
The Role of Myeloid Cells in Hepatotoxicity Related to Cancer Immunotherapy


Abstract
:Simple Summary
Abstract
1. Introduction
2. Liver Function during Homeostasis
Mechanisms of Liver Immune Tolerance
3. Hepatoxicity Related to Cancer Immunotherapies
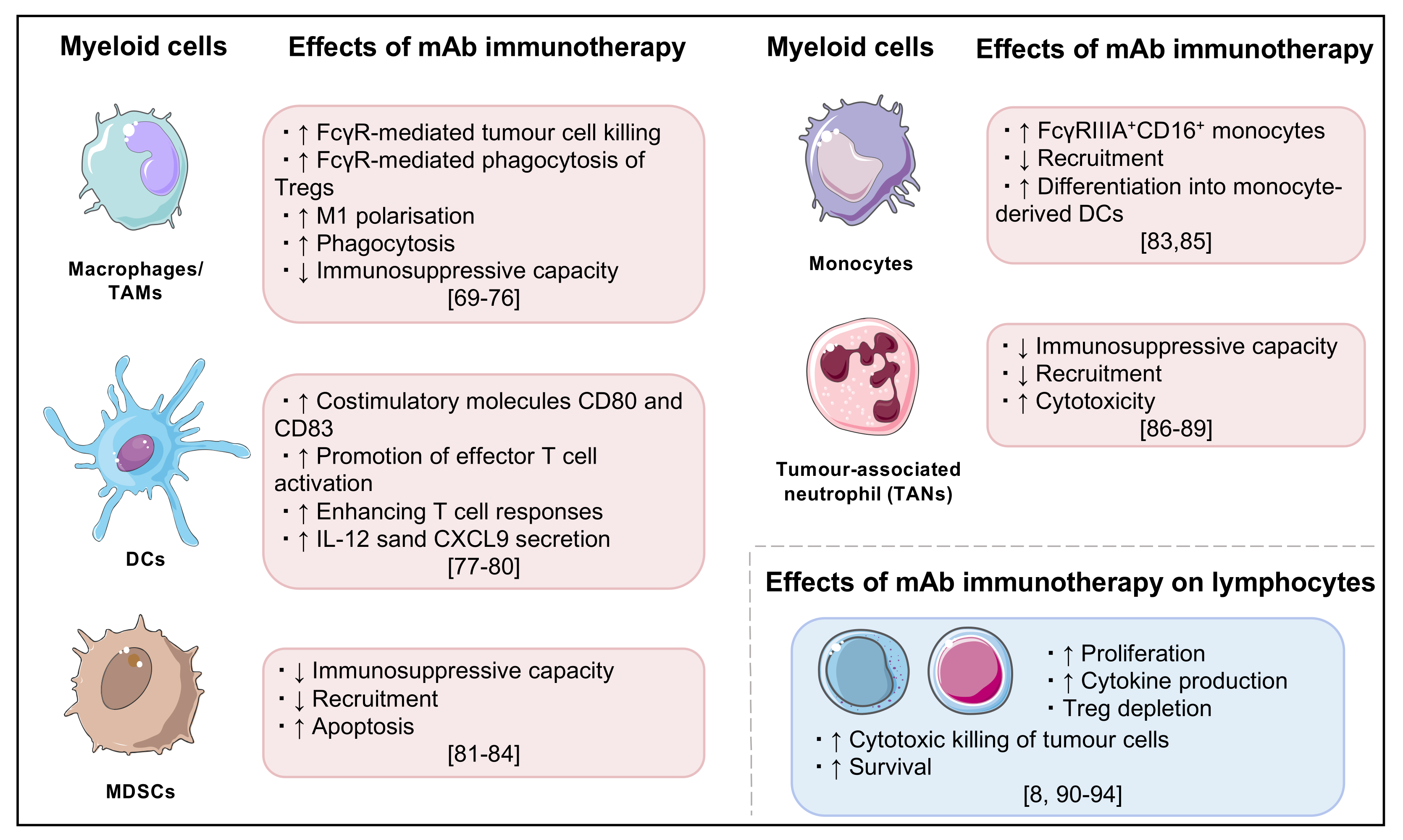
3.1. Breaking Hepatic Tolerance
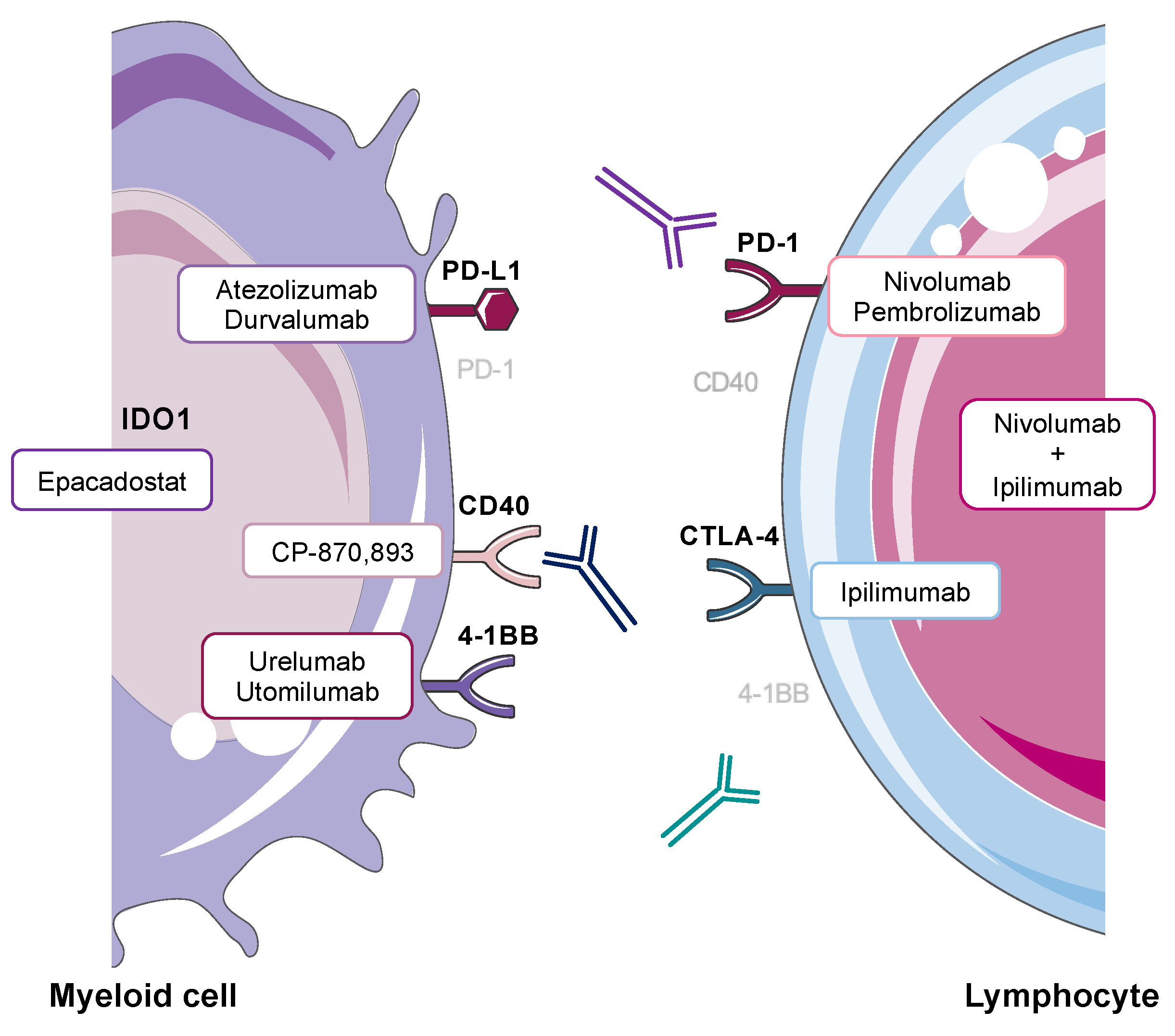
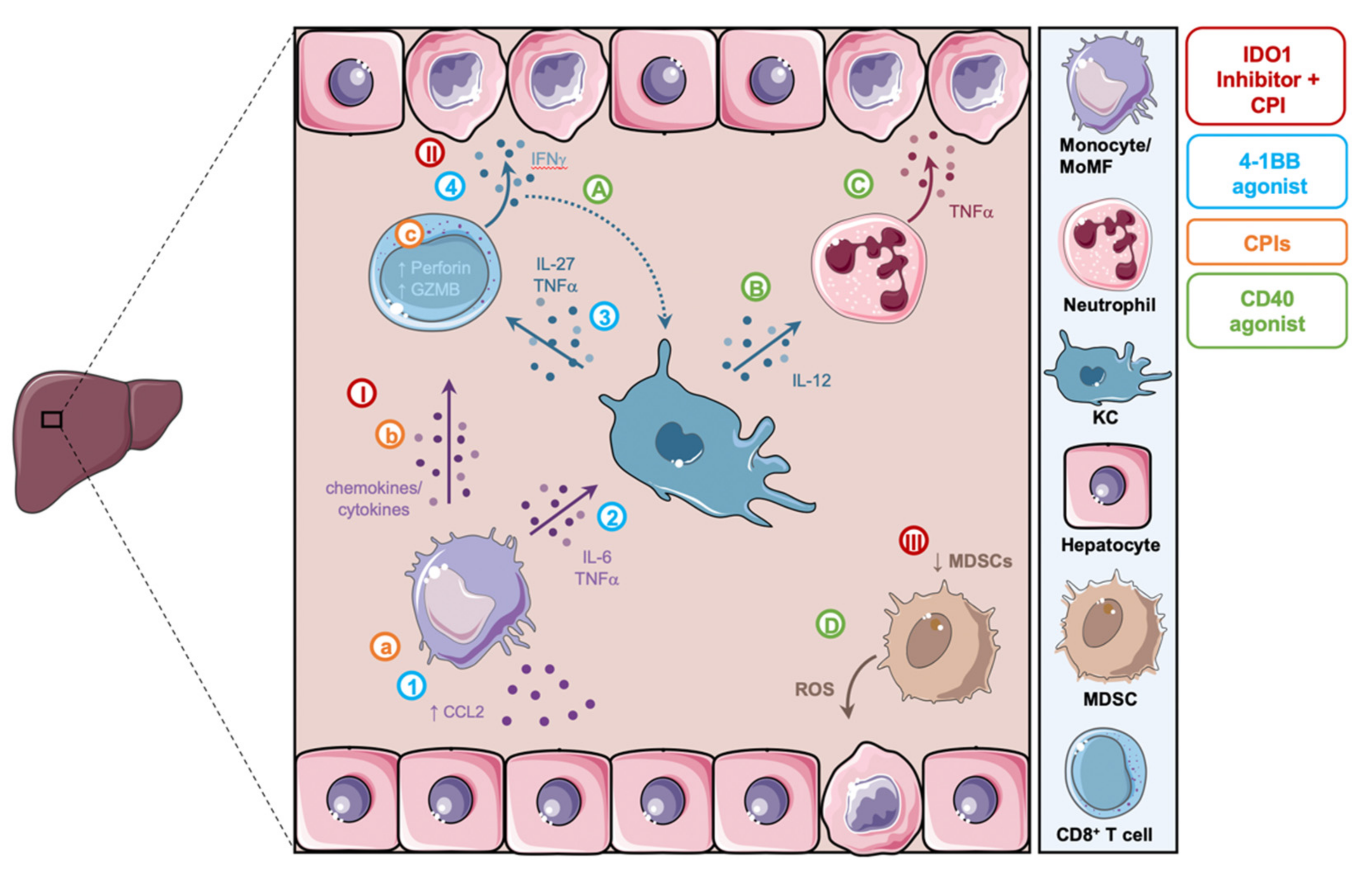
3.2. Checkpoint Inhibitors (CPIs)
3.3. Agonistic Anti-CD40
3.4. 4-1BB Activation
3.5. Combined Agents
3.5.1. Checkpoint Inhibitor Combination Therapy
3.5.2. Small Molecule Indoleamine 2,3-Dioxygenase 1 (IDO1) Inhibitors and CPIs
3.5.3. Immunotherapy in Combination with Conventional Chemotherapeutics
3.6. In Vivo Experimental Models of Immune-Related Adverse Events
4. Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.-J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-Year Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolchok, J.D.; Chiarion-Sileni, V.; Gonzalez, R.; Rutkowski, P.; Grob, J.-J.; Cowey, C.L.; Lao, C.D.; Wagstaff, J.; Schadendorf, D.; Ferrucci, P.F.; et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2017, 377, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Rüter, J.; Antonia, S.J.; Burris, H.A.; Huhn, R.D.; Vonderheide, R.H. Immune modulation with weekly dosing of an agonist CD40 antibody in a phase I study of patients with advanced solid tumors. Cancer Biol. Ther. 2010, 10, 983–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, K.; Wu, Y.-H.; Song, Y.; Yu, B. Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J. Hematol. Oncol. 2021, 14, 68. [Google Scholar] [CrossRef]
- Segal, N.H.; Logan, T.F.; Hodi, F.S.; McDermott, D.; Melero, I.; Hamid, O.; Schmidt, H.; Robert, C.; Chiarion-Sileni, V.; Ascierto, P.A.; et al. Results from an Integrated Safety Analysis of Urelumab, an Agonist Anti-CD137 Monoclonal Antibody. Clin. Cancer Res. 2017, 23, 1929–1936. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Wahab, N.; Shah, M.; Suarez-Almazor, M.E. Adverse Events Associated with Immune Checkpoint Blockade in Patients with Cancer: A Systematic Review of Case Reports. PLoS ONE 2016, 11, e0160221. [Google Scholar] [CrossRef]
- Fessas, P.; Possamai, L.A.; Clark, J.; Daniels, E.; Gudd, C.; Mullish, B.H.; Alexander, J.L.; Pinato, D.J. Immunotoxicity from checkpoint inhibitor therapy: Clinical features and underlying mechanisms. Immunology 2020, 159, 167–177. [Google Scholar] [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. N. Engl. J. Med. 2018, 378, 158–168. [Google Scholar] [CrossRef]
- Kottschade, L.A. Incidence and Management of Immune-Related Adverse Events in Patients Undergoing Treatment with Immune Checkpoint Inhibitors. Curr. Oncol. Rep. 2018, 20, 24. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Flaherty, K.T.; Khalil, M.; Stumacher, M.S.; Bajor, D.L.; Hutnick, N.A.; Sullivan, P.; Mahany, J.J.; Gallagher, M.; Kramer, A.; et al. Clinical Activity and Immune Modulation in Cancer Patients Treated With CP-870,893, a Novel CD40 Agonist Monoclonal Antibody. J. Clin. Oncol. 2007, 25, 876–883. [Google Scholar] [CrossRef]
- Haanen, J.B.A.G.; Carbonnel, F.; Robert, C.; Kerr, K.M.; Peters, S.; Larkin, J.; Jordan, K.; Committee, on behalf of the E.G. Management of toxicities from immunotherapy: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann. Oncol. 2017, 28, iv119–iv142. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; Chau, I.; Ernstoff, M.S.; Gardner, J.M.; Ginex, P.; et al. Management of Immune-Related Adverse Events in Patients Treated with Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1714–1768. [Google Scholar] [CrossRef] [PubMed]
- De Martin, E.; Michot, J.M.; Papouin, B.; Champiat, S.; Mateus, C.; Lambotte, O.; Roche, B.; Antonini, T.M.; Coilly, A.; Laghouati, S.; et al. Characterization of liver injury induced by cancer immunotherapy using immune checkpoint inhibitors. J. Hepatol. 2018, 68, 1181–1190. [Google Scholar] [CrossRef]
- Gudd, C.L.C.; Au, L.; Triantafyllou, E.; Shum, B.; Liu, T.; Nathwani, R.; Kumar, N.; Mukherjee, S.; Dhar, A.; Woollard, K.J.; et al. Activation and transcriptional profile of monocytes and CD8+ T cells are altered in checkpoint inhibitor-related hepatitis. J. Hepatol. 2021, 75, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Siwicki, M.; Gort-Freitas, N.A.; Messemaker, M.; Bill, R.; Gungabeesoon, J.; Engblom, C.; Zilionis, R.; Garris, C.; Gerhard, G.M.; Kohl, A.; et al. Resident Kupffer cells and neutrophils drive liver toxicity in cancer immunotherapy. Sci. Immunol. 2021, 6, eabi7083. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, H.P.; Arat, S.; Gao, J.; Wen, J.; Xia, S.; Kalabat, D.; Oziolor, E.; Virgen-Slane, R.; Affolter, T.; Ji, C. T cells and monocyte-derived myeloid cells mediate immunotherapy-related hepatitis in a mouse model. J. Hepatol. 2021, 75, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Bartkowiak, T.; Jaiswal, A.R.; Ager, C.R.; Chin, R.; Chen, C.H.; Budhani, P.; Ai, M.; Reilley, M.J.; Sebastian, M.M.; Hong, D.S.; et al. Activation of 4-1BB on liver myeloid cells triggers hepatitis via an interleukin-27–dependent pathway. Clin. Cancer Res. 2018, 24, 1138–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N. Engl. J. Med. 2015, 372, 2521–2532. [Google Scholar] [CrossRef]
