
TR3 Enhances AR Variant Production and Transactivation, Promoting Androgen Independence of Prostate Cancer Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Generation of Inducible TR3-Overexpressing CWR22Rv1 Cells
2.3. Next-Generation Sequencing (NGS) High Throughput RNA-Seq Analysis
2.4. Single-Strand RNA (ssRNA) Protection Assays
2.5. Cross-Linked RNA-Immunoprecipitation (CLIP)
2.6. ssRNA Oligo Pull-Down Assays
2.7. Xenograft Animal Model
2.8. Quantification and Statistical Analysis
3. Results
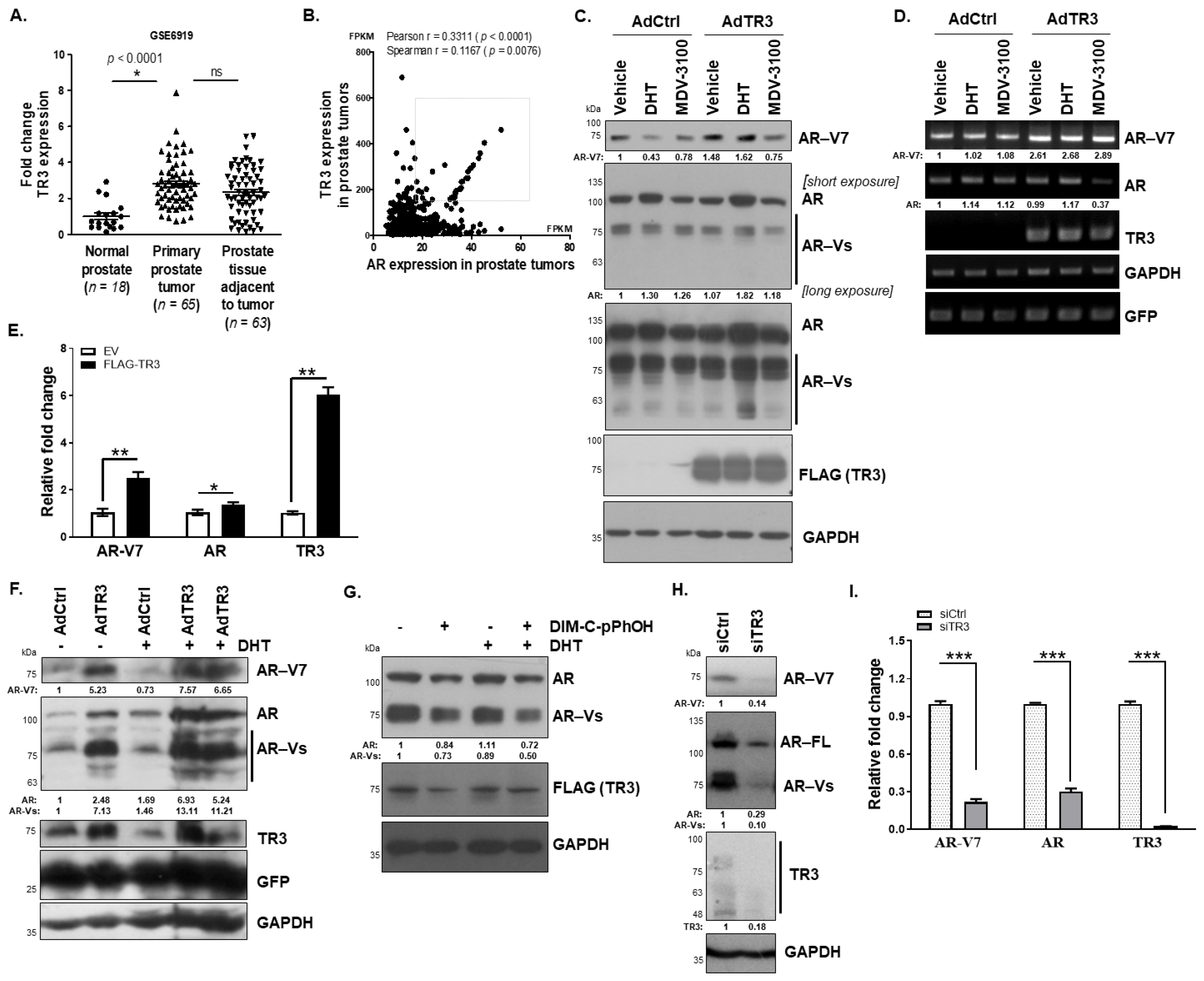
3.1. TR3 Regulates the Expression of AR and AR-Vs in Prostate Cancer
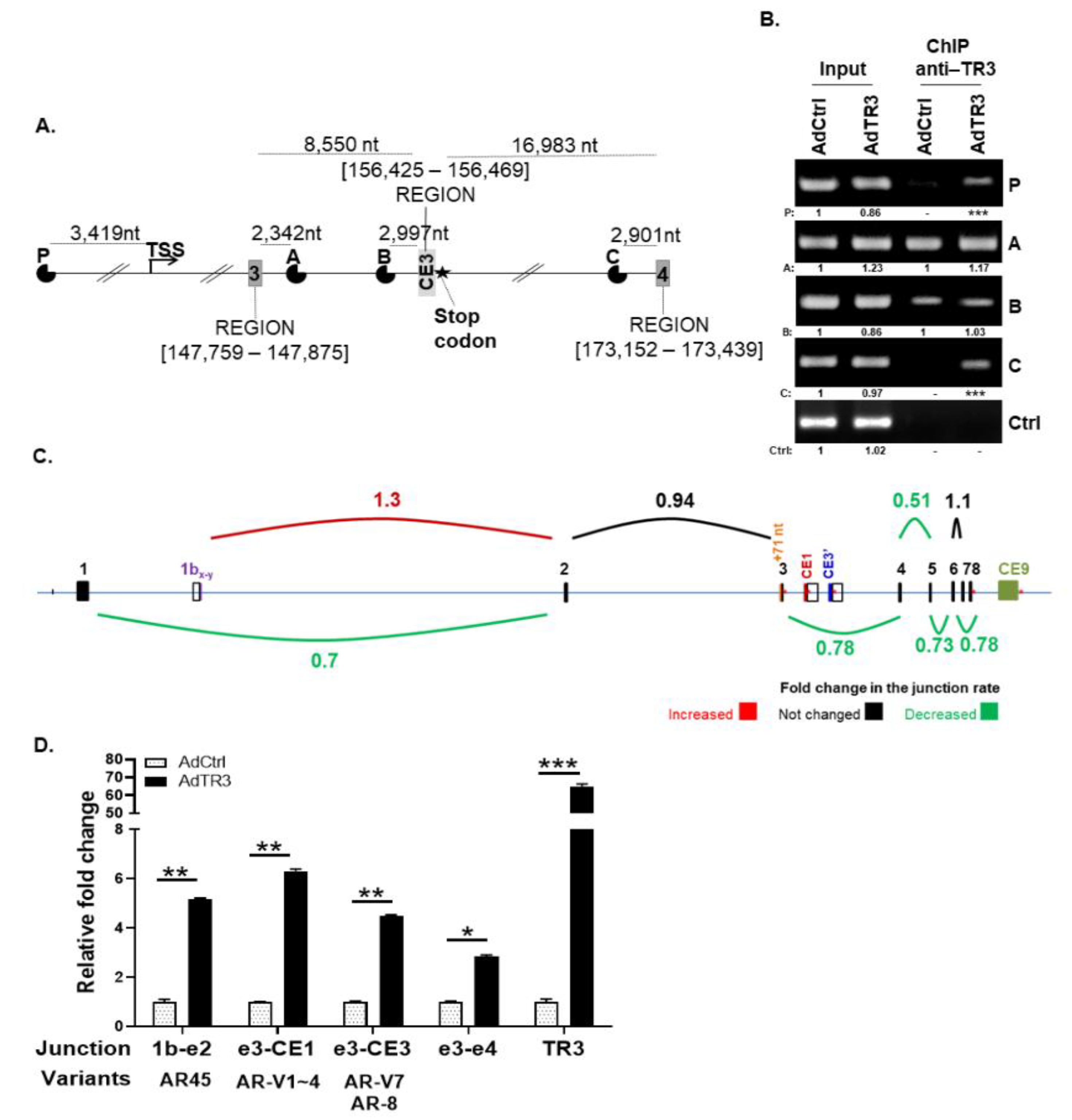
3.2. TR3 Regulates the Expression of AR Splicing Variants, Altering Splice Junction Rates between Exons
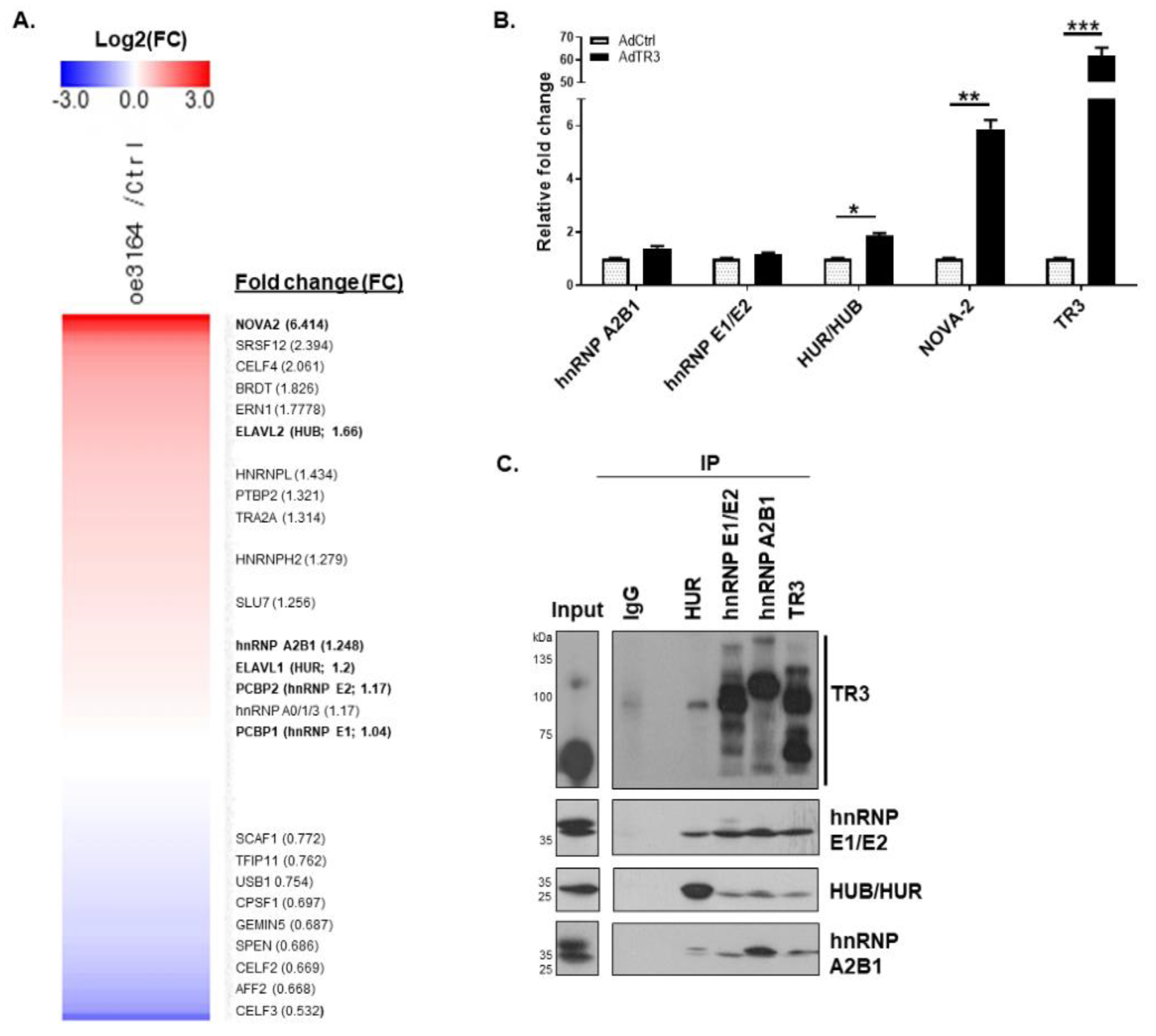
3.3. TR3 Physically Interacts with Some Splicing Factors Involved in AR Splicing Process
3.4. TR3 Overexpression Alters the Recruitment of Some Splicing Factors near Splicing Sites, Interacting with AR Pre-mRNA
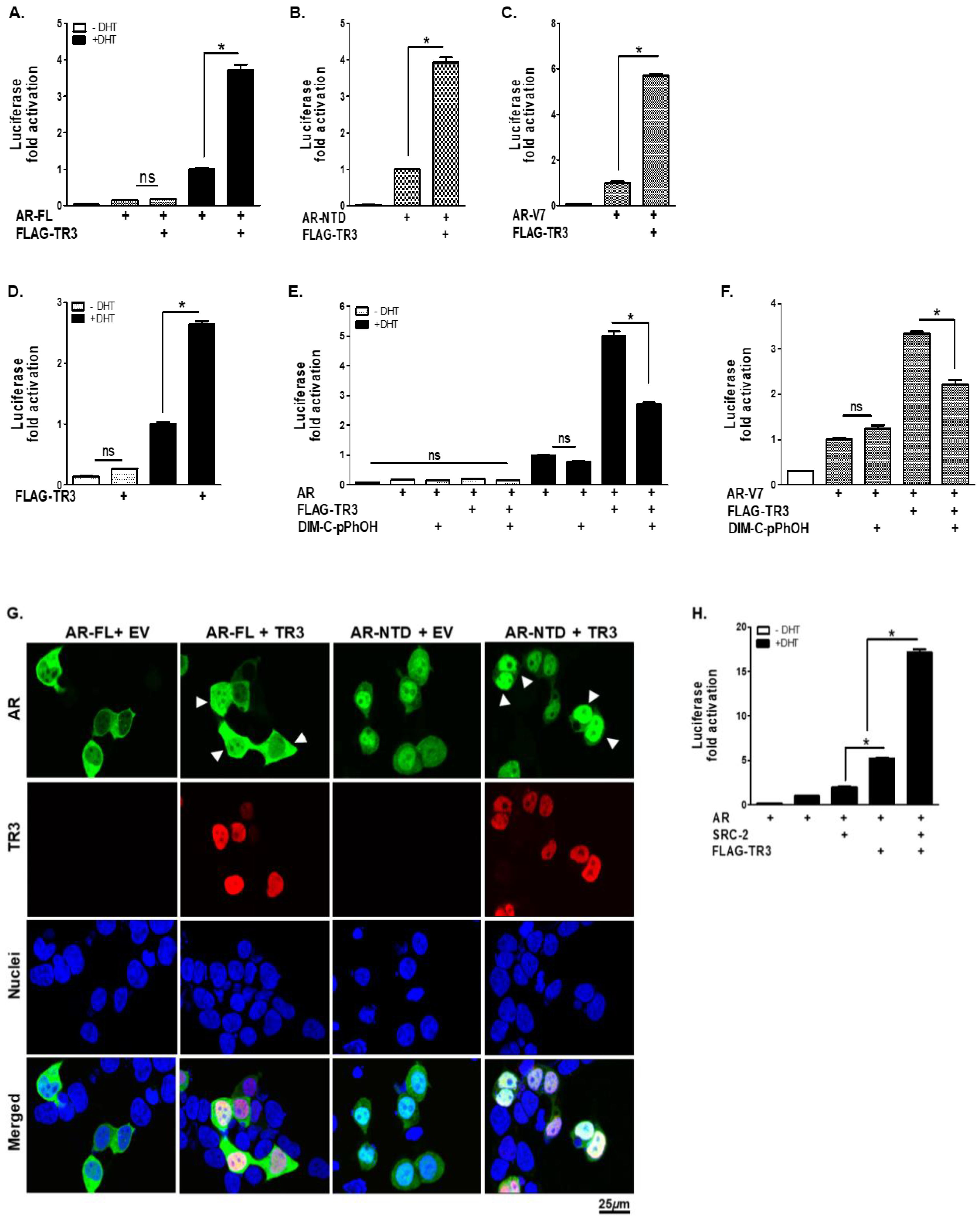
3.5. TR3 Enhances Androgen-Independent and Androgen-Dependent Transactivation of ARs
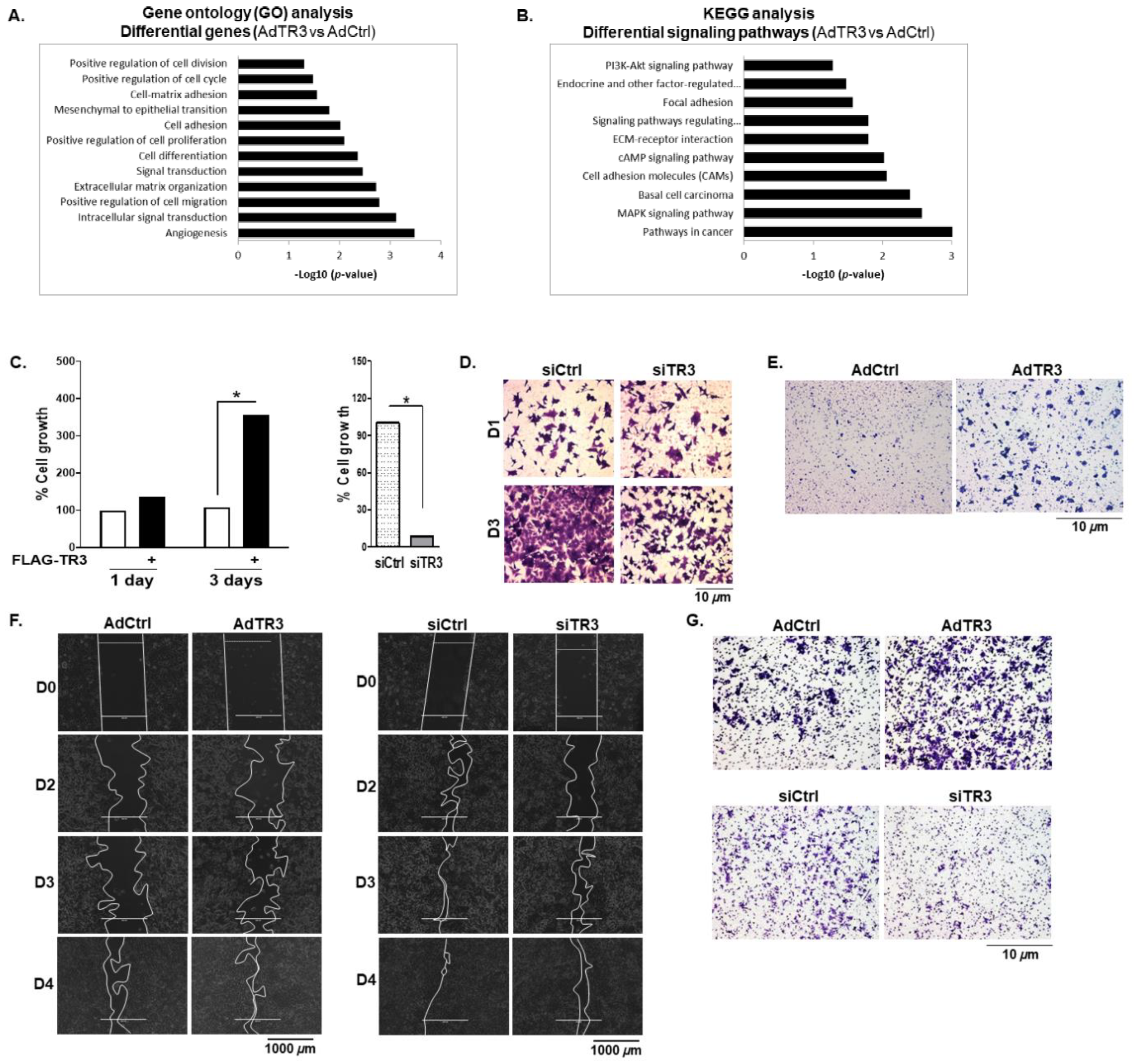
3.6. TR3 Overexpression Promotes the Proliferation and Mobility of Prostate Cancer Cells
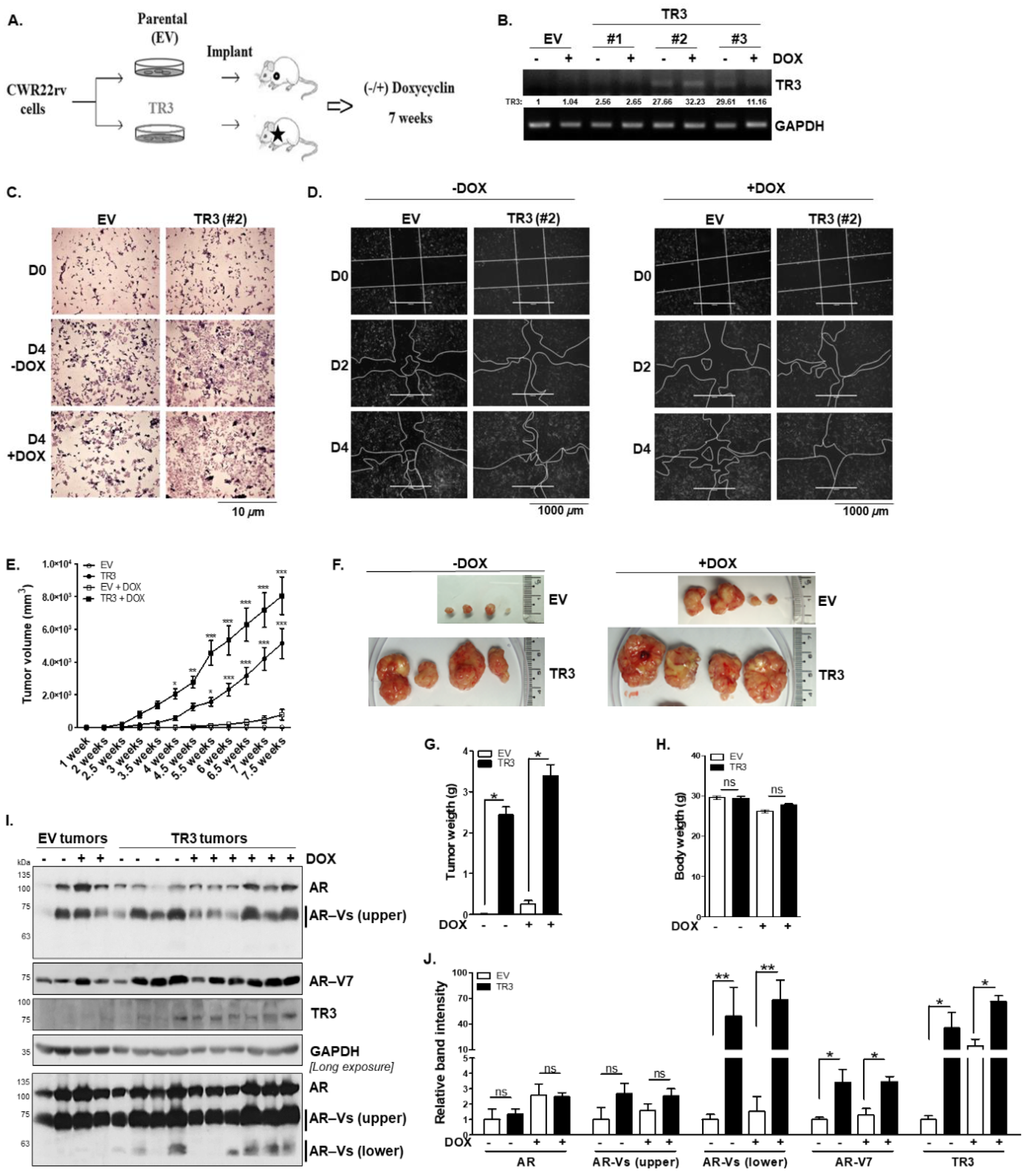
