1. Introduction
Molecular characterization of hematological malignances currently enables a comprehensive diagnostic and prognostic classification [
1] and can even be translated into therapeutic recommendations in some cases [
2]. The number of genetic biomarkers with clinical implications is constantly growing, and the latest World Health Organization (WHO) update for myeloid neoplasms and acute leukemia, published in 2016, already includes a substantial number of them [
3]. The most frequently disrupted genes in each of these pathologies have been reasonably well defined during the last decade. Regarding
de novo acute myeloid leukemia (AML), about 25 genes are recurrently mutated. The most commonly altered gene is
FLT3 (28%), followed by
NPM1 (27%) and
DNMT3A (26%). Among fusions, the
PML-
RARA in-frame rearrangement is identified in up to 9% of all samples, being the most prevalent [
4]. The prevalence of genetic alterations in AML varies in different series, depending on the disease subtype analyzed and the clinical context of the patients included in the studies [
5,
6]. Overall, the most mutated genes in MDS are
TET2 (20–25%),
DNMT3A (12–18%),
ASXL1 (15–25%) and
SF3B1 (20–30%) [
7,
8]; up to 30 genes are commonly altered [
7,
8]. Furthermore, myeloproliferative neoplasms are divided molecularly into two large groups, namely,
BCR-ABL1-positive chronic myeloid leukemia and Philadelphia chromosome-negative neoplasms. In the latter group, the three most clinically relevant genes analyzed in routine clinical care are
JAK2,
CALR and
MPL [
9]. The prevalence of mutations in each one depends on the entity under consideration (polycythema vera, essential thrombocythemia or primary myelofibrosis) [
9]. Finally, in the context of acute lymphoblastic leukemia (ALL), the mutational landscape is subject to the lineage (B or T) as well as to the age of the patients. The most widespread genetic alteration in adulthood ALL is the disruption of
IKZF1 (25–35% of cases), while it is altered in 12–17% of pediatric patients. In children, the
ETV6-RUNX1 fusion is the most common rearrangement (22%), while in adults it is the
BCR-ABL1 fusion (identified in 25% of cases) [
10]. One of the main challenges when studying the molecular profile of these hematological malignancies is the wide range of mandatory routine genetic analysis, including single-nucleotide variants (SNVs), copy number variants (CNVs), chromosomal rearrangements (SVs), as well as other structural changes, such as tandem duplications or variations in cell ploidy [
3,
7,
10,
11,
12,
13,
14]. This requires the performance of multiple techniques, such as the conventional karyotype, FISH, as well as different PCR approaches, some of them specific to each disease. Therefore, a number of different molecular techniques must be implemented in hospitals for an accurate characterization of hematological malignances, which is less cost-effective as the number of recommended biomarkers increases [
15].
Meanwhile, the rapid development of next-generation sequencing (NGS) technologies has boosted their implementation in daily clinical practice. The feasibility of NGS has been proved in different onco-hematological diseases, especially in the field of acute myeloid leukemia [
12]. NGS solutions provide a comprehensive approach to different biomarkers in a single assay. However, the detection of such a large number of genetic biomarkers of different natures is still a technological challenge. One of the main challenges of NGS lies in obtaining a CNV analysis that would correlate with conventional karyotype or SNP array. Numerous efforts are being made in this sense, and the first results are already available [
16].
Beyond obtaining reliable results at an affordable price, it is also important to generate an amount of data manageable by the stakeholders. Following this premise, sequencing the whole genome or exome generates a large amount of information, and most of these data may be misleading or useless for patient management in daily clinical practice.
Certainly, implementing NGS-based gene panels in clinical practice raises several challenges as well. Some of them are inherent to NGS technologies and could be solved with experience and knowledge. However, two particular challenges hamper the implementation of NGS solutions in the daily clinical practice of onco-hematology laboratories. First, the current international guidelines are based on previous experience; therefore, NGS, due to a lack of standardization, is not the main technology recommended for analyzing several biomarkers. Second, there are insufficient cost efficiency studies in this clinical field that would demonstrate the advantage of implementing NGS. Despite these challenges, NGS-based gene panels enable the identification of target genetic alterations in a single sample and in a single assay. Therefore, it may be a useful strategy for molecular diagnosis in onco-hematology that could replace or complement the conventional techniques.
Accordingly, we developed a DNA NGS-based gene panel that focuses on identifying the genetic markers recommended by the main clinical guidelines [
3,
4,
5,
6,
7,
8,
9]. The main objective of the study was to validate the sensitivity, specificity, repeatability and reproducibility of the Haematology OncoKitDx for the detection of clinically actionable variants, as well as to explore its clinical feasibility and utility.