- Eggermont, A.M.M.; Blank, C.U.; Mandala, M.; Long, G.V.; Atkinson, V.; Dalle, S.; Haydon, A.; Lichinitser, M.; Khattak, A.; Carlino, M.S.; et al. Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N. Engl. J. Med. 2018, 378, 1789–1801. [Google Scholar] [CrossRef]
- Wolchok, J.D.; Neyns, B.; Linette, G.; Negrier, S.; Lutzky, J.; Thomas, L.; Waterfield, W.; Schadendorf, D.; Smylie, M.; Guthrie, T., Jr.; et al. Ipilimumab monotherapy in patients with pretreated advanced melanoma: A randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol. 2010, 11, 155–164. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved Survival with Ipilimumab in Patients with Metastatic Melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatty, G.L.; Torigian, D.A.; Chiorean, E.G.; Saboury, B.; Brothers, A.; Alavi, A.; Troxel, A.B.; Sun, W.; Teitelbaum, U.R.; Vonderheide, R.H.; et al. A Phase I Study of an Agonist CD40 Monoclonal Antibody (CP-870,893) in Combination with Gemcitabine in Patients with Advanced Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2013, 19, 6286–6295. [Google Scholar] [CrossRef] [Green Version]
- Calne, R.Y.; Sells, R.A.; Pena, J.R.; Davis, D.R.; Millard, P.R.; Herbertson, B.M.; Binns, R.M.; Davies, D.A.L. Induction of Immunological Tolerance by Porcine Liver Allografts. Nature 1969, 223, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Orlando, G.; Soker, S.; Wood, K. Operational tolerance after liver transplantation. J. Hepatol. 2009, 50, 1247–1257. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Xu, S.; Han, Y.; Cao, X. Apoptotic cells attenuate fulminant hepatitis by priming Kupffer cells to produce interleukin-10 through membrane-bound TGF-β. Hepatology 2011, 53, 306–316. [Google Scholar] [CrossRef]
- Wu, K.; Kryczek, I.; Chen, L.; Zou, W.; Welling, T.H. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009, 69, 8067–8075. [Google Scholar] [CrossRef] [Green Version]
- Erhardt, A.; Biburger, M.; Papadopoulos, T.; Tiegs, G. IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology 2007, 45, 475–485. [Google Scholar] [CrossRef]
- Heymann, F.; Tacke, F. Immunology in the liver-from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Crispe, I.N. The Liver as a Lymphoid Organ. Annu. Rev. Immunol. 2009, 27, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Triantafyllou, E.; Pop, O.T.; Possamai, L.A.; Wilhelm, A.; Liaskou, E.; Singanayagam, A.; Bernsmeier, C.; Khamri, W.; Petts, G.; Dargue, R.; et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut 2018, 67, 333–347. [Google Scholar] [CrossRef]
- Mills, C.D.; Kincaid, K.; Alt, J.M.; Heilman, M.J.; Hill, A.M. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol. 2000, 164, 6166–6173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006, 26, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Tacke, F. Hepatic macrophages in homeostasis and liver diseases: From pathogenesis to novel therapeutic strategies. Cell. Mol. Immunol. 2016, 13, 316–327. [Google Scholar] [CrossRef] [Green Version]
- Hammerich, L.; Tacke, F. Emerging roles of myeloid derived suppressor cells in hepatic inflammation and fibrosis. World J. Gastrointest. Pathophysiol. 2015, 6, 43. [Google Scholar] [CrossRef]
- Tang, J.; Yan, Z.; Feng, Q.; Yu, L.; Wang, H. The Roles of Neutrophils in the Pathogenesis of Liver Diseases. Front. Immunol. 2021, 12, 625472. [Google Scholar] [CrossRef]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil Extracellular Traps Kill Bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Lukacs-Kornek, V.; Schuppan, D. Dendritic cells in liver injury and fibrosis: Shortcomings and promises. J. Hepatol. 2013, 59, 1124–1126. [Google Scholar] [CrossRef] [Green Version]
- Goddard, S.; Youster, J.; Morgan, E.; Adams, D.H. Interleukin-10 Secretion Differentiates Dendritic Cells from Human Liver and Skin. Am. J. Pathol. 2004, 164, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Jenne, C.N.; Zhuo, L.; Kimata, K.; Kubes, P. Kupffer cells and activation of endothelial TLR4 coordinate neutrophil adhesion within liver sinusoids during endotoxemia. Am. J. Physiol. Liver Physiol. 2013, 305, G797–G806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Wang, Z.; Zou, Y.; Lu, E.; Duan, J.; Yang, H.; Wu, Q.; Zhao, X.; Wang, Y.; You, L.; et al. Pretreatment with lipopolysaccharide attenuates diethylnitrosamine-caused liver injury in mice via TLR4-dependent induction of Kupffer cell M2 polarization. Immunol. Res. 2015, 62, 137–145. [Google Scholar] [CrossRef]
- Liu, S.; Gallo, D.J.; Green, A.M.; Williams, D.L.; Gong, X.; Shapiro, R.A.; Gambotto, A.A.; Humphris, E.L.; Vodovotz, Y.; Billiar, T.R. Role of toll-like receptors in changes in gene expression and NF-kappa B activation in mouse hepatocytes stimulated with lipopolysaccharide. Infect. Immun. 2002, 70, 3433–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, T.; Degawa, T.; Takii, T.; Hayashi, H.; Okamoto, T.; Inoue, J.; Onozaki, K. TRAF6-NF-kappaB pathway is essential for interleukin-1-induced TLR2 expression and its functional response to TLR2 ligand in murine hepatocytes. Immunology 2003, 109, 127–136. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Crispe, I.N. Immune tolerance in liver disease. Hepatology 2014, 60, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef]
