
Genetic and Clinical Characteristics of ARID1A Mutated Melanoma Reveal High Tumor Mutational Load without Implications on Patient Survival

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Clinical Samples
2.2. DNA Isolation
2.3. Targeted Sequencing
2.4. Statistical Analysis
3. Results
3.1. Patient Characteristics
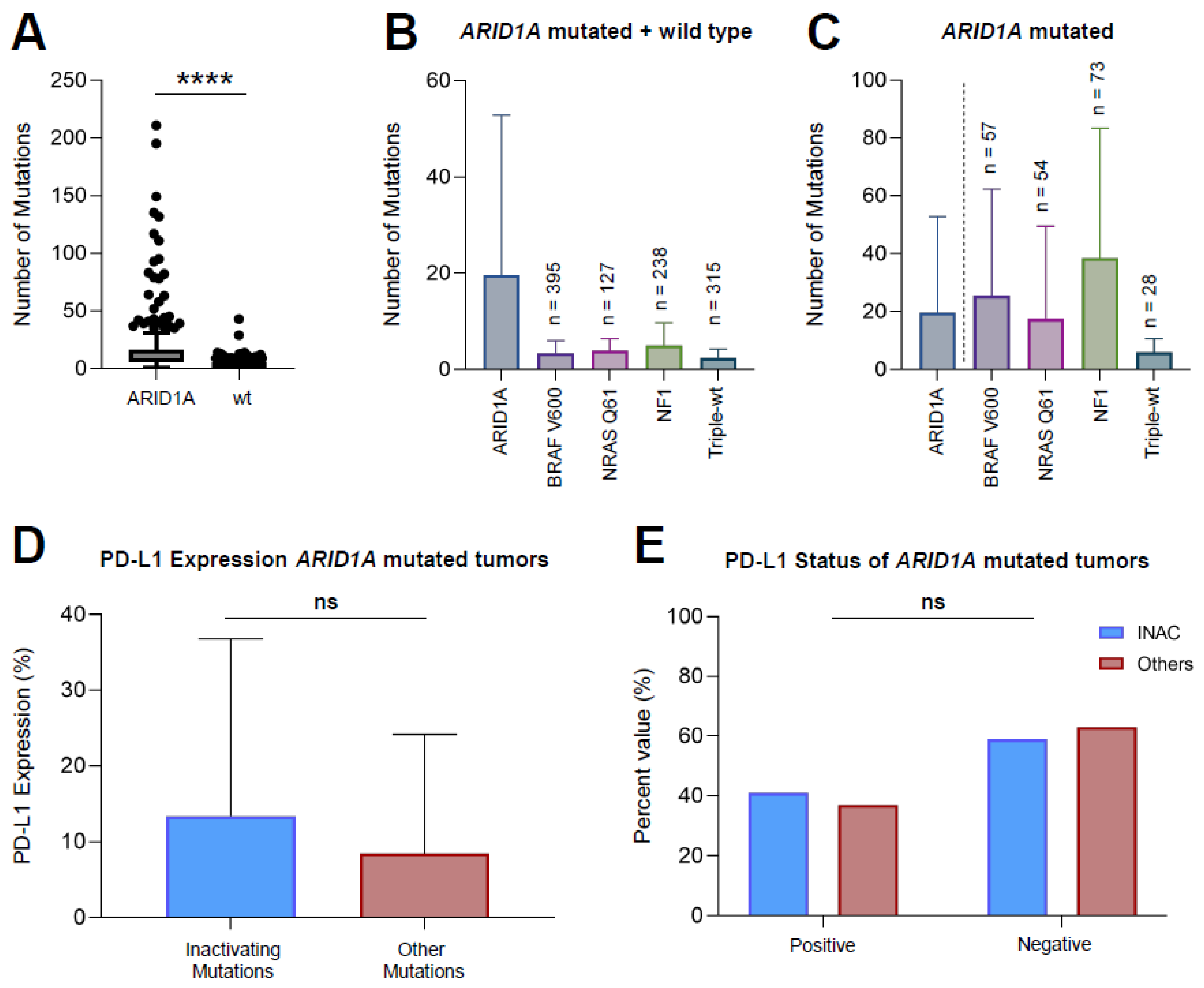
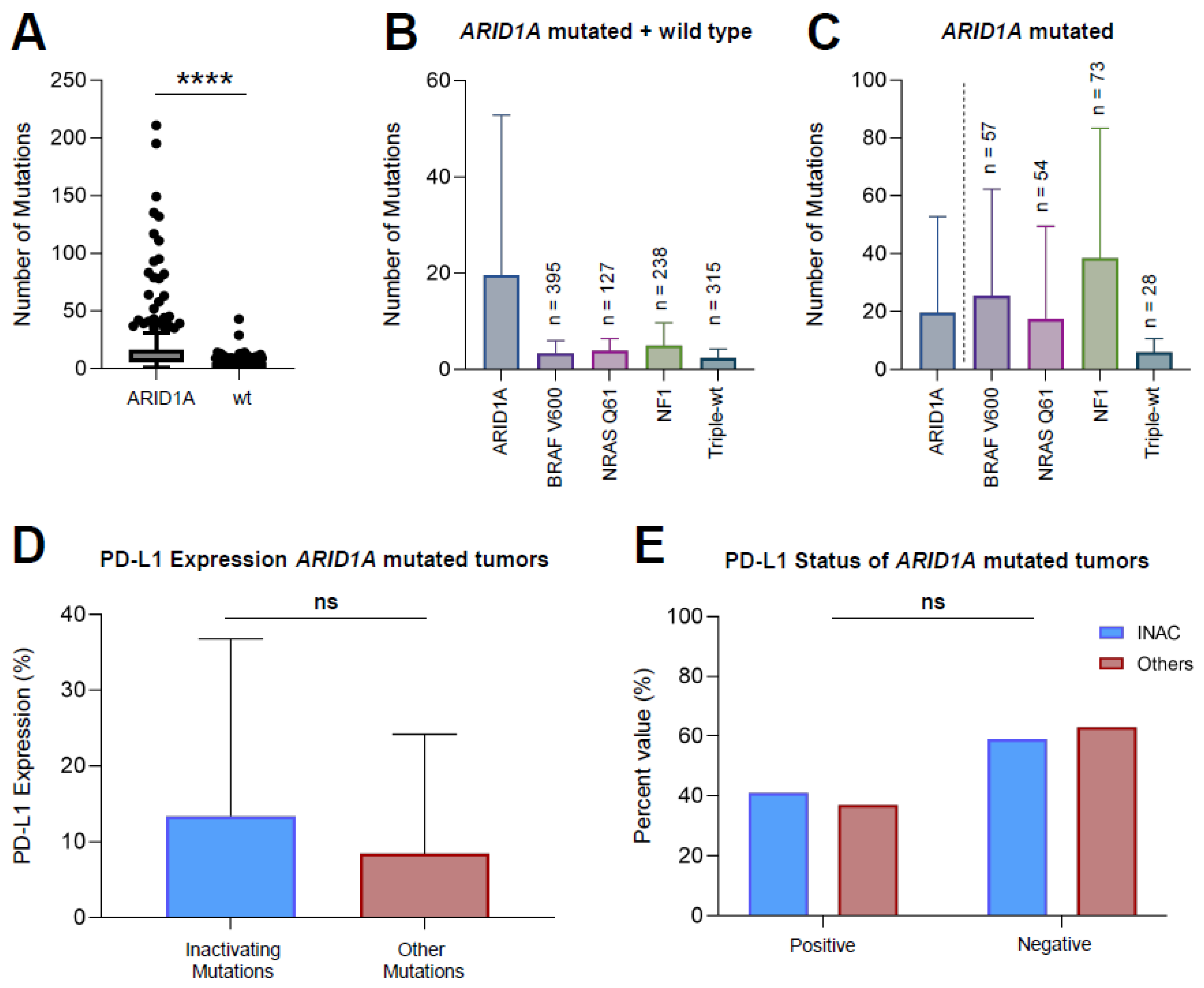
3.2. ARID1A Mutated Melanoma Harbors More Mutations Compared to ARID1A-wt Melanoma
3.3. Inactivating Mutations of ARID1A Do Not Lead to a Greater PD-L1 Expression Compared to Other Mutations
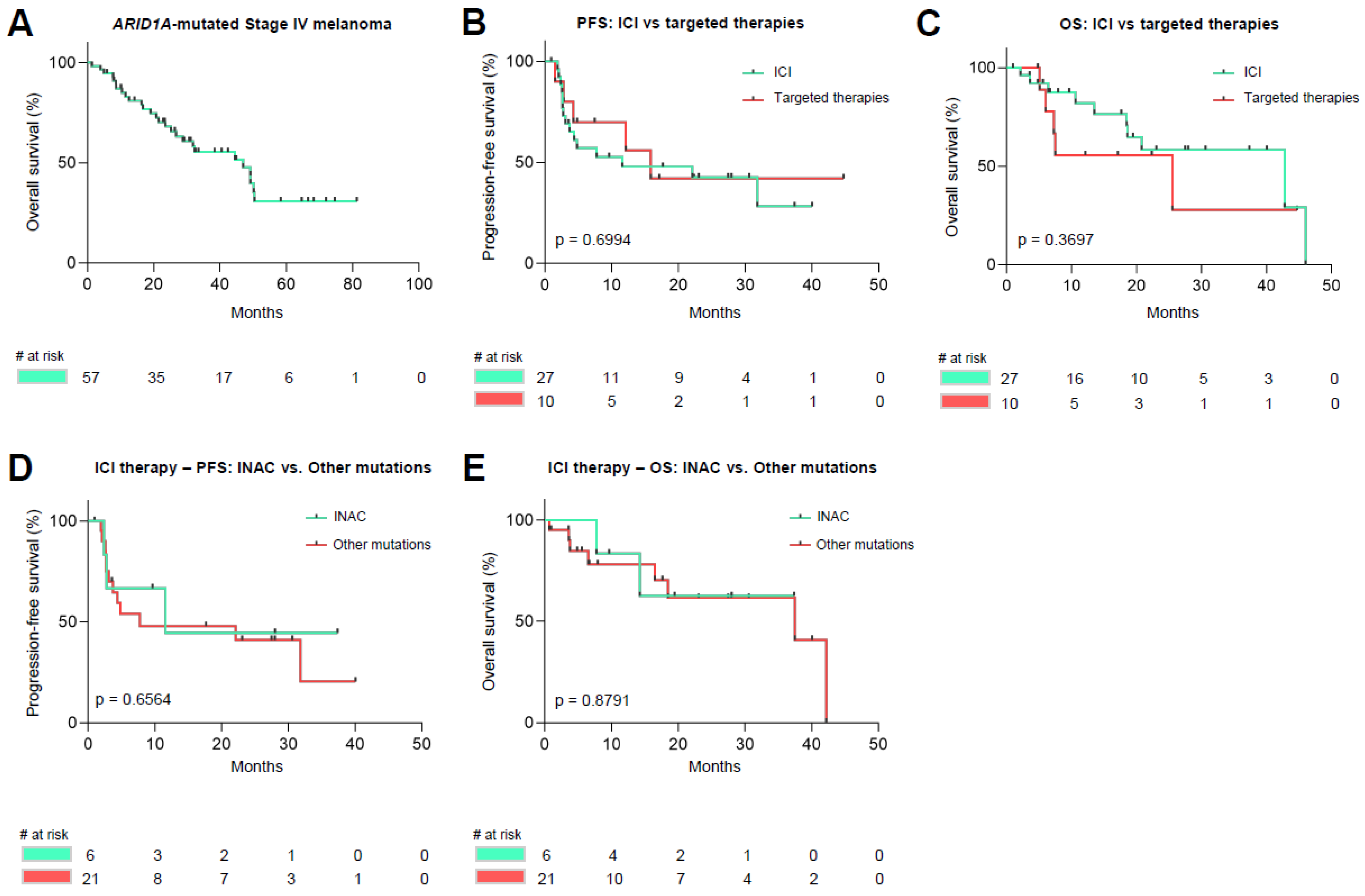
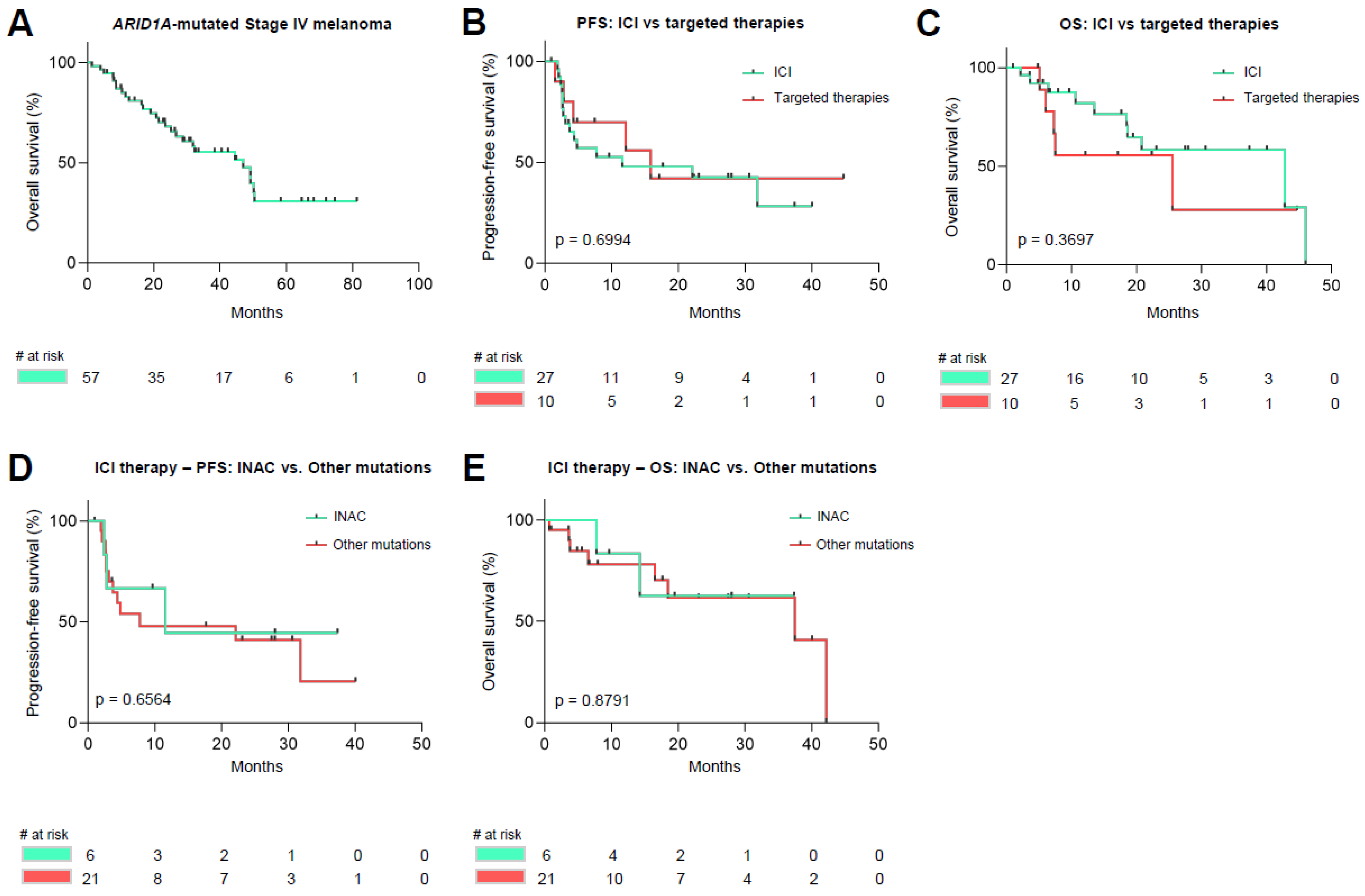
3.4. Survival Analysis of ARID1A Mutated Malignant Melanoma
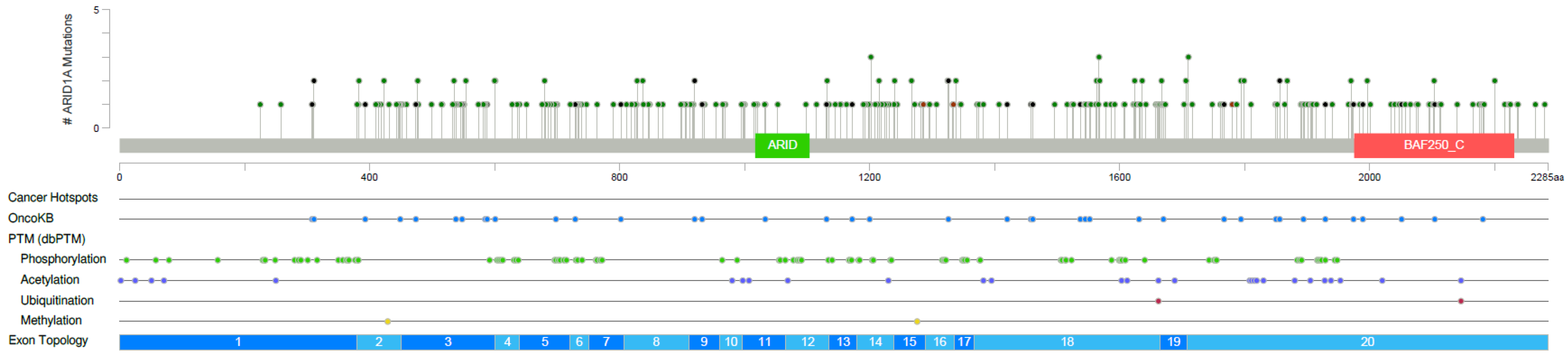
3.5. Distribution of UV-Induced Mutations amongst Melanoma Samples
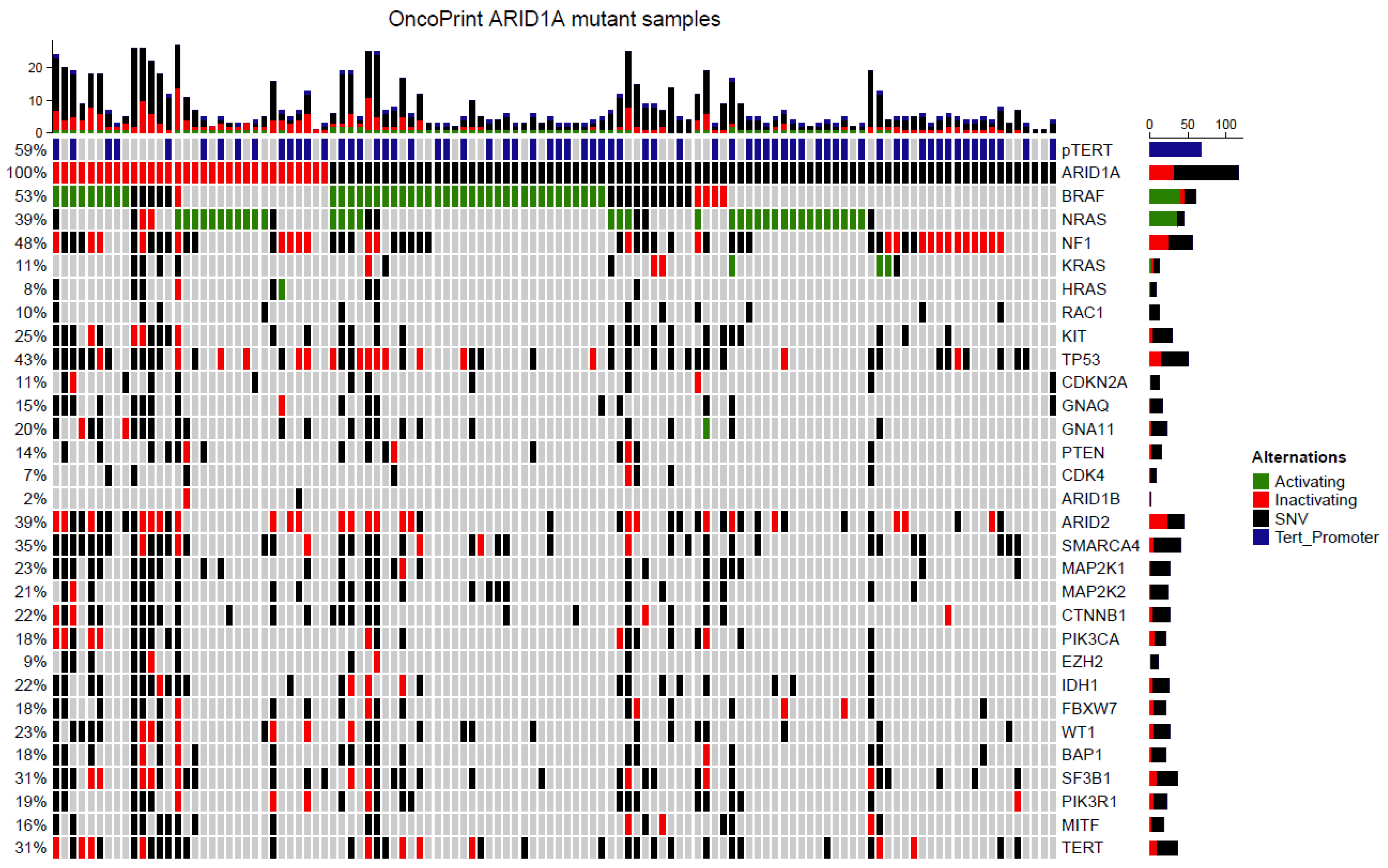
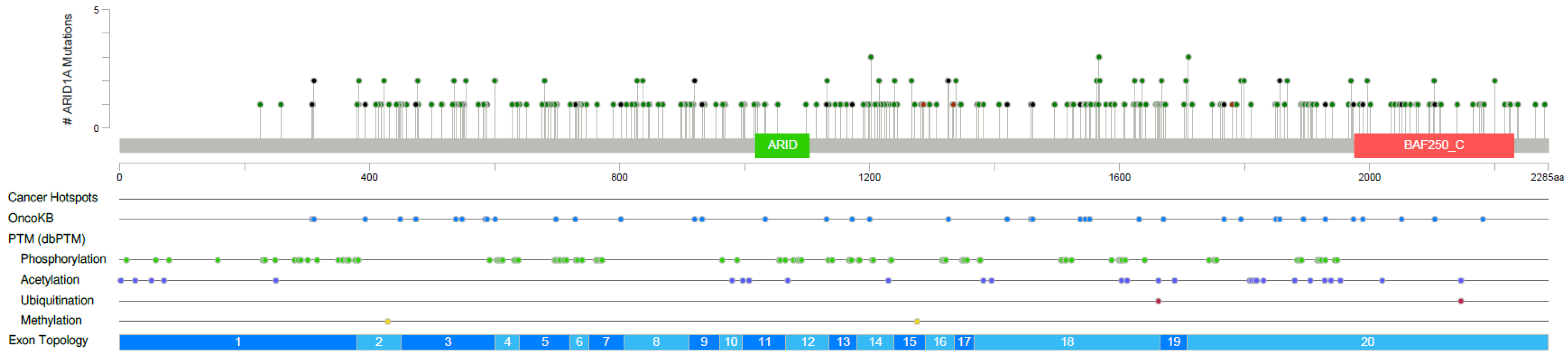
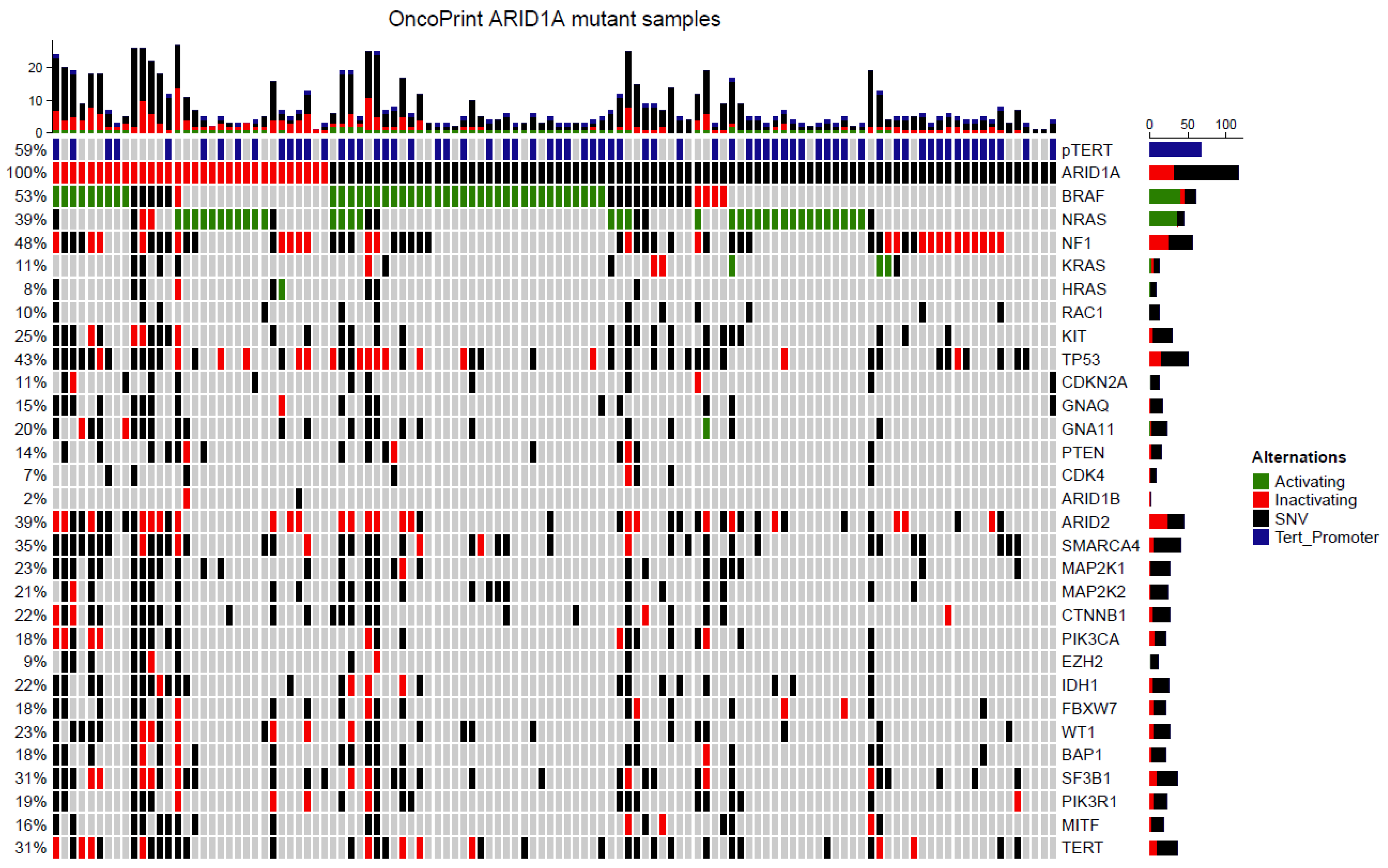
3.6. Targeted Next Generation Sequencing of ARID1A Mutated Melanoma
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ajithkumar, T.; Parkinson, C.; Fife, K.; Corrie, P.; Jefferies, S. Evolving treatment options for melanoma brain metastases. Lancet Oncol. 2015, 16, e486–e497. [Google Scholar] [CrossRef]
