
Impact of Lipid Metabolism on Antitumor Immune Response

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Lipid Metabolism in Myeloid Cells
2.1. Lipid Metabolism in Macrophages
2.2. Lipid Metabolism in Dendritic Cells
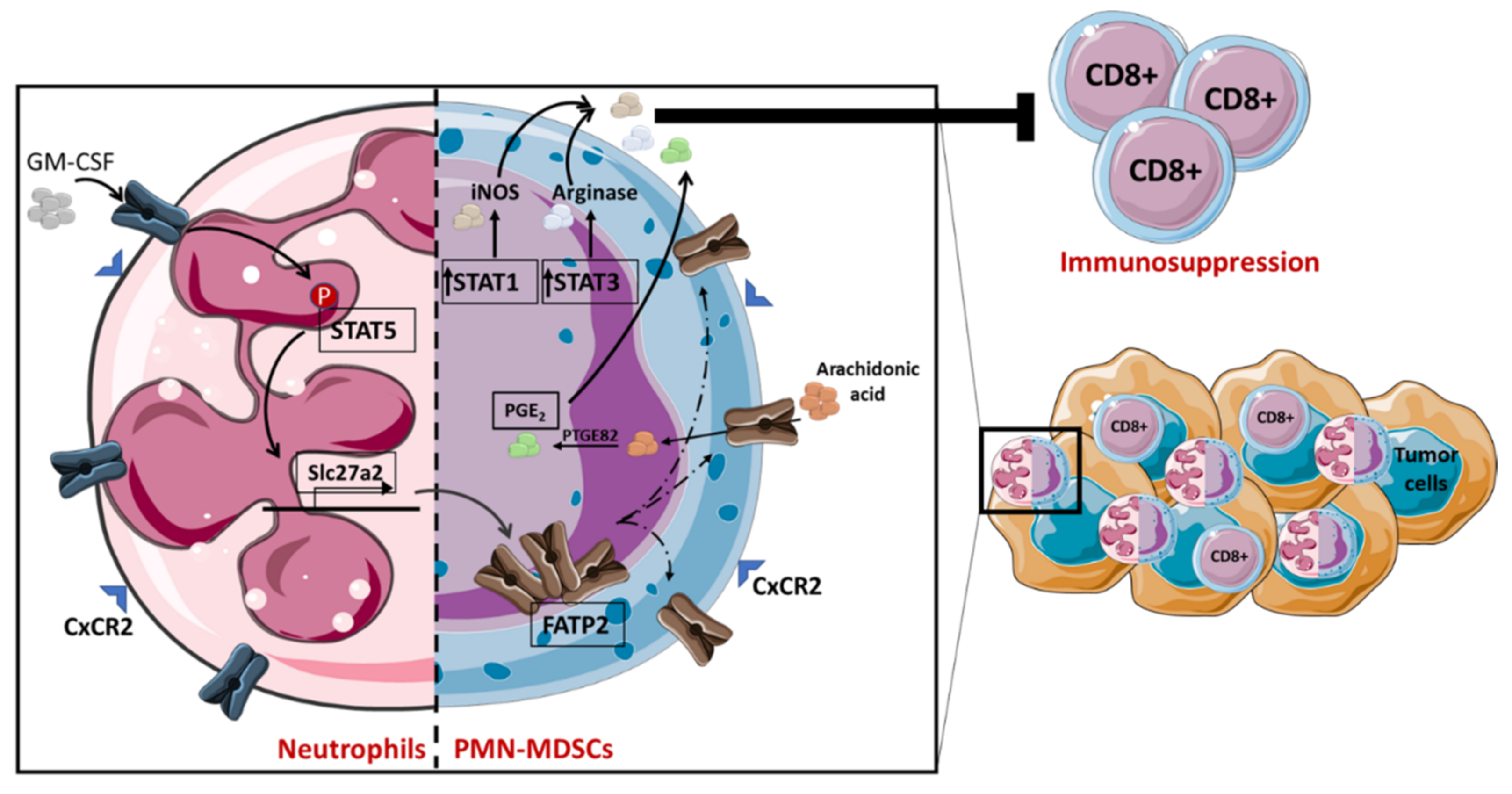
2.3. Lipid Metabolism in MDSC
