
Epigenetic Determinants of Racial Disparity in Breast Cancer: Looking beyond Genetic Alterations


Abstract
:Simple Summary
Abstract
1. Introduction
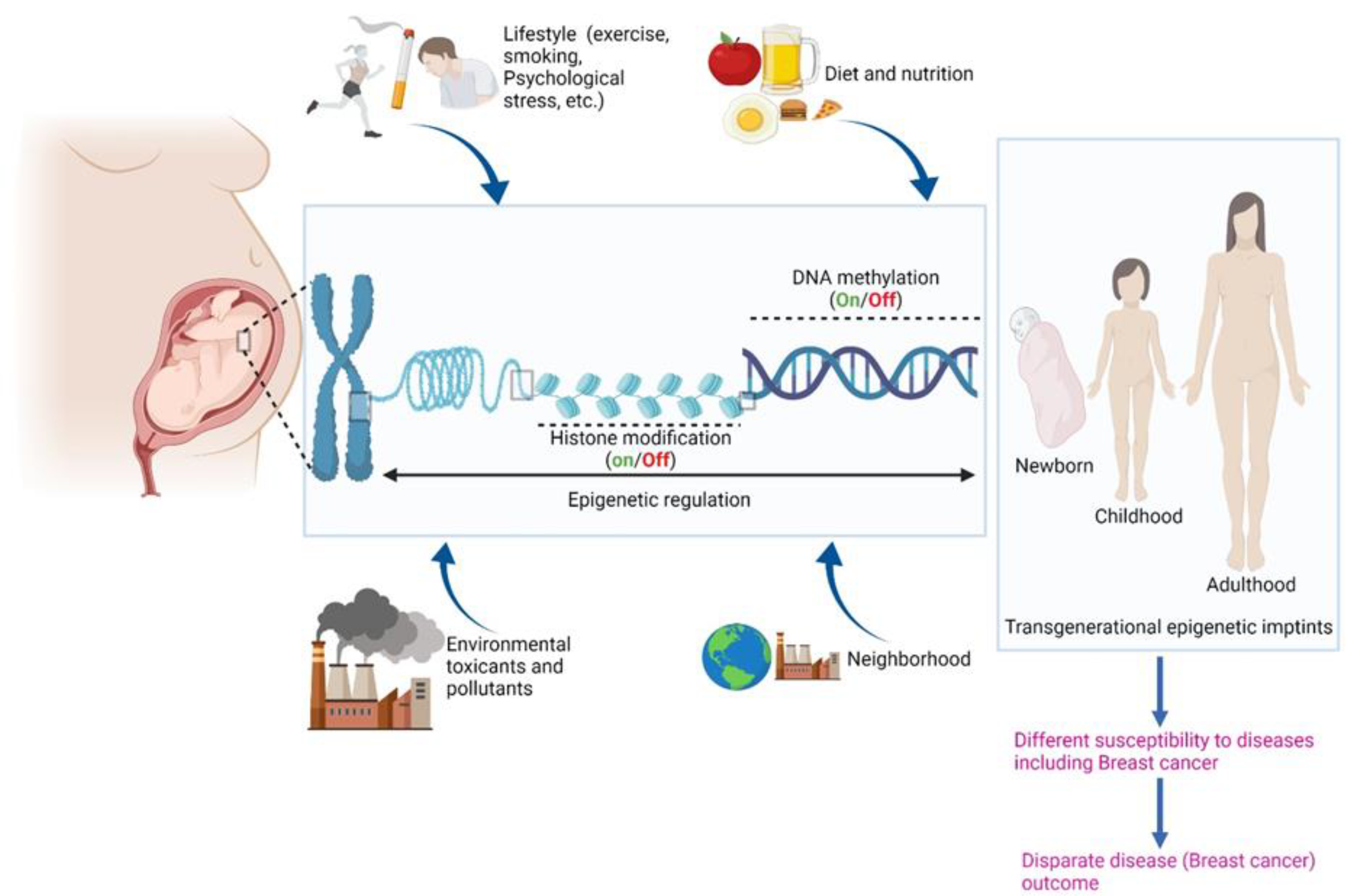
2. Epigenome: A Tutor of the Genome
3. Differences in DNA Methylation Patterns May Underpin Racial Disparity in Breast Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Suppressor Genes | Regular Function | Effect after Hypermethylation | Comments | References |
|---|---|---|---|---|
| BRCA1 |
|
|
| [31] |
| GSTP1 |
|
|
| [32] |
| NKX-2 NKX-5 |
|
|
| [33] |
| GARL2 |
|
|
| [33] |
| CDKN2A |
|
|
| [34] |
| RIL |
|
|
| [35] |
| PTEN |
|
|
| [36] |
| ARH1 |
|
|
| [37] |
| 14-3-3 sigma |
|
|
| [37] |
| RIZ1 |
|
|
| [37] |
| DAPK1 |
|
|
| [38] |
| Genes | Regular Function | Effect after Promoter Hypermethylation | Comments | References |
|---|---|---|---|---|
| CXCL12 |
|
|
| [39] |
| CXCR4 |
|
|
| [40] |
| TOX |
|
|
| [41] |
| SPOCK2 |
|
|
| [33] |
| DPY5 |
|
|
| [33] |
| PITX2 |
|
|
| [42] |
| TP53 |
|
|
| [43] |
| MDM2 |
|
|
| [28] |
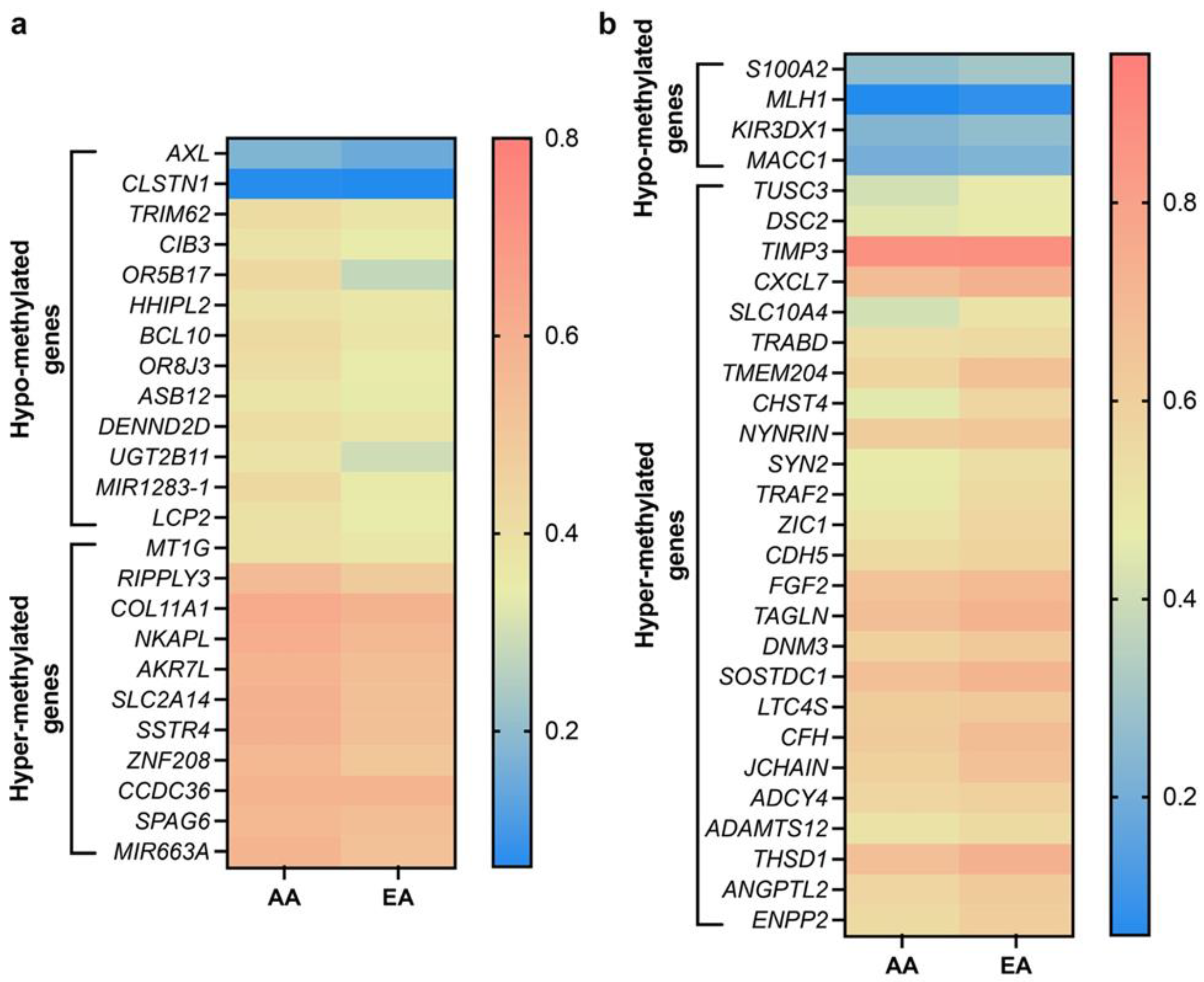
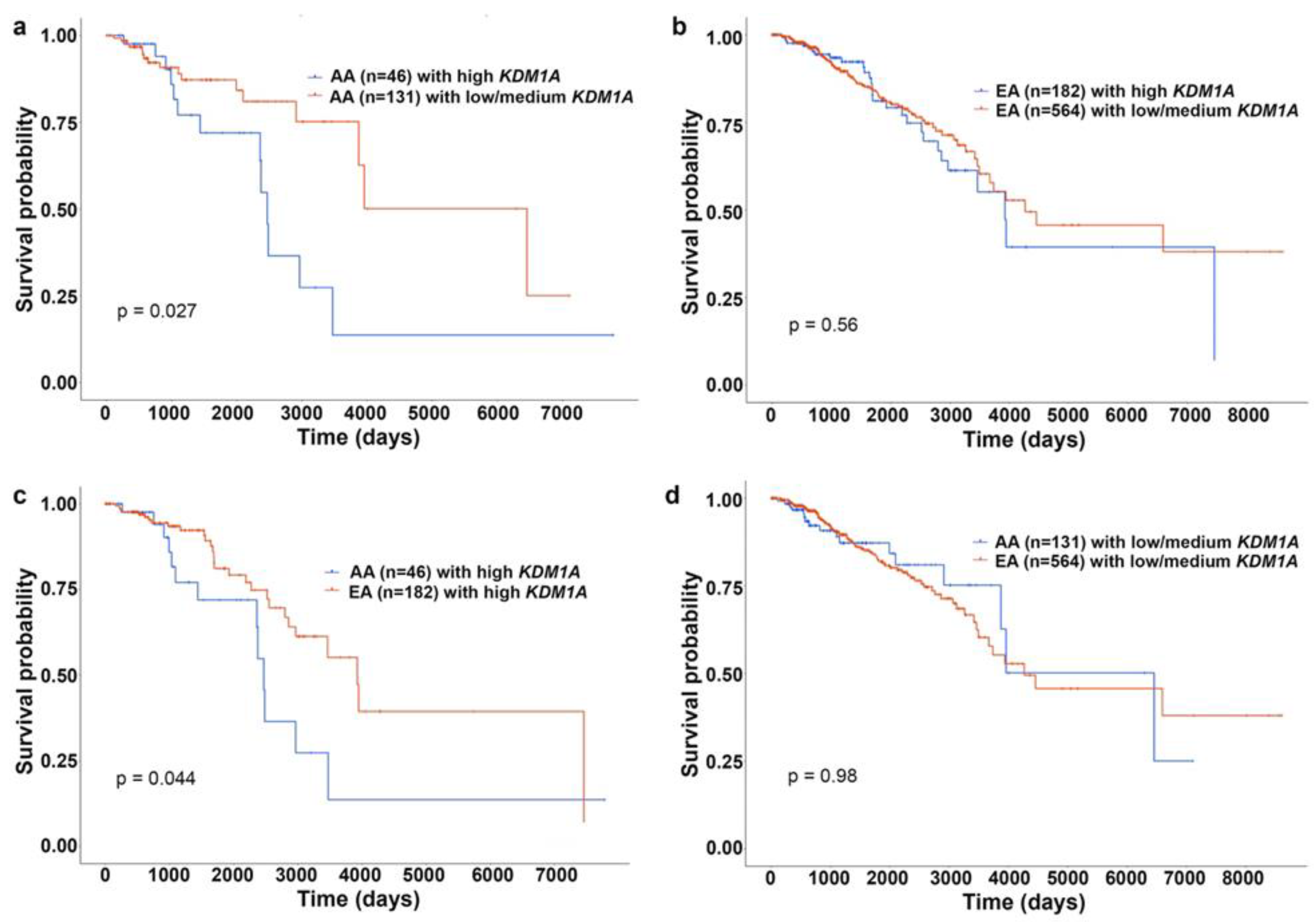
4. Differentially Methylated Genes in AA and EA Breast Cancer Patients: Unpublished Data from In Silico Analysis
5. Plausible Role of Histone Modifications in Breast Cancer Racial Disparity
6. RNA Modifications—Another Layer of Epigenetic Regulation
7. Role of Diet and Nutrition in Breast Cancer Racial Disparities through Epigenetic Modifications
8. Potential Role of Psychosocial Stress in Racial Disparities in Breast Cancer through Epigenetic Changes
9. Potential Role of Environmental Toxicants in Racial Disparities in Breast Cancer through Epigenetic Changes
9.1. Metallic Pollutants
9.2. Polycyclic Aromatic Hydrocarbons
9.3. Nitrogen Dioxide
10. Potential Role of Smoking in Racial Disparities in BC through Epigenetic Changes
11. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeSantis, C.E.; Fedewa, S.A.; Sauer, A.G.; Kramer, J.L.; Smith, R.A.; Jemal, A. Breast cancer statistics, 2015: Convergence of incidence rates between black and white women. CA Cancer J. Clin. 2016, 66, 31–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garlapati, C.; Joshi, S.; Sahoo, B.; Kapoor, S.; Aneja, R. The persisting puzzle of racial disparity in triple negative breast cancer: Looking through a new lens. Front. Biosci. 2019, 11, 75–88. [Google Scholar]
- Saini, G.; Joshi, S.; Garlapati, C.; Li, H.; Kong, J.; Krishnamurthy, J.; Reid, M.D.; Aneja, R. Polyploid giant cancer cell charac-terization: New frontiers in predicting response to chemotherapy in breast cancer. Semin. Cancer Biol. 2021. [Google Scholar]
- Ghimire, H.; Garlapati, C.; Janssen, E.A.M.; Krishnamurti, U.; Qin, G.; Aneja, R.; Perera, A.G.U. Protein Conformational Changes in Breast Cancer Sera Using Infrared Spectroscopic Analysis. Cancers 2020, 12, 1708. [Google Scholar] [CrossRef] [PubMed]
- Dunn, B.K.; Agurs-Collins, T.; Browne, D.; Lubet, R.; Johnson, K.A. Health disparities in breast cancer: Biology meets socio-economic status. Breast Cancer Res. Treat. 2010, 121, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Ogden, A.; Garlapati, C.; Li, X.; Turaga, R.C.; Oprea-Ilies, G.; Wright, N.; Bhattarai, S.; Mittal, K.; Wetherilt, C.S.; Krishnamurti, U.; et al. Multi-institutional study of nuclear KIFC1 as a biomarker of poor prognosis in African American women with triple-negative breast cancer. Sci. Rep. 2017, 7, 42289. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, M.C.; Grayson, J.; Lie, A.; Yu, C.H.; Shiao, S.-Y.P.K. Gene-environment interactions and predictors of breast cancer in family-based multi-ethnic groups. Oncotarget 2018, 9, 29019–29035. [Google Scholar] [CrossRef] [Green Version]
- Bailey, Z.D.; Krieger, N.; Agénor, M.; Graves, J.; Linos, N.; Bassett, M.T. Structural racism and health inequities in the USA: Evidence and interventions. Lancet 2017, 389, 1453–1463. [Google Scholar] [CrossRef]
- Nandi, A.; Glymour, M.M.; Subramanian, S.V. Association Among Socioeconomic Status, Health Behaviors, and All-Cause Mortality in the United States. Epidemiology 2014, 25, 170–177. [Google Scholar] [CrossRef]
- Williams, D.R.; Mohammed, S.A.; Shields, A.E. Understanding and effectively addressing breast cancer in African American women: Unpacking the social context. Cancer 2016, 122, 2138–2149. [Google Scholar] [CrossRef] [Green Version]
- Williams, D.R.; Collins, C. Racial residential segregation: A fundamental cause of racial disparities in health. Public Health Rep. 2001, 116, 404–416. [Google Scholar] [CrossRef]
- Taylor, T.R.; Williams, C.D.; Makambi, K.H.; Mouton, C.; Harrell, J.P.; Cozier, Y.; Palmer, J.R.; Rosenberg, L.; Adams-Campbell, L.L. Racial Discrimination and Breast Cancer Incidence in US Black Women: The Black Women’s Health Study. Am. J. Epidemiol. 2007, 166, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, M.S.; Labrèche, F.; Weichenthal, S.; Lavigne, E.; Valois, M.-F.; Hatzopoulou, M.; Van Ryswyk, K.; Shekarrizfard, M.; Villeneuve, P.; Crouse, D.; et al. The association between the incidence of postmenopausal breast cancer and concentrations at street-level of nitrogen dioxide and ultrafine particles. Environ. Res. 2017, 158, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, H.; Samadani, A.A. DNA Methylation and Cancer Development: Molecular Mechanism. Cell Biochem. Biophys. 2013, 67, 501–513. [Google Scholar] [CrossRef]
- Olden, K.; Olden, H.A.; Lin, Y.-S. The Role of the Epigenome in Translating Neighborhood Disadvantage into Health Dis-parities. Curr. Environ. Health Rep. 2015, 2, 163–170. [Google Scholar] [CrossRef] [Green Version]
- Lam, L.L.; Emberly, E.; Fraser, H.B.; Neumann, S.M.; Chen, E.; Miller, G.E.; Kobor, M.S. Factors underlying variable DNA methylation in a human community cohort. Proc. Natl. Acad. Sci. USA 2012, 109 (Suppl. 2), 17253–17260. [Google Scholar] [CrossRef] [Green Version]
- Bonetta, L. Epigenomics: The New Tool in Studying Complex Diseases. Nat. Educ. 2008, 1, 178. [Google Scholar]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Thayer, Z.M.; Kuzawa, C.W. Biological memories of past environments: Epigenetic pathways to health disparities. Epigenetics 2011, 6, 798–803. [Google Scholar] [CrossRef] [Green Version]
- Elsheikh, S.E.; Green, A.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; et al. Global Histone Modifications in Breast Cancer Correlate with Tumor Phenotypes, Prognostic Factors, and Patient Outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar] [CrossRef] [Green Version]