- Gellrich, F.F.; Schmitz, M.; Beissert, S.; Meier, F. Anti-PD-1 and Novel Combinations in the Treatment of Melanoma-An Update. J. Clin. Med. 2020, 9, 223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.H.; Aiba, S.; Brocker, E.B.; LeBoit, P.E.; et al. Distinct sets of genetic alterations in melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [Green Version]
- Nassar, K.W.; Tan, A.C. The mutational landscape of mucosal melanoma. Semin. Cancer Biol. 2020, 61, 139–148. [Google Scholar] [CrossRef]
- Rabbie, R.; Ferguson, P.; Wong, K.; Couturier, D.L.; Moran, U.; Turner, C.; Emanuel, P.; Haas, K.; Saunus, J.M.; Davidson, M.R.; et al. The mutational landscape of melanoma brain metastases presenting as the first visceral site of recurrence. Br. J. Cancer 2021, 124, 156–160. [Google Scholar] [CrossRef]
- Sidaway, P. Skin cancer: Mutational landscape of melanoma revealed. Nat. Rev. Clin. Oncol. 2017, 14, 393. [Google Scholar] [CrossRef]
- Li, J.; Wang, W.; Zhang, Y.; Cieslik, M.; Guo, J.; Tan, M.; Green, M.D.; Wang, W.; Lin, H.; Li, W.; et al. Epigenetic driver mutations in ARID1A shape cancer immune phenotype and immunotherapy. J. Clin. Investig. 2020, 130, 2712–2726. [Google Scholar] [CrossRef] [PubMed]
- Kadoch, C.; Hargreaves, D.C.; Hodges, C.; Elias, L.; Ho, L.; Ranish, J.; Crabtree, G.R. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat. Genet. 2013, 45, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, R.C.; Wang, T.L.; Shih, I.-M. The emerging roles of ARID1A in tumor suppression. Cancer Biol. Ther. 2014, 15, 655–664. [Google Scholar] [CrossRef] [Green Version]