3. Lipid Metabolism in Adaptive Immunity
3.1. Role of Lipid Metabolism in T-Cell Activation
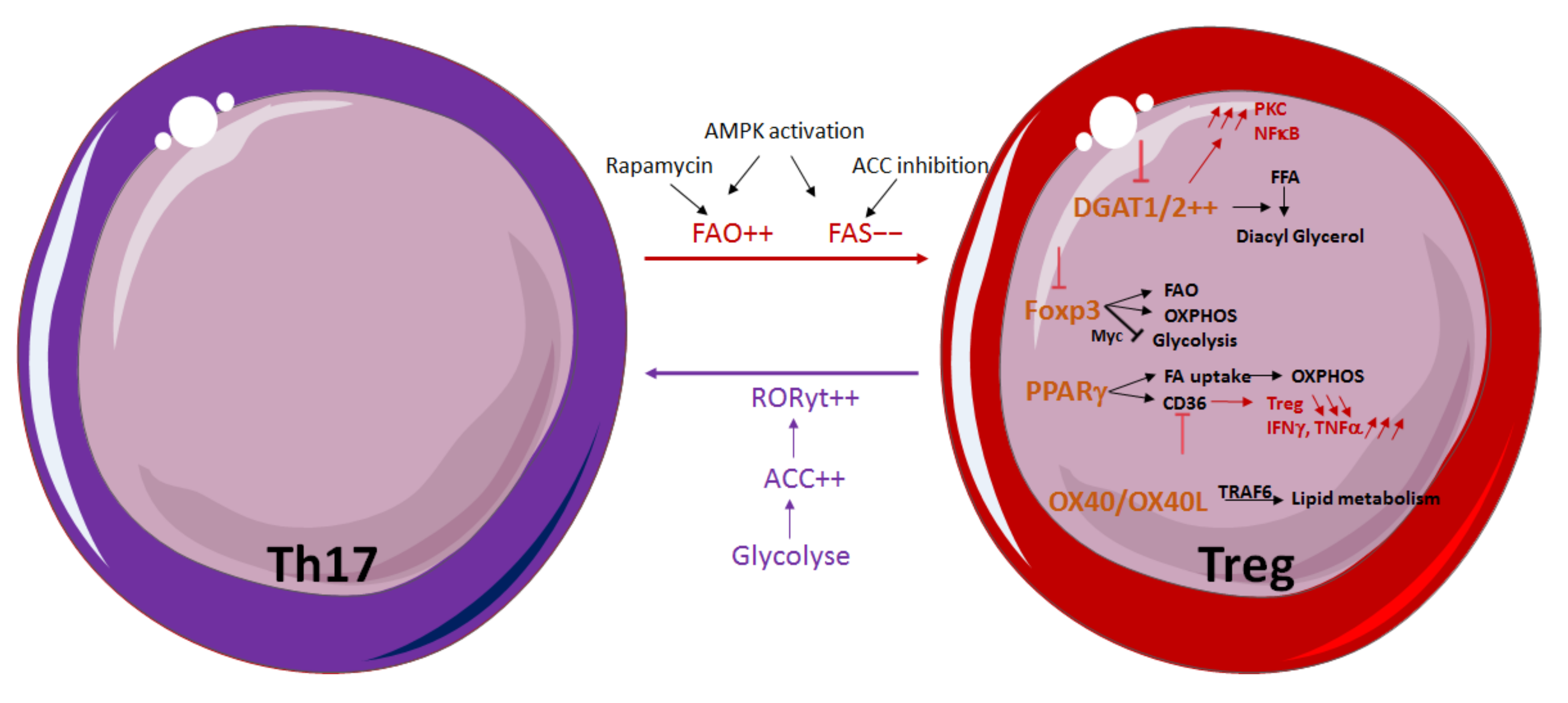
