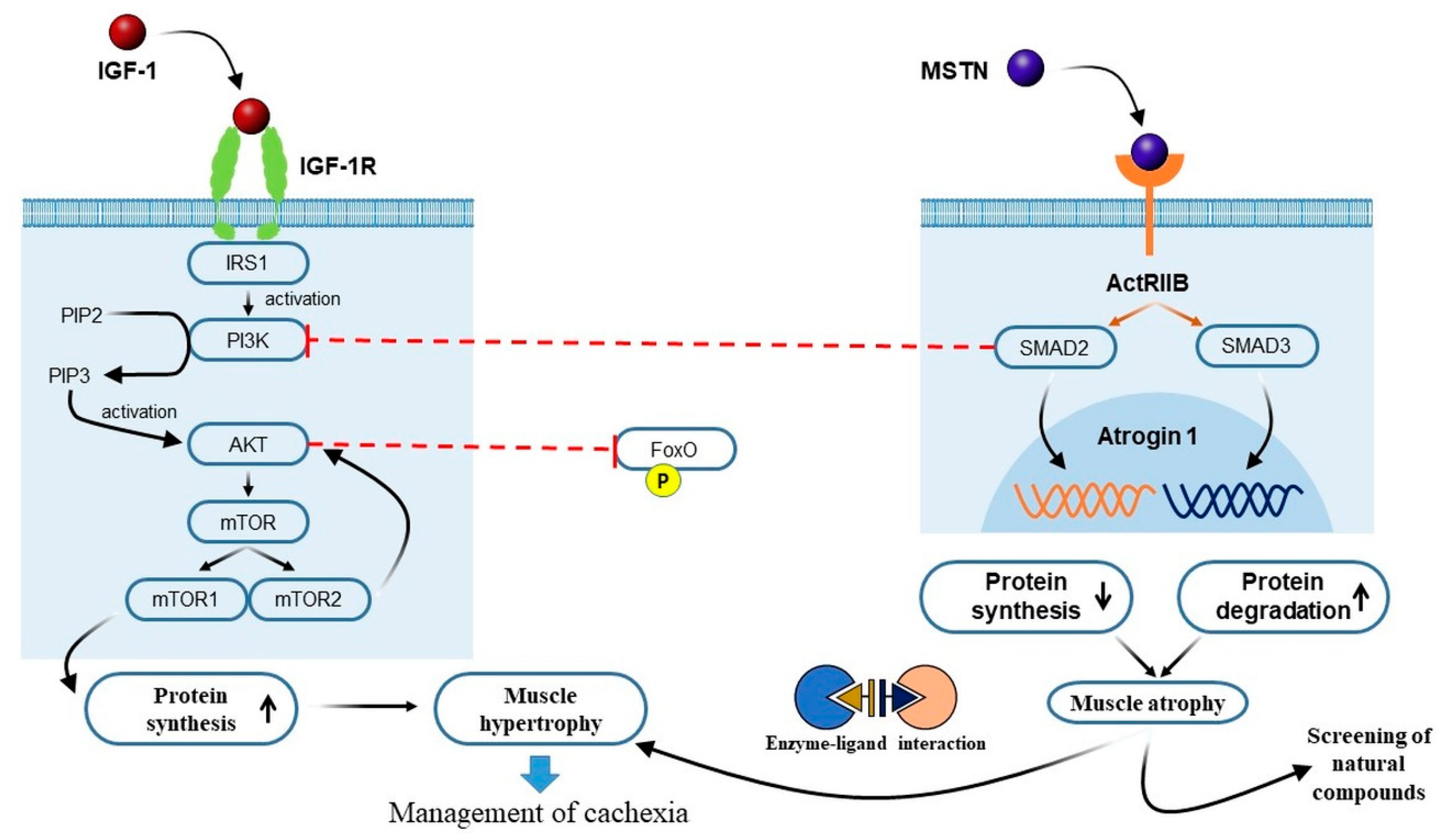
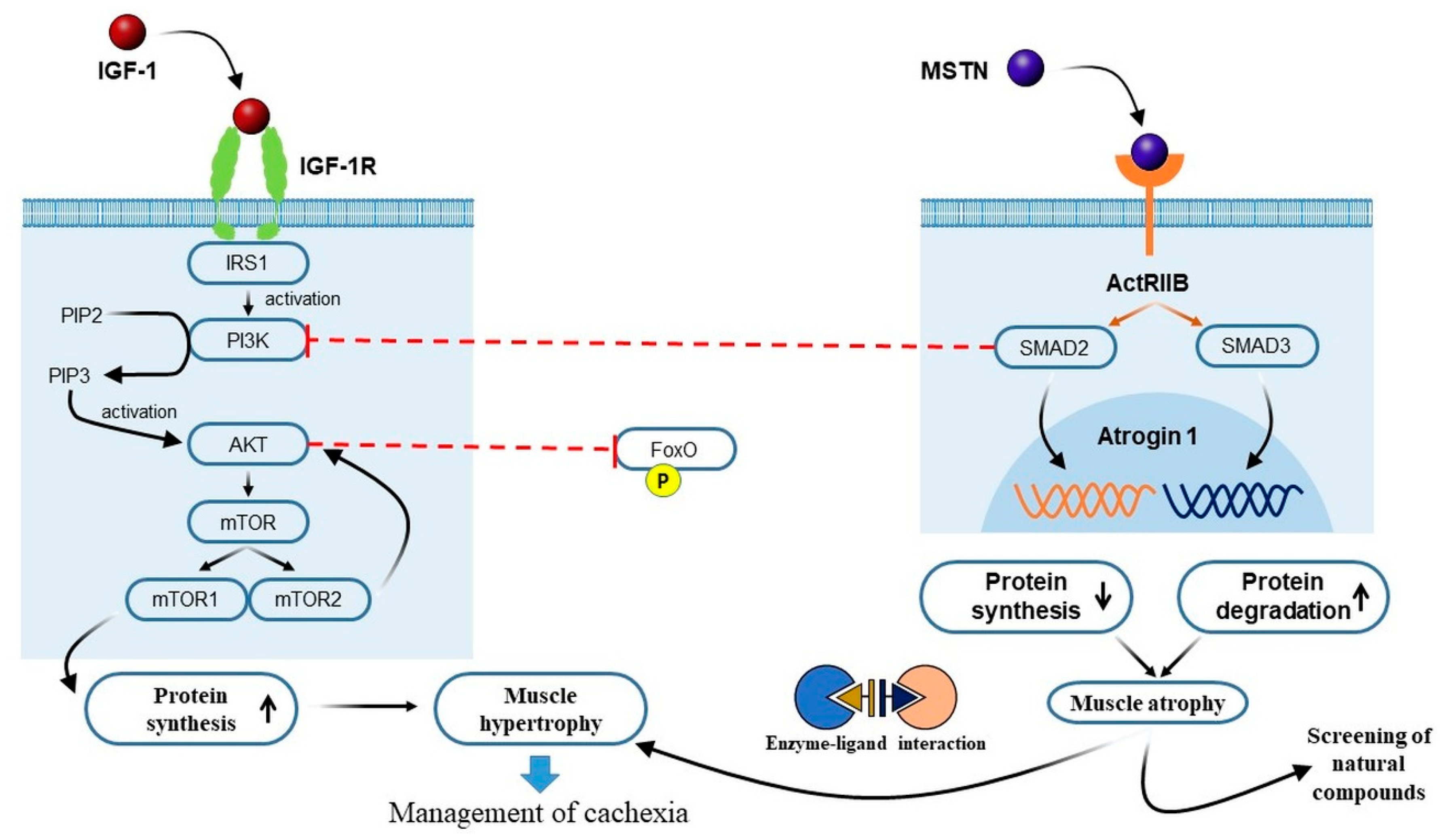
3.1. Crosstalk between IGF-1 and MSTN Signaling Pathways in Cancer Cachexia
3.2. The PI3K/Akt/mTOR Pathway and Cancer Cachexia
3.3. Roles of Peroxisome Proliferator-Activated Receptors in Cancer Cachexia
4. Treatment Options for Cancer Cachexia
4.1. Appetite Stimulation Using Anamorelin
4.2. Megestrol Acetate
4.3. Eicosapentaenoic Acid (EPA) Supplementation
4.4. Systemic Inflammation
4.5. TNF-α Inhibitors
4.6. IL-6 Inhibitors
4.7. Phytocannabinoids
4.8. Thalidomide
4.9. Corticosteroids
4.10. Targeting Myostatin and Activin
4.11. Metabolism Modulators
4.12. Non-Pharmacological Treatment Option
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Thibaut, M.M.; Sboarina, M.; Roumain, M.; Potgens, S.A.; Neyrinck, A.M.; Destree, F.; Gillard, J.; Leclercq, I.A.; Dachy, G.; Demoulin, J.B.; et al. Inflammation-induced cholestasis in cancer cachexia. J. Cachexia Sarcopenia Muscle 2021, 12, 70–90. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Nakamura, S.; Narimatsu, H. Nutritional Approach to Cancer Cachexia: A Proposal for Dietitians. Nutrients 2022, 14, 345. [Google Scholar] [CrossRef] [PubMed]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, T.; Ali, M.A.; Adigun, R. Anorexia and Cachexia; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Lee, E.J.; Jan, A.T.; Baig, M.H.; Ahmad, K.; Malik, A.; Rabbani, G.; Kim, T.; Lee, I.K.; Lee, Y.H.; Park, S.Y.; et al. Fibromodulin and regulation of the intricate balance between myoblast differentiation to myocytes or adipocyte-like cells. FASEB J. 2018, 32, 768–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.; Ahmad, K.; Shaikh, S.; Jan, A.T.; Seo, M.G.; Lee, E.J.; Choi, I. Dermatopontin in Skeletal Muscle Extracellular Matrix Regulates Myogenesis. Cells 2019, 8, 332. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.S.; Ahmad, K.; Lee, E.J.; Shaikh, S.; Choi, I. Computational Identification of Dithymoquinone as a Potential Inhibitor of Myostatin and Regulator of Muscle Mass. Molecules 2021, 26, 5407. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef] [Green Version]
- Baazim, H.; Antonio-Herrera, L.; Bergthaler, A. The interplay of immunology and cachexia in infection and cancer. Nat. Rev. Immunol. 2021, 1–13. [Google Scholar] [CrossRef]
- Dhanapal, R.; Saraswathi, T.; Govind, R.N. Cancer cachexia. J. Oral Maxillofac. Pathol. 2011, 15, 257–260. [Google Scholar] [CrossRef]
- Karagianni, V.T.; Papalois, A.E.; Triantafillidis, J.K. Nutritional status and nutritional support before and after pancreatectomy for pancreatic cancer and chronic pancreatitis. Indian J. Surg. Oncol. 2012, 3, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Jager-Wittenaar, H.; Dijkstra, P.U.; Dijkstra, G.; Bijzet, J.; Langendijk, J.A.; van der Laan, B.; Roodenburg, J.L.N. High prevalence of cachexia in newly diagnosed head and neck cancer patients: An exploratory study. Nutrition 2017, 35, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.; Kim, R.B.; Roh, J.L.; Lee, S.W.; Kim, S.B.; Choi, S.H.; Nam, S.Y.; Kim, S.Y. Prevalence and clinical significance of cancer cachexia based on time from treatment in advanced-stage head and neck squamous cell carcinoma. Head Neck 2017, 39, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Trobec, K.; von Haehling, S.; Anker, S.D.; Lainscak, M. Growth hormone, insulin-like growth factor 1, and insulin signaling-a pharmacological target in body wasting and cachexia. J. Cachexia Sarcopenia Muscle 2011, 2, 191–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmad, S.S.; Ahmad, K.; Lee, E.J.; Lee, Y.H.; Choi, I. Implications of Insulin-Like Growth Factor-1 in Skeletal Muscle and Various Diseases. Cells 2020, 9, 1773. [Google Scholar] [CrossRef]
- Yakovenko, A.; Cameron, M.; Trevino, J.G. Molecular therapeutic strategies targeting pancreatic cancer induced cachexia. World J. Gastrointest. Surg. 2018, 10, 95–106. [Google Scholar] [CrossRef]
- Morley, J.E.; Thomas, D.R.; Wilson, M.M. Cachexia: Pathophysiology and clinical relevance. Am. J. Clin. Nutr. 2006, 83, 735–743. [Google Scholar] [CrossRef] [Green Version]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Blagden, S.P.; Charman, S.C.; Sharples, L.D.; Magee, L.R.; Gilligan, D. Performance status score: Do patients and their oncologists agree? Br. J. Cancer 2003, 89, 1022–1027. [Google Scholar] [CrossRef]