- Okamura, R.; Kato, S.; Lee, S.; Jimenez, R.E.; Sicklick, J.K.; Kurzrock, R. ARID1A alterations function as a biomarker for longer progression-free survival after anti-PD-1/PD-L1 immunotherapy. J. Immunother. Cancer 2020, 8, e000438. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Wang, L.; Qu, J.; Zhou, N.; Hou, H.; Jiang, M.; Zhang, X. Effect and biomarker of immune checkpoint blockade therapy for ARID1A deficiency cancers. Biomed. Pharm. 2020, 130, 110626. [Google Scholar] [CrossRef]
- Keung, E.; Gershenwald, J. The eighth edition American Joint Committee on Cancer (AJCC) melanoma staging system: Implications for melanoma treatment and care. Expert Rev. Anticancer Ther. 2018, 18, 775–784. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Team, R. RStudio: Integrated Development for R; RStudio, Inc.: Boston, MA, USA, 2019. [Google Scholar]
- Petljak, M.; Alexandrov, L.B. Understanding mutagenesis through delineation of mutational signatures in human cancer. Carcinogenesis 2016, 37, 531–540. [Google Scholar] [CrossRef] [Green Version]
- Gos, A.; Jurkowska, M.; van Akkooi, A.; Robert, C.; Kosela-Paterczyk, H.; Koljenovic, S.; Kamsukom, N.; Michej, W.; Jeziorski, A.; Pluta, P.; et al. Molecular characterization and patient outcome of melanoma nodal metastases and an unknown primary site. Ann. Surg. Oncol. 2014, 21, 4317–4323. [Google Scholar] [CrossRef] [Green Version]
- Morgese, F.; Sampaolesi, C.; Torniai, M.; Conti, A.; Ranallo, N.; Giacchetti, A.; Serresi, S.; Onofri, A.; Burattini, M.; Ricotti, G.; et al. Gender Differences and Outcomes in Melanoma Patients. Oncol. Ther. 2020, 8, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaremba, A.; Murali, R.; Jansen, P.; Moller, I.; Sucker, A.; Paschen, A.; Zimmer, L.; Livingstone, E.; Brinker, T.J.; Hadaschik, E.; et al. Clinical and genetic analysis of melanomas arising in acral sites. Eur. J. Cancer 2019, 119, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Gao, B.; Zhou, Z.; Chen, T.; Xie, W.; Huang, M.; Li, W. Hepatocellular carcinoma with ARID1A mutation is associated with higher TMB and poor survival. J. Clin. Oncol. 2020, 38, e16667. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, J.; Wen, Q.; Zhu, X.; Li, D.; Liu, B.; Hu, J.; Wang, W.; Yao, M.; Wang, K. The landscape of ARID1A variants and correlation with tumor mutational burden in Chinese solid tumor patients. J. Clin. Oncol. 2019, 37, e14624. [Google Scholar] [CrossRef]