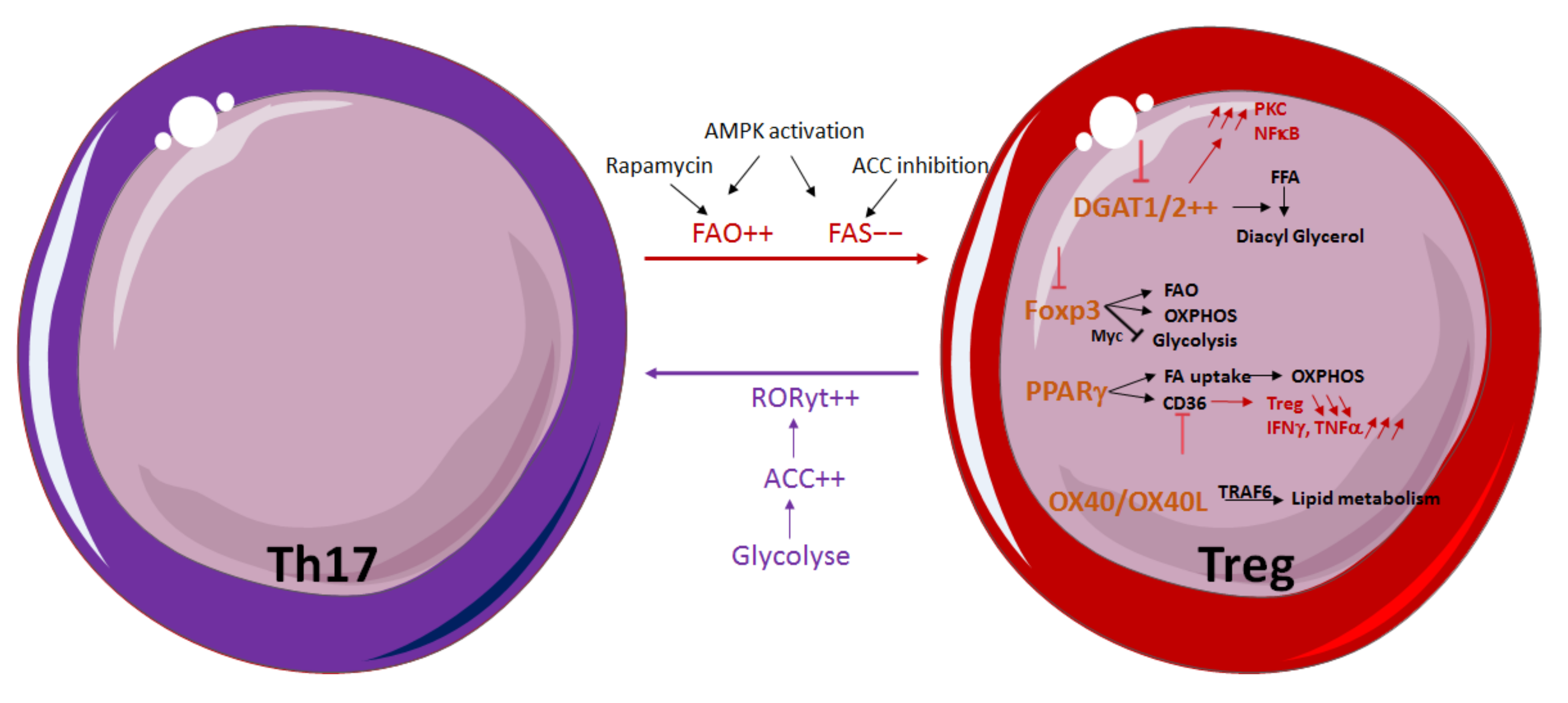
3.2. Influence of Lipid Metabolism on T-Cell Differentiation and Function
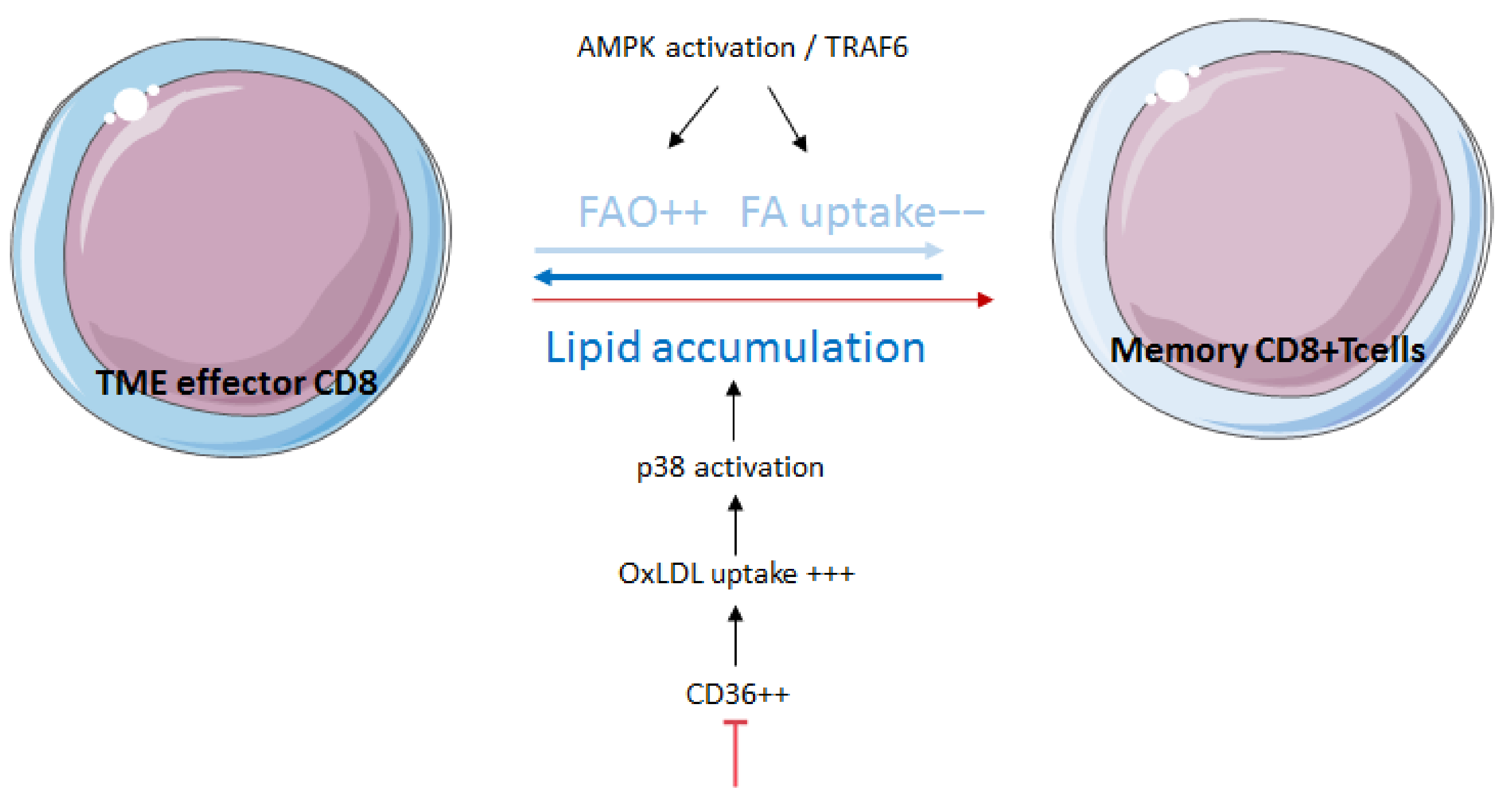