3.7. TR3 Overexpression Enhances In Vivo Tumorigenesis of Androgen-Independent Prostate Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AR | androgen receptor |
| ARE | androgen-response element |
| AR-Vs | androgen receptor variants |
| AR-FL | full-length AR |
| AR-NTD | AR N-terminal domain |
| CE3 | cryptic exon 3 |
| CLIP | cross-linked RNA-immunoprecipitation |
| CRPC | castration-resistant prostate cancer |
| DBD | DNA-binding domain |
| ELAVL1/2 | ELAV-like protein 1/2 |
| hnRNP | heterogeneous nuclear ribonucleoprotein |
| IGV | integrative Genomic Viewer |
| LDB | ligand-binding domain |
| NTD | NH2-terminal transactivation domain |
| pre-mRNA | precursor mRNA |
| PMN-MDSCs | mature myeloid-derived suppressor cells |
| RBPs | RNA-binding proteins |
| RNA pol II CTD | C-terminal domain of RNA polymerase II |
| RRM | RNA recognition motif |
| ssRNA oligo | thirty-mer ssRNA labeled with biotin at the 5′ end |
| TRE | TR3 response element |
References
- Tan, M.H.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.L. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, B.J.; Feldman, D. The development of androgen-independent prostate cancer. Nat. Rev. Cancer 2001, 1, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, C.C.; Plymate, S.R. The link between androgen receptor splice variants and castration-resistant prostate cancer. Horm. Cancer 2014, 5, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.L.; Xie, N.; Sun, S.; Plymate, S.; Mostaghel, E.; Dong, X. Mechanisms