2. Materials and Methods
2.1. Panel Design
The panel targets the main biomarkers for diagnosis, prognosis, therapy and resistance among the most frequent onco-hematological diseases, namely AML, myeloproliferative neoplasm (MPN), myelodysplastic syndrome (MDS) and ALL. It was designed according to the current World Health Organization classification [
3], the ELN (European Leukemia Network) working group recommendations [
11], the NCCN guidelines [
7,
10,
13,
14] and the Food & Drug Administration and European Medicines Agency drugs approval.
The DNA NGS-based panel Haematology OncoKitDx examines the coding regions of 78 genes by complete sequencing, looking for several types of genetic alterations, such as point mutations and small deletions and insertions. The included genes are
ARID5B,
ASXL1,
ASXL2,
ATRX,
BCOR,
BCORL1,
BLNK,
BRAF,
CALR,
CBL,
CDKN2A,
CDKN2B,
CEBPA,
CHIC2,
CREBBP,
CRLF2,
CSF3R,
CSNK1A1,
CUX1,
DDX3X,
DDX41,
DNMT3A,
EP300,
ETNK1,
ETV6,
EZH2,
FBXW7,
FLT3,
GATA1,
GATA2 (intron 4 included),
GATA3,
HAVCR2,
IDH1,
IDH2,
IKZF1,
IL7R,
JAK1,
JAK2,
JAK3,
KIT,
KMT2A,
KMT2C,
KRAS,
MPL,
NF1,
NFE2,
NOTCH1,
NPM1,
NR3C1,
NRAS,
P2RY8,
PAX5,
PHF6,
PIGA,
PPM1D,
PTEN,
PTK2B,
PTPN11,
RAD21,
RB1,
RUNX1,
SETBP1,
SF3B1,
SH2B3,
SMC1A,
SMC3,
SRP72,
SRSF2,
STAG1,
STAG2,
STAT5B,
TET2,
TP53,
TPMT,
TYK2,
U2AF1,
WT1 and
ZRSR2. A low-density SNP array of 996 single-nucleotide polymorphisms (SNPs) located throughout the entire genome was included in the design of the panel, and a custom bioinformatics pipeline was developed with the purpose of detecting CNVs throughout the entire genome, including gain and loss of whole chromosomes, chromosomal arms, genes or even exons. In addition, the panel was also designed to identify rearrangements by detecting the intronic breakpoints previously described in 27 genes:
ABL1,
ABL2,
BCR,
CBFA2T3,
CBFB,
CSF1R,
EPOR,
ETV6,
FGFR1,
FUS,
JAK2,
KMT2A,
MEF2D,
MNX1,
MYH11,
NPM1,
NUP214,
NUP98,
PDGFRA,
PDGFRB,
RARA,
RBM15,
RUNX1,
SET,
STIL,
TAL1 and
TCF3. This strategy makes it possible to detect fusions of these genes with any other partner throughout the genome. The Haematology OncoKitDx also tests for 14 relevant SNPs in pharmacogenetics according to the PharmGKB database (2A evidence or higher) [
17].
2.2. Sample Selection, Preparation, Sequencing, Bioinformatics Pipeline and Variant Classification
A total of 63 samples were selected for the analytical validation of the assay: 9 reference samples from Coriell Cell Repositories (3), Horizon Discovery (2), Incliva Biobank (2) and Agilent Technologies (2); and a total of 54 bone marrow samples from patients. Sensitivity and specificity for point variations and small indels detection were assessed in 19 samples. Of these, 12 samples were employed for CNV evaluation, 19 samples for fusions and large rearrangements analysis, 3 for pharmacogenetics testing and 11 samples for FLT3-ITD detection analysis. Both repeatability and reproducibility evaluation were conducted with samples previously analyzed in this technical validation.
In order to evaluate the assay’s feasibility in the clinical context, 170 bone marrow DNA samples were assessed for SNVs, INDELs, CNVs and fusions. Samples from six Spanish hospitals were selected based on their own scientific or clinical interest. Most samples (153/170) had been previously analyzed with different conventional techniques (RT-qPCR, FISH or karyotype) and/or by means of other commercial NGS panels (Sophia myeloid solution, Oncomine Myeloid Research Assay, Oncomine Childhood Cancer Research Assay or TruSight Myeloid Sequencing Panel). 17 of the 170 samples had not been previously studied by any molecular technique but were also included.