- Knolle, P.A.; Uhrig, A.; Hegenbarth, S.; Löser, E.; Schmitt, E.; Gerken, G.; Lohse, A.W. IL-10 down-regulates T cell activation by antigen-presenting liver sinusoidal endothelial cells through decreased antigen uptake via the mannose receptor and lowered surface expression of accessory molecules. Clin. Exp. Immunol. 1998, 114, 427–433. [Google Scholar] [CrossRef]
- Zheng, M.; Tian, Z. Liver-Mediated Adaptive Immune Tolerance. Front. Immunol. 2019, 10, 2525. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Tagami, T.; Yamazaki, S.; Uede, T.; Shimizu, J.; Sakaguchi, N.; Mak, T.W.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25(+)CD4(+) regulatory T cells constitutively expressing cytotoxic T lymphocyte-associated antigen 4. J. Exp. Med. 2000, 192, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Wang, D.; Zhang, G.; Guo, X. The Role Of PD-1/PD-L1 Axis in Treg Development and Function: Implications for Cancer Immunotherapy. OncoTargets Ther. 2019, 12, 8437–8445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar]
- Kido, M.; Watanabe, N.; Okazaki, T.; Akamatsu, T.; Tanaka, J.; Saga, K.; Nishio, A.; Honjo, T.; Chiba, T. Fatal autoimmune hepatitis induced by concurrent loss of naturally arising regulatory T cells and PD-1-mediated signaling. Gastroenterology 2008, 135, 1333–1343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Wang, G.; Lv, Y.; Wan, Y.Y.; Zheng, J. Inhibition of Cdk8/Cdk19 Activity Promotes Treg Cell Differentiation and Suppresses Autoimmune Diseases. Front. Immunol. 2019, 10, 1988. [Google Scholar] [CrossRef] [Green Version]
- Diehl, L.; Schurich, A.; Grochtmann, R.; Hegenbarth, S.; Chen, L.; Knolle, P.A. Tolerogenic maturation of liver sinusoidal endothelial cells promotes B7-homolog 1-dependent CD8+ T cell tolerance. Hepatology 2008, 47, 296–305. [Google Scholar] [CrossRef]
- Dong, H.; Zhu, G.; Tamada, K.; Flies, D.B.; van Deursen, J.M.A.; Chen, L. B7-H1 Determines Accumulation and Deletion of Intrahepatic CD8+ T Lymphocytes. Immunity 2004, 20, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Zeng, C.; Wen, W.; Morgans, A.K.; Pao, W.; Shu, X.-O.; Zheng, W. Disparities by Race, Age, and Sex in the Improvement of Survival for Major Cancers: Results from the National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) Program in the United States, 1990 to 2010. JAMA Oncol. 2015, 1, 88–96. [Google Scholar] [CrossRef]
- Patel, J.D.; Krilov, L.; Adams, S.; Aghajanian, C.; Basch, E.; Brose, M.S.; Carroll, W.L.; De Lima, M.; Gilbert, M.R.; Kris, M.G.; et al. Clinical cancer advances 2013: Annual report on progress against cancer from the American Society of Clinical Oncology. J. Clin. Oncol. 2014, 32, 129–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chester, C.; Sanmamed, M.F.; Wang, J.; Melero, I. Immunotherapy targeting 4-1BB: Mechanistic rationale, clinical results, and future strategies. Blood 2018, 131, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Kluger, H.; Callahan, M.K.; Postow, M.A.; Rizvi, N.A.; Lesokhin, A.M.; Segal, N.H.; Ariyan, C.E.; Gordon, R.A.; Reed, K.; et al. Nivolumab plus Ipilimumab in advanced melanoma. N. Engl. J. Med. 2013, 369, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonnans, C.; Thomas, G.; He, W.; Jung, B.; Chen, W.; Liao, M.; Heyen, J.; Buetow, B.; Pillai, S.; Matsumoto, D.; et al. CD40 agonist-induced IL-12p40 potentiates hepatotoxicity. J. Immunother. Cancer 2020, 8, e000624. [Google Scholar] [CrossRef] [PubMed]
- Bhave, P.; Buckle, A.; Sandhu, S.; Sood, S. Mortality due to immunotherapy related hepatitis. J. Hepatol. 2018, 69, 976–978. [Google Scholar] [CrossRef] [Green Version]
- Doherty, G.J.; Duckworth, A.M.; Davies, S.E.; Mells, G.F.; Brais, R.; Harden, S.V.; Parkinson, C.A.; Corrie, P.G. Severe steroid-resistant anti-PD1 T-cell checkpoint inhibitor-induced hepatotoxicity driven by biliary injury. ESMO Open 2017, 2, e000268. [Google Scholar] [CrossRef] [Green Version]
- Zen, Y.; Yeh, M.M. Hepatotoxicity of immune checkpoint inhibitors: A histology study of seven cases in comparison with autoimmune hepatitis and idiosyncratic drug-induced liver injury. Mod. Pathol. 2018, 31, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Johncilla, M.; Misdraji, J.; Pratt, D.S.; Agoston, A.T.; Lauwers, G.Y.; Srivastava, A.; Doyle, L.A. Ipilimumab-associated Hepatitis: Clinicopathologic Characterization in a Series of 11 Cases. Am. J. Surg. Pathol. 2015, 39, 1075–1084. [Google Scholar] [CrossRef]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J. V Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Pincetic, A.; Bournazos, S.; DiLillo, D.J.; Maamary, J.; Wang, T.T.; Dahan, R.; Fiebiger, B.-M.; Ravetch, J. V Type I and type II Fc receptors regulate innate and adaptive immunity. Nat. Immunol. 2014, 15, 707–716. [Google Scholar] [CrossRef]
- Uchida, J.; Hamaguchi, Y.; Oliver, J.A.; Ravetch, J.V.; Poe, J.C.; Haas, K.M.; Tedder, T.F. The innate mononuclear phagocyte network depletes B lymphocytes through Fc receptor-dependent mechanisms during anti-CD20 antibody immunotherapy. J. Exp. Med. 2004, 199, 1659–1669. [Google Scholar] [CrossRef]