- Mehrotra, J.; Ganpat, M.M.; Kanaan, Y.; Fackler, M.J.; McVeigh, M.; Lahti-Domenici, J.; Polyak, K.; Argani, P.; Naab, T.; Garrett, E.; et al. Estrogen Receptor/Progesterone Receptor-Negative Breast Cancers of Young African-American Women Have a Higher Frequency of Methylation of Multiple Genes than Those of Caucasian Women. Clin. Cancer Res. 2004, 10, 2052–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yan, L.; Davidson, N.E. DNA methylation in breast cancer. Endocr. Relat. Cancer 2001, 8, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.M.-K.; Yu, J. Promoter hypermethylation of tumour suppressor genes as potential biomarkers in colorectal cancer. Int. J. Mol. Sci. 2015, 16, 2472–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Dorsey, T.H.; Terunuma, A.; Kittles, R.A.; Ambs, S.; Kwabi-Addo, B. Relationship between Tumor DNA Methylation Status and Patient Characteristics in African-American and European-American Women with Breast Cancer. PLoS ONE 2012, 7, e37928. [Google Scholar] [CrossRef] [PubMed]
- Mehrotra, J.; Vali, M.; McVeigh, M.; Kominsky, S.L.; Fackler, M.J.; Lahti-Domenici, J.; Polyak, K.; Sacchi, N.; Garrett-Mayer, E.; Argani, P.; et al. Very high frequency of hypermethylated genes in breast cancer metastasis to the bone, brain, and lung. Clin. Cancer Res. 2004, 10, 3104–3109. [Google Scholar] [CrossRef] [Green Version]
- Klopocki, E.; Kristiansen, G.; Wild, P.J.; Klaman, I.; Castaños-Vélez, E.; Singer, G.; Stöhr, R.; Simon, R.; Sauter, G.; Leibiger, H.; et al. Loss of SFRP1 is associated with breast cancer progression and poor prognosis in early stage tumors. Int. J. Oncol. 2004, 25, 641–649. [Google Scholar] [CrossRef]
- Conway, K.; Edmiston, S.N.; Tse, C.-K.; Bryant, C.; Kuan, P.F.; Hair, B.Y.; Parrish, E.A.; May, R.; Swift-Scanlan, T. Racial Var-iation in Breast Tumor Promoter Methylation in the Carolina Breast Cancer Study. Cancer Epidemiol. Biomark. Prev. 2015, 24, 921–930. [Google Scholar] [CrossRef] [Green Version]
- Tanas, A.S.; Sigin, V.O.; Kalinkin, A.I.; Litviakov, N.V.; Slonimskaya, E.M.; Ibragimova, M.K.; Ignatova, E.O.; Simonova, O.A.; Kuznetsova, E.B.; Kekeeva, T.V.; et al. Genome-wide methylotyping resolves breast cancer epigenetic heterogeneity and suggests novel therapeutic perspectives. Epigenomics 2019, 11, 605–617. [Google Scholar] [CrossRef] [Green Version]
- Ambrosone, C.B.; Young, A.C.; Sucheston, L.E.; Wang, D.; Yan, L.; Liu, S.; Tang, L.; Hu, Q.; Freudenheim, J.L.; Shields, P.G.; et al. Genome-wide methylation patterns provide insight into differences in breast tumor biology between American women of African and European ancestry. Oncotarget 2013, 5, 237–248. [Google Scholar] [CrossRef] [Green Version]
- Song, M.-A.; Brasky, T.M.; Marian, C.; Weng, D.Y.; Taslim, C.; Dumitrescu, R.G.; A Llanos, A.; Freudenheim, J.L.; Shields, P.G. Racial differences in genome-wide methylation profiling and gene expression in breast tissues from healthy women. Epigenetics 2015, 10, 1177–1187. [Google Scholar] [CrossRef]
- Zhang, L.; Long, X. Association of BRCA1 promoter methylation with sporadic breast cancers: Evidence from 40 studies. Sci. Rep. 2015, 5, 17869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, C.; Wei, X.-M.; Zeng, X.-T.; Wang, F.-B.; Weng, H.; Long, X. Aberrant GSTP1 promoter methylation is associated with increased risk and advanced stage of breast cancer: A meta-analysis of 19 case-control studies. BMC Cancer 2015, 15, 920. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.; Kwabi-Addo, B.; Ittmann, M.; Jelinek, J.; Shen, L.; Yu, Y.; Issa, J.P.J. Identification of novel tumor markers in prostate, colon and breast cancer by unbiased methylation profiling. PLoS ONE 2008, 3, e2079. [Google Scholar] [CrossRef]
- Bae, Y.K.; Brown, A.; Garrett, E.; Bornman, D.; Fackler, M.J.; Sukumar, S.; Herman, J.G.; Gabrielson, E. Hypermethylation in Histologically Distinct Classes of Breast Cancer. Clin. Cancer Res. 2004, 10, 5998–6005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Shetty, P.B.; Feng, W.; Chenault, C.; Bast, R.C.; Issa, J.-P.J.; Hilsenbeck, S.G.; Yu, Y. Methylation of HIN-1, RASSF1A, RIL and CDH13 in breast cancer is associated with clinical characteristics, but only RASSF1A methylation is associated with outcome. BMC Cancer 2012, 12, 243. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Chen, J.; Mo, X. The association of PTEN hypermethylation and breast cancer: A meta-analysis. OncoTargets Ther. 2016, 9, 5643–5650. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Shen, L.; Wen, S.; Rosen, D.G.; Jelinek, J.; Hu, X.; Huan, S.; Huang, M.; Liu, J.; Sahin, A.A.; et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007, 9, R57. [Google Scholar] [CrossRef] [Green Version]
- Yadav, P.; Masroor, M.; Nandi, K.; Kaza, R.C.M.; Jain, S.K.; Khuarana, N.; Saxena, A. Promoter Methylation of BRCA1, DAPK1 and RASSF1A is Associated with Increased Mortality among Indian Women with Breast Cancer. Asian Pac. J. Cancer Prev. 2018, 19, 443–448. [Google Scholar]
- Wendt, M.K.; Cooper, A.N.; Dwinell, M.B. Epigenetic silencing of CXCL12 increases the metastatic potential of mammary carcinoma cells. Oncogene 2008, 27, 1461–1471. [Google Scholar] [CrossRef] [Green Version]
- Ramos, E.A.S.; Grochoski, M.; Prado, K.B.; Seniski, G.G.; Cavalli, I.J.; Ribeiro, E.M.S.F.; Camargo, A.A.; Costa, F.F.; Klassen, G. Epigenetic Changes of CXCR4 and Its Ligand CXCL12 as Prognostic Factors for Sporadic Breast Cancer. PLoS ONE 2011, 6, e29461. [Google Scholar] [CrossRef] [Green Version]
- Tessema, M.; Yingling, C.M.; Grimes, M.J.; Thomas, C.L.; Liu, Y.; Leng, S.; Joste, N.; Belinsky, S.A. Differential Epigenetic Regulation of TOX Subfamily High Mobility Group Box Genes in Lung and Breast Cancers. PLoS ONE 2012, 7, e34850. [Google Scholar] [CrossRef] [PubMed]
- Absmaier, M.; Napieralski, R.; Schuster, T.; Aubele, M.; Walch, A.; Magdolen, V.; Dorn, J.; Gross, E.; Harbeck, N.; Noske, A.; et al. PITX2 DNA-methylation predicts response to anthracycline-based adjuvant chemotherapy in triple-negative breast cancer patients. Int. J. Oncol. 2018, 52, 755–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, J.H.; Kim, S.J.; Noh, D.-Y.; Park, I.A.; Choe, K.J.; Yoo, O.J.; Kang, H.-S. Methylation in the p53 Promoter Is a Supple-mentary Route to Breast Carcinogenesis: Correlation between CpG Methylation in the p53 Promoter and the Mutation of the p53 Gene in the Progression from Ductal Carcinoma In Situ to Invasive Ductal Carcinoma. Lab. Investig. 2001, 81, 573–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrashekar, D.S.; Bashetl, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Va-rambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Azare, J.; Doane, A.; Leslie, K.; Chang, Q.; Berishaj, M.; Nnoli, J.; Mark, K.; Al-Ahmadie, H.; Gerald, W.; Hassimi, M.; et al. Stat3 Mediates Expression of Autotaxin in Breast Cancer. PLoS ONE 2011, 6, e27851. [Google Scholar] [CrossRef] [Green Version]