DNA sample concentration was measured by fluorometric quantification using the Qubit dsDNA BR Assay kit (Thermo Fisher Scientific, Waltham, MA, USA) and the Qubit dsDNA HS Assay kit (Invitrogen, Waltham, MA, USA). A dilution with 50–100 ng nuclease-free water in 7 µL was prepared to start the library preparation protocol. Enzymatic fragmentation of 50–100 ng of gDNA from each sample to an average of 175 bases was carried out with the SureSelect XT HS and XT Low input enzymatic fragmentation kit (Agilent). The library preparation was automated by means of the Magnis NGS Prep System and Magnis SureSelect XT HS kit (Agilent). To obtain indexed libraries by molecular barcoding, unique molecular indexes (UMIs) were employed and the Agencourt AMPure XP beads (Beckman Coulter, Brea, CA, USA) were used for purification. Libraries were universally amplified by PCR (9 or 10 cycles, depending on the amount of input DNA) with adapter-specific primers for the union of indexes, universal indexes and sequencing adapters. Indexed DNA fragments were then purified and library quality control was performed on the Agilent 2200 TapeStation. A mix of biotinylated capture probes, specifically designed for the Haematology OncoKitDx, was hybridized to regions of interest and then captured with streptavidin beads. Library enrichment involved post-capture PCR amplification (12 cycles) and purification with beads. Libraries were quantified using the Qubit dsDNA HS Assay kit (Invitrogen). A denaturation protocol was carried out prior to sequencing, using the NextSeq 500/550 Mid Output v2.5 and NextSeq 500/550 High Output v2.5 sequencing kits (Illumina) and Phix control (Illumina). Libraries were diluted to 1.5 pM. Pools were then loaded onto the NextSeq 550 system (Illumina) for massive library sequencing in “Stand-alone” mode with 2 × 75 paired-end reads following the manufacturer’s instructions. The bioinformatics pipeline was set up as previously reported by Martinez-Fernández P et al. [
18].
Classification of the identified variants using the DataGenomics software was done according to their functional and clinical evidence, as described by Martinez-Fernández P et al. [
18].
The assay’s analytical validity was also evaluated by establishing the percentage of samples carrying variants with diagnostic, prognostic and therapeutic implications according to guidelines (tier I variants) and high-impact literature (tier II variants) (Tier I and II biomarkers stated in
File S1). This percentage of samples carrying clinically relevant variants was established based on the identification of SNVs, SVs and gains and losses in target genes, but not on the presence of large CNVs. Large CNVs were considered clinically relevant in all cases. The percentage of clinically relevant variants were grouped into each of the four parameters of interest (diagnosis, prognosis, therapy and resistance), for each neoplasm aims to describe and summarize the collected data in a global way. As the panel includes the most frequently mutated genes and genes with the highest level of clinical evidence for each of the pathologies analyzed, the number of variants with clinical evidence at the diagnostic, prognostic, therapeutic and resistance-to-therapy levels is expected to be high and consistent with previous reports. This description is not intended to create the impression that the panel detects something other than what might be expected, based on the knowledge available on the analyzed pathology; it is simply to illustrate how the biomarkers that are expected to be found may be detected by a single test, in this case, by the NGS test.
2.3. Clinical Feasibility Evaluation
The genetic risk assigned by the Haematology OncoKitDx, according to the ELN-2017 recommendations for AML patients, was compared with the risk originally assigned by its reference hospital after performing a conventional karyotype and a commercial NGS panel. The patients were classified into three categories (Favorable, Intermediate and Adverse) based on the genetic risk according to ELN [
4]. Two extra categories were added (Favorable or Intermediate; Intermediate or Adverse) to classify those cases in which the
FLT3 allelic ratio could not be used to establish the definitive genetic risk group. A Cohen’s Kappa test was performed to establish the correlation between both approaches. This test quantifies the agreement between observers that independently classify the same n units into the same k nominal or ordinal categories. It adjusts the observed proportion of agreement and ranges from −P
c/(1 − P
c) to 1, where P
c is the expected agreement that results from chance. The strength of agreement in the Kappa statistic is <0 Poor; 0–0.2 Slight; 0.2–0.4 Fair; 0.4–0.6 Moderate; 0.6–0.8 Substantial; 0.8–1 Almost Perfect; 1 Perfect [
19]. Regarding myelodysplastic syndromes, the cytogenetic risk, assigned according to the IPSS-R guidelines by means of conventional karyotype, was compared with the cytogenetic risk assigned by the Haematology OncoKitDx panel. The level of agreement was established by Cohen’s Kappa [
19].
