
A Six-Gene Prognostic and Predictive Radiotherapy-Based Signature for Early and Locally Advanced Stages in Non-Small-Cell Lung Cancer


 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
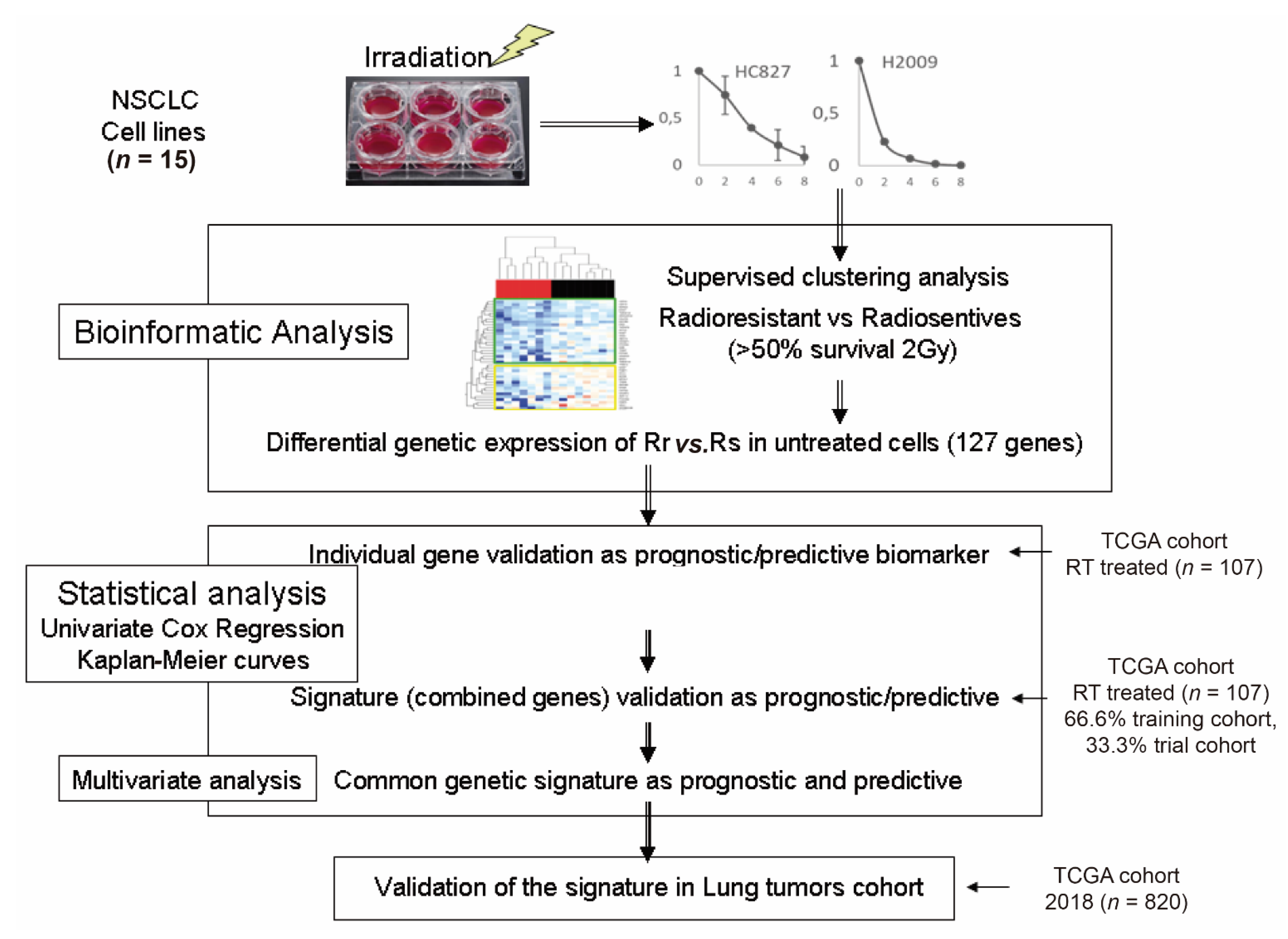
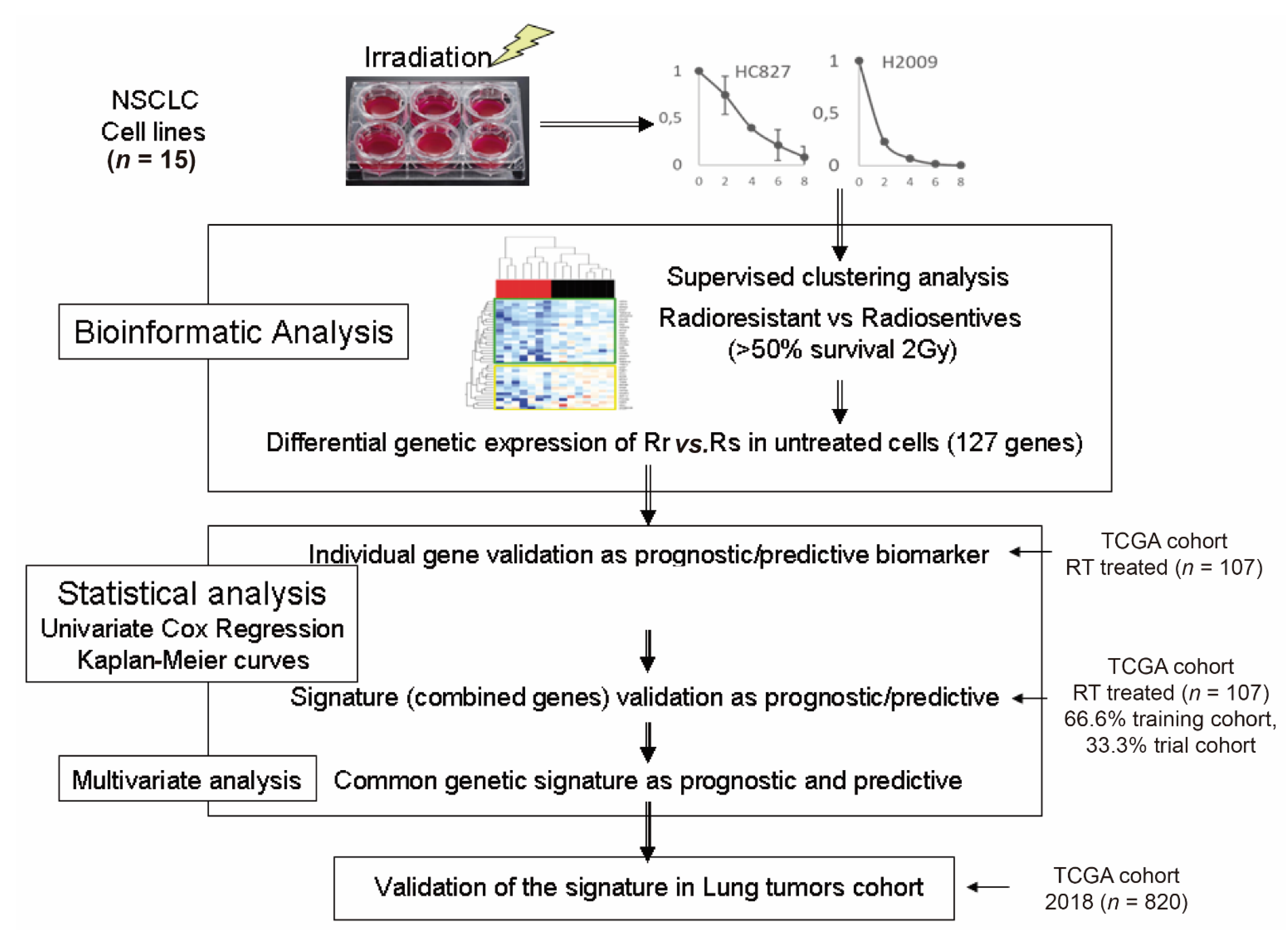
2. Materials and Methods
2.1. Cell Lines
2.2. Clonogenicity Test for the Response to Ionizing Radiation by Determining the SF2 Parameter