- Gül, N.; Babes, L.; Siegmund, K.; Korthouwer, R.; Bögels, M.; Braster, R.; Vidarsson, G.; ten Hagen, T.L.M.; Kubes, P.; van Egmond, M. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J. Clin. Investig. 2014, 124, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Simpson, T.R.; Li, F.; Montalvo-Ortiz, W.; Sepulveda, M.A.; Bergerhoff, K.; Arce, F.; Roddie, C.; Henry, J.Y.; Yagita, H.; Wolchok, J.D.; et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 2013, 210, 1695–1710. [Google Scholar] [CrossRef]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef]
- Gubin, M.M.; Esaulova, E.; Ward, J.P.; Malkova, O.N.; Runci, D.; Wong, P.; Noguchi, T.; Arthur, C.D.; Meng, W.; Alspach, E.; et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell 2018, 175, 1014–1030.e19. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.-Y.; Chen, Y.-L.; Wu, W.-Y.; Lin, H.-W.; Chiang, Y.-C.; Chang, C.-F.; Tai, Y.-J.; Hsu, H.-C.; Chen, C.-A.; Sun, W.-Z.; et al. Blockade of PD-L1 Enhances Cancer Immunotherapy by Regulating Dendritic Cell Maturation and Macrophage Polarization. Cancers 2019, 11, 1400. [Google Scholar] [CrossRef] [Green Version]
- Stecher, C.; Battin, C.; Leitner, J.; Zettl, M.; Grabmeier-Pfistershammer, K.; Höller, C.; Zlabinger, G.J.; Steinberger, P. PD-1 Blockade Promotes Emerging Checkpoint Inhibitors in Enhancing T Cell Responses to Allogeneic Dendritic Cells. Front. Immunol. 2017, 8, 572. [Google Scholar] [CrossRef]
- Wang, Y.; Xiang, Y.; Xin, V.W.; Wang, X.-W.; Peng, X.-C.; Liu, X.-Q.; Wang, D.; Li, N.; Cheng, J.-T.; Lyv, Y.-N.; et al. Dendritic cell biology and its role in tumor immunotherapy. J. Hematol. Oncol. 2020, 13, 107. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Qiu, X.; Zhang, Z.; Zhang, S.; Zhang, Y.; Liang, Y.; Guo, J.; Peng, H.; Chen, M.; Fu, Y.-X.; et al. PD-L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat. Commun. 2020, 11, 4835. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Sun, H.-W.; Yang, Y.-Y.; Chen, H.-T.; Yu, X.-J.; Wu, W.-C.; Xu, Y.-T.; Jin, L.-L.; Wu, X.-J.; Xu, J.; et al. Reprogramming immunosuppressive myeloid cells by activated T cells promotes the response to anti-PD-1 therapy in colorectal cancer. Signal Transduct. Target. Ther. 2021, 6, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Liu, T.; Zhu, W.; Xie, S.; Zhao, Z.; Feng, B.; Guo, H.; Yang, R. Targeting MDSC for Immune-Checkpoint Blockade in Cancer Immunotherapy: Current Progress and New Prospects. Clin. Med. Insights Oncol. 2021, 15, 11795549211035540. [Google Scholar] [CrossRef] [PubMed]
- Schetters, S.T.T.; Rodriguez, E.; Kruijssen, L.J.W.; Crommentuijn, M.H.W.; Boon, L.; Van den Bossche, J.; Den Haan, J.M.M.; Van Kooyk, Y. Monocyte-derived APCs are central to the response of PD1 checkpoint blockade and provide a therapeutic target for combination therapy. J. Immunother. Cancer 2020, 8, e000588. [Google Scholar] [CrossRef] [PubMed]
- De Cicco, P.; Ercolano, G.; Ianaro, A. The New Era of Cancer Immunotherapy: Targeting Myeloid-Derived Suppressor Cells to Overcome Immune Evasion. Front. Immunol. 2020, 11, 1680. [Google Scholar] [CrossRef]
- Romano, E.; Kusio-Kobialka, M.; Foukas, P.G.; Baumgaertner, P.; Meyer, C.; Ballabeni, P.; Michielin, O.; Weide, B.; Romero, P.; Speiser, D.E. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl. Acad. Sci. USA 2015, 112, 6140–6145. [Google Scholar] [CrossRef] [Green Version]
- Yajuk, O.; Baron, M.; Toker, S.; Zelter, T.; Fainsod-Levi, T.; Granot, Z. The PD-L1/PD-1 Axis Blocks Neutrophil Cytotoxicity in Cancer. Cells 2021, 10, 1510. [Google Scholar] [CrossRef]
- Zhang, Y.; Guoqiang, L.; Sun, M.; Lu, X. Targeting and exploitation of tumor-associated neutrophils to enhance immunotherapy and drug delivery for cancer treatment. Cancer Biol. Med. 2020, 17, 32–43. [Google Scholar] [CrossRef]
- Sun, L.; Clavijo, P.E.; Robbins, Y.; Patel, P.; Friedman, J.; Greene, S.; Das, R.; Silvin, C.; Van Waes, C.; Horn, L.A.; et al. Inhibiting myeloid-derived suppressor cell trafficking enhances T cell immunotherapy. JCI Insight 2019, 4, e126853. [Google Scholar] [CrossRef] [Green Version]
- Yamauchi, Y.; Safi, S.; Blattner, C.; Rathinasamy, A.; Umansky, L.; Juenger, S.; Warth, A.; Eichhorn, M.; Muley, T.; Herth, F.J.F.; et al. Circulating and Tumor Myeloid-derived Suppressor Cells in Resectable Non-Small Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 2018, 198, 777–787. [Google Scholar] [CrossRef]
- Yoshidome, H.; Kohno, H.; Shida, T.; Kimura, F.; Shimizu, H.; Ohtsuka, M.; Nakatani, Y.; Miyazaki, M. Significance of monocyte chemoattractant protein-1 in angiogenesis and survival in colorectal liver metastases. Int. J. Oncol. 2009, 34, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis-Marcisak, E.F.; Fitzgerald, A.A.; Kessler, M.D.; Danilova, L.; Jaffee, E.M.; Zaidi, N.; Weiner, L.M.; Fertig, E.J. A novel mechanism of natural killer cell response to anti-CTLA-4 therapy identified by integrative analysis of mouse and human tumors. bioRxiv 2020. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J. Exp. Med. 1996, 183, 2533–2540. [Google Scholar] [CrossRef]
- Leach, D.R.; Krummel, M.F.; Allison, J.P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 1996, 271, 1734. [Google Scholar]