- Peixoto da Silva, S.; Santos, J.M.O.; Costa, E.S.M.P.; Gil da Costa, R.M.; Medeiros, R. Cancer cachexia and its pathophysiology: Links with sarcopenia, anorexia and asthenia. J. Cachexia Sarcopenia Muscle 2020, 11, 619–635. [Google Scholar] [CrossRef]
- Roeland, E.J. Cancer cachexia: The elephant in the room? J. Cachexia Sarcopenia Muscle 2022, 13, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Elkina, Y.; von Haehling, S.; Anker, S.D.; Springer, J. The role of myostatin in muscle wasting: An overview. J. Cachexia Sarcopenia Muscle 2011, 2, 143–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trendelenburg, A.U.; Meyer, A.; Rohner, D.; Boyle, J.; Hatakeyama, S.; Glass, D.J. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am. J. Physiol. Cell Physiol. 2009, 296, C1258–C1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amirouche, A.; Durieux, A.C.; Banzet, S.; Koulmann, N.; Bonnefoy, R.; Mouret, C.; Bigard, X.; Peinnequin, A.; Freyssenet, D. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology 2009, 150, 286–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morissette, M.R.; Cook, S.A.; Buranasombati, C.; Rosenberg, M.A.; Rosenzweig, A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am. J. Physiol. Cell Physiol. 2009, 297, C1124–C1132. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [Green Version]
- Han, H.Q.; Mitch, W.E. Targeting the myostatin signaling pathway to treat muscle wasting diseases. Curr. Opin. Support. Palliat. Care 2011, 5, 334–341. [Google Scholar] [CrossRef]
- Dschietzig, T.B. Myostatin—From the Mighty Mouse to cardiovascular disease and cachexia. Clin. Chim. Acta 2014, 433, 216–224. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Anker, M.S.; von Haehling, S.; Springer, J. Blocking myostatin: Muscle mass equals muscle strength? J. Cachexia Sarcopenia Muscle 2020, 11, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Ahmad, S.S.; Lim, J.H.; Ahmad, K.; Shaikh, S.; Lee, Y.S.; Park, S.J.; Jin, J.O.; Lee, Y.H.; Choi, I. Interaction of Fibromodulin and Myostatin to Regulate Skeletal Muscle Aging: An Opposite Regulation in Muscle Aging, Diabetes, and Intracellular Lipid Accumulation. Cells 2021, 10, 2083. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Sartori, R.; Schirwis, E.; Blaauw, B.; Bortolanza, S.; Zhao, J.; Enzo, E.; Stantzou, A.; Mouisel, E.; Toniolo, L.; Ferry, A.; et al. BMP signaling controls muscle mass. Nat. Genet. 2013, 45, 1309–1318. [Google Scholar] [CrossRef]
- Martinez-Hackert, E.; Sundan, A.; Holien, T. Receptor binding competition: A paradigm for regulating TGF-β family action. Cytokine Growth Factor Rev. 2021, 57, 39–54. [Google Scholar] [CrossRef]
- Lee, S.J. Targeting the myostatin signaling pathway to treat muscle loss and metabolic dysfunction. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Freeman, L.M.; Rush, J.E.; Cunningham, S.M.; Yang, V.K.; Bulmer, B.J. Pilot study of a myostatin antagonist in dogs with cardiac cachexia. J. Vet. Cardiol. 2015, 17, 210–215. [Google Scholar] [CrossRef]
- Loncar, G.; Springer, J.; Anker, M.; Doehner, W.; Lainscak, M. Cardiac cachexia: Hic et nunc. J. Cachexia Sarcopenia Muscle 2016, 7, 246–260. [Google Scholar] [CrossRef]
- Rohm, M.; Zeigerer, A.; Machado, J.; Herzig, S. Energy metabolism in cachexia. EMBO Rep. 2019, 20, e47258. [Google Scholar] [CrossRef]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef]
- Schmitt, T.L.; Martignoni, M.E.; Bachmann, J.; Fechtner, K.; Friess, H.; Kinscherf, R.; Hildebrandt, W. Activity of the Akt-dependent anabolic and catabolic pathways in muscle and liver samples in cancer-related cachexia. J. Mol. Med. 2007, 85, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, S.; Ahmad, K.; Ahmad, S.S.; Lee, E.J.; Lim, J.H.; Beg, M.M.A.; Verma, A.K.; Choi, I. Natural Products in Therapeutic Management of Multineurodegenerative Disorders by Targeting Autophagy. Oxidative Med. Cell. Longev. 2021, 2021, 6347792. [Google Scholar] [CrossRef] [PubMed]