of the androgen receptor splicing in prostate cancer cells. Oncogene 2014, 33, 3140–3150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Alsagabi, M.; Fan, D.; Bova, G.S.; Tewfik, A.H.; Dehm, S.M. Intragenic rearrangement and altered RNA splicing of the androgen receptor in a cell-based model of prostate cancer progression. Cancer Res. 2011, 71, 2108–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, C.E.; Rubin, M.A. Genomic rearrangements in prostate cancer. Curr. Opin. Urol. 2015, 25, 71–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeap, B.B.; Voon, D.C.; Vivian, J.P.; McCulloch, R.K.; Thomson, A.M.; Giles, K.M.; Czyzyk-Krzeska, M.F.; Furneaux, H.; Wilce, M.C.; Wilce, J.A.; et al. Novel binding of HuR and poly(C)-binding protein to a conserved UC-rich motif within the 3’-untranslated region of the androgen receptor messenger RNA. J. Biol. Chem. 2002, 277, 27183–27192. [Google Scholar] [CrossRef] [Green Version]
- Nadiminty, N.; Tummala, R.; Liu, C.; Lou, W.; Evans, C.P.; Gao, A.C. NF-κB2/p52:c-Myc:hnRNPA1 Pathway Regulates Expression of Androgen Receptor Splice Variants and Enzalutamide Sensitivity in Prostate Cancer. Mol. Cancer Ther. 2015, 14, 1884. [Google Scholar] [CrossRef] [Green Version]