Furthermore, we established a diagnosis for AML samples (according to the 2016 WHO classification) based on the Haematology OncoKitDx results. The AML samples were divided into two groups: (1) samples with information about the diagnosis established at their centers or (2) samples referred as AML for which the precise diagnosis according to WHO 2016 established at their hospitals was not provided.
In the first group, the correspondence between the diagnosis of the center and that derived from Hematology OncokitDx was evaluated. The possible results were match or not match.
In the second group of samples, a diagnosis was assigned according to the current WHO classification based on molecular information derived from the Hematology OncoKitDx panel. The agreement between the diagnosis established by the different hospitals and the result of Haematology OncokitDx could not be assessed. Additionally, to evaluate the clinical usefulness of the panel in this group of patients, it was ascertained whether the molecular alterations detected were more consistent with the suspected diagnosis (AML) than with MDS, MPN, MDS/MPN or myeloid/lymphoid neoplasms.
To consider a molecular alteration or profile of alterations to be more consistent with AML versus MDS, MPN, MDS/MPN or myeloid/lymphoid neoplasms, at least one identified mutation had to meet one of these criteria:
- -
One or more research manuscripts from Q1 or Q2 journals (JCR 2021) reporting that the mutation is more common in AML than in other myeloid neoplasms (or it is typical of transformation to AML).
or
- -
One or more clinical guidelines stating that the mutation is more frequent in AML than in other myeloid neoplasms (or it is typical of transformation to AML).
or
- -
One or more reviews from Q1 or Q2 journals (JCR 2021) claiming that one mutation is more usual in AML than in other myeloid neoplasms (or it is typical of transformation to AML).
or
- -
Identifying a mutation that is pathognomonic for AML or exclusively described in AML.
Likewise, for the group of samples referred to as myeloproliferative neoplasms, the ability of the panel to differentiate the neoplasms between those BCR-ABL positive vs. BCR-ABL negative was evaluated. Additionally, in samples negative for BCR-ABL, it was analyzed whether the molecular alterations observed were consistent with MPN or, contrarily, were suggestive of MDS or MPN/MDS syndromes.
Finally, due to the small size of the ALL group cohort, it was decided to report the data with diagnostic and prognostic implications in a descriptive manner. The ability to assign a cytogenetic risk in B-ALL through the Haematology OncoKitDx was assessed as per the NCCN guidelines [
10].
Concerning the germline, a potential constitutional origin of variants with a VAF (variant allelic frequency) between 35% and 100% in the SRP72, CEBPA, DDX41, RUNX1, ETV6, GATA2, SRP72 and TP53 genes was reported. Likewise, possible CNVs in the chromosomal region 14q32.2 were investigated in pursuit of genomic duplications, including both ATG2B and GSKIP genes. We also sought for mutations in other genes with germline implications not, however, included in the WHO 2016 classification, such as NF1 or PTPN11.
4. Discussion
The present work shows the analytical validation and clinical feasibility of the NGS-based panel Haematology OncoKitDx. The panel has been designed to reach an adequate molecular diagnosis, transfer prognostic information and detect alterations that may respond to targeted therapies in patients with hematological neoplasms while examining a single DNA sample. The clinical classification of the variants was adapted to current international guidelines. This panel has been optimized to detect SNVs, rearrangements and CNVs, as well as some polymorphisms related to pharmacogenetics. Due to the relevance of cytogenetic alterations in onco-hematology, the panel was specially designed to detect large CNVs throughout the entire genome. It has been analytically validated on Illumina’s NextSeq550 System platform by analyzing commercial reference DNA samples.