- Szwed, A.; Kim, E.; Jacinto, E. Regulation and metabolic functions of mTORC1 and mTORC2. Physiol. Rev. 2021, 101, 1371–1426. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Li, R.J.; Liu, Y.; Liu, H.Q.; Li, J. Ketogenic diets and protective mechanisms in epilepsy, metabolic disorders, cancer, neuronal loss, and muscle and nerve degeneration. J. Food Biochem. 2020, 44, e13140. [Google Scholar] [CrossRef]
- Zheng, R.; Huang, S.; Zhu, J.; Lin, W.; Xu, H.; Zheng, X. Leucine attenuates muscle atrophy and autophagosome formation by activating PI3K/AKT/mTOR signaling pathway in rotator cuff tears. Cell Tissue Res. 2019, 378, 113–125. [Google Scholar] [CrossRef]
- Lee, S.; Kim, M.B.; Kim, C.; Hwang, J.K. Whole grain cereal attenuates obesity-induced muscle atrophy by activating the PI3K/Akt pathway in obese C57BL/6N mice. Food Sci. Biotechnol. 2018, 27, 159–168. [Google Scholar] [CrossRef]
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- He, Y.; Sun, M.M.; Zhang, G.G.; Yang, J.; Chen, K.S.; Xu, W.W.; Li, B. Targeting PI3K/Akt signal transduction for cancer therapy. Signal. Transduct. Target. Ther. 2021, 6, 425. [Google Scholar] [CrossRef]
- Manne, N.D.; Lima, M.; Enos, R.T.; Wehner, P.; Carson, J.A.; Blough, E. Altered cardiac muscle mTOR regulation during the progression of cancer cachexia in the ApcMin/+ mouse. Int. J. Oncol. 2013, 42, 2134–2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Wu, A.L.; Warnes, C.; Liu, J.; Zhang, C.; Kawasome, H.; Terada, N.; Boppart, M.D.; Schoenherr, C.J.; Chen, J. mTOR regulates skeletal muscle regeneration in vivo through kinase-dependent and kinase-independent mechanisms. Am. J. Physiol. Cell Physiol. 2009, 297, C1434–C1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Neels, J.G.; Grimaldi, P.A. Physiological functions of peroxisome proliferator-activated receptor beta. Physiol. Rev. 2014, 94, 795–858. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Wang, M.; Yang, K.; Chi, T.; Liao, Z.; Wei, P. PPAR-α Modulators as Current and Potential Cancer Treatments. Front. Oncol. 2021, 11, 707. [Google Scholar] [CrossRef]
- Manickam, R.; Duszka, K.; Wahli, W. PPARs and Microbiota in Skeletal Muscle Health and Wasting. Int. J. Mol. Sci. 2020, 21, 8056. [Google Scholar] [CrossRef]
- Manickam, R.; Wahli, W. Roles of Peroxisome Proliferator-Activated Receptor beta/delta in skeletal muscle physiology. Biochimie 2017, 136, 42–48. [Google Scholar] [CrossRef]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef]
- Phua, W.W.T.; Wong, M.X.Y.; Liao, Z.; Tan, N.S. An aPPARent Functional Consequence in Skeletal Muscle Physiology via Peroxisome Proliferator-Activated Receptors. Int. J. Mol. Sci. 2018, 19, 1425. [Google Scholar] [CrossRef] [Green Version]
- Castillero, E.; Nieto-Bona, M.P.; Fernandez-Galaz, C.; Martin, A.I.; Lopez-Menduina, M.; Granado, M.; Villanua, M.A.; Lopez-Calderon, A. Fenofibrate, a PPARα agonist, decreases atrogenes and myostatin expression and improves arthritis-induced skeletal muscle atrophy. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E790–E799. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, M.D.; Hwang, S.K.; Pauli, C.; Murphy, C.J.; Cheng, Z.; Hopkins, B.D.; Wu, D.; Loughran, R.M.; Emerling, B.M.; Zhang, G.; et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E743–E752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, R. PPARβ/δ in human cancer. Biochimie 2017, 136, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Miura, P.; Chakkalakal, J.V.; Boudreault, L.; Bélanger, G.; Hébert, R.L.; Renaud, J.M.; Jasmin, B.J. Pharmacological activation of PPARbeta/delta stimulates utrophin A expression in skeletal muscle fibers and restores sarcolemmal integrity in mature mdx mice. Hum. Mol. Genet. 2009, 18, 4640–4649. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Primers 2021, 7, 13. [Google Scholar] [CrossRef]
- Ahmad, K.; Shaikh, S.; Ahmad, S.S.; Lee, E.J.; Choi, I. Cross-Talk Between Extracellular Matrix and Skeletal Muscle: Implications for Myopathies. Front. Pharm. 2020, 11, 142. [Google Scholar] [CrossRef] [Green Version]
- Jiang, F.; Zhang, Z.; Zhang, Y.; Pan, X.; Yu, L.; Liu, S. L-Carnitine ameliorates cancer cachexia in mice partly via the carnitine palmitoyltransferase-associated PPAR-γ signaling pathway. Oncol. Res. Treat. 2015, 38, 511–516. [Google Scholar] [CrossRef]