- Stracke, L.M.; Clair, T.; Liotta, L.A. Autotaxin, tumor motility-stimulating exophosphodiesterase. Adv. Enzym. Regul. 1997, 37, 135–144. [Google Scholar] [CrossRef]
- Fan, Y.; Mu, J.; Huang, M.; Imani, S.; Wang, Y.; Lin, S.; Fan, J.; Wen, Q. Epigenetic identification of ADCY4 as a biomarker for breast cancer: An integrated analysis of adenylate cyclases. Epigenomics 2019, 11, 1561–1579. [Google Scholar] [CrossRef]
- Wang, H.; Yu, J.; Guo, Y.; Zhang, Z.; Liu, G.; Li, J.; Zhang, X.; Jin, T.; Wang, Z. Genetic variants in the ZNF208 gene are associated with esophageal cancer in a Chinese Han population. Oncotarget 2016, 7, 86829–86835. [Google Scholar] [CrossRef] [Green Version]
- Kumar, U.; Grigorakis, S.I.; Watt, H.L.; Sasi, R.; Snell, L.; Watson, P.; Chaudhari, S. Somatostatin receptors in primary human breast cancer: Quantitative analysis of mRNA for subtypes 1--5 and correlation with receptor protein expression and tumor pathology. Breast Cancer Res. Treat. 2005, 92, 175–186. [Google Scholar] [CrossRef]
- Halvorsen, A.R.; Helland, Å.; Fleischer, T.; Haug, K.M.; Alnaes, G.I.G.; Nebdal, D.; Syljuåsen, R.G.; Touleimat, N.; Busato, F.; Tost, J.; et al. Differential DNA methylation analysis of breast cancer reveals the impact of immune signaling in radiation therapy. Int. J. Cancer 2014, 135, 2085–2095. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Gong, H.; Wang, T.; Wang, J.; Han, Z.; Bai, G.; Han, S.; Yang, X.; Zhou, W.; Liu, T.; et al. SOSTDC1 inhibits bone metastasis in non-small cell lung cancer and may serve as a clinical therapeutic target. Int. J. Mol. Med. 2018, 42, 3424–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, K.A.; Blish, K.R.; Birse, C.E.; Triplette, M.A.; Kute, T.E.; Russell, G.B.; D’Agostino, R.B.; Miller, L.D.; Torti, F.M.; Torti, S.V. SOSTDC1 differentially modulates Smad and beta-catenin activation and is down-regulated in breast cancer. Breast Cancer Res. Treat. 2011, 129, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, D.; Kimler, B.F.; Nothnick, W.B.; Davis, M.K.; Fan, F.; Tawfik, O. Transgelin: A potentially useful diagnostic marker differentially expressed in triple-negative and non-triple-negative breast cancers. Hum. Pathol. 2015, 46, 876–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.; Cao, F.; Gao, X.-J.; Wang, H.-B.; Chen, F.; Cai, S.-J.; Zhang, C.; Hu, Y.-W.; Ma, J.; Gu, X.; et al. ZIC1 acts a tumor suppressor in breast cancer by targeting survivin. Int. J. Oncol. 2018, 53, 937–948. [Google Scholar] [CrossRef] [Green Version]
- Peramuhendige, P.; Marino, S.; Bishop, R.T.; De Ridder, D.; Khogeer, A.; Baldini, I.; Capulli, M.; Rucci, N.; Idris, A.I. TRAF2 in osteotropic breast cancer cells enhances skeletal tumour growth and promotes osteolysis. Sci. Rep. 2018, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Jin, F. Screening of differentially methylated genes in breast cancer and risk model construction based on TCGA database. Oncol. Lett. 2018, 16, 6407–6416. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Ren, Y.; Qian, C.; Liu, J.; Li, G.; Li, Z. Over-expression of CDX2 alleviates breast cancer by up-regulating microRNA let-7b and inhibiting COL11A1 expression. Cancer Cell Int. 2020, 20, 13. [Google Scholar] [CrossRef]
- Maneerat, Y.; Prasongsukarn, K.; Benjathummarak, S.; Dechkhajorn, W. PPBP and DEFA1/DEFA3 genes in hyperlipidaemia as feasible synergistic inflammatory biomarkers for coronary heart disease. Lipids Health Dis. 2017, 16, 80. [Google Scholar] [CrossRef] [Green Version]
- Su, C.-W.; Lin, C.-W.; Yang, W.-E.; Yang, S.-F. TIMP-3 as a therapeutic target for cancer. Ther. Adv. Med. Oncol. 2019, 11, 1758835919864247. [Google Scholar] [CrossRef] [Green Version]
- Kolegraff, K.; Nava, P.; Helms, M.N.; Parkos, C.A.; Nusrat, A. Loss of desmocollin-2 confers a tumorigenic phenotype to colonic epithelial cells through activation of Akt/β-catenin signaling. Mol. Biol. Cell 2011, 22, 1121–1134. [Google Scholar] [CrossRef]
- Kratochvílová, K.; Horak, P.; Ešner, M.; Souček, K.; Pils, D.; Anees, M.; Tomasich, E.; Dráfi, F.; Jurtíková, V.; Hampl, A.; et al. Tumor suppressor candidate 3 (TUSC3) prevents the epithelial-to-mesenchymal transition and inhibits tumor growth by modulating the endoplasmic reticulum stress response in ovarian cancer cells. Int. J. Cancer 2015, 137, 1330–1340. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.-F.; Fan, Y.-C.; Gao, S.; Yang, Y.; Zhang, J.-J.; Wang, K. MT1M and MT1G promoter methylation as biomarkers for hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 4723–4729. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Xie, X.; Li, L.; Tang, H.; Ye, X.; Chen, L.; Tang, W.; Gao, J.; Pan, L.; Zhang, X.; et al. Diagnostic and prognostic value of serum MACC1 in breast cancer patients. Oncotarget 2016, 7, 84408–84415. [Google Scholar] [CrossRef] [PubMed]
- Miaskowski, C.; Dodd, M.; Paul, S.M.; West, C.; Hamolsky, D.; Abrams, G.; Cooper, B.A.; Elboim, C.; Neuhaus, J.; Schmidt, B.L.; et al. Lymphatic and Angiogenic Candidate Genes Predict the Development of Secondary Lymphedema following Breast Cancer Surgery. PLoS ONE 2013, 8, e60164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Shidfar, A.; Ivancic, D.; Ranjan, M.; Liu, L.; Choi, M.-R.; Parimi, V.; Gursel, D.B.; Sullivan, M.E.; Najor, M.S.; et al. Overexpression of lipid metabolism genes and PBX1 in the contralateral breasts of women with estrogen receptor-negative breast cancer. Int. J. Cancer 2017, 140, 2484–2497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Hu, Y.; Ju, J.; Hou, L.; Li, Z.; Xiao, D.; Li, Y.; Yao, J.; Wang, C.; Zhang, Y.; et al. Downregulation of miR-522 suppresses proliferation and metastasis of non-small cell lung cancer cells by directly targeting DENN/MADD domain containing 2D. Sci. Rep. 2016, 6, 19346. [Google Scholar] [CrossRef] [Green Version]
- Ismail, I.H.; Dronyk, A.; Hu, X.; Hendzel, M.; Shaw, A.R. BCL10 is recruited to sites of DNA damage to facilitate DNA dou-ble-strand break repair. Cell Cycle 2016, 15, 84–94. [Google Scholar] [CrossRef] [Green Version]
- Quintás-Cardama, A.; Post, S.M.; Solis, L.M.; Xiong, S.; Yang, P.; Chen, N.; Wistuba, I.I.; Killary, A.M.; Lozano, G. Loss of the novel tumour suppressor and polarity gene Trim62 (Dear1) synergizes with oncogenic Ras in invasive lung cancer. J. Pathol. 2014, 234, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Harkness, E.F.; Barrow, E.; Newton, K.; Green, K.; Clancy, T.; Lalloo, F.; Hill, J.; Evans, D.G. Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: A cohort study. J. Med. Genet. 2015, 52, 553–556. [Google Scholar] [CrossRef]
