
Medullary Thyroid Carcinoma Mutational Spectrum Update and Signaling-Type Inference by Transcriptional Profiles: Literature Meta-Analysis and Study of Tumor Samples

 , , , ,
, , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. MTC Sample Collection
2.2. MTC Study Review and Meta-Analysis
2.3. Gene Expression Profiles in Our MTC Series
2.4. Functional Enrichment Analysis
2.5. Immune Genes and Scores
2.6. MEN2B-like and MEN2A-like Signaling Score
2.7. MEN2B-/MEN2A-like Score Validation in an Independent Dataset
2.8. Statistical Analysis
3. Results
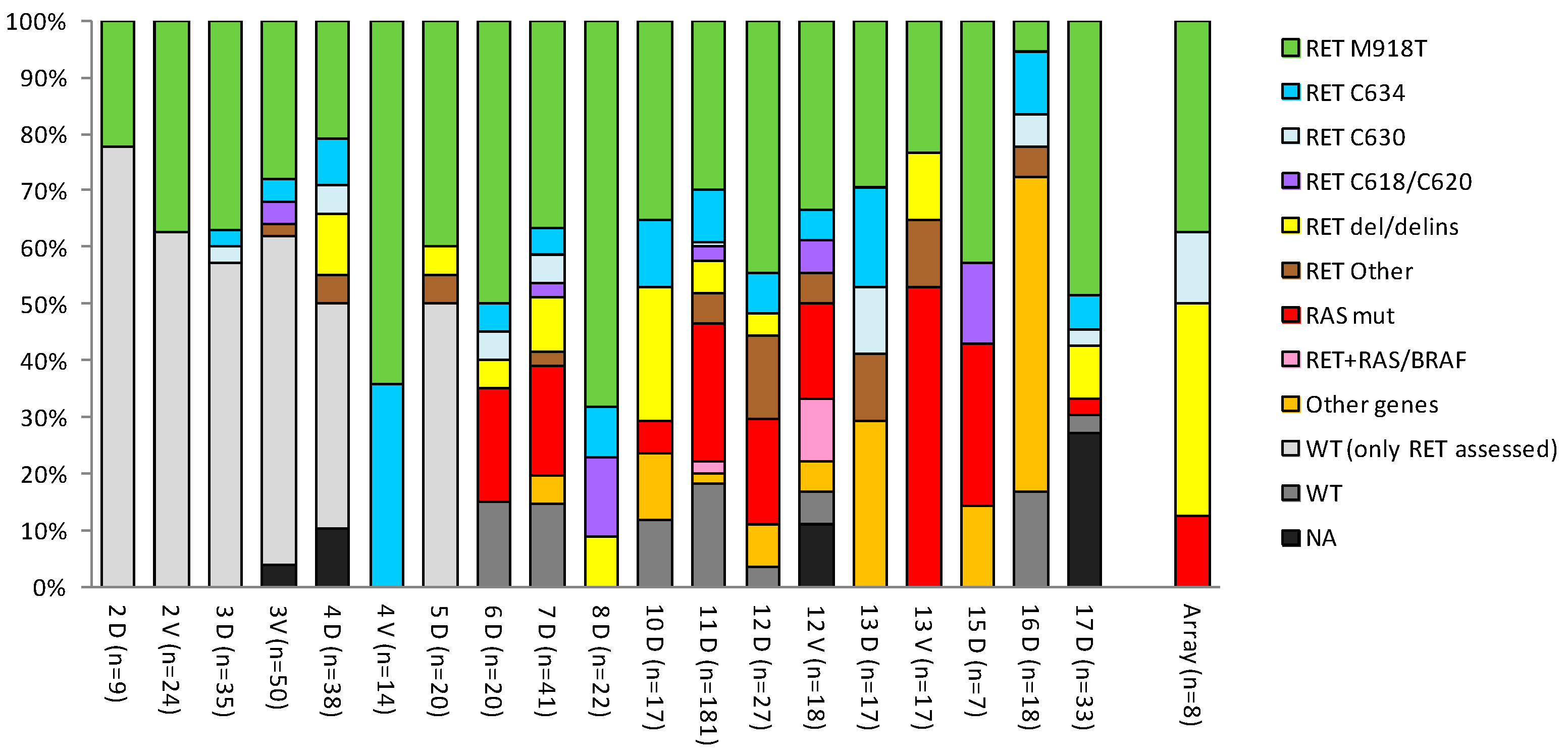
3.1. Overview of High-Throughput Studies in MTC
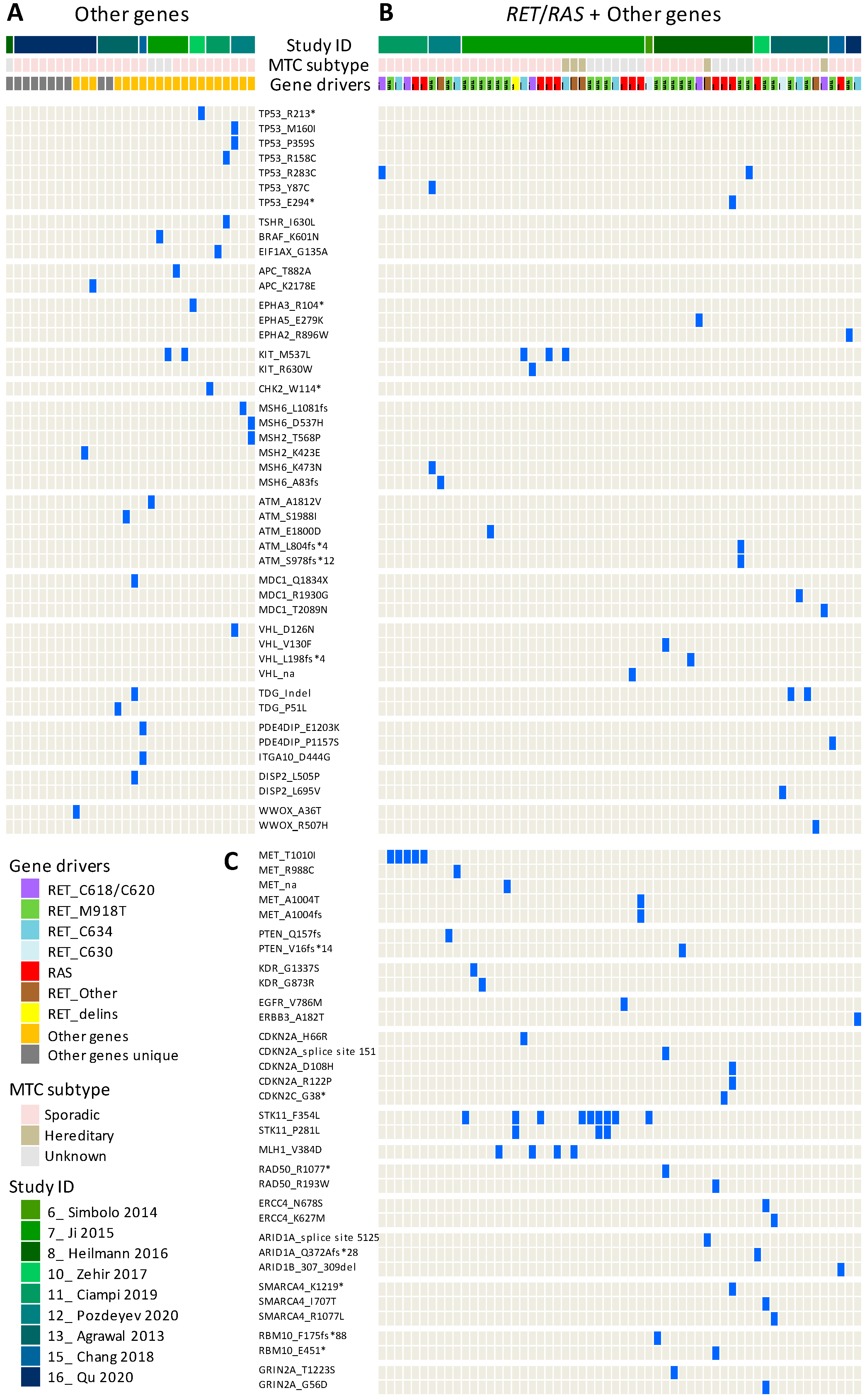
3.2. Meta-Analysis of MTC Gene Drivers Highlights a More Complex Mutational Landscape
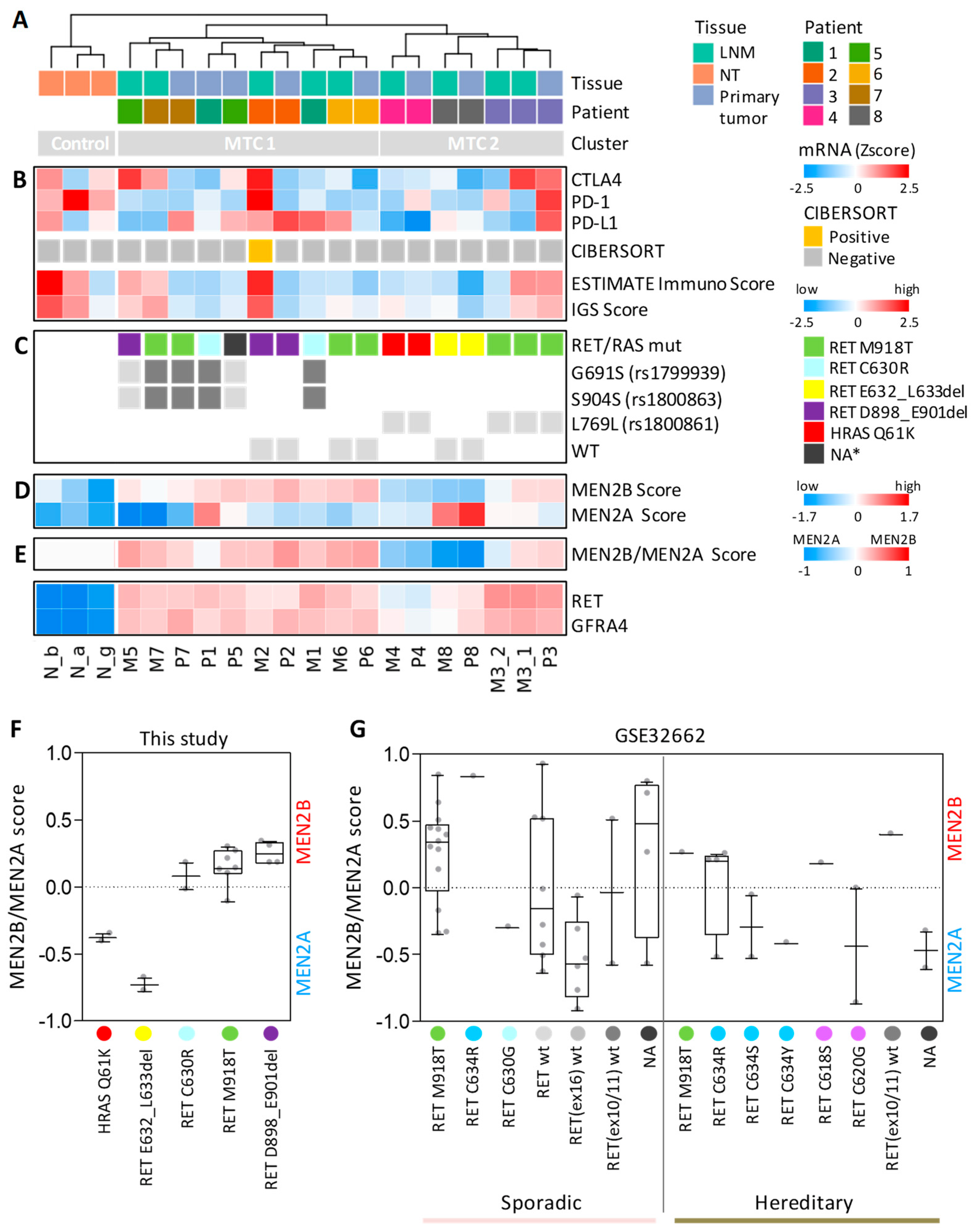
3.3. Gene Expression Profiles in Our Cohort of Sporadic MTCs Identify Specific Sample Stratification and Signaling Pathways
3.4. MTC Signaling Subtypes Are Inferred by an MEN2B/MEN2A-Related Gene Signature
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kondo, T.; Ezzat, S.; Asa, S.L. Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat. Rev. Cancer 2006, 6, 292–306. [Google Scholar] [CrossRef]
- Fagin, J.A.; Wells, S.A., Jr. Biologic and Clinical Perspectives on Thyroid Cancer. N. Engl. J. Med. 2016, 375, 1054–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabanillas, M.E.; McFadden, D.G.; Durante, C. Thyroid cancer. Lancet 2016, 388, 2783–2795. [Google Scholar] [CrossRef]
- Wells, S.A., Jr.; Asa, S.L.; Dralle, H.; Elisei, R.; Evans, D.B.; Gagel, R.F.; Lee, N.; Machens, A.; Moley, J.F.; Pacini, F.; et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015, 25, 567–610. [Google Scholar] [CrossRef] [PubMed]
- Bible, K.C.; Ryder, M. Evolving molecularly targeted therapies for advanced-stage thyroid cancers. Nat. Rev. Clin. Oncol. 2016, 13, 403–416. [Google Scholar] [CrossRef]
- Xing, M. Molecular pathogenesis and mechanisms of thyroid cancer. Nat. Rev. Cancer 2013, 13, 184–199. [Google Scholar] [CrossRef]
- Elisei, R.; Bottici, V.; Cappagli, V.; Ramone, T.; Tacito, A.; Ciampi, R.; Romei, C. Clinical utility of genetic diagnosis for sporadic and hereditary medullary thyroid carcinoma. Ann. Endocrinol. 2019, 80, 187–190. [Google Scholar] [CrossRef]
- Barletta, J.A.; Nosé, V.; Sadow, P.M. Genomics and Epigenomics of Medullary Thyroid Carcinoma: From Sporadic Disease to Familial Manifestations. Endocr. Pathol. 2021, 32, 35–43. [Google Scholar] [CrossRef]