- Gallo, S.; Arcidiacono, M.V.; Tisato, V.; Piva, R.; Penolazzi, L.; Bosi, C.; Feo, C.V.; Gafà, R.; Secchiero, P. Upregulation of the alternative splicing factor NOVA2 in colorectal cancer vasculature. OncoTargets Ther. 2018, 11, 6049–6056. [Google Scholar] [CrossRef] [Green Version]
- Safe, S.; Jin, U.H.; Hedrick, E.; Reeder, A.; Lee, S.O. Minireview: Role of orphan nuclear receptors in cancer and potential as drug targets. Mol. Endocrinol. 2014, 28, 157–172. [Google Scholar] [CrossRef]
- Chang, H.U.A.C. Antisense TR3 Orphan Receptor Can Increase Prostate Cancer Cell Viability with Etoposide Treatment. Endocrinology 1998, 139, 2329–2334. [Google Scholar]
- Lee, S.O.; Abdelrahim, M.; Yoon, K.; Chintharlapalli, S.; Papineni, S.; Kim, K.; Wang, H.; Safe, S. Inactivation of the orphan nuclear receptor TR3/Nur77 inhibits pancreatic cancer cell and tumor growth. Cancer Res. 2010, 70, 6824–6836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedrick, E.; Safe, S. Transforming Growth Factor β/NR4A1-Inducible Breast Cancer Cell Migration and Epithelial-to-Mesenchymal Transition Is p38α (Mitogen-Activated Protein Kinase 14) Dependent. Mol. Cell. Biol. 2017, 37, e00306-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Yuan, L.; Yu, W.; Sun, Y.; Jiang, D.; Wang, X.; Feng, X.; Wang, Z.; Xu, J.; Yang, R.; et al. A Regulation Loop between YAP and NR4A1 Balances Cell Proliferation and Apoptosis. Cell Rep. 2020, 33, 108284. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-C.; Hsieh, J.