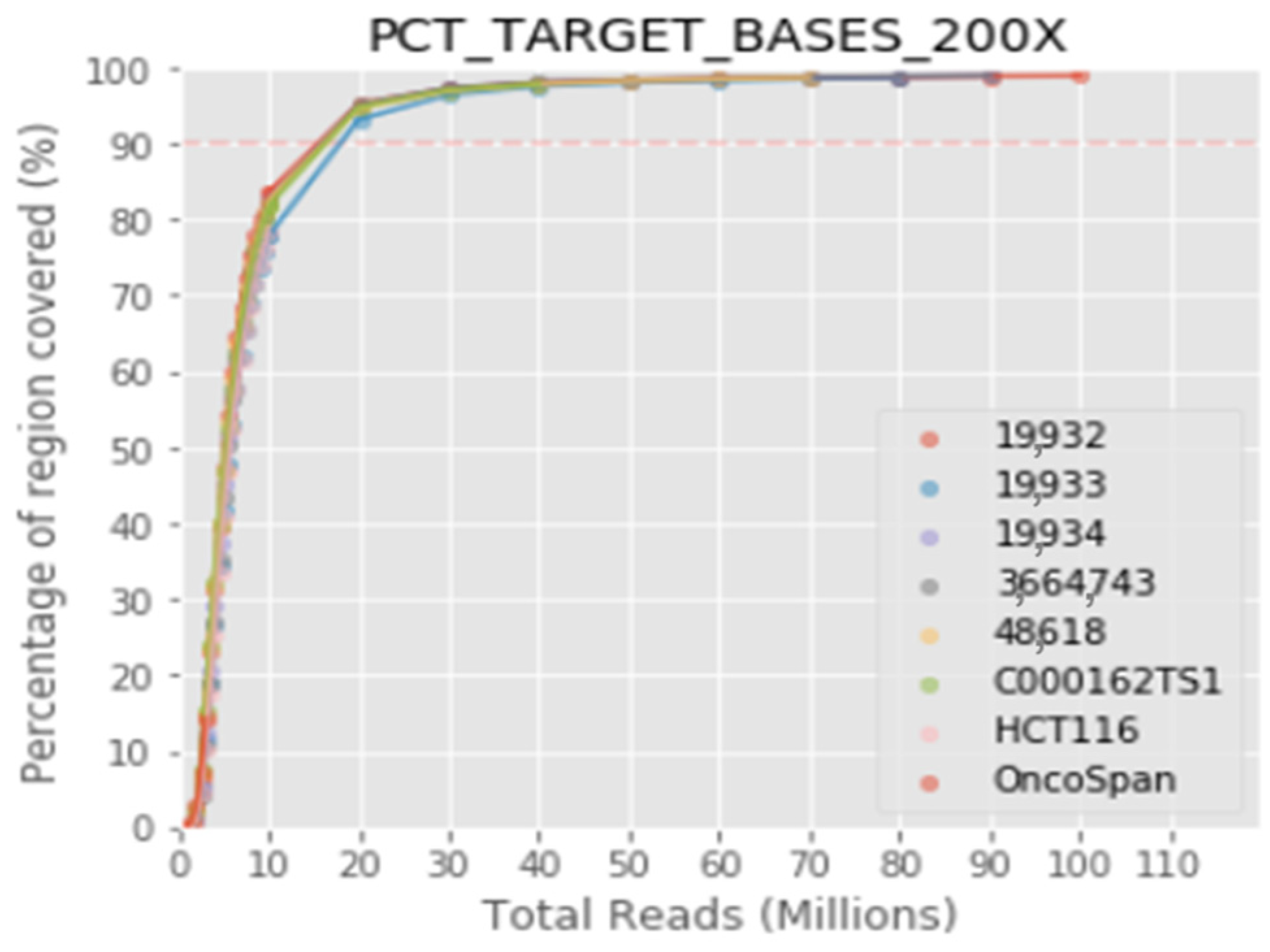
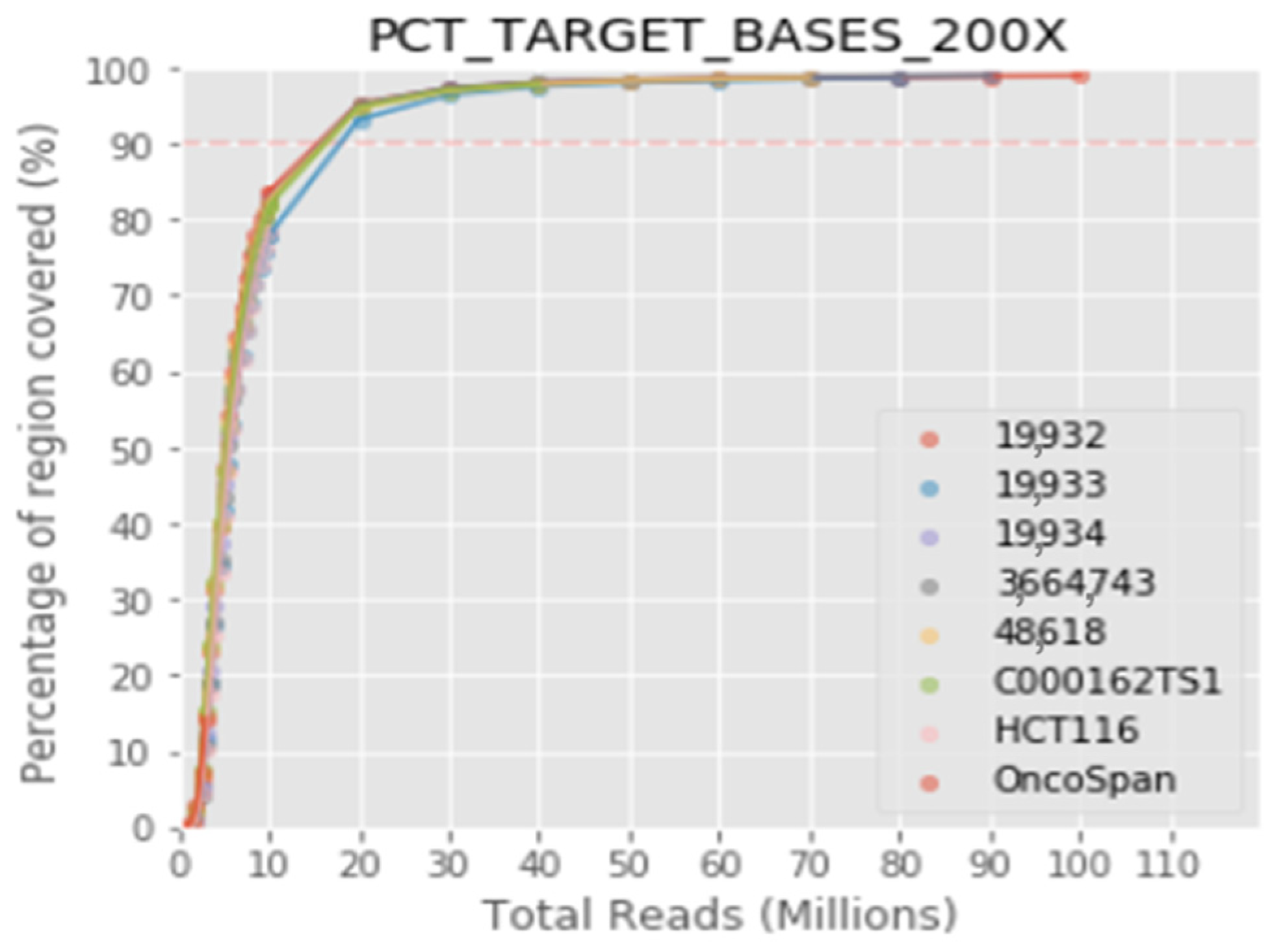
The protocol integrates highly sensitive capture of regions of interest with hybridization probes and molecular barcoding of each DNA fragment with a single adapter for high-performance massive sequencing (NGS). In this validation, the specific detection of the variants previously identified in the target genes has been verified, as well as the repeatability, reproducibility and detection limit of the technique. The results obtained highlight the robustness of the assay, with overall repeatability and reproducibility values greater than 99.9%, specificity more than 99% and sensibility greater than 99%. The limit of detection established for the bioinformatics pipeline was >2% for structural variants and small insertions/duplications. The limit of detection for CNV with respect to the total copies of a sample has been established at 20%. The validated bioinformatics pipeline achieves a mean coverage of 1400× and 99.3% of the target region acquires a 100× read depth.
From an analytical point of view, the robustness of the panel to detect variants with clinical implications has been confirmed. According to the information gathered in the main clinical guidelines, as well as in the previous literature [
3,
4,
5,
6,
7,
8,
9,
10,
13,
14], variants of diagnostic, prognostic, therapeutic and resistance to therapy value were identified in 78.8%, 81.8%, 37.1% and 30% of all patients, respectively. Furthermore, only 6% of all cases did not carry clinically relevant variants. This fact is neither a surprise, nor does it provide a specific scientific value, but it is congruent with the literature available. It simply shows that the main biomarkers included in clinical guidelines and internationally recognized articles can be adequately characterized by means of this NGS-based approach. This description is only intended to engage forward thinking on the cost-effectiveness of analyzing the high number of clinically relevant biomarkers with NGS solutions or several conventional techniques directed at specific markers. It is not intended to draw conclusions, but simply to initiate debate and raise the hypothesis that NGS technology might replace conventional techniques in onco-hematology, if its sensitivity and specificity continue to be demonstrated, the interpretation of the results is standardized and, of course, a favorable cost-effectiveness ratio is demonstrated.