- Romei, C.; Ciampi, R.; Elisei, R. A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat. Rev. Endocrinol. 2016, 12, 192–202. [Google Scholar] [CrossRef]
- Mulligan, L.M. RET revisited: Expanding the oncogenic portfolio. Nat. Rev. Cancer 2014, 14, 173–186. [Google Scholar] [CrossRef]
- Ciampi, R.; Romei, C.; Ramone, T.; Prete, A.; Tacito, A.; Cappagli, V.; Bottici, V.; Viola, D.; Torregrossa, L.; Ugolini, C.; et al. Genetic Landscape of Somatic Mutations in a Large Cohort of Sporadic Medullary Thyroid Carcinomas Studied by Next-Generation Targeted Sequencing. iScience 2019, 20, 324–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moura, M.M.; Cavaco, B.M.; Pinto, A.E.; Leite, V. High prevalence of RAS mutations in RET-negative sporadic medullary thyroid carcinomas. J. Clin. Endocrinol. Metab. 2011, 96, E863–E868. [Google Scholar] [CrossRef] [Green Version]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Oczko-Wojciechowska, M.; Czarniecka, A.; Gawlik, T.; Jarzab, B.; Krajewska, J. Current status of the prognostic molecular markers in medullary thyroid carcinoma. Endocr. Connect. 2020, 9, R251–R263. [Google Scholar] [CrossRef] [PubMed]
- Romeo, P.; Colombo, C.; Granata, R.; Calareso, G.; Gualeni, A.V.; Dugo, M.; De, C.L.; Rizzetti, M.G.; Zanframundo, A.; Aiello, A.; et al. Circulating miR-375 as a novel prognostic marker for metastatic medullary thyroid cancer patients. Endocr. Relat. Cancer 2018, 25, 217–231. [Google Scholar] [CrossRef] [Green Version]
- Ye, L.; Santarpia, L.; Cote, G.J.; El-Naggar, A.K.; Gagel, R.F. High resolution array-comparative genomic hybridization profiling reveals deoxyribonucleic acid copy number alterations associated with medullary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2008, 93, 4367–4372. [Google Scholar] [CrossRef] [Green Version]
- Iñiguez-Ariza, N.M.; Jasim, S.; Ryder, M.M.; Chintakuntlawar, A.V.; Morris, J.C.; Hilger, C.R.; Menefee, M.E.; Smallridge, R.C.; Karlin, N.J.; Alcaino, C.; et al. Foundation One Genomic Interrogation of Thyroid Cancers in Patients with Metastatic Disease Requiring Systemic Therapy. J. Clin. Endocrinol. Metab. 2020, 105, e2346–e2357. [Google Scholar] [CrossRef]