- Hua, T.N.; Oh, J.; Kim, S.; Antonio, J.M.; Vo, V.T.; Om, J.; Choi, J.-W.; Kim, J.-Y.; Jung, C.-W.; Park, M.-J. Peroxisome proliferator-activated receptor gamma as a theragnostic target for mesenchymal-type glioblastoma patients. Exp. Mol. Med. 2020, 52, 629–642. [Google Scholar] [CrossRef]
- Gendy, A.M.; Amin, M.M.; Al-Mokaddem, A.K.; Abd Ellah, M.F. Cilostazol mitigates mesenteric ischemia/reperfusion-induced lung lesion: Contribution of PPAR-γ, NF-κB, and STAT3 crosstalk. Life Sci. 2021, 266, 118882. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Li, Y.; Zhang, L.; Yu, S. Preclinical Investigation of Alpinetin in the Treatment of Cancer-Induced Cachexia via Activating PPARγ. Front. Pharmacol. 2021, 12, 1221. [Google Scholar] [CrossRef]
- Shen, Q.; Kuang, J.X.; Miao, C.X.; Zhang, W.L.; Li, Y.W.; Zhang, X.W.; Liu, X. Alantolactone ameliorates cancer cachexia-associated muscle atrophy mainly by inhibiting the STAT3 signaling pathway. Phytomedicine 2022, 95, 153858. [Google Scholar] [CrossRef] [PubMed]
- Tierney, M.T.; Aydogdu, T.; Sala, D.; Malecova, B.; Gatto, S.; Puri, P.L.; Latella, L.; Sacco, A. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 2014, 20, 1182–1186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, W.A.; Berardi, E.; Cardillo, V.M.; Acharyya, S.; Aulino, P.; Thomas-Ahner, J.; Wang, J.; Bloomston, M.; Muscarella, P.; Nau, P.; et al. NF-kappaB-mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J. Clin. Investig. 2013, 123, 4821–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaskiewicz, A.; Domoradzki, T.; Pajak, B. Targeting the JAK2/STAT3 Pathway-Can We Compare It to the Two Faces of the God Janus? Int. J. Mol. Sci. 2020, 21, 8261. [Google Scholar] [CrossRef] [PubMed]
- Watchorn, T.M.; Waddell, I.; Dowidar, N.; Ross, J.A. Proteolysis-inducing factor regulates hepatic gene expression via the transcription factors NF-(kappa)B and STAT3. FASEB J. 2001, 15, 562–564. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jeong, A.J.; Ye, S.K. Highlighted STAT3 as a potential drug target for cancer therapy. BMB Rep. 2019, 52, 415–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Z.; Cabell, L.A.; Schaefer, T.S.; McMurray, J.S. Identification of a high-affinity phosphopeptide inhibitor of Stat3. Bioorg. Med. Chem. Lett. 2003, 13, 633–636. [Google Scholar] [CrossRef]
- Handle, F.; Puhr, M.; Schaefer, G.; Lorito, N.; Hoefer, J.; Gruber, M.; Guggenberger, F.; Santer, F.R.; Marques, R.B.; van Weerden, W.M.; et al. The STAT3 Inhibitor Galiellalactone Reduces IL6-Mediated AR Activity in Benign and Malignant Prostate Models. Mol. Cancer Ther. 2018, 17, 2722–2731. [Google Scholar] [CrossRef] [Green Version]
- Assi, H.H.; Paran, C.; VanderVeen, N.; Savakus, J.; Doherty, R.; Petruzzella, E.; Hoeschele, J.D.; Appelman, H.; Raptis, L.; Mikkelsen, T.; et al. Preclinical characterization of signal transducer and activator of transcription 3 small molecule inhibitors for primary and metastatic brain cancer therapy. J. Pharm. Exp. Ther. 2014, 349, 458–469. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Shen, L.; Zhan, Z.; Liu, Y.; Zhang, C.; Guo, R.; Luo, Y.; Xie, Z.; Feng, Y.; Wu, G. The long noncoding RNA MALAT1 modulates adipose loss in cancer-associated cachexia by suppressing adipogenesis through PPAR-γ. Nutr. Metab. 2021, 18, 27. [Google Scholar] [CrossRef]
- Kim, J.; Piao, H.-L.; Kim, B.-J.; Yao, F.; Han, Z.; Wang, Y.; Xiao, Z.; Siverly, A.N.; Lawhon, S.E.; Ton, B.N. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018, 50, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Han, J.; Wang, H.; Meng, Q.; Chen, L.; Liu, Y.; Feng, Y.; Wu, G. Cachexia-related long noncoding RNA, CAAlnc1, suppresses adipogenesis by blocking the binding of HuR to adipogenic transcription factor mRNAs. Int. J. Cancer 2019, 145, 1809–1821. [Google Scholar] [CrossRef] [PubMed]
- Moore-Carrasco, R.; Figueras, M.; Ametller, E.; López-Soriano, F.J.; Argilés, J.M.; Busquets, S. Effects of the PPARγ agonist GW1929 on muscle wasting in tumour-bearing mice. Oncol. Rep. 2008, 19, 253–256. [Google Scholar] [CrossRef] [PubMed]
- Marceca, G.P.; Londhe, P.; Calore, F. Management of Cancer Cachexia: Attempting to Develop New Pharmacological Agents for New Effective Therapeutic Options. Front. Oncol. 2020, 10, 298. [Google Scholar] [CrossRef] [Green Version]