2.3. RNA Extraction from Cells
2.4. cDNA Microarrays
2.5. Transcriptome Analysis
2.6. Patient Cohort
2.7. Statistical Packages and Analysis
2.8. Obtaining Gene Signatures
3. Results
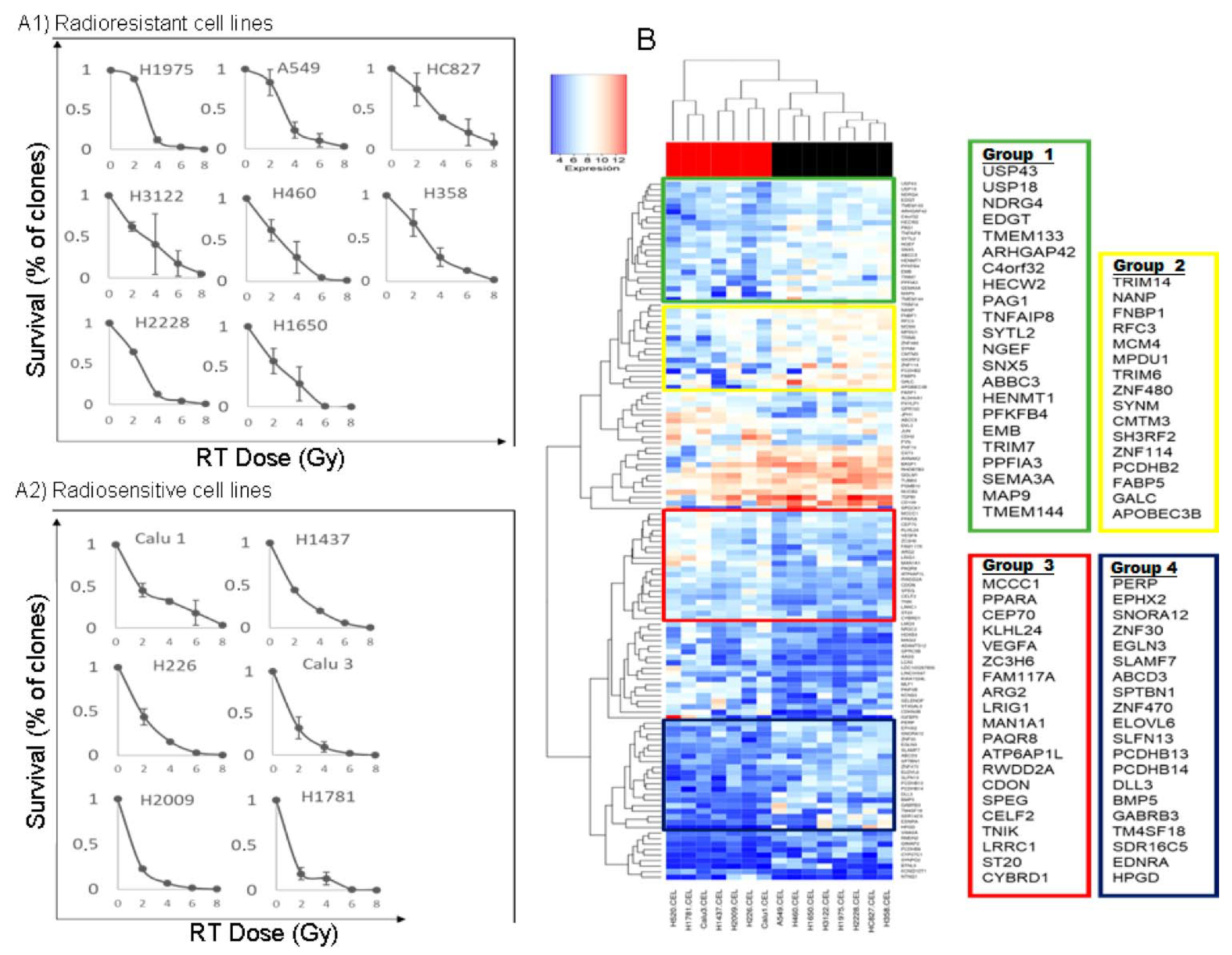
3.1. Survival Values at 2 Gy and Survival Curves of All Cell Lines
3.2. Identification of Genes with Differential Expression between Radiosensitive and Radioresistant Lines
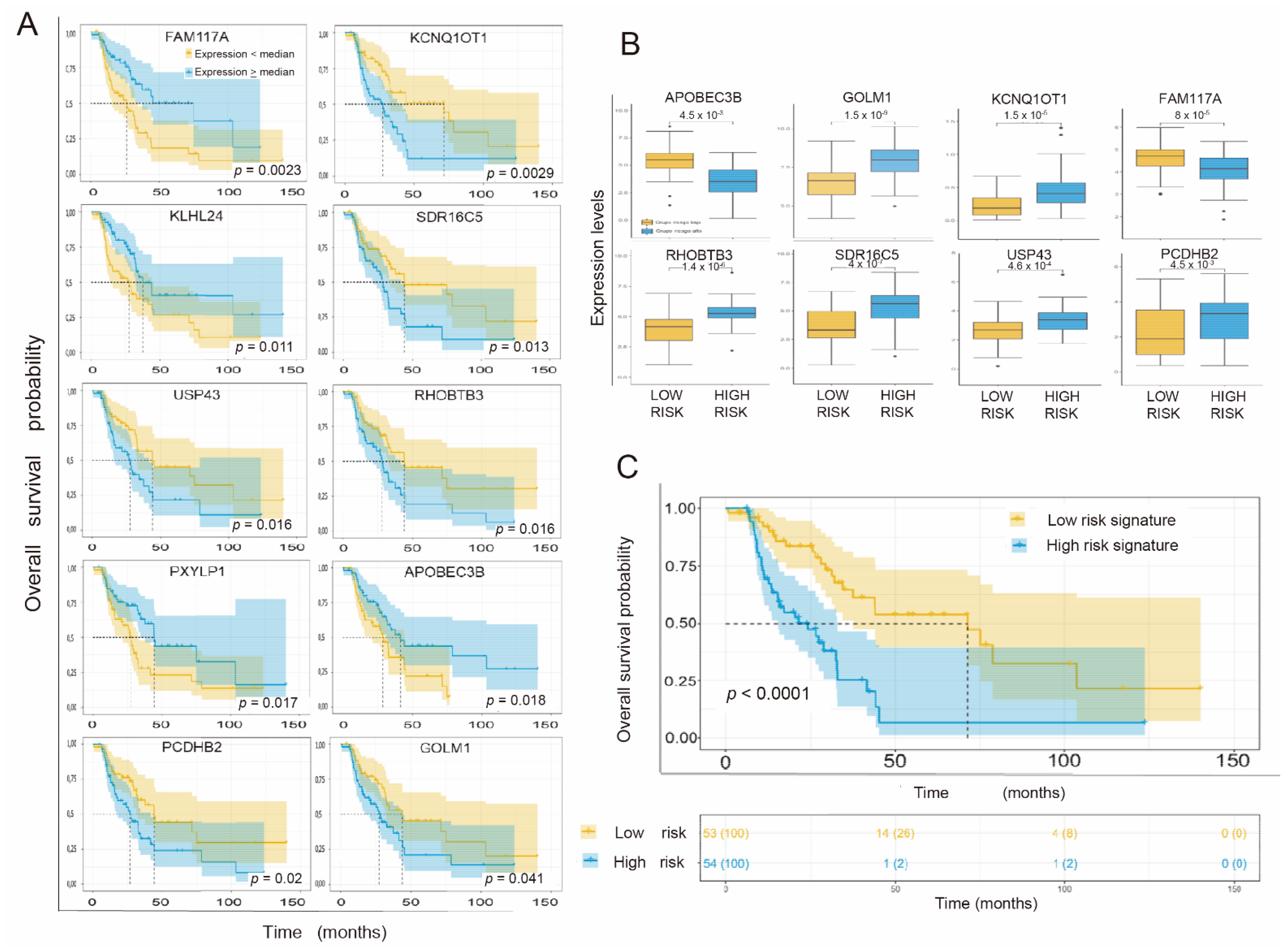