- Nikiforova, M.N.; Wald, A.I.; Roy, S.; Durso, M.B.; Nikiforov, Y.E. Targeted next-generation sequencing panel (ThyroSeq) for detection of mutations in thyroid cancer. J. Clin. Endocrinol. Metab. 2013, 98, E1852–E1860. [Google Scholar] [CrossRef] [Green Version]
- Du, P.; Kibbe, W.A.; Lin, S.M. lumi: A pipeline for processing Illumina microarray. Bioinformatics 2008, 24, 1547–1548. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.A.; Cai, C.; Langfelder, P.; Geschwind, D.H.; Kurian, S.M.; Salomon, D.R.; Horvath, S. Strategies for aggregating gene expression data: The collapseRows R function. BMC Bioinform. 2011, 12, 322. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdòttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martà nez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Treviño, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Pozdeyev, N.; Erickson, T.A.; Zhang, L.; Ellison, K.; Rivard, C.J.; Sams, S.; Hirsch, F.R.; Haugen, B.R.; French, J.D. Comprehensive Immune Profiling of Medullary Thyroid Cancer. Thyroid 2020, 30, 1263–1279. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Watson, M.A.; DeBenedetti, M.K.; Hiraki, Y.; Moley, J.F.; Milbrandt, J. Expression profiles provide insights into early malignant potential and skeletal abnormalities in multiple endocrine neoplasia type 2B syndrome tumors. Cancer Res. 2004, 64, 3907–3913. [Google Scholar] [CrossRef] [Green Version]
- Rusinek, D.; Pfeifer, A.; Cieslicka, M.; Kowalska, M.; Pawlaczek, A.; Krajewska, J.; Szpak-Ulczok, S.; Tyszkiewicz, T.; Halczok, M.; Czarniecka, A.; et al. TERT Promoter Mutations and Their Impact on Gene Expression Profile in Papillary Thyroid Carcinoma. Cancers 2020, 12, 1597. [Google Scholar] [CrossRef]
- Fernandez, N.F.; Gundersen, G.W.; Rahman, A.; Grimes, M.L.; Rikova, K.; Hornbeck, P.; Ma’ayan, A. Clustergrammer, a web-based heatmap visualization and analysis tool for high-dimensional biological data. Sci. Data 2017, 4, 170151. [Google Scholar] [CrossRef]
- Khomtchouk, B.B.; Hennessy, J.R.; Wahlestedt, C. shinyheatmap: Ultra fast low memory heatmap web interface for big data genomics. PLoS ONE 2017, 12, e0176334. [Google Scholar] [CrossRef] [Green Version]
- Maliszewska, A.; Leandro-Garcia, L.J.; Castelblanco, E.; Macià, A.; De Cubas, A.; Goméz-Lòpez, G.; Inglada-Pérez, L.; Álvarez-Escolà, C.; De la Vega, L.; Letòn, R.; et al. Differential gene expression of medullary thyroid carcinoma reveals specific markers associated with genetic conditions. Am. J. Pathol 2013, 182, 350–362. [Google Scholar] [CrossRef]