- Avancini, A.; Trestini, I.; Tregnago, D.; Lanza, M.; Menis, J.; Belluomini, L.; Milella, M.; Pilotto, S. A multimodal approach to cancer-related cachexia: From theory to practice. Expert Rev. Anticancer Ther. 2021, 21, 819–826. [Google Scholar] [CrossRef]
- Maddocks, M.; Hopkinson, J.; Conibear, J.; Reeves, A.; Shaw, C.; Fearon, K.C. Practical multimodal care for cancer cachexia. Curr. Opin. Support. Palliat. Care 2016, 10, 298–305. [Google Scholar] [CrossRef] [Green Version]
- Argiles, J.M.; Lopez-Soriano, F.J.; Stemmler, B.; Busquets, S. Novel targeted therapies for cancer cachexia. Biochem. J. 2017, 474, 2663–2678. [Google Scholar] [CrossRef]
- Suzuki, H.; Asakawa, A.; Amitani, H.; Nakamura, N.; Inui, A. Cancer cachexia--pathophysiology and management. J. Gastroenterol. 2013, 48, 574–594. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, G.; Maccio, A.; Madeddu, C.; Serpe, R.; Massa, E.; Dessi, M.; Panzone, F.; Contu, P. Randomized phase III clinical trial of five different arms of treatment in 332 patients with cancer cachexia. Oncologist 2010, 15, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Myrianthefs, P.M.; Batistaki, C. Cancer cachexia and immunomodulation. J. BUON 2005, 10, 181–188. [Google Scholar]
- Inui, A. Cancer anorexia-cachexia syndrome: Current issues in research and management. CA Cancer J. Clin. 2002, 52, 72–91. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.C.; Bachmann, J.; Prokopchuk, O.; Friess, H.; Martignoni, M.E. Molecular pathways leading to loss of skeletal muscle mass in cancer cachexia--Can findings from animal models be translated to humans? BMC Cancer 2016, 16, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argiles, J.M.; Lopez-Soriano, F.J.; Busquets, S. Mediators of cachexia in cancer patients. Nutrition 2019, 66, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Dev, R.; Wong, A.; Hui, D.; Bruera, E. The Evolving Approach to Management of Cancer Cachexia. Oncology 2017, 31, 23–32. [Google Scholar] [PubMed]
- Porporato, P.E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, e200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, S.; Kaibara, A.; Ishibashi, N.; Shirouzu, K. Glutamine supplementation in cancer patients. Nutrition 2001, 17, 766–768. [Google Scholar] [CrossRef]
- Schlemmer, M.; Suchner, U.; Schapers, B.; Duerr, E.M.; Alteheld, B.; Zwingers, T.; Stehle, P.; Zimmer, H.G. Is glutamine deficiency the link between inflammation, malnutrition, and fatigue in cancer patients? Clin. Nutr. 2015, 34, 1258–1265. [Google Scholar] [CrossRef]
- Bruggeman, A.R.; Kamal, A.H.; LeBlanc, T.W.; Ma, J.D.; Baracos, V.E.; Roeland, E.J. Cancer Cachexia: Beyond Weight Loss. J. Oncol. Pract. 2016, 12, 1163–1171. [Google Scholar] [CrossRef]
- Zhang, L.; Pan, J.; Dong, Y.; Tweardy, D.J.; Dong, Y.; Garibotto, G.; Mitch, W.E. Stat3 activation links a C/EBPdelta to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 2013, 18, 368–379. [Google Scholar] [CrossRef] [Green Version]
- Lira, F.S.; Antunes Bde, M.; Seelaender, M.; Rosa Neto, J.C. The therapeutic potential of exercise to treat cachexia. Curr. Opin. Support. Palliat. Care 2015, 9, 317–324. [Google Scholar] [CrossRef]
- Temel, J.S.; Abernethy, A.P.; Currow, D.C.; Friend, J.; Duus, E.M.; Yan, Y.; Fearon, K.C. Anamorelin in patients with non-small-cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): Results from two randomised, double-blind, phase 3 trials. Lancet Oncol. 2016, 17, 519–531. [Google Scholar] [CrossRef]
- Wakabayashi, H.; Arai, H.; Inui, A. The regulatory approval of anamorelin for treatment of cachexia in patients with non-small cell lung cancer, gastric cancer, pancreatic cancer, and colorectal cancer in Japan: Facts and numbers. J. Cachexia Sarcopenia Muscle 2021, 12, 14–16. [Google Scholar] [CrossRef] [PubMed]