-T.; Gleave, M.E.; Brown, N.M.; Pathak, S.; Chung, L.W.K. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: Role of bone stromal cells. Int. J. Cancer 1994, 57, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, G.N.; Anezinis, P.E.; Chang, S.-M.; Zhau, H.E.; Kim, E.E.; Hopwood, V.L.; Pathak, S.; von Eschenbach, A.C.; Chung, L.W. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994, 54, 2577–2581. [Google Scholar] [PubMed]
- Gagliardi, M.; Matarazzo, M.R. RIP: RNA Immunoprecipitation. Methods Mol. Biol. 2016, 1480, 73–86. [Google Scholar] [CrossRef]
- Heitjan, D.F.; Manni, A.; Santen, R.J. Statistical Analysis of In Vivo Tumor Growth Experiments. Cancer Res. 1993, 53, 6042–6050. [Google Scholar]
- Hanna, R.N.; Carlin, L.M.; Hubbeling, H.G.; Nackiewicz, D.; Green, A.M.; Punt, J.A.; Geissmann, F.; Hedrick, C.C. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C-monocytes. Nat. Immunol. 2011, 12, 778–785. [Google Scholar] [CrossRef] [Green Version]
- Nogués, G.; Kadener, S.; Cramer, P.; de la Mata, M.; Fededa, J.P.; Blaustein, M.; Srebrow, A.; Kornblihtt, A.R. Control of alternative pre-mRNA splicing by RNA Pol II elongation: Faster is not always better. IUBMB Life 2003, 55, 235–241. [Google Scholar] [CrossRef]