- Thielmann, C.M.; Chorti, E.; Matull, J.; Murali, R.; Zaremba, A.; Lodde, G.; Jansen, P.; Richter, L.; Kretz, J.; Moller, I.; et al. NF1-mutated melanomas reveal distinct clinical characteristics depending on tumour origin and respond favourably to immune checkpoint inhibitors. Eur. J. Cancer 2021, 159, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.; Sinha, S.; Cheng, K.; Madan, S.; Erez, A.; Ryan, B.M.; Schäffer, A.A.; Aldape, K.; Ruppin, E. Using a Recently Approved Tumor Mutational Burden Biomarker to Stratify Patients for Immunotherapy May Introduce a Sex Bias. JCO Precis. Oncol. 2021, 5, 1147–1150. [Google Scholar] [CrossRef] [PubMed]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Li, M.; Jiang, Z.; Wang, X. ARID1A Mutations Are Associated with Increased Immune Activity in Gastrointestinal Cancer. Cells 2019, 8, 678. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.P.; Kurzrock, R. PD-L1 Expression as a Predictive Biomarker in Cancer Immunotherapy. Mol. Cancer Ther. 2015, 14, 847–856. [Google Scholar] [CrossRef] [Green Version]
- Placke, J.M.; Soun, C.; Bottek, J.; Herbst, R.; Terheyden, P.; Utikal, J.; Pfohler, C.; Ulrich, J.; Kreuter, A.; Pfeiffer, C.; et al. Digital Quantification of Tumor PD-L1 Predicts Outcome of PD-1-Based Immune Checkpoint Therapy in Metastatic Melanoma. Front. Oncol. 2021, 11, 741993. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.I.; Wolchok, J.D.; Robert, C.; Hwu, W.J.; Weber, J.S.; Ribas, A.; Hodi, F.S.; Joshua, A.M.; Kefford, R.; Hersey, P.; et al. Programmed Death-Ligand 1 Expression and Response to the Anti-Programmed Death 1 Antibody Pembrolizumab in Melanoma. J. Clin. Oncol. 2016, 34, 4102–4109. [Google Scholar] [CrossRef] [PubMed]
- Morrison, C.; Pabla, S.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Dressman, D.; Papanicolau-Sengos, A.; Burgher, B.; Andreas, J.; Giamo, V.; et al. Predicting response to checkpoint inhibitors in melanoma beyond PD-L1 and mutational burden. J. Immunother. Cancer 2018, 6, 32. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. CheckMate 067: 6.5-year outcomes in patients (pts) with advanced melanoma. J. Clin. Oncol. 2021, 39, 9506. [Google Scholar] [CrossRef]
- Alaiwi, S.A.; Nassar, A.H.; Xie, W.; Bakouny, Z.; Berchuck, J.E.; Braun, D.A.; Baca, S.C.; Nuzzo, P.V.; Flippot, R.; Mouhieddine, T.H.; et al. Mammalian SWI/SNF Complex Genomic Alterations and Immune Checkpoint Blockade in Solid Tumors. Cancer Immunol. Res. 2020, 8, 1075–1084. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable, n (%) | |
|---|---|
| Age | |
| Median | 61 |
| Range | 22–94 |
| ≤60 | 54 (46.6) |
| >60 | 62 (53.4) |
| Sex | |
| Female | 40 (34.5) |
| Male | 76 (65.5) |
| Mutated Oncogene | |
| BRAF V600E | 61 (52.6) |
| NRAS Q61 | 45 (38.8) |