- Peeraphatdit, T.; Wang, J.; Odenwald, M.A.; Hu, S.; Hart, J.; Charlton, M.R. Hepatotoxicity from Immune Checkpoint Inhibitors: A Systematic Review and Management Recommendation. Hepatology 2020, 72, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1–Selected Patients with NSCLC. N. Engl. J. Med. 2020, 383, 1328–1339. [Google Scholar] [CrossRef]
- Weber, J.; Thompson, J.A.; Hamid, O.; Minor, D.; Amin, A.; Ron, I.; Ridolfi, R.; Assi, H.; Maraveyas, A.; Berman, D.; et al. A randomized, double-blind, placebo-controlled, phase II study comparing the tolerability and efficacy of ipilimumab administered with or without prophylactic budesonide in patients with unresectable stage III or IV melanoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5591–5598. [Google Scholar] [CrossRef] [Green Version]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and Ipilimumab versus Ipilimumab in Untreated Melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ran, Y.; Wang, K.; Zhu, Y.; Li, J. Incidence and risk of hepatic toxicities with PD-1 inhibitors in cancer patients: A meta-analysis. Drug Des. Dev. Ther. 2016, 10, 3153–3161. [Google Scholar] [CrossRef] [Green Version]
- Tiegs, G.; Lohse, A.W. Immune tolerance: What is unique about the liver. J. Autoimmun. 2010, 34, 1–6. [Google Scholar] [CrossRef]
- Bogdanos, D.P.; Gao, B.; Gershwin, M.E. Liver immunology. Compr. Physiol. 2013, 3, 567–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horst, A.K.; Neumann, K.; Diehl, L.; Tiegs, G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell. Mol. Immunol. 2016, 13, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Jenne, C. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef]
- Metushi, I.G.; Hayes, M.A.; Uetrecht, J. Treatment of PD-1-/- mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology 2015, 61, 1332–1342. [Google Scholar] [CrossRef] [PubMed]
- Medina-Echeverz, J.; Ma, C.; Duffy, A.G.; Eggert, T.; Hawk, N.; Kleiner, D.E.; Korangy, F.; Greten, T.F. Systemic agonistic anti-CD40 treatment of tumor-bearing mice modulates hepatic myeloid-suppressive cells and causes immune-mediated liver damage. Cancer Immunol. Res. 2015, 3, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Affolter, T.; Llewellyn, H.P.; Bartlett, D.W.; Zong, Q.; Xia, S.; Torti, V.; Ji, C. Inhibition of immune checkpoints PD-1, CTLA-4, and IDO1 coordinately induces immune-mediated liver injury in mice. PLoS ONE 2019, 14, e0217276. [Google Scholar] [CrossRef] [PubMed]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Shi, Y.; El-Far, M.; Hill, B.J.; Noto, A.; Ancuta, P.; Peretz, Y.; et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar] [CrossRef] [Green Version]
- Bally, A.P.R.; Lu, P.; Tang, Y.; Austin, J.W.; Scharer, C.D.; Ahmed, R.; Boss, J.M. NF-κB regulates PD-1 expression in macrophages. J. Immunol. 2015, 194, 4545–4554. [Google Scholar] [CrossRef] [Green Version]
- Suzman, D.L.; Pelosof, L.; Rosenberg, A.; Avigan, M.I. Hepatotoxicity of immune checkpoint inhibitors: An evolving picture of risk associated with a vital class of immunotherapy agents. Liver Int. 2018, 38, 976–987. [Google Scholar] [CrossRef] [Green Version]
- Fujimura, T.; Sato, Y.; Tanita, K.; Kambayashi, Y.; Otsuka, A.; Fujisawa, Y.; Yoshino, K.; Matsushita, S.; Funakoshi, T.; Hata, H.; et al. Serum levels of soluble CD163 and CXCL5 may be predictive markers for immune-related adverse events in patients with advanced melanoma treated with nivolumab: A pilot study. Oncotarget 2018, 9, 15542–15551. [Google Scholar] [CrossRef] [Green Version]
- McElroy, A.K.; Shrivastava-Ranjan, P.; Harmon, J.R.; Martines, R.B.; Silva-Flannery, L.; Flietstra, T.D.; Kraft, C.S.; Mehta, A.K.; Lyon, G.M.; Varkey, J.B.; et al. Macrophage Activation Marker Soluble CD163 Associated with Fatal and Severe Ebola Virus Disease in Humans. Emerg. Infect. Dis. 2019, 25, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, A.; Horiike, N.; Akbar, S.M.F.; Michitaka, K.; Matsuyama, T.; Onji, M. Soluble CD163 in patients with liver diseases: Very high levels of soluble CD163 in patients with fulminant hepatic failure. J. Gastroenterol. 2005, 40, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Grewal, I.S.; Flavell, R.A. CD40 and CD154 in cell-mediated immunity. Annu. Rev. Immunol. 1998, 16, 111–135. [Google Scholar] [CrossRef] [PubMed]
- Caux, C.; Massacrier, C.; Vanbervliet, B.; Dubois, B.; Van Kooten, C.; Durand, I.; Banchereau, J. Activation of human dendritic cells through CD40 cross-linking. J. Exp. Med. 1994, 180, 1263–1272. [Google Scholar] [CrossRef] [PubMed]
- Diehl, L.; den Boer, A.T.; Schoenberger, S.P.; van der Voort, E.I.H.; Schumacher, T.N.M.; Melief, C.J.M.; Offringa, R.; Toes, R.E.M. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat. Med. 1999, 5, 774–779. [Google Scholar] [CrossRef] [PubMed]
- French, R.R.; Chan, H.T.C.; Tutt, A.L.; Glennie, M.J. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat. Med. 1999, 5, 548–553. [Google Scholar] [CrossRef]
- Vonderheide, R.H.; Burg, J.M.; Mick, R.; Trosko, J.A.; Li, D.; Shaik, M.N.; Tolcher, A.W.; Hamid, O. Phase I study of the CD40 agonist antibody CP-870,893 combined with carboplatin and paclitaxel in patients with advanced solid tumors. Oncoimmunology 2013, 2, e23033. [Google Scholar] [CrossRef] [Green Version]