- McCracken, S.; Fong, N.; Yankulov, K.; Ballantyne, S.; Pan, G.; Greenblatt, J.; Patterson, S.D.; Wickens, M.; Bentley, D.L. The C-terminal domain of RNA polymerase II couples mRNA processing to transcription. Nature 1997, 385, 357–361. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Boyne, A.R.; Millhouse, S.R.; Manley, J.L. The RNA polymerase II C-terminal domain promotes splicing activation through recruitment of a U2AF65-Prp19 complex. Genes Dev. 2011, 25, 972–983. [Google Scholar] [CrossRef] [Green Version]
- Ahrens-Fath, I.; Politz, O.; Geserick, C.; Haendler, B. Androgen receptor function is modulated by the tissue-specific AR45 variant. FEBS J. 2005, 272, 74–84. [Google Scholar] [CrossRef]
- Cao, S.; Zhan, Y.; Dong, Y. Emerging data on androgen receptor splice variants in prostate cancer. Endocr.-Relat. Cancer 2016, 23, T199–T210. [Google Scholar] [CrossRef] [PubMed]
- Vinayagam, A.; Stelzl, U.; Foulle, R.; Plassmann, S.; Zenkner, M.; Timm, J.; Assmus, H.E.; Andrade-Navarro, M.A.; Wanker, E.E. A directed protein interaction network for investigating intracellular signal transduction. Sci. Signal. 2011, 4, rs8. [Google Scholar] [CrossRef] [PubMed]
- Craig, N.L. Molecular Biology: Principles of Genome Function; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Pienta, K.J.; Bradley, D. Mechanisms underlying the development of androgen-independent prostate cancer. Clin. Cancer Res. 2006, 12, 1665–1671. [Google Scholar] [CrossRef] [Green Version]