- Kojima, M.; Hosoda, H.; Date, Y.; Nakazato, M.; Matsuo, H.; Kangawa, K. Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 1999, 402, 656–660. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.D.; Nogueiras, R.; Andermann, M.L.; Andrews, Z.B.; Anker, S.D.; Argente, J.; Batterham, R.L.; Benoit, S.C.; Bowers, C.Y.; Broglio, F.; et al. Ghrelin. Mol. Metab. 2015, 4, 437–460. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Kanemaru, A.; Fukushima, T.; Yamamoto, K.; Tanaka, H.; Haruyama, Y.; Itoh, H.; Matsumoto, N.; Kangawa, K.; Nakazato, M.; et al. Ghrelin administration suppresses inflammation-associated colorectal carcinogenesis in mice. Cancer Sci. 2015, 106, 1130–1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takayama, K.; Katakami, N.; Yokoyama, T.; Atagi, S.; Yoshimori, K.; Kagamu, H.; Saito, H.; Takiguchi, Y.; Aoe, K.; Koyama, A.; et al. Anamorelin (ONO-7643) in Japanese patients with non-small cell lung cancer and cachexia: Results of a randomized phase 2 trial. Support. Care Cancer 2016, 24, 3495–3505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uysal, P.; Afsar, C.U.; Sozer, V.; Inanc, B.; Agaoglu, F.; Gural, Z.; Fazlioglu, N.Y.; Cuhadaroglu, C.; Uzun, H. Evaluation of the relationship between serum ghrelin levels and cancer cachexia in patients with locally advanced nonsmall-cell lung cancer treated with chemoradiotherapy. J. Cancer Res. Ther. 2020, 16, 855–859. [Google Scholar] [CrossRef] [PubMed]
- Pascual Lopez, A.; Roque i Figuls, M.; Urrutia Cuchi, G.; Berenstein, E.G.; Almenar Pasies, B.; Balcells Alegre, M.; Herdman, M. Systematic review of megestrol acetate in the treatment of anorexia-cachexia syndrome. J. Pain Symptom Manag. 2004, 27, 360–369. [Google Scholar] [CrossRef]
- Ruiz-Garcia, V.; Lopez-Briz, E.; Carbonell-Sanchis, R.; Bort-Marti, S.; Gonzalvez-Perales, J.L. Megestrol acetate for cachexia-anorexia syndrome. A systematic review. J. Cachexia Sarcopenia Muscle 2018, 9, 444–452. [Google Scholar] [CrossRef]
- Dewey, A.; Baughan, C.; Dean, T.; Higgins, B.; Johnson, I. Eicosapentaenoic acid (EPA, an omega-3 fatty acid from fish oils) for the treatment of cancer cachexia. Cochrane Database Syst. Rev. 2007, 2007, CD004597. [Google Scholar] [CrossRef]
- Damsbo-Svendsen, S.; Ronsholdt, M.D.; Lauritzen, L. Fish oil-supplementation increases appetite in healthy adults. A randomized controlled cross-over trial. Appetite 2013, 66, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Di Girolamo, F.G.; Situlin, R.; Mazzucco, S.; Valentini, R.; Toigo, G.; Biolo, G. Omega-3 fatty acids and protein metabolism: Enhancement of anabolic interventions for sarcopenia. Curr. Opin. Clin. Nutr. Metab. Care 2014, 17, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Giles, K.; Guan, C.; Jagoe, T.R.; Mazurak, V. Diet composition as a source of variation in experimental animal models of cancer cachexia. J. Cachexia Sarcopenia Muscle 2016, 7, 110–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, H.J.; Patel, B.M. TNF-alpha and cancer cachexia: Molecular insights and clinical implications. Life Sci. 2017, 170, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Singh, R. Cytokines and Chemokines in Cancer Cachexia and Its Long-Term Impact on COVID-19. Cells 2022, 11, 579. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. Interleukin (IL-6) Immunotherapy. Cold Spring Harb. Perspect. Biol. 2018, 10, a028456. [Google Scholar] [CrossRef]
- Advani, S.M.; Advani, P.G.; VonVille, H.M.; Jafri, S.H. Pharmacological management of cachexia in adult cancer patients: A systematic review of clinical trials. BMC Cancer 2018, 18, 1174. [Google Scholar] [CrossRef] [Green Version]
- Gerriets, V.; Bansal, P.; Goyal, A.; Khaddour, K. Tumor Necrosis Factor Inhibitors; StatPearls: Treasure Island, FL, USA, 2021. [Google Scholar]
- Miyamoto, Y.; Hanna, D.L.; Zhang, W.; Baba, H.; Lenz, H.J. Molecular Pathways: Cachexia Signaling-A Targeted Approach to Cancer Treatment. Clin. Cancer Res. 2016, 22, 3999–4004. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, T.; Kishimoto, T. The biology and medical implications of interleukin-6. Cancer Immunol. Res. 2014, 2, 288–294. [Google Scholar] [CrossRef] [Green Version]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 Reprograms Host Metabolism to Suppress Anti-tumor Immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef]
- Choy, E.H.; De Benedetti, F.; Takeuchi, T.; Hashizume, M.; John, M.R.; Kishimoto, T. Translating IL-6 biology into effective treatments. Nat. Rev. Rheumatol. 2020, 16, 335–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Jin, H.; Chen, Y.; Huang, T.; Mi, Y.; Zou, Z. Cancer cachexia: Molecular mechanism and pharmacological management. Biochem. J. 2021, 478, 1663–1688. [Google Scholar] [CrossRef] [PubMed]