- Hoechst, B.; Ormandy, L.A.; Ballmaier, M.; Lehner, F.; Krüger, C.; Manns, M.P.; Greten, T.F.; Korangy, F. A New Population of Myeloid-Derived Suppressor Cells in Hepatocellular Carcinoma Patients Induces CD4+CD25+Foxp3+ T Cells. Gastroenterology 2008, 135, 234–243. [Google Scholar] [CrossRef]
- Kapanadze, T.; Gamrekelashvili, J.; Ma, C.; Chan, C.; Zhao, F.; Hewitt, S.; Zender, L.; Kapoor, V.; Felsher, D.W.; Manns, M.P.; et al. Regulation of accumulation and function of myeloid derived suppressor cells in different murine models of hepatocellular carcinoma. J. Hepatol. 2013, 59, 1007–1013. [Google Scholar] [CrossRef] [Green Version]
- Connolly, M.K.; Mallen-St. Clair, J.; Bedrosian, A.S.; Malhotra, A.; Vera, V.; Ibrahim, J.; Henning, J.; Pachter, H.L.; Bar-Sagi, D.; Frey, A.B.; et al. Distinct populations of metastases-enabling myeloid cells expand in the liver of mice harboring invasive and preinvasive intra-abdominal tumor. J. Leukoc. Biol. 2010, 87, 713–725. [Google Scholar] [CrossRef]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef] [PubMed]
- Steele, C.W.; Karim, S.A.; Leach, J.D.G.; Bailey, P.; Upstill-Goddard, R.; Rishi, L.; Foth, M.; Bryson, S.; McDaid, K.; Wilson, Z.; et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell 2016, 29, 832–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melero, I.; Shuford, W.W.; Newby, S.A.; Aruffo, A.; Ledbetter, J.A.; Hellström, K.E.; Mittler, R.S.; Chen, L. Monoclonal antibodies against the 4-1BB T-cell activation molecule eradicate established tumors. Nat. Med. 1997, 3, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Hammers, H.J.; Plimack, E.R.; Infante, J.R.; Rini, B.I.; McDermott, D.F.; Lewis, L.D.; Voss, M.H.; Sharma, P.; Pal, S.K.; Razak, A.R.A.; et al. Safety and Efficacy of Nivolumab in Combination with Ipilimumab in Metastatic Renal Cell Carcinoma: The CheckMate 016 Study. J. Clin. Oncol. 2017, 35, 3851–3858. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Karamchandani, D.M.; Chetty, R. Immune checkpoint inhibitor-induced gastrointestinal and hepatic injury: Pathologists’ perspective. J. Clin. Pathol. 2018, 71, 665–671. [Google Scholar] [CrossRef]
- Everett, J.; Srivastava, A.; Misdraji, J. Fibrin Ring Granulomas in Checkpoint Inhibitor-induced Hepatitis. Am. J. Surg. Pathol. 2017, 41, 134–137. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Smith, C.; Thomas, S.; Mandik-Nayak, L.; Laury-Kleintop, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [Google Scholar] [CrossRef]
- Godin-Ethier, J.; Hanafi, L.-A.; Piccirillo, C.A.; Lapointe, R. Indoleamine 2,3-Dioxygenase Expression in Human Cancers: Clinical and Immunologic Perspectives. Clin. Cancer Res. 2011, 17, 6985–6991. [Google Scholar] [CrossRef] [Green Version]
- Meireson, A.; Devos, M.; Brochez, L. IDO Expression in Cancer: Different Compartment, Different Functionality? Front. Immunol. 2020, 11, 531491. [Google Scholar] [CrossRef]
- Sumpter, T.L.; Dangi, A.; Matta, B.M.; Huang, C.; Stolz, D.B.; Vodovotz, Y.; Thomson, A.W.; Gandhi, C.R. Hepatic Stellate Cells Undermine the Allostimulatory Function of Liver Myeloid Dendritic Cells via STAT3-Dependent Induction of IDO. J. Immunol. 2012, 189, 3848–3858. [Google Scholar] [CrossRef] [PubMed]
- Nagano, J.; Shimizu, M.; Hara, T.; Shirakami, Y.; Kochi, T.; Nakamura, N.; Ohtaki, H.; Ito, H.; Tanaka, T.; Tsurumi, H.; et al. Effects of Indoleamine 2,3-Dioxygenase Deficiency on High-Fat Diet-Induced Hepatic Inflammation. PLoS ONE 2013, 8, e73404. [Google Scholar] [CrossRef]
- Ito, H.; Hoshi, M.; Ohtaki, H.; Taguchi, A.; Ando, K.; Ishikawa, T.; Osawa, Y.; Hara, A.; Moriwaki, H.; Saito, K.; et al. Ability of IDO To Attenuate Liver Injury in α-Galactosylceramide–Induced Hepatitis Model. J. Immunol. 2010, 185, 4554–4560. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Thomas, L.; Bondarenko, I.; O’Day, S.; Weber, J.; Garbe, C.; Lebbe, C.; Baurain, J.-F.; Testori, A.; Grob, J.-J.; et al. Ipilimumab plus Dacarbazine for Previously Untreated Metastatic Melanoma. N. Engl. J. Med. 2011, 364, 2517–2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langer, C.J.; Gadgeel, S.M.; Borghaei, H.; Papadimitrakopoulou, V.A.; Patnaik, A.; Powell, S.F.; Gentzler, R.D.; Martins, R.G.; Stevenson, J.P.; Jalal, S.I.; et al. Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: A randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol. 2016, 17, 1497–1508. [Google Scholar] [CrossRef]
- Paz-Ares, L.; Luft, A.; Vicente, D.; Tafreshi, A.; Gümüş, M.; Mazières, J.; Hermes, B.; Çay Şenler, F.; Csőszi, T.; Fülöp, A.; et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2040–2051. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodríguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef]
- West, H.; McCleod, M.; Hussein, M.; Morabito, A.; Rittmeyer, A.; Conter, H.J.; Kopp, H.-G.; Daniel, D.; McCune, S.; Mekhail, T.; et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): A multicentre, randomised, open-label, phase 3 tria. Lancet Oncol. 2019, 20, 924–937. [Google Scholar] [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 2078–2092. [Google Scholar] [CrossRef] [PubMed]
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Ann. Oncol. 2019, 30, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef]