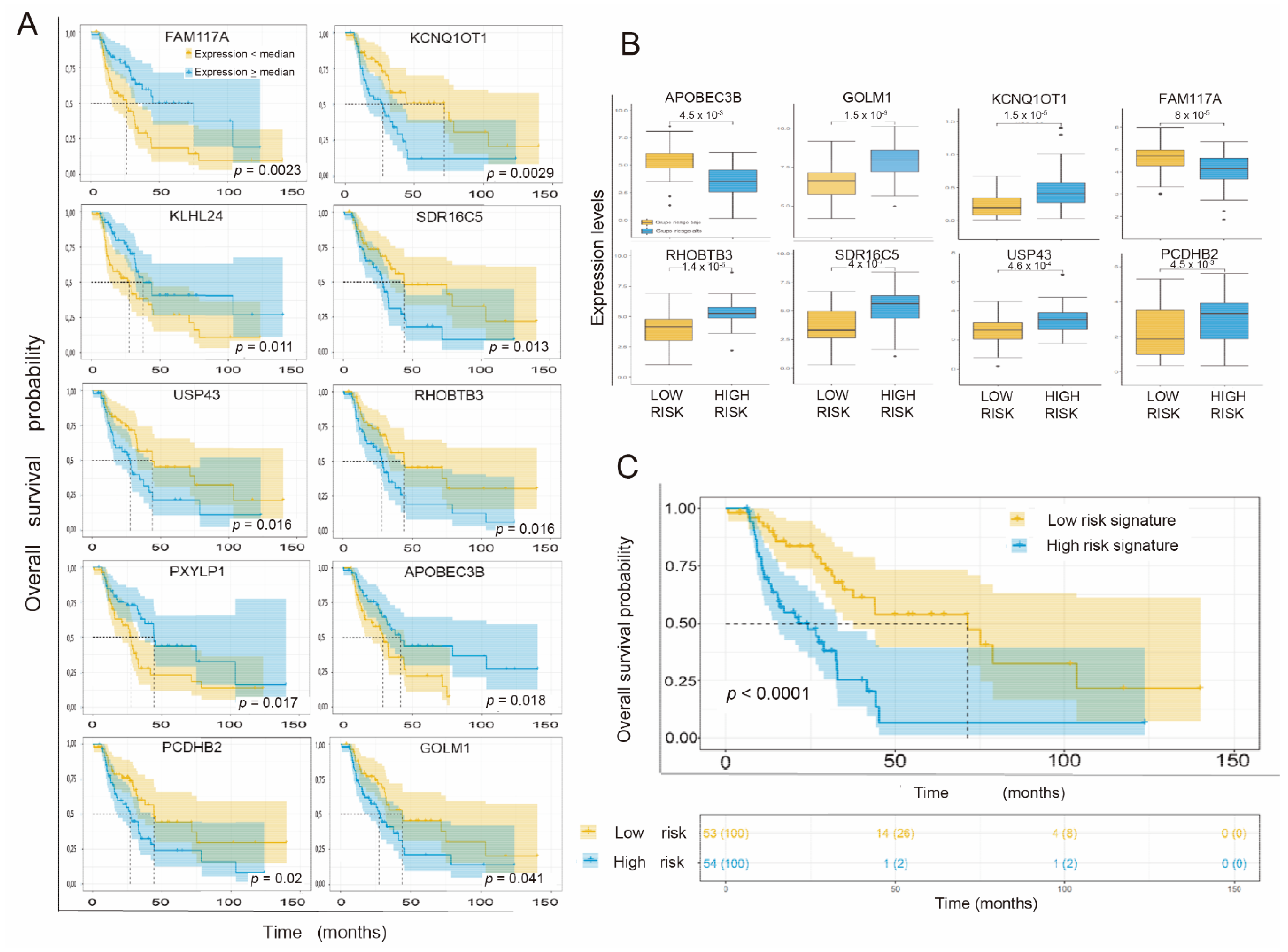
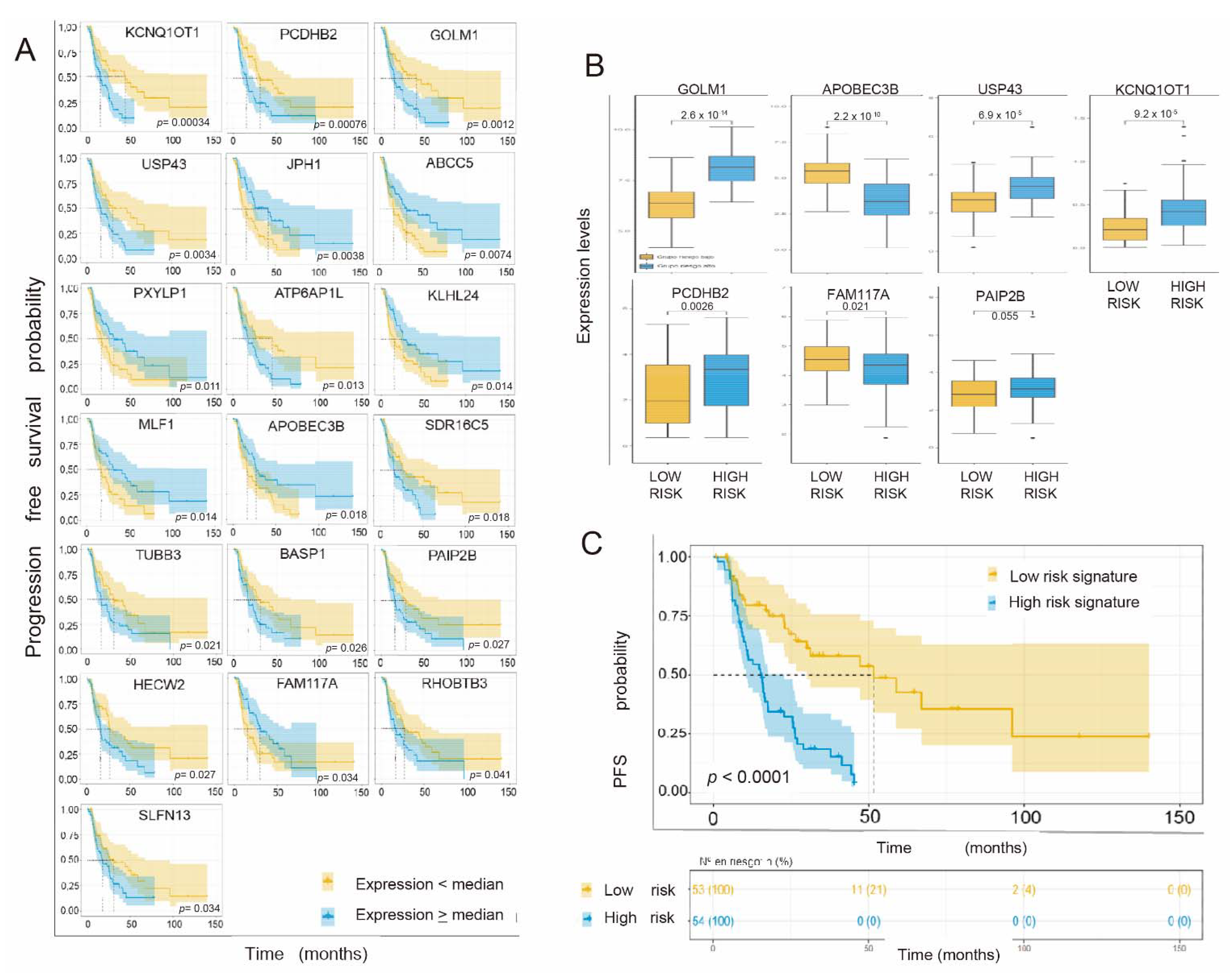
3.3. Individual Validation of Genes with Differential Expression as Prognostic Biomarkers in the TCGA Cohort of Radiation-Treated Patients
3.4. Obtaining the Prognostic Signature
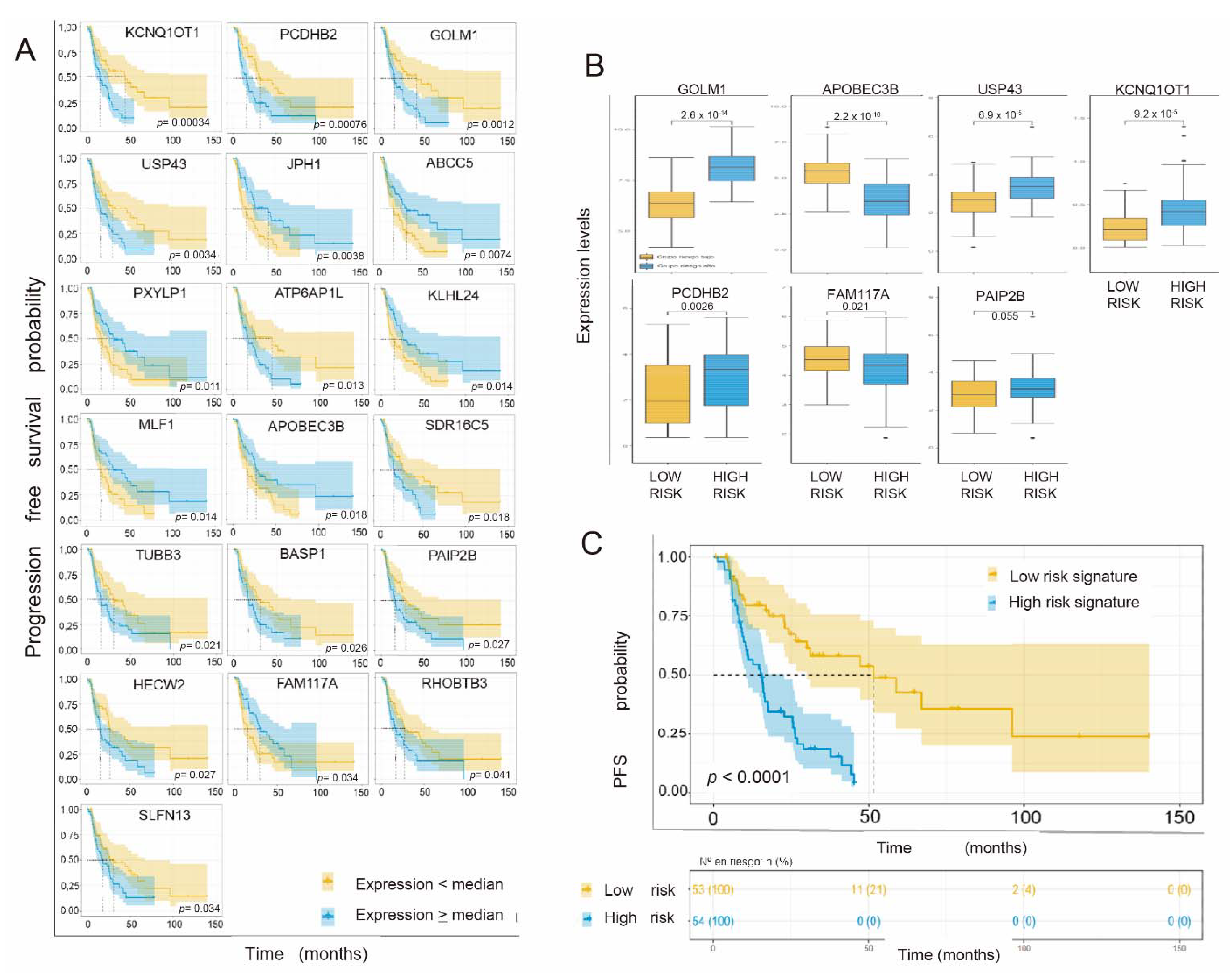
3.5. Individual Validation of Genes with Differential Expression between Radiosensitive and Radioresistant Lines as