- Roeland, E.J.; Bohlke, K.; Baracos, V.E.; Bruera, E.; Del Fabbro, E.; Dixon, S.; Fallon, M.; Herrstedt, J.; Lau, H.; Platek, M.; et al. Management of Cancer Cachexia: ASCO Guideline. J. Clin. Oncol. 2020, 38, 2438–2453. [Google Scholar] [CrossRef] [PubMed]
- Nigro, E.; Formato, M.; Crescente, G.; Daniele, A. Cancer Initiation, Progression and Resistance: Are Phytocannabinoids from Cannabis sativa L. Promising Compounds? Molecules 2021, 26, 2668. [Google Scholar] [CrossRef] [PubMed]
- Pellati, F.; Borgonetti, V.; Brighenti, V.; Biagi, M.; Benvenuti, S.; Corsi, L. Cannabis sativa L. and Nonpsychoactive Cannabinoids: Their Chemistry and Role against Oxidative Stress, Inflammation, and Cancer. Biomed. Res. Int. 2018, 2018, 1691428. [Google Scholar] [CrossRef] [Green Version]
- D’Amato, R.J.; Loughnan, M.S.; Flynn, E.; Folkman, J. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. USA 1994, 91, 4082–4085. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Guo, F.; Zhu, X.; He, X.; Xie, L. Thalidomide and its analogues: A review of the potential for immunomodulation of fibrosis diseases and opthalmopathy. Exp. Ther. Med. 2017, 14, 5251–5257. [Google Scholar] [CrossRef]
- Dale, B.; Cheng, M.; Park, K.S.; Kaniskan, H.U.; Xiong, Y.; Jin, J. Advancing targeted protein degradation for cancer therapy. Nat. Rev. Cancer 2021, 21, 638–654. [Google Scholar] [CrossRef]
- Reid, J.; Mills, M.; Cantwell, M.; Cardwell, C.R.; Murray, L.J.; Donnelly, M. Thalidomide for managing cancer cachexia. Cochrane Database Syst Rev. 2012, 2021, CD008664. [Google Scholar] [CrossRef]
- Hasselgren, P.O.; Alamdari, N.; Aversa, Z.; Gonnella, P.; Smith, I.J.; Tizio, S. Corticosteroids and muscle wasting: Role of transcription factors, nuclear cofactors, and hyperacetylation. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 423–428. [Google Scholar] [CrossRef]
- Braun, T.P.; Marks, D.L. The regulation of muscle mass by endogenous glucocorticoids. Front. Physiol. 2015, 6, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rybalka, E.; Timpani, C.A.; Debruin, D.A.; Bagaric, R.M.; Campelj, D.G.; Hayes, A. The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle. Cells 2020, 9, 2657. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Fontes-Oliveira, C.C.; Toledo, M.; Lopez-Soriano, F.J.; Busquets, S. Cachexia: A problem of energetic inefficiency. J. Cachexia Sarcopenia Muscle 2014, 5, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Senesse, P.; Gioulbasanis, I.; Antoun, S.; Bozzetti, F.; Deans, C.; Strasser, F.; Thoresen, L.; Jagoe, R.T.; Chasen, M.; et al. Diagnostic criteria for the classification of cancer-associated weight loss. J. Clin. Oncol. 2015, 33, 90–99. [Google Scholar] [CrossRef]
- Crawford, J. What are the criteria for response to cachexia treatment? Ann. Palliat. Med. 2019, 8, 43–49. [Google Scholar] [CrossRef]
- Tisdale, M.J. Reversing cachexia. Cell 2010, 142, 511–512. [Google Scholar] [CrossRef] [Green Version]
- Aiello, D.; Patel, K.; Lasagna, E. The myostatin gene: An overview of mechanisms of action and its relevance to livestock animals. Anim. Genet. 2018, 49, 505–519. [Google Scholar] [CrossRef] [Green Version]
- Molfino, A.; Amabile, M.I.; Imbimbo, G.; Rizzo, V.; Pediconi, F.; Catalano, C.; Emiliani, A.; Belli, R.; Ramaccini, C.; Parisi, C.; et al. Association between Growth Differentiation Factor-15 (GDF-15) Serum Levels, Anorexia and Low Muscle Mass among Cancer Patients. Cancers 2020, 13, 99. [Google Scholar] [CrossRef]
- Khamoui, A.V.; Kim, J.S. Candidate mechanisms underlying effects of contractile activity on muscle morphology and energetics in cancer cachexia. Eur. J. Cancer Care 2012, 21, 143–157. [Google Scholar] [CrossRef]
- Grande, A.J.; Silva, V.; Sawaris Neto, L.; Teixeira Basmage, J.P.; Peccin, M.S.; Maddocks, M. Exercise for cancer cachexia in adults. Cochrane Database Syst. Rev. 2021, 3, CD010804. [Google Scholar] [CrossRef]
- Laurent, M.R.; Dubois, V.; Claessens, F.; Verschueren, S.M.; Vanderschueren, D.; Gielen, E.; Jardi, F. Muscle-bone interactions: From experimental models to the clinic? A critical update. Mol. Cell. Endocrinol. 2016, 432, 14–36. [Google Scholar] [CrossRef] [PubMed]