- Das, A.T.; Tenenbaum, L.; Berkhout, B. Tet-On Systems For Doxycycline-inducible Gene Expression. Curr. Gene Ther. 2016, 16, 156–167. [Google Scholar] [CrossRef] [Green Version]
- Brightwell, G.; Poirier, V.; Cole, E.; Ivins, S.; Brown, K.W. Serum-dependent and cell cycle-dependent expression from a cytomegalovirus-based mammalian expression vector. Gene 1997, 194, 115–123. [Google Scholar] [CrossRef]
- Takayama, K.-I.; Suzuki, T.; Fujimura, T.; Yamada, Y.; Takahashi, S.; Homma, Y.; Suzuki, Y.; Inoue, S. Dysregulation of spliceosome gene expression in advanced prostate cancer by RNA-binding protein PSF. Proc. Natl. Acad. Sci. USA 2017, 114, 10461–10466. [Google Scholar] [CrossRef] [Green Version]
- Dong, X.; Sweet, J.; Challis, J.R.; Brown, T.; Lye, S.J. Transcriptional activity of androgen receptor is modulated by two RNA splicing factors, PSF and p54nrb. Mol. Cell. Biol. 2007, 27, 4863–4875. [Google Scholar] [CrossRef] [Green Version]
- Emili, A.; Shales, M.; McCracken, S.; Xie, W.; Tucker, P.W.; Kobayashi, R.; Blencowe, B.J.; Ingles, C.J. Splicing and transcription-associated proteins PSF and p54nrb/NonO bind to the RNA polymerase II CTD. RNA 2002, 8, 1102–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambout, X.; Dequiedt, F.; Maquat, L.E. Beyond Transcription: Roles of Transcription Factors in Pre-mRNA Splicing. Chem. Rev. 2018, 118, 4339–4364. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.T.; Song, C.H.; Kim, K.J.; Lee, K. A new compound targets the AF-1 of androgen receptor and decreases its activity and protein levels in prostate cancer cells. Am. J. Cancer Res. 2020, 10, 4607–4623. [Google Scholar] [PubMed]
- Luo, J.; Deng, Z.-L.; Luo, X.; Tang, N.; Song, W.-X.; Chen, J.; Sharff, K.A.; Luu, H.H.; Haydon, R.C.; Kinzler, K.W.; et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat. Protoc. 2007, 2, 1236–12247. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tran, T.T.; Lee, K. TR3 Enhances AR Variant Production and Transactivation, Promoting Androgen Independence of Prostate Cancer Cells. Cancers 2022, 14, 1911. https://doi.org/10.3390/cancers14081911
Tran TT, Lee K. TR3 Enhances AR Variant Production and Transactivation, Promoting Androgen Independence of Prostate Cancer Cells. Cancers. 2022; 14(8):1911. https://doi.org/10.3390/cancers14081911
Chicago/Turabian StyleTran, Tuyen Thanh, and Keesook Lee. 2022. "TR3 Enhances AR Variant Production and Transactivation, Promoting Androgen Independence of Prostate Cancer Cells" Cancers 14, no. 8: 1911. https://doi.org/10.3390/cancers14081911
APA StyleTran, T. T., & Lee, K. (2022). TR3 Enhances AR Variant Production and Transactivation, Promoting Androgen Independence of Prostate Cancer Cells. Cancers, 14(8), 1911. https://doi.org/10.3390/cancers14081911