a Predictive Biomarker of Response in the TCGA Cohort of Interest
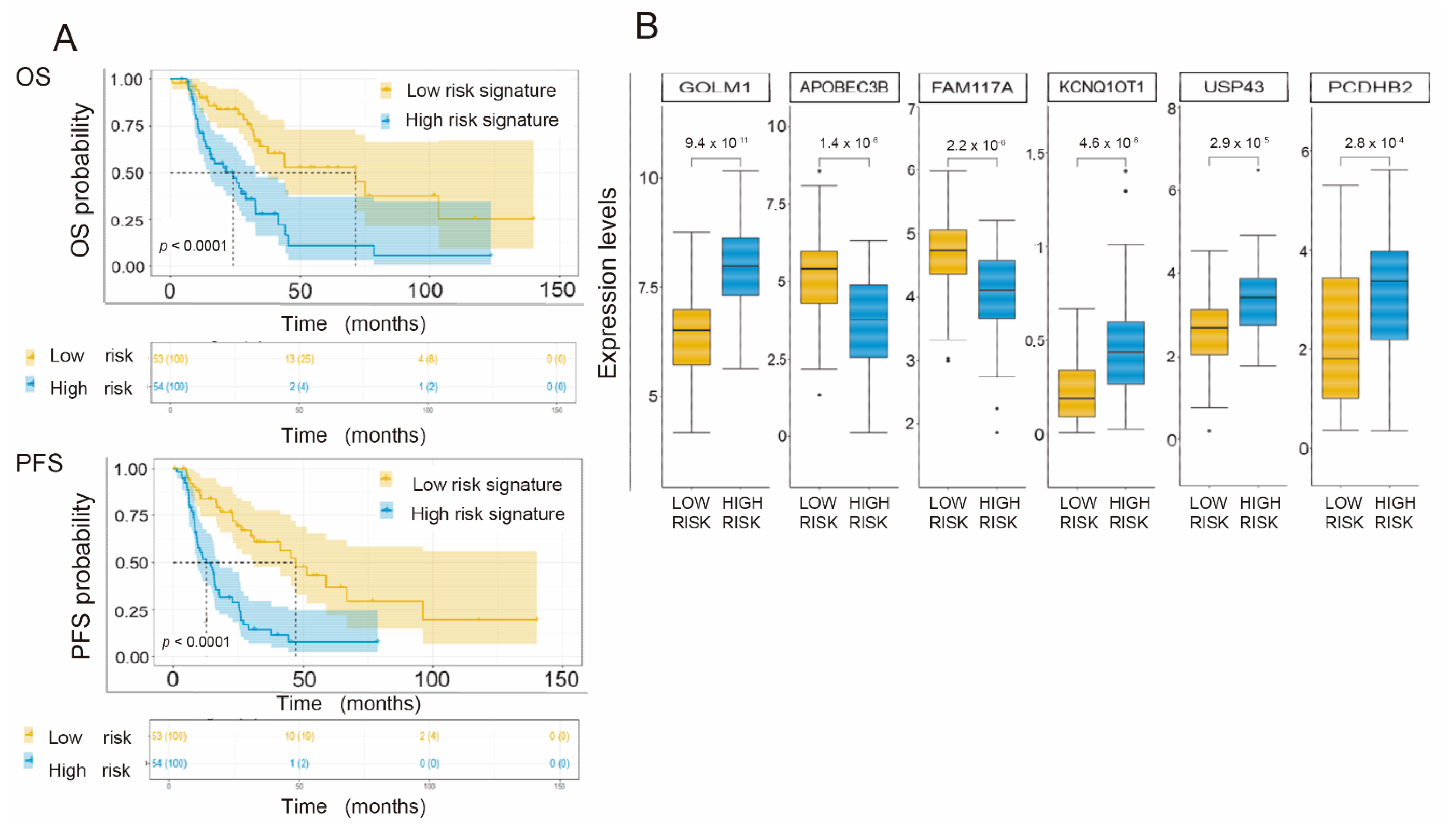
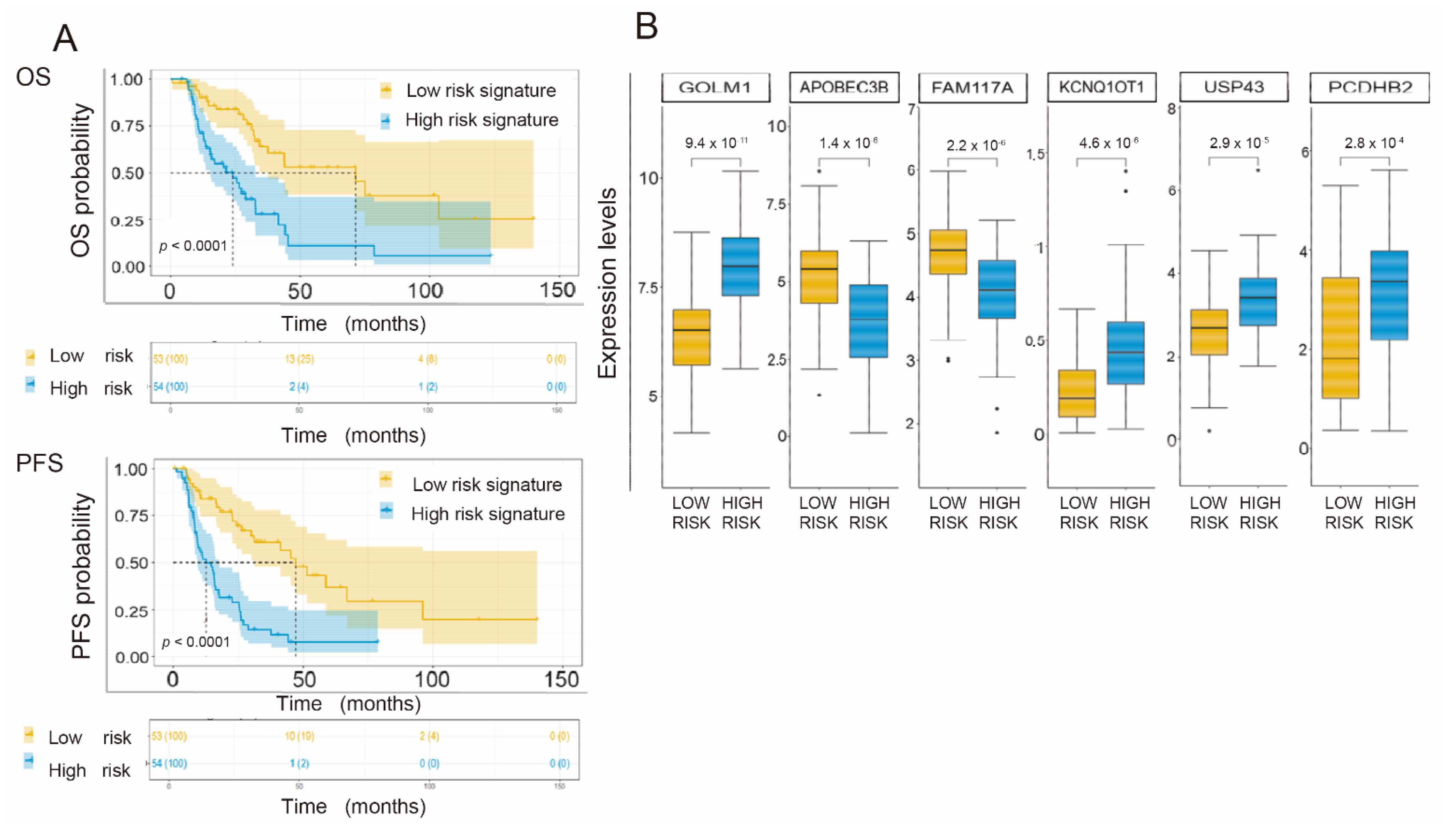
3.6. Generation of a Common Gene Signature for Global Survival Prediction and Progression-Free Survival
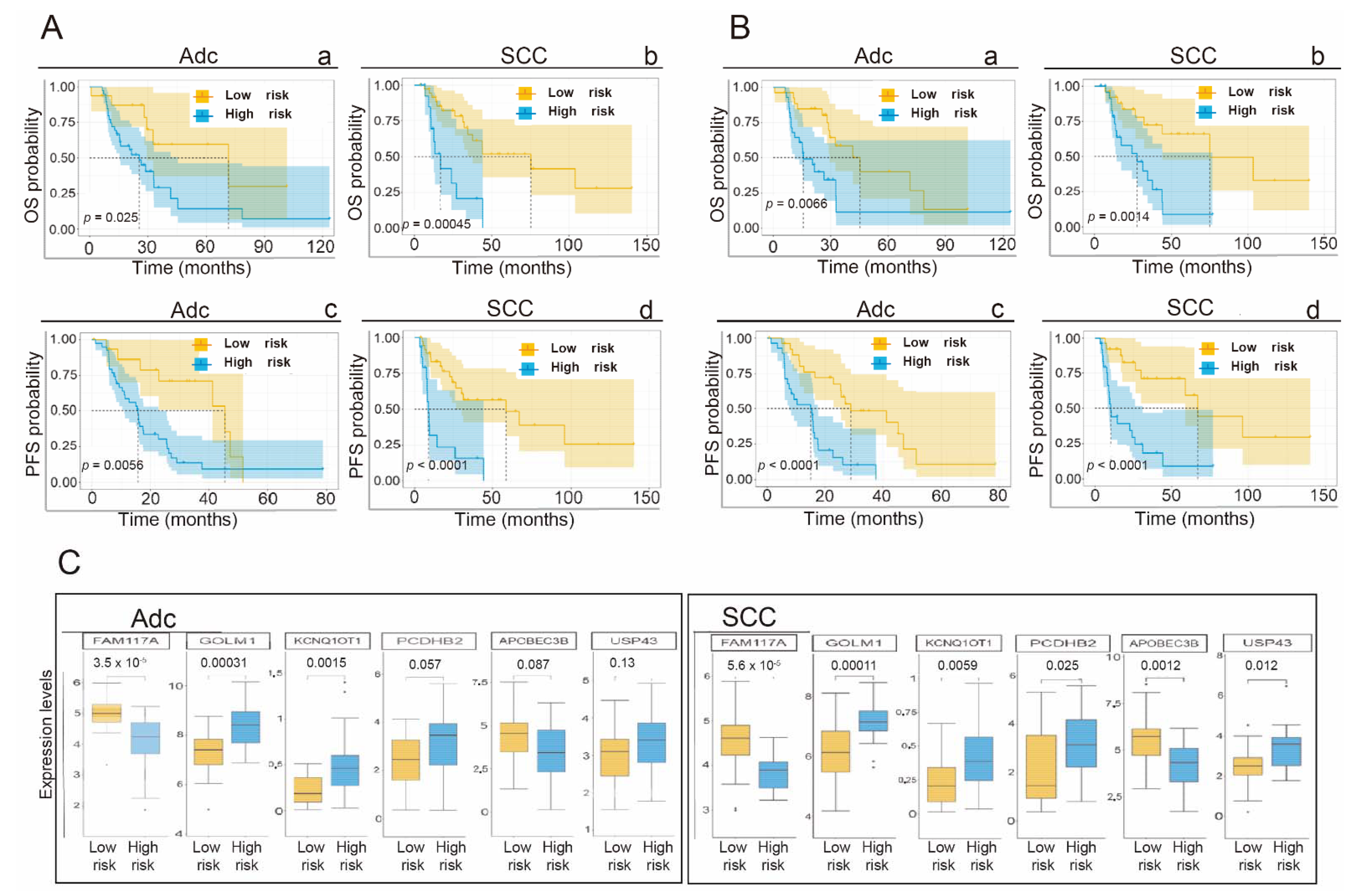
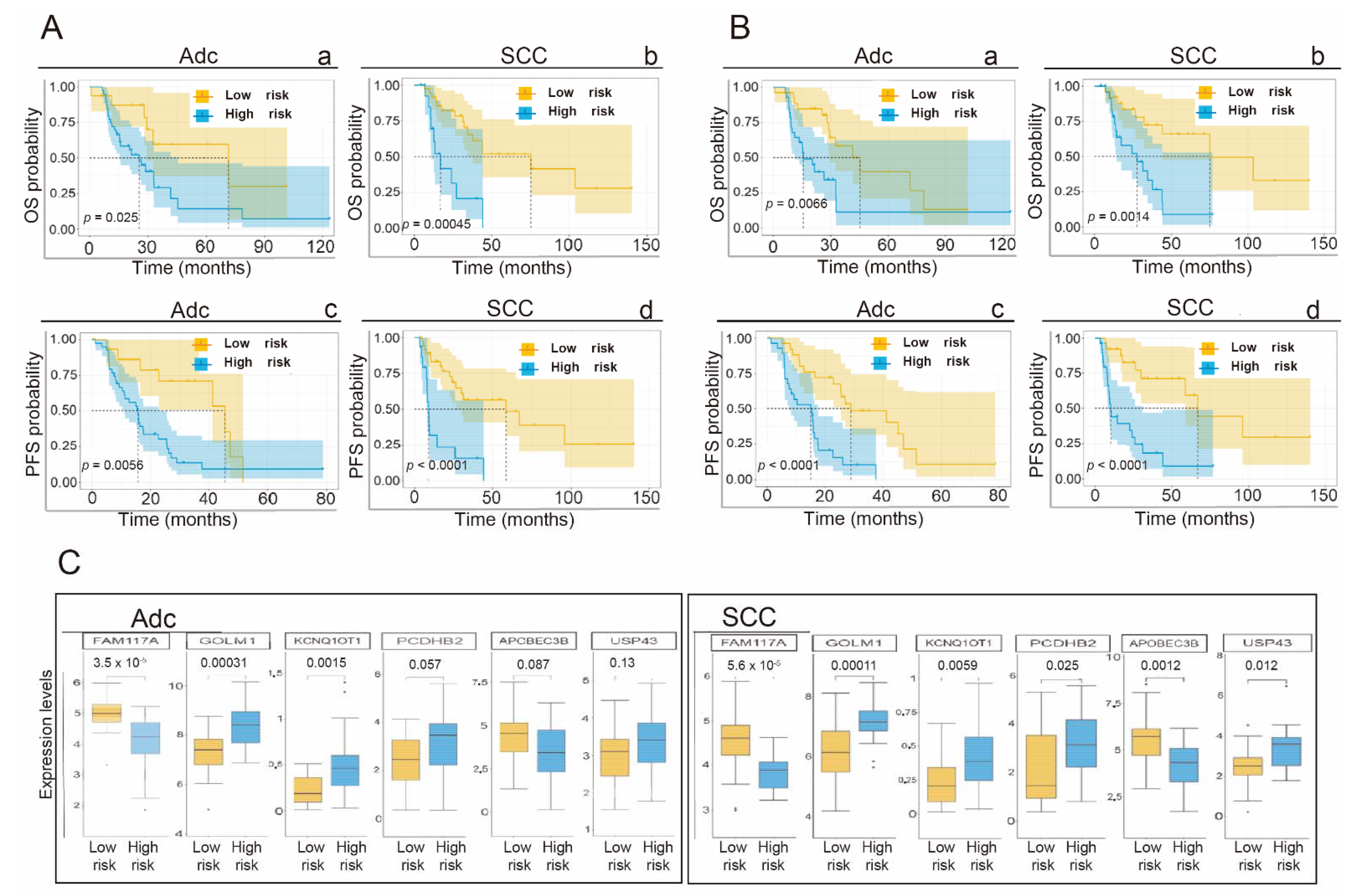
3.7. Application of the Gene Signature for Prediction of OS and PFS Depending on the Histological Subtype
3.8. Univariate and Multivariate Analysis of Clinical and Pathological Variables, Their Impact on Overall Survival and PFS and Their Influence on the Proposed Gene Signature
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- National Comprehensive Cancer Network (NCCN). NCCN Clinical Practice Guidelines in Oncology. In Non-Small Cell Lung Cancer; Version 1.2020; NCCN: Plymouth Meeting, PA, USA.