| NF1 | 48 (41.4) |
| ARID1A | 116 (100) |
| Primary Tumor | |
| Cutaneous | 97 (83.6) |
| Mucosal | 3 (2.6) |
| Occult | 16 (13.8) |
| Subtype of Cutaneous Tumors | |
| SSM | 25 (21.6) |
| NMM | 35 (30.2) |
| ALM | 11 (9.5) |
| LMM | 2 (1.7) |
| Desmoplastic | 4 (3.4) |
| Spitzoid | 2 (1.7) |
| Unknown | 18 (15.5) |
| Ulceration | |
| Present | 49 (42.2) |
| Missing | 37 (31.9) |
| Unknown | 30 (25.9) |
| Sentinel Lymph Node Biopsy | |
| Positive | 24 (20.7) |
| Negative | 44 (37.9) |
| Not performed | 48 (41.4) |
| PD-L1 | |
| Positive | 31 (26.7) |
| Negative | 56 (48.3) |
| Unknown | 29 (25.0) |
| Tumor Thickness | |
| <1 mm | 9 (7.8) |
| 1–2 mm | 24 (20.7) |
| 2–4 mm | 27 (23.3) |
| >4 mm | 31 (26.7) |
| Unknown | 24 (20.7) |
| Tumor Location | |
| Trunk | 36 (37.1) |
| Lower Extremity | 26 (26.8) |
| Upper Extremity | 8 (8.2) |
| Head and Neck | 27 (27.8) |
| Variable (n, %) | INAC (n = 32) | Others (n = 84) | p-Value |
|---|---|---|---|
| PD-L1 positive (>5%) | 9 (28.1) | 23 (27.4) | 0.71 |
| PD-L1 negative (<5%) | 13 (40.6) | 40 (47.6) | |
| Not tested | 10 (31.3) | 21 (25.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thielmann, C.M.; Matull, J.; Roth, S.; Placke, J.-M.; Chorti, E.; Zaremba, A.; Lodde, G.; Jansen, P.; Krefting, F.; Kretz, J.; et al. Genetic and Clinical Characteristics of ARID1A Mutated Melanoma Reveal High Tumor Mutational Load without Implications on Patient Survival. Cancers 2022, 14, 2090. https://doi.org/10.3390/cancers14092090