- Heymsfield, S.B.; Coleman, L.A.; Miller, R.; Rooks, D.S.; Laurent, D.; Petricoul, O.; Praestgaard, J.; Swan, T.; Wade, T.; Perry, R.G.; et al. Effect of Bimagrumab vs. Placebo on Body Fat Mass Among Adults With Type 2 Diabetes and Obesity: A Phase 2 Randomized Clinical Trial. JAMA Netw. Open 2021, 4, e2033457. [Google Scholar] [CrossRef] [PubMed]
- Rooks, D.S.; Laurent, D.; Praestgaard, J.; Rasmussen, S.; Bartlett, M.; Tanko, L.B. Effect of bimagrumab on thigh muscle volume and composition in men with casting-induced atrophy. J. Cachexia Sarcopenia Muscle 2017, 8, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Lord, S.R.; Studenski, S.A.; Warden, S.J.; Fielding, R.A.; Recknor, C.P.; Hochberg, M.C.; Ferrari, S.L.; Blain, H.; Binder, E.F.; et al. Myostatin antibody (LY2495655) in older weak fallers: A proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. 2015, 3, 948–957. [Google Scholar] [CrossRef]
- Cao, Z.; Scott, A.M.; Hoogenraad, N.J.; Osellame, L.D. Mediators and clinical treatment for cancer cachexia: A systematic review. JCSM Rapid Commun. 2021, 4, 166–186. [Google Scholar] [CrossRef]
- Alves, C.R.; da Cunha, T.F.; da Paixao, N.A.; Brum, P.C. Aerobic exercise training as therapy for cardiac and cancer cachexia. Life Sci. 2015, 125, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Jee, H.; Chang, J.E.; Yang, E.J. Positive Prehabilitative Effect of Intense Treadmill Exercise for Ameliorating Cancer Cachexia Symptoms in a Mouse Model. J. Cancer 2016, 7, 2378–2387. [Google Scholar] [CrossRef] [Green Version]
- Moreira, V.M.; da Silva Franco, C.C.; Prates, K.V.; Gomes, R.M.; de Moraes, A.M.P.; Ribeiro, T.A.; Martins, I.P.; Previate, C.; Pavanello, A.; Matiusso, C.C.I.; et al. Aerobic Exercise Training Attenuates Tumor Growth and Reduces Insulin Secretion in Walker 256 Tumor-Bearing Rats. Front. Physiol. 2018, 9, 465. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Treatment Options | Level Decrease | Level Increase | References |
|---|---|---|---|
| Omega-3 fatty acids | decrease TNF-α and IL-1 | recover the ability of nutrition | [91,92,93,94,95] |
| Glucocorticoids | prevent the synthesis/discharge of proinflammatory cytokines | ||
| Non-steroidal anti-inflammatory drugs | reduce inflammation | reduce muscle wasting | |
| Drugs (cytokine inhibition) | |||
| Glutamine supplementation | can reduce muscle wasting in cancer patients | [96,97,98] | |
| Megestrol, Dronabinol | increase weight | [99] | |
| Appetite stimulation (cannabinoids or erythropoietin) | ameliorate cachexia | [93,94,100] | |
| Anti-dopaminergics (like metoclopramide | |||
| Muscle creation stimulation (branched-chain amino acids | |||
| Exercise (strength and aerobic training) | reduces proinflammatory cytokine levels | increases anti-inflammatory cytokine levels | [101] |
| Ghrelin agonists | therapeutic targeted approaches that reduce wasting in cancer patients | [21] | |
| Androgen receptor agonists | |||
| β-blockers | |||
| anti-MSTN peptides | |||
| Ghrelin analogs | reduce systemic inflammation and muscle catabolism | increase food intake and aid lean body mass retention | [102] |
| MSTN blockade | reduces inflammation and muscle wasting | [102] | |
| Blockade of Stat3 | reduces muscle atrophy and inflammatory cytokine expression | [100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahmad, S.S.; Ahmad, K.; Shaikh, S.; You, H.J.; Lee, E.-Y.; Ali, S.; Lee, E.J.; Choi, I. Molecular Mechanisms and Current Treatment Options for Cancer Cachexia. Cancers 2022, 14, 2107. https://doi.org/10.3390/cancers14092107
Ahmad SS, Ahmad K, Shaikh S, You HJ, Lee E-Y, Ali S, Lee EJ, Choi I. Molecular Mechanisms and Current Treatment Options for Cancer Cachexia. Cancers. 2022; 14(9):2107. https://doi.org/10.3390/cancers14092107
Chicago/Turabian StyleAhmad, Syed Sayeed, Khurshid Ahmad, Sibhghatulla Shaikh, Hye Jin You, Eun-Young Lee, Shahid Ali, Eun Ju Lee, and Inho Choi. 2022. "Molecular Mechanisms and Current Treatment Options for Cancer Cachexia" Cancers 14, no. 9: 2107. https://doi.org/10.3390/cancers14092107
APA StyleAhmad, S. S., Ahmad, K., Shaikh, S., You, H. J., Lee, E.-Y., Ali, S., Lee, E. J., & Choi, I. (2022). Molecular Mechanisms and Current Treatment Options for Cancer Cachexia. Cancers, 14(9), 2107. https://doi.org/10.3390/cancers14092107