- Ameur, N.; Lacroix, L.; Roucan, S.; Roux, V.; Broutin, S.; Talbot, M.; Dupuy, C.; Caillou, B.; Schlumberger, M.; Bidart, J.M. Aggressive inherited and sporadic medullary thyroid carcinomas display similar oncogenic pathways. Endocr. Relat. Cancer 2009, 16, 1261–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oczko-Wojciechowska, M.; Swierniak, M.; Krajewska, J.; Kowalska, M.; Kowal, M.; Stokowy, T.; Wojtas, B.; Rusinek, D.; Pawlaczek, A.; Czarniecka, A.; et al. Differences in the transcriptome of medullary thyroid cancer regarding the status and type of RET gene mutations. Sci. Rep. 2017, 7, 42074. [Google Scholar] [CrossRef] [PubMed]
- Simbolo, M.; Mian, C.; Barollo, S.; Fassan, M.; Mafficini, A.; Neves, D.; Scardoni, M.; Pennelli, G.; Rugge, M.; Pelizzo, M.R.; et al. High-throughput mutation profiling improves diagnostic stratification of sporadic medullary thyroid carcinomas. Virchows Arch. 2014, 465, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.H.; Oh, Y.L.; Hong, M.; Yun, J.W.; Lee, H.W.; Kim, D.; Ji, Y.; Kim, D.H.; Park, W.Y.; Shin, H.T.; et al. Identification of Driving ALK Fusion Genes and Genomic Landscape of Medullary Thyroid Cancer. PLoS Genet. 2015, 11, e1005467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heilmann, A.M.; Subbiah, V.; Wang, K.; Sun, J.X.; Elvin, J.A.; Chmielecki, J.; Sherman, S.I.; Murthy, R.; Busaidy, N.L.; Subbiah, I.; et al. Comprehensive Genomic Profiling of Clinically Advanced Medullary Thyroid Carcinoma. Oncology 2016, 90, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Vanden, B.P.; Schrock, A.B.; Anderson, P.M.; Morris, J.C., III; Heilmann, A.M.; Holmes, O.; Wang, K.; Johnson, A.; Waguespack, S.G.; Ou, S.I.; et al. Pediatric, Adolescent, and Young Adult Thyroid Carcinoma Harbors Frequent and Diverse Targetable Genomic Alterations, Including Kinase Fusions. Oncologist 2017, 22, 255–263. [Google Scholar] [CrossRef] [Green Version]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Agrawal, N.; Jiao, Y.; Sausen, M.; Leary, R.; Bettegowda, C.; Roberts, N.J.; Bhan, S.; Ho, A.S.; Khan, Z.; Bishop, J.; et al. Exomic sequencing of medullary thyroid cancer reveals dominant and mutually exclusive oncogenic mutations in RET and RAS. J. Clin. Endocrinol. Metab. 2013, 98, E364–E369. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Li, L.; Ye, L.; Jiang, X.; Shen, L.; Gao, Z.; Fang, W.; Huang, F.; Su, T.; Zhou, Y.; et al. Exome sequencing reveals mutant genes with low penetrance involved in MEN2A-associated tumorigenesis. Endocr. Relat. Cancer 2015, 22, 23–33. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.S.; Chang, C.C.; Huang, H.Y.; Lin, C.Y.; Yeh, K.T.; Chang, J.G. Detection of Molecular Alterations in Taiwanese Patients with Medullary Thyroid Cancer Using Whole-Exome Sequencing. Endocr. Pathol. 2018, 29, 324–331. [Google Scholar] [CrossRef]
- Qu, N.; Shi, X.; Zhao, J.J.; Guan, H.; Zhang, T.T.; Wen, S.S.; Liao, T.; Hu, J.Q.; Liu, W.Y.; Wang, Y.L.; et al. Genomic and Transcriptomic Characterization of Sporadic Medullary Thyroid Carcinoma. Thyroid 2020, 30, 1025–1036. [Google Scholar] [CrossRef] [PubMed]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozdeyev, N.; Gay, L.; Sokol, E.S.; Hartmaier, R.J.; Deaver, K.E.; Davis, S.N.; French, J.D.; Vanden, B.P.; LaBarbera, D.V.; Tan, A.C.; et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, B.; Fuchs, T.; Dogan, S.; Landa, I.; Katabi, N.; Fagin, J.A.; Tuttle, R.M.; Sherman, E.; Gill, A.J.; Ghossein, R. Dissecting Anaplastic Thyroid Carcinoma: A Comprehensive Clinical, Histologic, Immunophenotypic, and Molecular Study of 360 Cases. Thyroid 2020, 30, 1505–1517. [Google Scholar] [CrossRef] [PubMed]
- Tarabichi, M.; Saiselet, M.; Tresallet, C.; Hoang, C.; Larsimont, D.; Andry, G.; Maenhaut, C.; Detours, V. Revisiting the transcriptional analysis of primary tumours and associated nodal metastases with enhanced biological and statistical controls: Application to thyroid cancer. Br. J. Cancer 2015, 112, 1665–1674. [Google Scholar] [CrossRef] [Green Version]
- Saiselet, M.; Gacquer, D.; Spinette, A.; Craciun, L.; Caussin-Petrucci, M.; Andry, G.; Detours, V.; Maenhaut, C. New global analysis of the microRNA transcriptome of primary tumors and lymph node metastases of papillary thyroid cancer. BMC Genom. 2015, 16, 828. [Google Scholar] [CrossRef]
- Colombo, C.; Minna, E.; Gargiuli, C.; Muzza, M.; Dugo, M.; De, C.L.; Pogliaghi, G.; Tosi, D.; Bulfamante, G.; Greco, A.; et al. The molecular and gene/miRNA expression profiles of radioiodine resistant papillary thyroid cancer. J. Exp. Clin. Cancer Res. 2020, 39, 245. [Google Scholar] [CrossRef]
- Minna, E.; Department of Research, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy. MAB21L2 gene expression in normal thyroid vs. follicular cell derived thyroid cancer. unpublished data. 2021. [Google Scholar]