The clinical feasibility of the panel has been first evaluated when classifying AML patients based on the ELN 2017 genetic risk classification. From this point of view, the results obtained in the group of 33 patients are promising. It was possible to assign a genetic risk group in cases where the karyotype could not be obtained and the results were consistent between both strategies (Commercial NGS panel plus Karyotype vs. Haematology OncoKitDx) for the remaining cases. The Cohen’s Kappa test results (K = 0.94) backs the agreement between both approximations. The only non-concordant case was due to a non-detection of two real mutations by the sequencing performed in its hospital. In a preliminary way, the DNA panel Haematology OncoKitDx might be considered a useful tool for the classification of patients by genetic risk group, at least when the karyotype cannot be obtained. However, these promising results should be confirmed in a longer series.
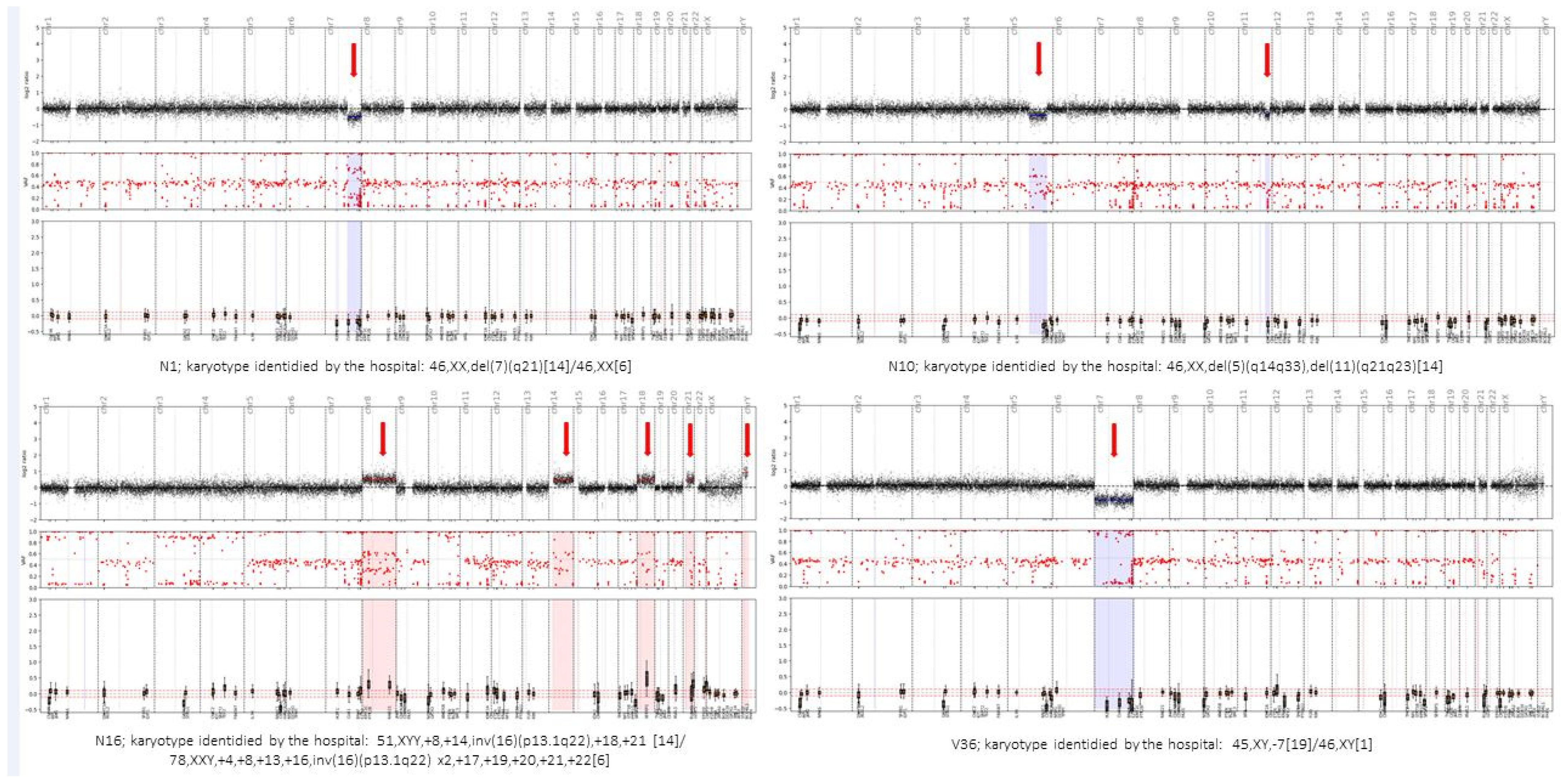
On another note, the Haematology OncoKitDx panel seems to be a reliable and efficient tool for establishing a WHO diagnosis in AML patients. This is supported by the fact that we have established the same WHO diagnosis as the hospitals in 97.4% of the compared AML samples. Meanwhile, to arrive at the final diagnosis, the centers used an NGS panel (with the preparation of two sequencing libraries: DNA + RNA and, at least, a conventional karyotype), while in the present study, we established the diagnosis only based on the results derived from the Haematology OncoKitDx panel. The only discrepancy was due to an event not included in the first version of the panel, which will foreseeably be detected in the second one. Concerning patients with myelodysplastic syndromes, the incidence of mutations in the main genes is consistent with that previously described in the literature. It demonstrates adequate correlation between the conventional karyotype and the CNV plot obtained by the panel to assign an IPSS-R cytogenetic risk group. The Cohen’s Kappa coefficient (K = 0.71) result was positive. However, the discrepancies found in three patients highlight some of the limitations of each of the techniques used. In patient number 17, Haematology OncoKitDx identified two CNVs not reported by the conventional karyotype. The sensitivity to detect CNVs by means of a conventional karyotype is less than the sensitivity achieved by Haematology OncoKitDx (our panel has been validated for detecting CNVs of 0.5 Kb). In order to classify the patients according to IPSS-R cytogenetic risk group, the minimum CNV size counts for the categorization should be established. On the other hand, the balanced translocations observed by conventional karyotype are not detected by means of a CNV plot obtained from next-generation sequencing. These events are detected as gene fusions by NGS techniques; therefore, this fusion must be considered as a cytogenetic event that