- Goyette, M.A.; Duhamel, S.; Aubert, L.; Pelletier, A.; Savage, P.; Thibault, M.-P.; Johnson, R.M.; Carmeliet, P.; Basik, M.; Gaboury, L.; et al. The Receptor Tyrosine Kinase AXL Is Required at Multiple Steps of the Metastatic Cascade during HER2-Positive Breast Cancer Progression. Cell Rep. 2018, 23, 1476–1490. [Google Scholar] [CrossRef]
- Wicki, R.; Franz, C.; Scholl, F.A.; Heizmann, C.W.; Schäfer, B.W. Repression of the candidate tumor suppressor gene S100A2 in breast cancer is mediated by site-specific hypermethylation. Cell Calcium 1997, 22, 243–254. [Google Scholar] [CrossRef]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006, 75, 243–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumitrescu, R.G.; Verma, M. Cancer Epigenetics: Methods and Protocols; Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2021; Volume 863. [Google Scholar]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichol, J.N.; Dupéré-Richer, D.; Ezponda, T.; Licht, J.D.; Miller, W.H., Jr. H3K27 methylation: A focal point of epigenetic deregulation in cancer. Adv. Cancer Res. 2016, 131, 59–95. [Google Scholar] [PubMed] [Green Version]
- Holm, K.; Grabau, D.; Lövgren, K.; Aradottir, S.; Gruvberger-Saal, S.; Howlin, J.; Saal, L.H.; Ethier, S.P.; Bendahl, P.-O.; Stål, O.; et al. Global H3K27 trimethylation and EZH2 abundance in breast tumor subtypes. Mol. Oncol. 2012, 6, 494–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezponda, T.; Licht, J.D. Molecular Pathways: Deregulation of Histone H3 Lysine 27 Methylation in Cancer—Different Paths, Same Destination. Clin. Cancer Res. 2014, 20, 5001–5008. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Lee, H.; Yoon, J.-G.; Madan, A.; Wayner, E.; Tonning, S.; Hothi, P.; Schroeder, B.; Ulasov, I.; Foltz, G.; et al. Global analysis of H3K4me3 and H3K27me3 profiles in glioblastoma stem cells and identification of SLC17A7 as a bivalent tumor suppressor gene. Oncotarget 2015, 6, 5369. [Google Scholar] [CrossRef] [Green Version]
- Yamane, K.; Tateishi, K.; Klose, R.J.; Fang, J.; Fabrizio, L.A.; Erdjument-Bromage, H.; Taylor-Papadimitriou, J.; Tempst, P.; Zhang, Y. PLU-1 Is an H3K4 Demethylase Involved in Transcriptional Repression and Breast Cancer Cell Proliferation. Mol. Cell 2007, 25, 801–812. [Google Scholar] [CrossRef]
- Mungamuri, S.K.; Murk, W.; Grumolato, L.; Bernstein, E.; Aaronson, S.A. Chromatin modifications sequentially enhance ErbB2 expression in ErbB2-positive breast cancers. Cell Rep. 2013, 5, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Slattery, M.L.; John, E.M.; Stern, M.C.; Herrick, J.; Lundgreen, A.; Giuliano, A.R.; Hines, L.; Baumgartner, K.B.; Torres-Mejia, G.; Wolff, R.K. Associations with growth factor genes (FGF1, FGF2, PDGFB, FGFR2, NRG2, EGF, ERBB2) with breast cancer risk and survival: The Breast Cancer Health Disparities Study. Breast Cancer Res. Treat. 2013, 140, 587–601. [Google Scholar] [CrossRef] [Green Version]
- Messier, T.L.; Gordon, J.A.R.; Boyd, J.R.; Tye, C.; Browne, G.; Stein, J.L.; Lian, J.B.; Stein, G.S. Histone H3 lysine 4 acetylation and methylation dynamics define breast cancer subtypes. Oncotarget 2016, 7, 5094–5109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verigos, J.; Karakaidos, P.; Kordias, D.; Papoudou-Bai, A.; Evangelou, Z.; Harissis, H.V.; Klinakis, A.; Magklara, A. The Histone Demethylase LSD1/ΚDM1A Mediates Chemoresistance in Breast Cancer via Regulation of a Stem Cell Program. Cancers 2019, 11, 1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosseini, A.; Minucci, S. A comprehensive review of lysine-specific demethylase 1 and its roles in cancer. Epigenomics 2017, 9, 1123–1142. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, R.; Dell’Aversana, C.; De Marchi, T.; Rotili, D.; Liu, N.Q.; Novakovic, B.; Boccella, S.; Di Maro, S.; Cosconati, S.; Baldi, A.; et al. Inhibition of histone demethylases LSD1 and UTX regulates ERα signaling in breast cancer. Cancers 2019, 11, 2027. [Google Scholar] [CrossRef] [Green Version]
- Boulding, T.; McCuaig, R.D.; Tan, A.; Hardy, K.; Wu, F.; Dunn, J.; Kalimutho, M.; Sutton, C.R.; Forwood, J.; Bert, A.G.; et al. LSD1 activation promotes inducible EMT programs and modulates the tumour microenvironment in breast cancer. Sci. Rep. 2018, 8, 73. [Google Scholar] [CrossRef]
- Lim, S.; Janzer, A.; Becker, A.; Zimmer, A.; Schüle, R.; Buettner, R.; Kirfel, J. Lysine-specific demethylase 1 (LSD1) is highly expressed in ER-negative breast cancers and a biomarker predicting aggressive biology. Carcinogenesis 2010, 31, 512–520. [Google Scholar] [CrossRef]
- Therneau, M.T.; Grambsch, P.M.; Fleming, T. A Package for Survival Analysis in S; Mayo Foundation: Rochester, MN, USA, 1994. [Google Scholar]
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Survminer: Drawing Survival Curves Using ’ggplot2’; R Package Version 0.3; R Core Team: Vienna, Austria, 2017. [Google Scholar]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Ahmad, A.; Azim, S.; Zubair, H.; Khan, M.A.; Singh, S.; Carter, J.E.; Rocconi, R.P.; Singh, A.P. Epigenetic basis of cancer health disparities: Looking beyond genetic differences. Biochim. Biophys. Acta. Rev. Cancer 2017, 1868, 16–28. [Google Scholar] [CrossRef]
- Wei, J.W.; Huang, K.; Yang, C.; Kang, C.S. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [Green Version]
- Nassar, F.; Talhouk, R.; Zgheib, N.K.; Tfayli, A.; El Sabban, M.; El Saghir, N.S.; Boulos, F.; Jabbour, M.N.; Chalala, C.; Boustany, R.-M.; et al. microRNA Expression in Ethnic Specific Early Stage Breast Cancer: An Integration and Comparative Analysis. Sci. Rep. 2017, 7, 16829. [Google Scholar] [CrossRef] [Green Version]
- Lewis, A.K.; Jordan, H.R.; Tollefsbol, T.O. Effects of SAHA and EGCG on Growth Potentiation of Triple-Negative Breast Cancer Cells. Cancers 2018, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munsell, M.F.; Sprague, B.L.; Berry, D.A.; Chisholm, G.; Trentham-Dietz, A. Body mass index and breast cancer risk according to postmenopausal estrogen-progestin use and hormone receptor status. Epidemiol. Rev. Rev. 2014, 36, 114–136. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Cruzata, L.; Zhang, W.; McDonald, J.A.; Tsai, W.Y.; Valdovinos, C.; Falci, L.; Wang, Q.; Crew, K.D.; Santella, R.M.; Hershman, D.L.; et al. Dietary modifications, weight loss, and changes in metabolic markers affect global DNA methylation in Hispanic, African American, and Afro-Caribbean breast cancer survivors. J. Nutr. 2015, 145, 783–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naushad, S.M.; Hussain, T.; Al-Attas, O.S.; Prayaga, A.; Digumarti, R.R.; Gottumukkala, S.R.; Kutala, V.K. Molecular insights into the association of obesity with breast cancer risk: Relevance to xenobiotic metabolism and CpG island methylation of tumor suppressor genes. Mol. Cell. Biochem. 2014, 392, 273–280. [Google Scholar] [CrossRef]