- The Cancer Genome Atlas Research Network. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Bongiovanni, M.; Rebecchini, C.; Saglietti, C.; Bulliard, J.L.; Marino, L.; de Leval, L.; Sykiotis, G.P. Very low expression of PD-L1 in medullary thyroid carcinoma. Endocr. Relat. Cancer 2017, 24, L35–L38. [Google Scholar] [CrossRef]
- Shi, X.; Li, C.W.; Tan, L.C.; Wen, S.S.; Liao, T.; Zhang, Y.; Chen, T.Z.; Ma, B.; Yu, P.C.; Lu, Z.W.; et al. Immune Co-inhibitory Receptors PD-1, CTLA-4, TIM-3, LAG-3, and TIGIT in Medullary Thyroid Cancers: A Large Cohort Study. J. Clin. Endocrinol. Metab. 2021, 106, 120–132. [Google Scholar] [CrossRef]
- Aiello, A.; Department of Pathology, Fondazione IRCCS Istituto Nazionale dei Tumori, Milan, Italy. RET exon16 Sanger sequencing in patient matched primary and lymph node metastatic MTC tissues. unpublished data. 2021. [Google Scholar]
- Musholt, T.J.; Hanack, J.; Brehm, C.; von Wasielewsk, R.; Musholt, P.B. Searching for non-RET molecular alterations in medullary thyroid carcinoma: Expression analysis by mRNA differential display. World J. Surg. 2005, 29, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Moerch, U.; Nielsen, H.S.; Lundsgaard, D.; Oleksiewicz, M.B. Flow sorting from organ material by intracellular markers. Cytom. Part A J. Int. Soc. Anal. Cytol. 2007, 71, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Method of the Year 2020: Spatially resolved transcriptomics. Nat. Methods 2021, 18, 9–14. [CrossRef] [PubMed]
- Machens, A.; Frank-Raue, K.; Lorenz, K.; Rondot, S.; Raue, F.; Dralle, H. Clinical relevance of RET variants G691S, L769L, S836S and S904S to sporadic medullary thyroid cancer. Clin. Endocrinol. 2012, 76, 691–697. [Google Scholar] [CrossRef]
- Skalniak, A.; Trofimiuk-Müldner, M.; Przybylik-Mazurek, E.; Hubalewska-Dydejczyk, A. Modifier Role of Common RET Variants in Sporadic Medullary Thyroid Carcinoma. Int. J. Mol. Sci. 2021, 22, 11794. [Google Scholar] [CrossRef]
- Colombo, C.; Minna, E.; Rizzetti, M.G.; Romeo, P.; Lecis, D.; Persani, L.; Mondellini, P.; Pierotti, M.A.; Greco, A.; Fugazzola, L.; et al. The modifier role of RET-G691S polymorphism in hereditary medullary thyroid carcinoma: Functional characterization and expression/penetrance studies. Orphanet. J. Rare Dis. 2015, 10, 25. [Google Scholar] [CrossRef] [Green Version]