should be accounted for when assigning an IPSS-R risk group. A limitation of DNA-based NGS would be when the rearrangement observed by conventional karyotype results in a gene fusion that is not among the targets of the panel. Although all clinically significant fusions have been included, some fusions may occur at an unusual breakpoint and not be detected by DNA-based NGS panels. This could be the case with patient number 3. This panel captures ETV6 introns number 2, 3, 4 and 5 (NM_001987). The second version will include introns 6 and 7 as well. Finally, derived chromosomes resulting from unbalanced chromosomal events are accounted by conventional cytogenetics as a single event. However, in some cases, such as that of patient number 6, there is a gain and a loss of material from the same chromosome. Therefore, two events would be counted per NGS, while conventional karyotype would count it only as one. This may not affect the assignment of the risk group according to the IPSS-R, but it could affect other classification systems.
The analysis performed in the myeloproliferative neoplasms cohort highlights the importance of performing NGS studies for an accurate diagnosis of the different myeloid neoplasms. Since the detection of FIP1L1-PDGFRA, identified in two of the ten patients negative for BCR-ABL1, may not be detected by conventional cytogenetics, other techniques are usually essential (reverse transcriptase-polymerase chain reaction [RT-PCR], with nested RT or FISH analysis) (WHO classification). However, NGS technology may be a good strategy to test for the four most important alterations in myeloproliferative neoplasms (namely JAK2, CALR, MPL mutations and BCR-ABL1 fusions), together with biomarkers diagnostic of myeloid/lymphoid neoplasms with rearrangements of PDGFRA, PDGFRB, FGFR1, and PCM1-JAK2 or inherent to myelodysplastic/myeloproliferative neoplasms (MDS/MPN) diseases. By integrating all the information derived from NGS panels, a more precise diagnostic characterization is expected to be obtained in myeloid neoplasms.
Regarding acute lymphoblastic leukemia, although the series included in the validation was small, the validation has once again revealed the possibility of integrating the detection of point mutations, fusions, and CNVs of both single gene (IKZF1) or certain locus (CDKN2A/B) and large chromosomal aberrations (ploidy). This strategy has made it possible to analyze the main biomarkers with diagnostic and prognostic implications in ALL through a single analysis.
The germline implications of some of the target genes make it necessary to consider the possible constitutional origin of a large number of variants. In our study, more than 18% of all patients carried at least one variant consistent with a germline origin. The inclusion of these genes in the panel is essential, since they constitute separate entities according to the WHO 2016 classification.


 ,
, 


{kind=link}
{kind=link}
{kind=link}