Thielmann CM, Matull J, Roth S, Placke J-M, Chorti E, Zaremba A, Lodde G, Jansen P, Krefting F, Kretz J, et al. Genetic and Clinical Characteristics of ARID1A Mutated Melanoma Reveal High Tumor Mutational Load without Implications on Patient Survival. Cancers. 2022; 14(9):2090. https://doi.org/10.3390/cancers14092090
Chicago/Turabian StyleThielmann, Carl Maximilian, Johanna Matull, Sebastian Roth, Jan-Malte Placke, Eleftheria Chorti, Anne Zaremba, Georg Lodde, Philipp Jansen, Frederik Krefting, Julia Kretz, and et al. 2022. "Genetic and Clinical Characteristics of ARID1A Mutated Melanoma Reveal High Tumor Mutational Load without Implications on Patient Survival" Cancers 14, no. 9: 2090. https://doi.org/10.3390/cancers14092090
APA StyleThielmann, C. M., Matull, J., Roth, S., Placke, J.-M., Chorti, E., Zaremba, A., Lodde, G., Jansen, P., Krefting, F., Kretz, J., Möller, I., Sucker, A., Paschen, A., Livingstone, E., Zimmer, L., Ugurel, S., Schadendorf, D., Hadaschik, E., & Griewank, K. G. (2022). Genetic and Clinical Characteristics of ARID1A Mutated Melanoma Reveal High Tumor Mutational Load without Implications on Patient Survival. Cancers, 14(9), 2090. https://doi.org/10.3390/cancers14092090