- Lantieri, F.; Caroli, F.; Ceccherini, I.; Griseri, P. The involvement of the RET variant G691S in medullary thyroid carcinoma: Conflicting results of meta-analyses need to be reconciled. Int. J. Cancer 2013, 133, 2808–2819. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Reference | Approach | Platform | Subtype * | Specimen * | Tissue * | Patient (n) * | Sample (n) * | Data Deposition ** |
|---|---|---|---|---|---|---|---|---|---|
| 1 | Jain 2004 [26] | Microarray | Hg-U95Av2 GeneChips (Affymetrix) | H | Frozen | P/M | 19 | 25 1 | No |
| 2 | Ameur 2009 [31] | Microarray | Custom-designed (Agilent Technologies) | S/H | Frozen | P | 13 | 13 | Yes (arrayexpress) 2 |
| 3 | Maliszewska 2013 [30] | Microarray | Whole Human Genome Array 4 × 44K (Agilent Technologies) | S/H | Frozen | MTC 3 | 49 | 52 4 | Yes (GSE32662) |
| 4 | Oczko-W. 2017 [32] | Microarray | GeneChip Gene 1.0 ST arrays (Affymetrix) | S/H | Frozen | MTC 3 | 60 | 60 | No |
| 5 | Ye 2008 [16] | CNA | Human Genome hybridization 244K platform (Agilent Technologies) | S/H | Frozen | P | 30 | 30 | No |
| 6 | Simbolo 2014 [33] | Targeted NGS | Ion AmpliSeq Cancer Hotspot Panel v2 (Life Technologies) 5 | S | FFPE | MTC 3 | 20 | 20 | No |
| 7 | Ji 2015 [34] | Targeted NGS | Ion AmpliSeq Cancer Hotspot Panel v2 (Life Technologies) 5 | S/H/U 6 | FFPE | MTC 3 | 84 | 84 | No |
| 8 | Heilmann 2016 [35] | Targeted NGS | FoundationOne panel 7 | S/H/U 8 | FFPE | P/M 8 | 34 | 34 | No |
| 9 | Vanden Borre 2017 [36] | Targeted NGS | FoundationOne panel 7 | U 9 | FFPE | P/M 9 | 14 | 14 | No |
| 10 | Zehir 2017 [37] | Targeted NGS | MSK-IMPACT panel 10 | S 11 | FFPE | P/M 11 | 17 | 17 | Yes (cBioportal) 10 |
| 11 | Ciampi 2019 [11] | Targeted NGS | Custom panel (Thermo Fisher) 12 | S | Frozen/FFPE | P/M 13 | 181 | 181 | Yes (COSP47106) 12 |
| 12 | Pozdeyev 2020 [25] | Targeted NGS; RNAseq | ThyroSeq v3 14, TruSEQ RNA Exome (Illumina) | S/H | FFPE | P/M 15 | 27 | 3015 | No |
| 13 | Agrawal 2013 [38] | WES | SureSelect paired-end v2.0 human exome (Agilent Technologies) | S | Frozen | MTC 3 | 17 | 17 | No |
| 14 | Cai 2015 [39] | WES | 44M human exome array (NimbleGenEZ) | H | Frozen | MTC 3 | 4 | 4 | No |
| 15 | Chang 2018 [40] | WES | TruSeq Exome (Illumina) | S | NA | MTC 3 | 7 | 7 | No |
| 16 | Qu 2020 [41] | WES; RNAseq | AI whole Exome CNV (iGeneTech); VAHTS RNA-Seq Library (Vazyme) 16 | S | Frozen | P | 18 | 18 | No |