- Wu, J.; Miao, J.; Ding, Y.; Zhang, Y.; Huang, X.; Zhou, X.; Tang, R. MiR-4458 inhibits breast cancer cell growth, migration, and invasiveness by targeting CPSF4. Biochem. Cell Biol. 2019, 97, 722–730. [Google Scholar] [CrossRef]
- Zinn, R.L.; Pruitt, K.; Eguchi, S.; Baylin, S.B.; Herman, J.G. hTERT is expressed in cancer cell lines despite promoter DNA methylation by preservation of unmethylated DNA and active chromatin around the transcription start site. Cancer Res. 2007, 67, 194–201. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.S.; Kala, R.; Tollefsbol, T.O. Mechanisms for the Inhibition of Colon Cancer Cells by Sulforaphane through Epigenetic Modulation of MicroRNA-21 and Human Telomerase Reverse Transcriptase (hTERT) Down-regulation. Curr. Cancer Drug Targets 2018, 18, 97–106. [Google Scholar] [CrossRef]
- Deeb, D.; Gao, X.; Liu, Y.B.; Zhang, Y.; Shaw, J.; Valeriote, F.A.; Gautam, S.C. Inhibition of hTERT in pancreatic cancer cells by pristimerin involves suppression of epigenetic regulators of gene transcription. Oncol. Rep. 2017, 37, 1914–1920. [Google Scholar] [CrossRef]
- Barlési, F.; Giaccone, G.; Gallegos-Ruiz, M.I.; Loundou, A.; Span, S.W.; Lefesvre, P.; Kruyt, F.A.; Rodriguez, J.A. Global histone modifications predict prognosis of resected non–small-cell lung cancer. J. Clin. Oncol. 2007, 25, 4358–4364. [Google Scholar] [CrossRef]
- Sapienza, C.; Issa, J.-P. Diet, nutrition, and cancer epigenetics. Annu. Rev. Nutr. 2016, 36, 665–681. [Google Scholar] [CrossRef]
- Kim, Y. Folate and carcinogenesis: Evidence, mechanisms, and implications. J. Nutr. Biochem. 1999, 10, 66–88. [Google Scholar] [CrossRef]
- Mikol, Y.B.; Hoover, K.L.; Creasia, D.; Poirier, L.A. Hepatocarcinogenesis in rats fed methyl-deficient, amino acid-defined diets. Carcinogenesis 1983, 4, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Larsson, C.S.; Giovannucci, E.; Wolk, A. Folate and Risk of Breast Cancer: A Meta-analysis. JNCI J. Natl. Cancer Inst. 2007, 99, 64–76. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Wang, K.; Ye, F.; Lei, L.; Zhou, Y.; Chen, J.; Zhao, G.; Chang, H. Folate intake and the risk of breast cancer: An up-to-date meta-analysis of prospective studies. Eur. J. Clin. Nutr. 2019, 73, 1657–1660. [Google Scholar] [CrossRef] [PubMed]
- Key, J.; Hodgson, S.; Omar, R.Z.; Jensen, T.K.; Thompson, S.G.; Boobis, A.R.; Davies, D.S.; Elliott, P. Meta-analysis of studies of alcohol and breast cancer with consideration of the methodological issues. Cancer Causes Control 2006, 17, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Davidson, N.E.; Hunter, S.; Yang, X.; Payne-Wilks, K.; Roland, C.L.; Phillips, D.; Bentley, C.; Dai, M.; Williams, S.M. Methyl-group dietary intake and risk of breast cancer among African-American women: A case–control study by methylation status of the estrogen receptor alpha genes. Cancer Causes Control 2003, 14, 827–836. [Google Scholar] [CrossRef]
- Christensen, B.C.; Kelsey, K.T.; Zheng, S.; Houseman, E.A.; Marsit, C.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.; Karagas, M.R.; Kushi, L.; et al. Breast cancer DNA methylation profiles are associated with tumor size and alcohol and folate intake. PLoS Genet. 2010, 6, e1001043. [Google Scholar] [CrossRef] [Green Version]
- Alinejad, V.; Dolati, S.; Motallebnezhad, M.; Yousefi, M. The role of IL17B-IL17RB signaling pathway in breast cancer. Biomed. Pharmacother. 2017, 88, 795–803. [Google Scholar] [CrossRef]
- Niraula, S.; Ocana, A.; Ennis, M.; Goodwin, P.J. Body size and breast cancer prognosis in relation to hormone receptor and menopausal status: A meta-analysis. Breast Cancer Res. Treat. 2012, 134, 769–781. [Google Scholar] [CrossRef]
- Cao, Y.; Hou, L.; Wang, W. Dietary total fat and fatty acids intake, serum fatty acids and risk of breast cancer: A meta-analysis of prospective cohort studies. Int. J. Cancer 2016, 138, 1894–1904. [Google Scholar] [CrossRef]
- Tao, M.-H.; Marian, C.; Nie, J.; Ambrosone, C.; Krishnan, S.S.; Edge, S.B.; Trevisan, M.; Shields, P.G.; Freudenheim, J.L. Body mass and DNA promoter methylation in breast tumors in the Western New York Exposures and Breast Cancer Study1–3. Am. J. Clin. Nutr. 2011, 94, 831–838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, I.N.; Story, M.T.; Nelson, M.C. Neighborhood environments: Disparities in access to healthy foods in the U.S. Am. J. Prev. Med. 2009, 36, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Zenk, S.N.; Schulz, A.J.; Israel, B.A.; James, S.A.; Bao, S.; Wilson, M.L. Fruit and vegetable access differs by community racial composition and socioeconomic position in Detroit, Michigan. Ethn. Dis. 2006, 16, 275–280. [Google Scholar] [PubMed]
- Fong, A.J.; Lafaro, K.; Ituarte, P.H.G.; Fong, Y. Association of Living in Urban Food Deserts with Mortality from Breast and Colorectal Cancer. Ann. Surg. Oncol. 2021, 28, 1311–1319. [Google Scholar] [CrossRef]
- Forshee, A.R.; Storey, M.L.; Ritenbaugh, C. Breast cancer risk and lifestyle differences among premenopausal and post-menopausal African-American women and white women. Cancer 2003, 97 (Suppl. 1), 280–288. [Google Scholar] [CrossRef]
- Yedjou, C.G.; Sims, J.N.; Miele, L.; Noubissi, F.; Lowe, L.; Fonseca, D.D.; Alo, R.A.; Payton, M.; Tchounwou, P.B. Health and Racial Disparity in Breast Cancer. Adv. Exp. Med. Biol. 2019, 1152, 31. [Google Scholar]
- Stolley, M.R.; Sharp, L.; Wells, A.M.; Simon, N.; Schiffer, L. Health Behaviors and Breast Cancer: Experiences of Urban African American Women. Health Educ. Behav. 2006, 33, 604–624. [Google Scholar] [CrossRef]
- Michael, Y.L.; Carlson, N.E.; Chlebowski, R.T.; Aickin, M.; Weihs, K.L.; Ockene, J.K.; Bowen, D.J.; Ritenbaugh, C. Influence of stressors on breast cancer incidence in the Women’s Health Initiative. Health Psychol. Off. J. Div. Health Psychol. Am. Psychol. Assoc. 2009, 28, 137–146. [Google Scholar]
- Lin, Y.; Wang, C.; Zhong, Y.; Huang, X.; Peng, L.; Shan, G.; Wang, K.; Sun, Q. Striking life events associated with primary breast cancer susceptibility in women: A meta-analysis study. J. Exp. Clin. Cancer Res. 2013, 32, 53. [Google Scholar] [CrossRef] [Green Version]
- Chiriac, V.-F.; Baban, A.; Dumitrascu, D.L. Psychological stress and breast cancer incidence: A systematic review. Clujul Med. 2018, 91, 18–26. [Google Scholar] [CrossRef] [Green Version]