- Dimeloe, S.; Frick, C.; Fischer, M.; Gubser, P.M.; Razik, L.; Bantug, G.R.; Ravon, M.; Langenkamp, A.; Hess, C. Human regulatory T cells lack the cyclophosphamide-extruding transporter ABCB1 and are more susceptible to cyclophosphamide-induced apoptosis. Eur. J. Immunol. 2014, 44, 3614–3620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsubo, D.; Yamashita, K.; Fujita, M.; Nishi, M.; Kimura, Y.; Hasegawa, H.; Suzuki, S.; Kakeji, Y. Early-phase Treatment by Low-dose 5-Fluorouracil or Primary Tumor Resection Inhibits MDSC-mediated Lung Metastasis Formation. Anticancer Res. 2015, 35, 4425–4431. [Google Scholar] [PubMed]
- Schaer, D.A.; Geeganage, S.; Amaladas, N.; Lu, Z.H.; Rasmussen, E.R.; Sonyi, A.; Chin, D.; Capen, A.; Li, Y.; Meyer, C.M.; et al. The Folate Pathway Inhibitor Pemetrexed Pleiotropically Enhances Effects of Cancer Immunotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 7175–7188. [Google Scholar] [CrossRef]
- Wanderley, C.W.; Colón, D.F.; Luiz, J.P.M.; Oliveira, F.F.; Viacava, P.R.; Leite, C.A.; Pereira, J.A.; Silva, C.M.; Silva, C.R.; Silva, R.L.; et al. Paclitaxel Reduces Tumor Growth by Reprogramming Tumor-Associated Macrophages to an M1 Profile in a TLR4-Dependent Manner. Cancer Res. 2018, 78, 5891–5900. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [Green Version]
- Dosset, M.; Vargas, T.R.; Lagrange, A.; Boidot, R.; Végran, F.; Roussey, A.; Chalmin, F.; Dondaine, L.; Paul, C.; Lauret Marie-Joseph, E.; et al. PD-1/PD-L1 pathway: An adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology 2018, 7, e1433981. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Hamanishi, J.; Matsumura, N.; Abiko, K.; Murat, K.; Baba, T.; Yamaguchi, K.; Horikawa, N.; Hosoe, Y.; Murphy, S.K.; et al. Chemotherapy Induces Programmed Cell Death-Ligand 1 Overexpression via the Nuclear Factor-κB to Foster an Immunosuppressive Tumor Microenvironment in Ovarian Cancer. Cancer Res. 2015, 75, 5034–5045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournel, L.; Wu, Z.; Stadler, N.; Damotte, D.; Lococo, F.; Boulle, G.; Ségal-Bendirdjian, E.; Bobbio, A.; Icard, P.; Trédaniel, J.; et al. Cisplatin increases PD-L1 expression and optimizes immune check-point blockade in non-small cell lung cancer. Cancer Lett. 2019, 464, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Li, W.; Hu, J.; Zhu, E.C.; Su, Q. Hepatotoxicity in patients with solid tumors treated with PD-1/PD-L1 inhibitors alone, PD-1/PD-L1 inhibitors plus chemotherapy, or chemotherapy alone: Systematic review and meta-analysis. Eur. J. Clin. Pharmacol. 2020, 76, 1345–1354. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.; Uetrecht, J. The Combination of Anti-CTLA-4 and PD1-/- Mice Unmasks the Potential of Isoniazid and Nevirapine to Cause Liver Injury. Chem. Res. Toxicol. 2015, 28, 2287–2291. [Google Scholar] [CrossRef]
- Adam, K.; Iuga, A.; Tocheva, A.S.; Mor, A. A novel mouse model for checkpoint inhibitor-induced adverse events. PLoS ONE 2021, 16, e0246168. [Google Scholar] [CrossRef]
- Park, R.; Lopes, L.; Saeed, A. Anti-PD-1/L1-associated immune-related adverse events as harbinger of favorable clinical outcome: Systematic review and meta-analysis. Clin. Transl. Oncol. 2021, 23, 100–109. [Google Scholar] [CrossRef]
- Cathcart-Rake, E.J.; Sangaralingham, L.R.; Henk, H.J.; Shah, N.D.; Riaz, I.B.; Mansfield, A.S. A Population-based Study of Immunotherapy-related Toxicities in Lung Cancer. Clin. Lung Cancer 2020, 21, 421–427.e2. [Google Scholar] [CrossRef]
- Chang, C.-Y.; Park, H.; Malone, D.C.; Wang, C.-Y.; Wilson, D.L.; Yeh, Y.-M.; Van Boemmel-Wegmann, S.; Lo-Ciganic, W.-H. Immune Checkpoint Inhibitors and Immune-Related Adverse Events in Patients with Advanced Melanoma: A Systematic Review and Network Meta-analysis. JAMA Netw. Open 2020, 3, e201611. [Google Scholar] [CrossRef]
- MacParland, S.A.; Liu, J.C.; Ma, X.-Z.; Innes, B.T.; Bartczak, A.M.; Gage, B.K.; Manuel, J.; Khuu, N.; Echeverri, J.; Linares, I.; et al. Single cell RNA sequencing of human liver reveals distinct intrahepatic macrophage populations. Nat. Commun. 2018, 9, 4383. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Qu, Y.; Xia, P.; Chen, Y.; Zhu, X.; Zhang, J.; Wang, G.; Tian, Y.; Ying, J.; Fan, Z. Transdifferentiation of tumor infiltrating innate lymphoid cells during progression of colorectal cancer. Cell Res. 2020, 30, 610–622. [Google Scholar] [CrossRef]
- Peng, S.; Hebert, L.L.; Eschbacher, J.M.; Kim, S. Single-Cell RNA Sequencing of a Postmenopausal Normal Breast Tissue Identifies Multiple Cell Types That Contribute to Breast Cancer. Cancers 2020, 12, 3639. [Google Scholar] [CrossRef] [PubMed]