| 17 | This study | Microarray | HumanHT-12 WG-DASL V4.0 (Illumina) | S | FFPE | P/M | 11 | 20 17 | Yes (GSE196264) |
| Patient ID | Sex | Age (at dx) | TNM | Stage | ETE | Array ID (Primary Tumor) | Array ID (LNM) | RET/RAS Mutation | RET Polymorphisms |
|---|---|---|---|---|---|---|---|---|---|
| 1 | M | 61 | pT1aN1a | III | no | P 1 | M 1 | RET C630R | rs1799939; rs1800863 ** |
| 2 | M | 60 | pT3N1b | IVa | yes | P 2 | M 2 | RET D898_E901del a | wt |
| 3 | M | 56 | pT2mN1b | IVa | no | P 3 | M 3.1; M 3.2 * | RET M918T b | rs1800861 |
| 4 | F | 52 | pT4N1b | IVa | yes | P 4 | M 4 | HRAS Q61K | rs1800861 |
| 5 | M | 50 | pT3mN1b | IVa | yes | P 5 | M 5 * | RET D898_E901del c | rs1799939; rs1800863 |
| 6 | M | 28 | pT3mN1b | IVa | yes | P 6 | M 6 | RET M918T | wt |
| 7 | F | 49 | pT3NX | III | yes | P 7 | M 7 * | RET M918T | rs1799939; rs1800863 ** |
| 8 | M | 41 | pT3N1b | IVa | no | P 8 | M 8 | RET E632_L633del | wt |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minna, E.; Romeo, P.; Dugo, M.; De Cecco, L.; Aiello, A.; Pistore, F.; Carenzo, A.; Greco, A.; Borrello, M.G. Medullary Thyroid Carcinoma Mutational Spectrum Update and Signaling-Type Inference by Transcriptional Profiles: Literature Meta-Analysis and Study of Tumor Samples. Cancers 2022, 14, 1951. https://doi.org/10.3390/cancers14081951
Minna E, Romeo P, Dugo M, De Cecco L, Aiello A, Pistore F, Carenzo A, Greco A, Borrello MG. Medullary Thyroid Carcinoma Mutational Spectrum Update and Signaling-Type Inference by Transcriptional Profiles: Literature Meta-Analysis and Study of Tumor Samples. Cancers. 2022; 14(8):1951. https://doi.org/10.3390/cancers14081951
Chicago/Turabian StyleMinna, Emanuela, Paola Romeo, Matteo Dugo, Loris De Cecco, Antonella Aiello, Federico Pistore, Andrea Carenzo, Angela Greco, and Maria Grazia Borrello. 2022. "Medullary Thyroid Carcinoma Mutational Spectrum Update and Signaling-Type Inference by Transcriptional Profiles: Literature Meta-Analysis and Study of Tumor Samples" Cancers 14, no. 8: 1951. https://doi.org/10.3390/cancers14081951
APA StyleMinna, E., Romeo, P., Dugo, M., De Cecco, L., Aiello, A., Pistore, F., Carenzo, A., Greco, A., & Borrello, M. G. (2022). Medullary Thyroid Carcinoma Mutational Spectrum Update and Signaling-Type Inference by Transcriptional Profiles: Literature Meta-Analysis and Study of Tumor Samples. Cancers, 14(8), 1951. https://doi.org/10.3390/cancers14081951