- McEwen, B.S. Stress, Adaptation, and Disease: Allostasis and Allostatic Load. Ann. N Y. Acad. Sci. 1998, 840, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Romero, L.M.; Butler, L.K. Endocrinology of Stress. Int. J. Comp. Psychol. 2007, 20, 89–95. [Google Scholar]
- Akinyemiju, T.; Wilson, L.E.; Deveaux, A.; Aslibekyan, S.; Cushman, M.; Gilchrist, S.; Safford, M.; Judd, S.; Howard, V. Asso-ciation of Allostatic Load with All-Cause and Cancer Mortality by Race and Body Mass Index in the REGARDS Cohort. Cancers 2020, 12, 1695. [Google Scholar] [CrossRef] [PubMed]
- Mathew, A.; Doorenbos, A.Z.; Li, H.; Jang, M.K.; Park, C.G.; Bronas, U.G. Allostatic Load in Cancer: A Systematic Review and Mini Meta-Analysis. Biol. Res. Nurs. 2020, 23, 1099800420969898. [Google Scholar] [CrossRef]
- Sephton, S.E.; Sapolsky, R.M.; Kraemer, H.C.; Spiegel, D. Diurnal Cortisol Rhythm as a Predictor of Breast Cancer Survival. JNCI J. Natl. Cancer Inst. 2000, 92, 994–1000. [Google Scholar] [CrossRef]
- Dai, S.; Mo, Y.; Wang, Y.; Xiang, B.; Liao, Q.; Zhou, M.; Li, X.; Li, Y.; Xiong, W.; Li, G.; et al. Chronic Stress Promotes Cancer Development. Front. Oncol. 2020, 10, 10. [Google Scholar] [CrossRef]
- Xing, C.Y.; Doose, M.; Qin, B.; Lin, Y.; Plascak, J.J.; Omene, C.; He, C.; Demissie, K.; Hong, C.-C.; Bandera, E.V.; et al. Predi-agnostic Allostatic Load as a Predictor of Poorly Differentiated and Larger Sized Breast Cancers among Black Women in the Women’s Circle of Health Follow-Up Study. Cancer Epidemiol. Prev. Biomark. 2020, 29, 216–224. [Google Scholar] [CrossRef]
- Sternthal, J.M.; Slopen, N.; Williams, D.R. Racial Disparities in Health. Du Bois Rev. Soc. Sci. Res. Race 2011, 8, 95–113. [Google Scholar] [CrossRef]
- Tomfohr, M.L.; Pung, M.A.; Dimsdale, J.E. Mediators of the relationship between race and allostatic load in African and White Americans. Health Psychol. 2016, 35, 322–332. [Google Scholar] [CrossRef]
- Geronimus, A.T.; Hicken, M.; Keene, D.; Bound, J. “Weathering” and age patterns of allostatic load scores among blacks and whites in the United States. Am. J. Public Health 2006, 96, 826–833. [Google Scholar] [CrossRef]
- Kroenke, C.H.; Kubzansky, L.D.; Schernhammer, E.S.; Holmes, M.D.; Kawachi, I. Social networks, social support, and survival after breast cancer diagnosis. J. Clin. Oncol. 2006, 24, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, P.; Boyd, P.T.; Blacklow, R.S.; Jackson, J.S.; Greenberg, R.S.; Austin, D.F.; Chen, V.W.; Edwards, B.K. The relationship between social ties and survival among black and white breast cancer patients. National Cancer Institute Black/White Cancer Survival Study Group. Cancer Epidemiol. Prev. Biomark. 1994, 3, 253–259. [Google Scholar]
- Weinstock, M.; Matlina, E.; Maor, G.I.; Rosen, H.; McEwen, B.S. Prenatal stress selectively alters the reactivity of the hypo-thalamic-pituitary adrenal system in the female rat. Brain Res. 1992, 595, 195–200. [Google Scholar] [CrossRef]
- Louvart, H.; Maccari, S.; Darnaudery, M. Prenatal stress affects behavioral reactivity to an intense stress in adult female rats. Brain Res. 2005, 1031, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.G.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef]
- Shields, A.E.; Wise, L.A.; Ruiz-Narvaez, E.A.; Seddighzadeh, B.; Byun, H.; Cozier, Y.C.; Rosenberg, L.; Palmer, J.R.; Baccarelli, A.A. Childhood abuse, promoter methylation of leukocyte NR3C1 and the potential modifying effect of emotional support. Epigenomics 2016, 8, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nesset, A.K.; Perri, A.M.; Mueller, C.R. Frequent promoter hypermethylation and expression reduction of the glucocorticoid receptor gene in breast tumors. Epigenetics 2014, 9, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Melas, P.A.; Wei, Y.; Wong, C.C.Y.; Sjöholm, L.K.; Åberg, E.; Mill, J.; Schalling, M.; Forsell, Y.; Lavebratt, C. Genetic and ep-igenetic associations of MAOA and NR3C1 with depression and childhood adversities. Int. J. Neuropsychopharmacol. 2013, 16, 1513–1528. [Google Scholar] [CrossRef] [Green Version]
- Pan, D.; Kocherginsky, M.; Conzen, S.D. Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res. 2011, 71, 6360–6370. [Google Scholar] [CrossRef] [Green Version]
- Needham, B.L.; Smith, J.A.; Zhao, W.; Wang, X.; Mukherjee, B.; Kardia, S.L.R.; Shively, C.A.; Seeman, T.E.; Liu, Y.; Roux, A.V.D. Life course socioeconomic status and DNA methylation in genes related to stress reactivity and inflammation: The multiethnic study of atherosclerosis. Epigenetics 2015, 10, 958–969. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.A.; Zhao, W.; Wang, X.; Ratliff, S.M.; Mukherjee, B.; Kardia, S.L.R.; Liu, Y.; Roux, A.V.D.; Needham, B.L. Neighborhood characteristics influence DNA methylation of genes involved in stress response and inflammation: The Multi-Ethnic Study of Atherosclerosis. Epigenetics 2017, 12, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lou, Z.; Wang, L. The role of FKBP5 in cancer aetiology and chemoresistance. Br. J. Cancer 2011, 104, 19–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gwynne, W.D.; Hallett, R.M.; Girgis-Gabardo, A.; Bojovic, B.; Dvorkin-Gheva, A.; Aarts, C.; Dias, K.; Bane, A.; Hassell, J.A. Serotonergic system antagonists target breast tumor initiating cells and synergize with chemotherapy to shrink human breast tumor xenografts. Oncotarget 2017, 8, 32101–32116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hix, L.M.; Shi, Y.H.; Brutkiewicz, R.R.; Stein, P.L.; Wang, C.-R.; Zhang, M. CD1d-Expressing Breast Cancer Cells Modulate NKT Cell-Mediated Antitumor Immunity in a Murine Model of Breast Cancer Metastasis. PLoS ONE 2011, 6, e20702. [Google Scholar] [CrossRef] [PubMed]
- Kuehnemuth, B.; Piseddu, I.; Wiedemann, G.; Lauseker, M.; Kuhn, C.; Hofmann, S.; Schmoeckel, E.; Endres, S.; Mayr, D.; Jeschke, U.; et al. CCL1 is a major regulatory T cell attracting factor in human breast cancer. BMC Cancer 2018, 18, 1278. [Google Scholar] [CrossRef]
- Sahay, D.; Terry, M.B.; Miller, R. Is breast cancer a result of epigenetic responses to traffic-related air pollution? A review of the latest evidence. Epigenomics 2019, 11, 701–714. [Google Scholar] [CrossRef]
- Lewis-Michl, E.L.; Melius, J.M.; Kallenbach, L.R.; Ju, C.L.; Talbot, T.O.; Orr, M.F. Breast cancer risk and residence near industry or traffic in Nassau and Suffolk Counties, Long Island, New York. Arch. Environ. Health Int. J. 1996, 51, 255–265. [Google Scholar] [CrossRef]
- Wei, Y.; Davis, J.; Bina, W.F. Ambient air pollution is associated with the increased incidence of breast cancer in US. Int. J. Environ. Health Res. 2012, 22, 12–21. [Google Scholar] [CrossRef]
- Arcaro, K.F. Antiestrogenicity of environmental polycyclic aromatic hydrocarbons in human breast cancer cells. Toxicology 1999, 133, 115–127. [Google Scholar] [CrossRef]