- O’Rourke, N.; Roqué i Figuls, M.; Bernadó, N.F.; Macbeth, F. Concurrent chemoradiotherapy in non-small cell lung cancer. Cochrane Database Syst. Rev. 2010, 6, CD002140. [Google Scholar] [CrossRef] [PubMed]
- Aupérin, A.; Le Péchoux, C.; Pignon, J.P.; Koning, C.; Jeremic, B.; Clamon, G.; Einhorn, L.; Ball, D.; Trovo, M.G.; Groen, H.J.M.; et al. Concomitant radio-chemotherapy based on platin compounds in patients with locally advanced non-small cell lung cancer (NSCLC): A meta-analysis of individual data from 1764 patients. Ann. Oncol. 2006, 17, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Conibear, J.; on behalf of AstraZeneca UK Limited. Rationale for concurrent chemoradiotherapy for patients with stage III non-small-cell lung cancer. Br. J. Cancer 2020, 123, 10–17. [Google Scholar] [CrossRef]
- Le Chevalier, T.; Arriagada, R.; Quoix, E.; Ruffle, P.; Martin, M.; Tarayre, M.; Marie-José, L.-T.; Douillard, J.-Y.; Laplanche, A. Radiotherapy alone versus combined chemotherapy and radiotherapy in nonresectable non-small-cell lung cancer: First analysis of a randomized trial in 353 patients. J. Natl. Cancer Inst. 1991, 83, 417–423. [Google Scholar] [CrossRef]
- Curran, W.J., Jr.; Paulus, R.; Langer, C.J.; Komaki, R.; Lee, J.S.; Hauser, S.; Movsas, B.; Wasserman, T.; Rosenthal, S.A.; Gore, E.; et al. Sequential vs. concurrent chemoradiation for stage III non-small cell lung cancer: Randomized phase III trial RTOG 9410. J. Natl. Cancer Inst. 2011, 103, 1452–1460. [Google Scholar] [CrossRef] [Green Version]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I.; Singh, B.; Heelan, R.; Rusch, V.; Fulton, L.; et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl. Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar] [CrossRef] [Green Version]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Paez, J.G.; Jänne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, H.; Kobayashi, S.; Costa, D. EGFR exon 20 insertion mutations in non-small-cell lung cancer: Preclinical data and clinical implications. Lancet Oncol. 2012, 13, e23–e31. [Google Scholar] [CrossRef]
- Pao, W.; Wang, T.Y.; Riely, G.J.; Miller, V.A.; Pan, Q.; Ladanyi, M.; Zakowski, M.F.; Heelan, R.T.; Kris, M.G.; Varmus, H.E. KRAS mutations and primary resistance of lung adenocarcinomas to gefitinib or erlotinib. PLoS Med. 2005, 2, e17. [Google Scholar] [CrossRef] [Green Version]
- Bean, J.; Brennan, C.; Shih, J.-Y.; Riely, G.; Viale, A.; Wang, L.; Chitale, D.; Motoi, N.; Szoke, J.; Broderick, S.; et al. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. USA 2007, 104, 20932–20937. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.-M.; Zhao, X.; Christensen, J.; et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Eberhard, D.A.; Johnson, B.E.; Amler, L.C.; Goddard, A.D.; Heldens, S.L.; Herbst, R.S.; Ince, W.L.; Jänne, P.A.; Januario, T.; Johnson, D.H.; et al. Mutations in the epidermal growth factor receptor and in KRAS are predictive and prognostic indicators in patients with non-small-cell lung cancer treated with chemotherapy alone and in combination with erlotinib. J. Clin. Oncol. 2005, 23, 5900–5909. [Google Scholar] [CrossRef]
- Das, A.K.; Sato, M.; Story, M.D.; Peyton, M.; Graves, R.; Redpath, S.; Girard, L.; Gazdar, A.F.; Shay, J.W.; Minna, J.D.; et al. Non-small-cell lung cancers with kinase domain mutations in the epidermal growth factor receptor are sensitive to ionizing radiation. Cancer Res. 2006, 66, 9601–9608. [Google Scholar] [CrossRef] [Green Version]
- Raben, D.; Helfrich, B.; Bunn, P.A., Jr. Targeted therapies for non-small-cell lung cancer: Biology, rationale, and preclinical results from a radiation oncology perspective. Int. J. Radiat. Oncol. Biol. Physic. 2004, 59 (Suppl. 2), S27–S38. [Google Scholar] [CrossRef]
- Bergqvist, M.; Brattström, D.; Gullbo, J.; Hesselius, P.; Brodin, O.; Wagenius, G. p53 status and its in vitro relationship to radiosensitivity and chemosensitivity in lung cancer. Anticancer Res. 2003, 23, 1207–1212. [Google Scholar]
- Hayes, D.N.; Monti, S.; Parmigiani, G.; Gilks, C.B.; Naoki, K.; Bhattacharjee, A.; Socinski, M.A.; Perou, C.; Meyerson, M. Gene expression profiling reveals reproducible human lung adenocarcinoma subtypes in multiple independent patient cohorts. J. Clin. Oncol. 2006, 24, 5079–5090. [Google Scholar] [CrossRef] [Green Version]
- Travis, W.D.; Brambilla, E.; Riely, G.J. New pathologic classification of lung cancer: Relevance for clinical practice and clinical trials. J. Clin. Oncol. 2013, 31, 992–1001. [Google Scholar] [CrossRef] [Green Version]
- Choong, N.W.; Salgia, R.; Vokes, E.E. Key signaling pathways and targets in lung cancer therapy. Clin. Lung Cancer 2007, 8 (Suppl. 2), S52–S60. [Google Scholar] [CrossRef]
- Kris, M.G.; Johnson, B.E., 3rd; Kwiatkowski, D.J.; Iafrate, A.J.; Wistuba, I.I.; Aronson, S.L.; Engelman, J.A.; Shyr, Y.; Khuri, F.R.; Rudin, C.M.; et al. Identification of driver mutations in tumor specimens from 1000 patients with lung adenocarcinoma: The NCI’s lung cancer mutation consortium (LCMC). J. Clin. Oncol. 2011, 29, CRA7506. [Google Scholar] [CrossRef]
- Beer, D.G.; Kardia, S.L.; Huang, C.C.; Giordano, T.J.; Levin, A.M.; Misek, D.E.; Lin, L.; Chen, G.; Gharib, T.G.; Thomas, D.G.; et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat. Med. 2002, 8, 816–824. [Google Scholar] [CrossRef] [PubMed]
- Director’s Challenge Consortium for the Molecular Classification of Lung Adenocarcinoma; Shedden, K.; Taylor, J.M.; Enkemann, S.A.; Tsao, M.S.; Yeatman, T.J.; Gerald, W.L.; Eschrich, S.; Jurisica, I.; Giordano, T.J.; et al. Gene expression-based survival prediction in lung adenocarcinoma: A multi-site, blinded validation study. Nat. Med. 2008, 14, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Larsen, J.E.; Pavey, S.; Passmore, L.H.; Bowman, R.V.; Hayward, N.K.; Fong, K. Gene expression signature predicts recurrence in lung adenocarcinoma. Clin. Cancer Res. 2007, 13, 2946–2954. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Tang, H.; Sun, Z.; Bungum, A.O.; Edell, E.S.; Lingle, W.L.; Stoddard, S.M.; Zhang, M.; Jen, J.; Yang, P.; et al. Network-based approach identified cell cycle genes as predictor of overall survival in lung adenocarcinoma patients. Lung Cancer 2013, 80, 91–98. [Google Scholar] [CrossRef] [Green Version]
- Tomida, S.; Takeuchi, T.; Shimada, Y.; Arima, C.; Matsuo, K.; Mitsudomi, T.; Yatabe, Y.; Takahashi, T. Relapse-related molecular signature in lung adenocarcinomas identifies patients with dismal prognosis. J. Clin. Oncol. 2009, 27, 2793–2799. [Google Scholar] [CrossRef]
- Bianchi, F.; Nuciforo, P.; Vecchi, M.; Bernard, L.; Tizzoni, L.; Marchetti, A.; Buttitta, F.; Felicioni, L.; Nicassio, F.; Di Fiore, P.P. Survival prediction of stage I lung adenocarcinomas by expression of 10 genes. J. Clin. Investig. 2007, 117, 3436–3444. [Google Scholar] [CrossRef] [Green Version]
- Raz, D.J.; Ray, M.R.; Kim, J.Y.; Ricchetti, R.; Taron, M.; Skrzypski, M.; Segal, M.; Gandara, D.R.; Rosell, R.; Jablons, D.M. A multigene assay is prognostic of survival in patients with early-stage lung adenocarcinoma. Clin. Cancer Res. 2008, 14, 5565–5570. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.-Y.; Park, E.S.; Kim, S.B.; Kim, S.C.; Sohn, B.H.; Chu, I.-S.; Jeong, W.; Mills, G.B.; Byers, L.A.; Lee, J.-S. Development and validation of a prognostic gene-expression signature for lung adenocarcinoma. PLoS ONE 2012, 7, e44225. [Google Scholar] [CrossRef]
- Akagi, I.; Okayama, H.; Schetter, A.J.; Robles, A.; Kohno, T.; Bowman, E.D.; Kazandjian, D.; Welsh, J.A.; Oue, N.; Saito, M.; et al. Combination of protein coding and noncoding gene expression as a robust prognostic classifier in stage I lung adenocarcinoma. Cancer Res. 2013, 73, 3821–3832. [Google Scholar] [CrossRef] [Green Version]