- Aliya, S.; Lee, H.; Alhammadi, M.; Umapathi, R.; Huh, Y.S. An Overview on Single-Cell Technology for Hepatocellular Carcinoma Diagnosis. Int. J. Mol. Sci. 2022, 23, 1402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, D.; Peng, M.; Tang, L.; Ouyang, J.; Xiong, F.; Guo, C.; Tang, Y.; Zhou, Y.; Liao, Q.; et al. Single-cell RNA sequencing in cancer research. J. Exp. Clin. Cancer Res. 2021, 40, 81. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Xu, Z.; Marignani, P.A. Single-cell RNA sequencing for the identification of early-stage lung cancer biomarkers from circulating blood. NPJ Genom. Med. 2021, 6, 87. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Chen, J.; Yang, L.; Cao, M.; Yu, Y.; Zhang, R.; Quan, H.; Jiang, Q.; Hua, Y.; Wei, W.; et al. Single-Cell Sequencing-Enabled Hexokinase 2 Assay for Noninvasive Bladder Cancer Diagnosis and Screening by Detecting Rare Malignant Cells in Urine. Anal. Chem. 2020, 92, 16284–16292. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Liu, J.; Ye, Y.; Pan, L.; Deng, H.; Wang, Y.; Yang, Y.; Diao, L.; Lin, S.H.; Mills, G.B.; et al. Multi-omics prediction of immune-related adverse events during checkpoint immunotherapy. Nat. Commun. 2020, 11, 4946. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Z.; Wu, D.; Chen, L.; Xie, L. Single-Cell RNA-Seq Analysis Reveals Microenvironmental Infiltration of Plasma Cells and Hepatocytic Prognostic Markers in HCC With Cirrhosis. Front. Oncol. 2020, 10, 596318. [Google Scholar] [CrossRef]
- Fustero-Torre, C.; Jiménez-Santos, M.J.; García-Martín, S.; Carretero-Puche, C.; García-Jimeno, L.; Ivanchuk, V.; Di Domenico, T.; Gómez-López, G.; Al-Shahrour, F. Beyondcell: Targeting cancer therapeutic heterogeneity in single-cell RNA-seq data. Genome Med. 2021, 13, 187. [Google Scholar] [CrossRef]
- Iivanainen, S.; Ekstrom, J.; Virtanen, H.; Kataja, V.V.; Koivunen, J.P. Electronic patient-reported outcomes and machine learning in predicting immune-related adverse events of immune checkpoint inhibitor therapies. BMC Med. Inform. Decis. Mak. 2021, 21, 205. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNγ and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef]
- Dudley, M.E.; Wunderlich, J.R.; Robbins, P.F.; Yang, J.C.; Hwu, P.; Schwartzentruber, D.J.; Topalian, S.L.; Sherry, R.; Restifo, N.P.; Hubicki, A.M.; et al. Cancer Regression and Autoimmunity in Patients After Clonal Repopulation with Antitumor Lymphocytes. Science 2002, 298, 850–854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Hepatic Myeloid Subset | Key Features | References |
|---|---|---|
| Monocytes/ Monocyte-derived macrophages (MoMF) |
| [32,33] |
| Kupffer cells (KC) |
| [34,35] |
| Myeloid-derived suppressor cells (MDSC) |
| [36] |
| Neutrophils |
| [37,38] |
| Dendritic cells (DC) |
| [39,40] |
| Immunotherapy Regimen | Any Grade, % (Number of Patients) | Grade 3–5, % (Number of Patients) | Reference |
|---|---|---|---|
| Anti-PD-1/ Anti-PD-L1 | 1.8 (5/277) 1.8 (9/509) 8 (25/313) 16.1 (46/286) | 1.8 (5/277) 1.4 (7/509) 3 (9/313) 4.2 (12/286) | Robert et al., 2015 [18] Eggermont et al., 2018 [19] Larkin et al., 2019 [1] Herbst et al., 2020 [96] |
| Anti-CTLA-4 | 1.2 (3/256) 26.4 (19/72) 3.8 (5/131) 15.5 (9/58) 7 (23/311) | 0.4 (1/256) 0 (0/72) 0 (0/131) 10.3 (6/58) 2 (5/311) | Robert et al., 2015 [18] Wolchok et al., 2010 [20] Hodi et al., 2010 [21] Weber et al., 2009 [97] Larkin et al., 2019 [1] |
| Combination anti-CTLA-4 and anti-PD-1 | 23 (12/53) 17.6 (55/313) 22.3 (21/94) 33 (103/313) | 15 (8/53) 8.3 (26/313) 10.6 (10/94) 20 (62/313) | Wolchok et al., 2013 [63] Larkin et al., 2015 [22] Postow et al., 2015 [98] Larkin et al., 2019 [1] |
| Immunotherapy Mechanism | Direct Effect on Hepatic Myeloid Cells and Their Crosstalk with Other Subsets | Reference |
|---|---|---|
| Checkpoint inhibitors (e.g., anti-PD-(L)1, anti-CTLA-4, anti-PD-1 + anti-CTLA-4) | Activation and liver homing of monocytes/MoMF, monocyte interaction with CD8+ T cells | Gudd et al., 2021 [14] |
| Agonistic anti-CD40 | Activation of KCs, recruitment and activation of neutrophils, reduced suppressive capacity of MDSCs | Medina-Echeverz et al., 2015 [105] Siwicki et al., 2021 [15] Bonnans et al., 2020 [64] |
| 4-1BB activation | Liver homing of monocytes, activation of KCs, activation of CD8+ T cells by KCs | Bartkowiak et al., 2018 [17] |
| IDO1 inhibitors in combination with CPIs (e.g., anti-CTLA-4, anti-PD-(L)1, anti-CTLA-4 + anti-PD-1) | Activation of MoMF, promotion of CD8+ T cell activation by MoMF, reduction of MDSCs | Affolter et al., 2019 [106] Llewellyn et al., 2021 [16] |
| Immunotherapy | Chemotherapy | Reference |
|---|---|---|
| Ipilimumab (anti-CTLA-4) | Dacarbazine | Robert et al., 2011 [134] |
| Pembrolizumab (anti-PD-1) | Carboplatin, pemetrexed, nab-paclitaxel | Gandhi et al., 2018 [141] Langer et al., 2016 [135] Paz-Ares et al., 2018 [136] |
| Atezolizumab (anti-PD-L1) | Carboplatin, nab-paclitaxel, etoposide | Horn et al., 2018 [137] Schmid et al., 2018 [138] Socinski et al., 2018 [139] West et al., 2019 [140] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gudd, C.L.C.; Possamai, L.A. The Role of Myeloid Cells in Hepatotoxicity Related to Cancer Immunotherapy. Cancers 2022, 14, 1913. https://doi.org/10.3390/cancers14081913
Gudd CLC, Possamai LA. The Role of Myeloid Cells in Hepatotoxicity Related to Cancer Immunotherapy. Cancers. 2022; 14(8):1913. https://doi.org/10.3390/cancers14081913
Chicago/Turabian StyleGudd, Cathrin L. C., and Lucia A. Possamai. 2022. "The Role of Myeloid Cells in Hepatotoxicity Related to Cancer Immunotherapy" Cancers 14, no. 8: 1913. https://doi.org/10.3390/cancers14081913
APA StyleGudd, C. L. C., & Possamai, L. A. (2022). The Role of Myeloid Cells in Hepatotoxicity Related to Cancer Immunotherapy. Cancers, 14(8), 1913. https://doi.org/10.3390/cancers14081913