- Choe, S.-Y.; Kim, S.-J.; Kim, H.-G.; Lee, J.H.; Choi, Y.; Lee, H.; Kim, Y. Evaluation of estrogenicity of major heavy metals. Sci. Total Environ. 2003, 312, 15–21. [Google Scholar] [CrossRef]
- Natarajan, R.; Aljaber, D.; Au, D.; Thai, C.; Sanchez, A.; Nunez, A.; Resto, C.; Chavez, T.; Jankowska, M.M.; Benmarhnia, T.; et al. Environmental exposures during puberty: Window of breast cancer risk and epigenetic damage. Int. J. Environ. Res. Public Health 2020, 17, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohai, P.; Lantz, P.M.; Morenoff, J.; House, J.S.; Mero, R.P. Racial and socioeconomic disparities in residential proximity to polluting industrial facilities: Evidence from the Americans’ Changing Lives Study. Am. J. Public Health 2009, 99 (Suppl. 3), S649–S656. [Google Scholar] [CrossRef] [PubMed]
- Woo, B.; Kravitz-Wirtz, N.; Sass, V.; Crowder, K.; Teixeira, S.; Takeuchi, D.T. Residential Segregation and Racial/Ethnic Dis-parities in Ambient Air Pollution. Race Soc. Probl. 2019, 11, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, J.; Zandbergen, P.A. Children at risk: Measuring racial/ethnic disparities in potential exposure to air pollution at school and home. J. Epidemiol. Community Health 2007, 61, 1074–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmuel, S.; White, A.J.; Sandler, D.P. Residential Exposure to Vehicular Traffic-Related Air Pollution During Childhood and Breast Cancer Risk. Environ. Res. 2017, 159, 257–263. [Google Scholar] [CrossRef]
- White, A.J.; O’Brien, K.M.; Niehoff, N.M.; Carroll, R.; Sandler, D.P. Metallic Air Pollutants and Breast Cancer Risk in a Na-tionwide Cohort Study. Epidemiology 2019, 30, 20–28. [Google Scholar] [CrossRef]
- Kresovich, J.K.; Erdal, S.; Chen, H.Y.; Gann, P.H.; Argos, M.; Rauscher, G.H. Metallic air pollutants and breast cancer heter-ogeneity. Environ. Res. 2019, 177, 108639. [Google Scholar] [CrossRef]
- Liu, R.; Nelson, D.; Hurley, S.; Hertz, A.; Reynolds, P. Residential Exposure to Estrogen Disrupting Hazardous Air Pollutants and Breast Cancer Risk: The California Teachers Study. Epidemiology 2015, 26, 365–373. [Google Scholar] [CrossRef]
- Ionescu, J.G.; Novotny, J.; Stejskal, V.; Lätsch, A.; Blaurock-Busch, E.; Eisenmann-Klein, M. Increased levels of transition metals in breast cancer tissue. Neuro Endocrinol. Lett. 2006, 27 (Suppl. 1), 36–39. [Google Scholar]
- Hanna, C.W.; Bloom, M.S.; Robinson, W.; Kim, D.; Parsons, P.; Saal, F.S.V.; Taylor, J.A.; Steuerwald, A.J.; Fujimoto, V.Y. DNA methylation changes in whole blood is associated with exposure to the environmental contaminants, mercury, lead, cadmium and bisphenol A, in women undergoing ovarian stimulation for IVF. Hum. Reprod. 2012, 27, 1401–1410. [Google Scholar] [CrossRef]
- Yu, K.-D.; Fan, L.; Di, G.-H.; Yuan, W.-T.; Zheng, Y.; Huang, W.; Chen, A.-X.; Yang, C.; Wu, J.; Shen, Z.-Z.; et al. Genetic variants in GSTM3 gene within GSTM4-GSTM2-GSTM1-GSTM5-GSTM3 cluster influence breast cancer susceptibility depending on GSTM1. Breast Cancer Res. Treat. 2010, 121, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Sadikovic, B.; Rodenhiser, D.I. Benzopyrene exposure disrupts DNA methylation and growth dynamics in breast cancer cells. Toxicol. Appl. Pharmacol. 2006, 216, 458–468. [Google Scholar] [CrossRef] [PubMed]
- White, A.J.; Chen, J.; Teitelbaum, S.L.; McCullough, L.E.; Xu, X.; Cho, Y.H.; Conway, K.; Beyea, J.; Stellman, S.D.; Steck, S.E.; et al. Sources of polycyclic aromatic hydrocarbons are associated with gene-specific promoter methylation in women with breast cancer. Environ. Res. 2016, 145, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, M.S.; Vazquez, A.; Kulkarni, D.A.; Kerrigan, J.E.; Atwal, G.; Metsugi, S.; Toppmeyer, D.L.; Levine, A.J.; Hirshfield, K.M. Polymorphic variants in TSC1 and TSC2 and their association with breast cancer phenotypes. Breast Cancer Res. Treat. 2011, 125, 861–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callahan, C.L.; Bonner, M.R.; Nie, J.; Han, D.; Wang, Y.; Tao, M.-H.; Shields, P.G.; Marian, C.; Eng, K.H.; Trevisan, M.; et al. Lifetime exposure to ambient air pollution and methylation of tumor suppressor genes in breast tumors. Environ. Res. 2018, 161, 418–424. [Google Scholar] [CrossRef]
- Keramatinia, A.; Hassanipour, S.; Nazarzadeh, M.; Wurtz, M.; Monfared, A.B.; Khayyamzadeh, M.; Bidel, Z.; Mhrvar, N.; Mosavi-Jarrahi, A. Correlation Between Nitrogen Dioxide as an Air Pollution Indicator and Breast Cancer: A Systematic Review and Meta-Analysis. Asian Pac. J. Cancer Prev. 2016, 17, 419–424. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, M.S.; Villeneuve, P.J.; Crouse, D.; To, T.; Weichenthal, S.A.; Wall, C.; Miller, A.B. Associations between incident breast cancer and ambient concentrations of nitrogen dioxide from a national land use regression model in the Canadian National Breast Screening Study. Environ. Int. 2019, 133, 105182. [Google Scholar] [CrossRef]
- Plusquin, M.; Guida, F.; Polidoro, S.; Vermeulen, R.; Raaschou-Nielsen, O.; Campanella, G.; Hoek, G.; Kyrtopoulos, S.; Georgiadis, P.; Naccarati, A.; et al. DNA methylation and exposure to ambient air pollution in two prospective cohorts. Environ. Int. 2017, 108, 127–136. [Google Scholar] [CrossRef]
- Chukkapalli, S.; Amessou, M.; Dilly, A.K.; Dekhil, H.; Zhao, J.; Liu, Q.; Bejna, A.; Thomas, R.D.; Bandyopadhyay, S.; Bismar, T.A.; et al. Role of the EphB2 receptor in autophagy, apoptosis and invasion in human breast cancer cells. Exp. Cell Res. 2014, 320, 233–246. [Google Scholar] [CrossRef]
- Husa, A.-M.; Magić, Ž.; Larsson, M.; Fornander, T.; Pérez-Tenorio, G. EPH/ephrin profile and EPHB2 expression predicts patient survival in breast cancer. Oncotarget 2016, 7, 21362–21380. [Google Scholar] [CrossRef] [Green Version]
- Hecht, S.S. Tobacco smoke carcinogens and breast cancer. Environ. Mol. Mutagen. 2002, 39, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Band, P.R.; Le, N.D.; Fang, R.; Deschamps, M. Carcinogenic and endocrine disrupting effects of cigarette smoke and risk of breast cancer. Lancet 2002, 360, 1044–1049. [Google Scholar] [CrossRef]
- Macacu, A.; Autier, P.; Boniol, M.; Boyle, P. Active and passive smoking and risk of breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2015, 154, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabat, G.C.; Kim, M.; Phipps, A.I.; Li, C.I.; Messina, C.R.; Wactawski-Wende, J.; Kuller, L.; Simon, M.S.; Yasmeen, S.; Was-sertheil-Smoller, S.; et al. Smoking and alcohol consumption in relation to risk of triple-negative breast cancer in a cohort of postmenopausal women. Cancer Causes Control CCC 2011, 22, 775–783. [Google Scholar] [CrossRef]