- Raponi, M.; Zhang, Y.; Yu, J.; Chen, G.; Lee, G.; Taylor, J.M.; Macdonald, J.; Thomas, D.; Moskaluk, C.; Wang, Y.; et al. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006, 66, 7466–7472. [Google Scholar] [CrossRef] [Green Version]
- Larsen, J.E.; Pavey, S.J.; Passmore, L.H.; Bowman, R.; Clarke, B.E.; Hayward, N.K.; Fong, K.M. Expression profiling defines a recurrence signature in lung squamous cell carcinoma. Carcinogenesis 2007, 28, 760–766. [Google Scholar] [CrossRef] [Green Version]
- Skrzypski, M.; Jassem, E.; Taron, M.; Sanchez, J.J.; Mendez, P.; Rzyman, W.; Gulida, G.; Raz, D.; Jablons, D.; Provencio, M.; et al. Three-gene expression signature predicts survival in early-stage squamous cell carcinoma of the lung. Clin. Cancer Res. 2008, 14, 4794–4799. [Google Scholar] [CrossRef] [Green Version]
- Landi, M.T.; Zhao, Y.; Rotunno, M.; Koshiol, J.; Liu, H.; Bergen, A.; Rubagotti, M.; Goldstein, A.M.; Linnoila, I.; Marincola, F.M.; et al. MicroRNA expression differentiates histology and predicts survival of lung cancer. Clin. Cancer Res. 2010, 16, 430–441. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Aerts, J.; den Hamer, B.; van Ijcken, W.; den Bakker, M.; Riegman, P.; Van Der Leest, C.; Van Der Spek, P.; Foekens, J.A.; Hoogsteden, H.C.; et al. Gene expression-based classification of non-small cell lung carcinomas and survival prediction. PLoS ONE 2010, 5, e10312. [Google Scholar] [CrossRef]
- Mitra, R.; Lee, J.; Jo, J.; Milani, M.; McClintick, J.N.; Edenberg, H.; Kesler, K.A.; Rieger, K.M.; Badve, S.; Cummings, O.W.; et al. Prediction of postoperative recurrence-free survival in non-small cell lung cancer by using an internationally validated gene expression model. Clin. Cancer Res. 2011, 17, 2934–2946. [Google Scholar] [CrossRef] [Green Version]
- Roepman, P.; Jassem, J.; Smit, E.F.; Muley, T.; Niklinski, J.; Van De Velde, T.; Witteveen, A.T.; Rzyman, W.; Floore, A.; Burgers, S.; et al. An immune response enriched 72-gene prognostic profile for early-stage non-small-cell lung cancer. Clin. Cancer Res. 2009, 15, 284–290. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Xiao, G.; Coombes, K.; Behrens, C.; Solis, L.M.; Raso, G.; Girard, L.; Erickson, H.S.; Roth, J.; Heymach, J.V.; et al. Robust gene expression signature from formalin-fixed paraffin-embedded samples predicts prognosis of non-small-cell lung cancer patients. Clin. Cancer Res. 2011, 17, 5705–5714. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.-S.; Son, D.-S.; Kim, S.-H.; Lee, J.; Jo, J.; Han, J.; Kim, H.; Lee, H.J.; Choi, H.Y.; Jung, Y.; et al. Prediction of recurrence-free survival in postoperative non-small cell lung cancer patients by using an integrated model of clinical information and gene expression. Clin. Cancer Res. 2008, 14, 7397–7404. [Google Scholar] [CrossRef] [Green Version]
- Boutros, P.C.; Lau, S.K.; Pintilie, M.; Liu, N.; Shepherd, F.A.; Der, S.D.; Tsao, M.-S.; Penn, L.Z.; Jurisica, I. Prognostic gene signatures for non-small-cell lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 2824–2828. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.-T.; Hsu, Y.-L.; Fulp, W.J.; Coppola, D.; Haura, E.B.; Yeatman, T.J.; Cress, W.D. Prognostic and predictive value of a malignancy-risk gene signature in early-stage non-small cell lung cancer. J. Natl. Cancer Inst. 2011, 103, 1859–1870. [Google Scholar] [CrossRef] [Green Version]
- Guo, N.L.; Wan, Y.-W.; Tosun, K.; Lin, H.; Msiska, Z.; Flynn, D.C.; Remick, S.C.; Vallyathan, V.; Dowlati, A.; Shi, X.; et al. Confirmation of gene expression-based prediction of survival in non-small cell lung cancer. Clin. Cancer Res. 2008, 14, 8213–8220. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Lemon, W.; Liu, P.-Y.; Yi, Y.; Morrison, C.; Yang, P.; Sun, Z.; Szőke, J.; Gerald, W.L.; Watson, M.; et al. A gene expression signature predicts survival of patients with stage I non-small cell lung cancer. PLoS Med. 2006, 3, e467. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Yu, S.-L.; Chen, C.-H.; Chang, G.-C.; Chen, C.-Y.; Yuan, A.; Cheng, C.-L.; Wang, C.-H.; Terng, H.-J.; Kao, S.-F.; et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. N. Engl. J. Med. 2007, 356, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.; Boutros, P.C.; Pintilie, M.; Blackhall, F.H.; Zhu, C.-Q.; Strumpf, D.; Johnston, M.R.; Darling, G.; Keshavjee, S.; Waddell, T.K.; et al. Three-gene prognostic classifier for early-stage non small-cell lung cancer. J. Clin. Oncol. 2007, 25, 5562–5569. [Google Scholar] [CrossRef]
- Zhu, C.; Ding, K.; Strumpf, D.; Weir, B.A.; Meyerson, M.; Pennell, N.; Thomas, R.K.; Naoki, K.; Ladd-Acosta, C.; Liu, N.; et al. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 4417–4424. [Google Scholar] [CrossRef]
- Tang, H.; Xiao, G.; Behrens, C.; Schiller, J.; Allen, J.; Chow, C.-W.; Suraokar, M.; Corvalan, A.; Mao, J.-H.; White, M.A.; et al. A 12-gene set predicts survival benefits from adjuvant chemotherapy in non-small cell lung cancer patients. Clin. Cancer Res. 2013, 19, 1577–1586. [Google Scholar] [CrossRef] [Green Version]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef]
- Torres-Roca, J.F.; Eschrich, S.; Zhao, H.; Bloom, G.; Sung, J.; McCarthy, S.; Cantor, A.B.; Scuto, A.; Li, C.; Zhang, S.; et al. Prediction of radiation sensitivity using a gene expression classifier. Cancer Res. 2005, 65, 7169–7176. [Google Scholar] [CrossRef] [Green Version]
- Eschrich, S.; Zhang, H.; Zhao, H.; Boulware, D.; Lee, J.-H.; Bloom, G.; Torres-Roca, J.F. Systems biology modeling of the radiation sensitivity network: A biomarker discovery platform. Int. J. Radiat. Oncol. Biol. Phys. 2009, 75, 497–505. [Google Scholar] [CrossRef] [Green Version]
- Strom, T.; Hoffe, S.E.; Fulp, W.; Frakes, J.; Coppola, D.; Springett, G.M.; Malafa, M.P.; Harris, C.L.; Eschrich, S.A.; Torres-Roca, J.F.; et al. Radiosensitivity index predicts for survival with adjuvant radiation in resectable pancreatic cancer. Radiother. Oncol. 2015, 117, 159–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amundson, S.A.; Do, K.T.; Vinikoor, L.C.; Lee, R.A.; Koch-Paiz, C.A.; Ahn, J.; Reimers, M.; Chen, Y.; Scudiero, D.A.; Weinstein, J.N.; et al. Integrating global gene expression and radiation survival parameters across the 60 cell lines of the National Cancer Institute Anticancer Drug Screen. Cancer Res. 2008, 68, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, K.A.; Berglund, A.E.; Welsh, E.A.; Naghavi, A.O.; Kim, Y.; Yu, M.; Robinson, T.J.; Eschrich, S.A.; Johnstone, P.A.; Torres-Roca, J.F. The radiosensitivity of brain metastases based upon primary histology utilizing a multigene index of tumor radiosensitivity. Neuro-Oncology 2017, 19, 1145–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, B.-S.; Han, W.; Kim, I.A. Tumor mutation burden, immune checkpoint crosstalk and radiosensitivity in single-cell RNA sequencing data of breast cancer. Radiother. Oncol. 2020, 142, 202–209. [Google Scholar] [CrossRef]
- Scott, J.G.; Berglund, A.; Schell, M.J.; Mihaylov, I.; Fulp, W.J.; Yue, B.; Welsh, E.; Caudell, J.J.; Ahmed, K.; Strom, T.S.; et al. A genome-based model for adjusting radiotherapy dose (GARD): A retrospective, cohort-based study. Lancet Oncol. 2017, 18, 202–211, Erratum in Lancet Oncol. 2017, 18, e65. [Google Scholar] [CrossRef]
- Hammond, S.M. An Overview of MicroRNAs. Adv. Drug Deliv. Rev. 2015, 87, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Boeri, M.; Verri, C.; Conte, D.; Roz, L.; Modena, P.; Facchinetti, F.; Calabrò, E.; Croce, C.M.; Pastorino, U.; Sozzi, G. MicroRNA signatures in tissues and plasma predict development and prognosis of computed tomography detected lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 3713–3718. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Hawkins, P.G.; Bi, N.; Dess, R.T.; Tewari, M.; Hearn, J.W.; Hayman, J.A.; Kalemkerian, G.P.; Lawrence, T.S.; Haken, R.K.T.; et al. Serum MicroRNA Signature Predicts Response to High-Dose Radiation Therapy in Locally Advanced Non-Small Cell Lung Cancer. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 107–114. [Google Scholar] [CrossRef]