- Kawai, M.; Malone, K.E.; Tang, M.T.C.; Li, C.I. Active smoking and risk of estrogen receptor positive and triple-negative breast cancer among women 20–44 years of age. Cancer 2014, 120, 1026–1034. [Google Scholar] [CrossRef] [Green Version]
- Bérubé, S.; Lemieux, J.; Moore, L.; Maunsell, E.; Brisson, J. Smoking at time of diagnosis and breast cancer-specific survival: New findings and systematic review with meta-analysis. Breast Cancer Res. BCR 2014, 16, R42. [Google Scholar] [CrossRef] [Green Version]
- Parada, H.; Sun, X.; Tse, C.-K.; Olshan, A.F.; Troester, M.A.; Conway, K. Active smoking and survival following breast cancer among African-American and non-African-American women in the Carolina Breast Cancer Study. Cancer Causes Control CCC 2017, 28, 929–938. [Google Scholar] [CrossRef]
- Jamal, A.; King, B.A.; Neff, L.J.; Whitmill, J.; Babb, S.D.; Graffunder, C.M. Current Cigarette Smoking Among Adults—United States, 2005–2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 1205–1211. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Jia, M.; Zhang, Y.; Breitling, L.P.; Brenner, H. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: A systematic review of DNA methylation studies. Clin. Epigenetics 2015, 7, 113. [Google Scholar] [CrossRef] [Green Version]
- Shenker, N.S.; Polidoro, S.; van Veldhoven, K.; Sacerdote, C.; Ricceri, F.; Birrell, M.A.; Belvisi, M.G.; Brown, R.; Vineis, P.; Flanagan, J.M. Epigenome-wide association study in the European Prospective Investigation into Cancer and Nutrition (EPIC-Turin) identifies novel genetic loci associated with smoking. Hum. Mol. Genet. 2013, 22, 843–851. [Google Scholar] [CrossRef] [Green Version]
- Conway, K.; Edmiston, S.N.; Parrish, E.; Bryant, C.; Tse, C.-K.; Swift-Scanlan, T.; McCullough, L.E.; Kuan, P.F. Breast tumor DNA methylation patterns associated with smoking in the Carolina Breast Cancer Study. Breast Cancer Res. Treat. 2017, 163, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-X.; Knyazev, P.G.; Cheburkin, Y.V.; Sharma, K.; Knyazev, Y.P.; Őrfi, L.; Szabadkai, I.; Daub, H.; Kéri, G.; Ullrich, A. AXL Is a Potential Target for Therapeutic Intervention in Breast Cancer Progression. Cancer Res. 2008, 68, 1905–1915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breton, C.V.; Byun, H.-M.; Wenten, M.; Pan, F.; Yang, A.; Gilliland, F.D. Prenatal tobacco smoke exposure affects global and gene-specific DNA methylation. Am. J. Respir. Crit. Care Med. 2009, 180, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, H.E.; Crott, J.W.; Ghandour, H.; E Dallal, G.; Choi, S.-W.; Keyes, M.K.; Jang, H.; Liu, Z.; Nadeau, M.; Johnston, A.; et al. Chronic cigarette smoking is associated with diminished folate status, altered folate form distribution, and increased genetic damage in the buccal mucosa of healthy adults. Am. J. Clin. Nutr. 2006, 83, 835–841. [Google Scholar] [CrossRef]
- Vardavas, C.I.; Linardakis, M.K.; Hatzis, C.M.; Malliaraki, N.; Saris, W.H.; Kafatos, A.G. Smoking status in relation to serum folate and dietary vitamin intake. Tob. Induc. Dis. 2008, 4, 8. [Google Scholar] [CrossRef] [Green Version]
- Shrubsole, M.J.; Jin, F.; Dai, Q.; O Shu, X.; Potter, J.; Hebert, J.R.; Gao, Y.T.; Zheng, W. Dietary Folate Intake and Breast Cancer Risk: Results from the Shanghai Breast Cancer Study. Cancer Res. 2001, 61, 7136–7141. [Google Scholar] [CrossRef] [Green Version]

| Tumor Suppressor Genes | Regular Function | Effect after Hypermethylation | Comments | References |
|---|---|---|---|---|
| RARB |
|
|
| [24] |
| CDH13 |
|
|
| [24] |
| RASSF1 |
|
|
| [24] |
| SCGB3A |
|
|
| [21] |
| TWIST1 |
|
|
| [21,25] |
| CCND1 |
|
|
| [21] |
| SFRP1 |
|
|
| [26] |
| TUSC3 |
|
|
| [27] |
| TES |
|
|
| [27] |
| Genes | p Value | Hypermethylation Higher in | Reported Function in Cancer | Reference |
|---|---|---|---|---|
| ENPP2 | 0.00456 | EA |
| [45,46] |
| SPAG6 | 0.0060803 | AA |
| NA |
| ADAMTS12 | 0.03684 | EA |
| [45,46] |
| CCDC36 | 0.00326 | AA |
| NA |
| ADCY4 | 0.00514 | EA |
| [47] |
| JCHAIN | 0.0104 | EA |
| NA |
| ZNF208 | 0.00058 | AA |
| [48] |
| CFH | 0.0073846 | EA |
| [45,46] |
| SSTR4 | 9.94 × 10−5 | AA |
| [49] |
| LTC4S | 0.01162 | EA |
| [50] |
| SLC2A14 | 2.00 × 10−5 | AA |
| [45,46] |
| SOSTDC1 | 0.00166 | EA |
| [51,52] |
| DNM3 | 0.0118419 | EA |
| [51,52] |
| TAGLN | 2.97 × 10−6 | EA |
| [53] |
| FGF2 | 0.01886 | EA |
| [51,52] |
| AKR7L | 0.00012 | AA |
| NA |
| CDH5 | 0.03354 | EA |
| [51,52] |
| ZIC1 | 0.00666 | EA |
| [54] |
| TANK/TRAF2 | 0.01394 | EA |
| [55] |
| NKAPL | 3.67 × 10−6 | AA |
| [56] |
| SYN2 | 0.00164 | EA |
| NA |
| NYNRIN | 0.00022 | AA |
| NA |
| CHST4 | 0.00358 | EA |
| NA |
| COL11A1 | 0.039 | AA |
| [57] |
| TMEM204 | 3.70 × 10−6 | EA |
| NA |
| TRABD | 0.01552 | EA |
| NA |
| SLC10A4 | 0.01166 | EA |
| NA |
| RIPPLY3 | 0.00072 | AA |
| NA |
| PPBP/CXCL7 | 0.0364 | EA |
| [58] |
| TIMP3 | 0.03268 | EA |
| [59] |
| DSC2 | 0.00672 | EA |
| [60] |
| TUSC3 | 3.47 × 10−8 | EA |
| [61] |
| MT1G | 0.0378 | AA |
| [62] |
| Genes | p Value | Hypomethylation Higher in | Reported Function in Cancer | Reference |
|---|---|---|---|---|
| MACC1 | 0.00758 | EA |
| [63] |
| KIR3DX1 | 0.00452 | EA |
| NA |
| LCP2 | 0.01498 | AA |
| [64] |
| MIR1283-1 | 0.0185 | AA |
| NA |
| UGT2B11 | 3.97 × 10−6 | AA |
| [65] |
| DENND2D | 0.01004 | AA |
| [66] |
| ASB12 | 0.032 | AA |
| The human protein atlas |
| OR8J3 | 0.03028 | AA |
| The human protein atlas |
| BCL10 | 0.04469 | AA |
| [67] |
| HHIPL2 | 0.0261 | AA |
| [51,52] |
| OR5B17 | 2.03 × 10−5 | AA |
| NA |
| CIB3 | 0.00180324 | AA |
| NA |
| TRIM62 | 0.0007 | AA |
| [68] |
| CLSTN1 | 0.027603 | AA |
| [51,52] |
| MLH1 | 0.04104 | EA |
| [69] |
| AXL | 8.29 × 10−5 | AA |
| [70] |
| S100A2 | 0.0004414 | EA |
| [71] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Joshi, S.; Garlapati, C.; Aneja, R. Epigenetic Determinants of Racial Disparity in Breast Cancer: Looking beyond Genetic Alterations. Cancers 2022, 14, 1903. https://doi.org/10.3390/cancers14081903
Joshi S, Garlapati C, Aneja R. Epigenetic Determinants of Racial Disparity in Breast Cancer: Looking beyond Genetic Alterations. Cancers. 2022; 14(8):1903. https://doi.org/10.3390/cancers14081903
Chicago/Turabian StyleJoshi, Shriya, Chakravarthy Garlapati, and Ritu Aneja. 2022. "Epigenetic Determinants of Racial Disparity in Breast Cancer: Looking beyond Genetic Alterations" Cancers 14, no. 8: 1903. https://doi.org/10.3390/cancers14081903
APA StyleJoshi, S., Garlapati, C., & Aneja, R. (2022). Epigenetic Determinants of Racial Disparity in Breast Cancer: Looking beyond Genetic Alterations. Cancers, 14(8), 1903. https://doi.org/10.3390/cancers14081903