- Dehing-Oberije, C.; Aerts, H.; Yu, S.; De Ruysscher, D.; Menheere, P.; Hilvo, M.; van der Weide, H.; Rao, B.; Lambin, P. Development and validation of a prognostic model using blood biomarker information for prediction of survival of non-small-cell lung cancer patients treated with combined chemotherapy and radiation or radiotherapy alone (NCT00181519, NCT00573040, and NCT00572325). Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 360–368. [Google Scholar]
- Postmus, P.E.; Kerr, K.M.; Oudkerk, M.; Senan, S.; Waller, D.A.; Vansteenkiste, J.; Escriu, C.; Peters, S.; ESMO Guidelines Committee. Early and locally advanced non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2017, 28 (Suppl. 4), iv1–iv21. [Google Scholar] [CrossRef]
- Detterbeck, F.C.; Boffa, D.J.; Kim, A.W.; Tanoue, L.T. The eighth edition lung cancer stage classification. Chest 2017, 151, 193–203. [Google Scholar] [CrossRef]
- Subramanian, J.; Simon, R. Gene expression-based prognostic signatures in lung cancer: Ready for clinical use? J. Natl. Cancer Inst. 2010, 102, 464–474. [Google Scholar] [CrossRef]
- Tomida, S.; Koshikawa, K.; Yatabe, Y.; Harano, T.; Ogura, N.; Mitsudomi, T.; Some, M.; Yanagisawa, K.; Takahashi, T.; Osada, H.; et al. Gene expression-based, individualized outcome prediction for surgically treated lung cancer patients. Oncogene 2004, 23, 5360–5370. [Google Scholar] [CrossRef] [Green Version]
- Zuo, S.; Wei, M.; Zhang, H.; Chen, A.; Wu, J.; Wei, J.; Dong, J. A robust six-gene prognostic signature for prediction of both disease-free and overall survival in non-small cell lung cancer. J. Transl. Med. 2019, 17, 152. [Google Scholar] [CrossRef]
- He, R.; Zuo, S. A Robust 8-Gene Prognostic Signature for Early-Stage Non-small Cell Lung Cancer. Front. Oncol. 2019, 9, 693. [Google Scholar] [CrossRef] [Green Version]
- Torres-Roca, J.F. A molecular assay of tumor radiosensitivity: A roadmap towards biology-based personalized radiation therapy. Pers. Med. 2012, 9, 547–557. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.A.; Chinnaiyan, P.; Fulp, W.J.; Eschrich, S.; Torres-Roca, J.F.; Caudell, J.J. The radiosensitivity index predicts for overall survival in glioblastoma. Oncotarget 2015, 6, 34414–34422. [Google Scholar] [CrossRef] [Green Version]
- Fowler, J.F. 21 years of biologically effective dose. Br. J. Radiol. 2010, 83, 554–568. [Google Scholar] [CrossRef]
- Miles, E.F.; Lee, W.R. Hypofractionation for prostate cancer: A critical review. Semin. Radiat. Oncol. 2008, 18, 41–47. [Google Scholar] [CrossRef]
- Ahmed, K.A.; Scott, J.G.; Arrington, J.A.; Naghavi, A.O.; Grass, G.D.; Perez, B.A.; Caudell, J.J.; Berglund, A.E.; Welsh, E.A.; Eschrich, S.A.; et al. Radiosensitivity of Lung Metastases by Primary Histology and Implications for Stereotactic Body Radiation Therapy Using the Genomically Adjusted Radiation Dose. J. Thorac. Oncol. 2018, 13, 1121–1127. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, K.A.; Grass, G.D.; Orman, A.G.; Liveringhouse, C.; Montejo, M.E.; Soliman, H.H.; Han, H.S.; Czerniecki, B.J.; Torres-Roca, J.F.; Diaz, R. Personalizing Radiation Treatment Delivery in the Management of Breast Cancer. Int. J. Breast Cancer 2018, 2018, 6729802. [Google Scholar] [CrossRef] [Green Version]
- Le, Q.-T.; Chen, E.; Salim, A.; Cao, H.; Kong, C.S.; Whyte, R.; Donington, J.; Cannon, W.; Wakelee, H.; Tibshirani, R.; et al. An evaluation of tumor oxygenation and gene expression in patients with early stage non-small cell lung cancers. Clin. Cancer Res. 2006, 12, 1507–1514. [Google Scholar] [CrossRef] [Green Version]
- Pujol, J.-L.; Molinier, O.; Ebert, W.; Daurès, J.-P.; Barlesi, F.; Buccheri, G.; Paesmans, M.; Quoix, E.; Moro-Sibilot, D.; Szturmowicz, M.; et al. CYFRA 21-1 is a prognostic determinant in non-small-cell lung cancer: Results of a meta-analysis in 2063 patients. Br. J. Cancer 2004, 90, 2097–2105. [Google Scholar] [CrossRef]
- Swinson, D.E.; Jones, J.L.; Richardson, D.; Wykoff, C.; Turley, H.; Pastorek, J.; Taub, N.; Harris, A.; O’Byrne, K.J. Carbonic anhydrase IX expression, a novel surrogate marker of tumor hypoxia, is associated with a poor prognosis in non-small-cell lung cancer. J. Clin. Oncol. 2003, 21, 473–482. [Google Scholar] [CrossRef]
- Tumati, V.; Kumar, S.; Yu, L.; Chen, B.; Choy, H.; Saha, D. Effect of PF-02341066 and radiation on non-small cell lung cancer cells. Oncol. Rep. 2013, 29, 1094–1100. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.; Lee, J.-S.; Lee, Y.-S.; Lee, K. Radiosensitivity of Cancer Cells Is Regulated by Translationally Controlled Tumor Protein. Cancers 2019, 11, 386. [Google Scholar] [CrossRef] [Green Version]
- Girdhani, S.; Rodriguez, A.; Dua, R.; Parry, R. Correlation of radiosensitivity of lung carcinoma to CD137L endogenous expression levels. J. Clin. Oncol. 2019, 37, e14229. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | SF2 | SD |
|---|---|---|
| Radioresistant | ||
| H1975 | 0.891 | 0.0104 |
| A549 | 0.832 | 0.162 |
| HC827 | 0.745 | 0.2 |
| H358 | 0.676 | - |
| H2228 | 0.646 | 0.056 |
| H3122 | 0.622 | 0.056 |
| H460 | 0.617 | 0.129 |
| H1650 | 0.570 | 0.156 |
| Radiosensitive | ||
| Calu1 | 0.454 | 0.079 |
| H520 | 0.490 | - |
| H1437 | 0.448 | - |
| H226 | 0.430 | 0.09 |
| Calu3 | 0.325 | 0.131 |
| H2009 | 0.228 | 0.024 |
| H1781 | 0.180 | 0.067 |
| Variables | Beta | ET | Wald | HR | 95% CI for HR | p-Value | |
|---|---|---|---|---|---|---|---|
| Inferior | Superior | ||||||
| Age at diagnosis | −0.25 | 0.27 | 0.88 | 0.78 | 0.46 | 1.32 | 0.35 |
| Year of diagnosis | 0.46 | 0.31 | 2.28 | 1.59 | 0.87 | 2.91 | 0.13 |
| Sex | −0.21 | 0.27 | 0.65 | 0.81 | 0.48 | 1.36 | 0.42 |
| Histological subtype | 0.38 | 0.27 | 1.97 | 1.46 | 0.86 | 2.48 | 0.16 |
| T component | −0.38 | 0.29 | 1.71 | 0.68 | 0.39 | 1.21 | 0.19 |
| Nodal load | −0.24 | 0.27 | 0.80 | 0.78 | 0.46 | 1.34 | 0.37 |
| Tumoral stage (AJCC) | −0.52 | 0.27 | 3.73 | 0.60 | 0.35 | 1.01 | 0.05 |
| Genetic signature OS | 1.10 | 0.28 | 15.16 | 2.99 | 1.72 | 5.19 | <0.0001 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peinado-Serrano, J.; Quintanal-Villalonga, Á.; Muñoz-Galvan, S.; Verdugo-Sivianes, E.M.; Mateos, J.C.; Ortiz-Gordillo, M.J.; Carnero, A. A Six-Gene Prognostic and Predictive Radiotherapy-Based Signature for Early and Locally Advanced Stages in Non-Small-Cell Lung Cancer. Cancers 2022, 14, 2054. https://doi.org/10.3390/cancers14092054
Peinado-Serrano J, Quintanal-Villalonga Á, Muñoz-Galvan S, Verdugo-Sivianes EM, Mateos JC, Ortiz-Gordillo MJ, Carnero A. A Six-Gene Prognostic and Predictive Radiotherapy-Based Signature for Early and Locally Advanced Stages in Non-Small-Cell Lung Cancer. Cancers. 2022; 14(9):2054. https://doi.org/10.3390/cancers14092054
Chicago/Turabian StylePeinado-Serrano, Javier, Álvaro Quintanal-Villalonga, Sandra Muñoz-Galvan, Eva M. Verdugo-Sivianes, Juan C. Mateos, María J. Ortiz-Gordillo, and Amancio Carnero. 2022. "A Six-Gene Prognostic and Predictive Radiotherapy-Based Signature for Early and Locally Advanced Stages in Non-Small-Cell Lung Cancer" Cancers 14, no. 9: 2054. https://doi.org/10.3390/cancers14092054
APA StylePeinado-Serrano, J., Quintanal-Villalonga, Á., Muñoz-Galvan, S., Verdugo-Sivianes, E. M., Mateos, J. C., Ortiz-Gordillo, M. J., & Carnero, A. (2022). A Six-Gene Prognostic and Predictive Radiotherapy-Based Signature for Early and Locally Advanced Stages in Non-Small-Cell Lung Cancer. Cancers, 14(9), 2054. https://doi.org/10.3390/cancers14092054