3.3. Fatty-Acid Oxidation Influences Memory and Exhaustion of T Cells
3.4. Influence of Lipid Metabolism on B Cells
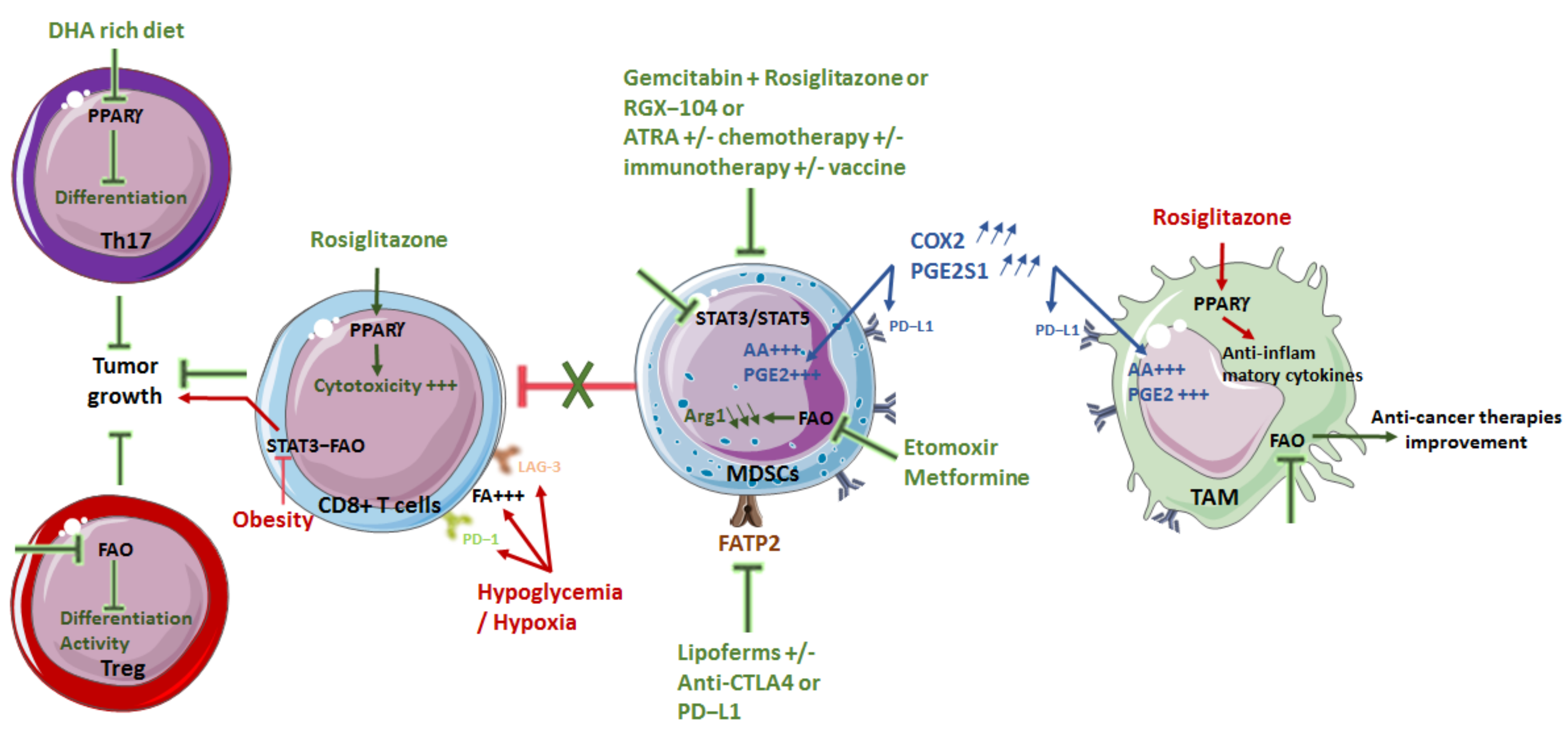
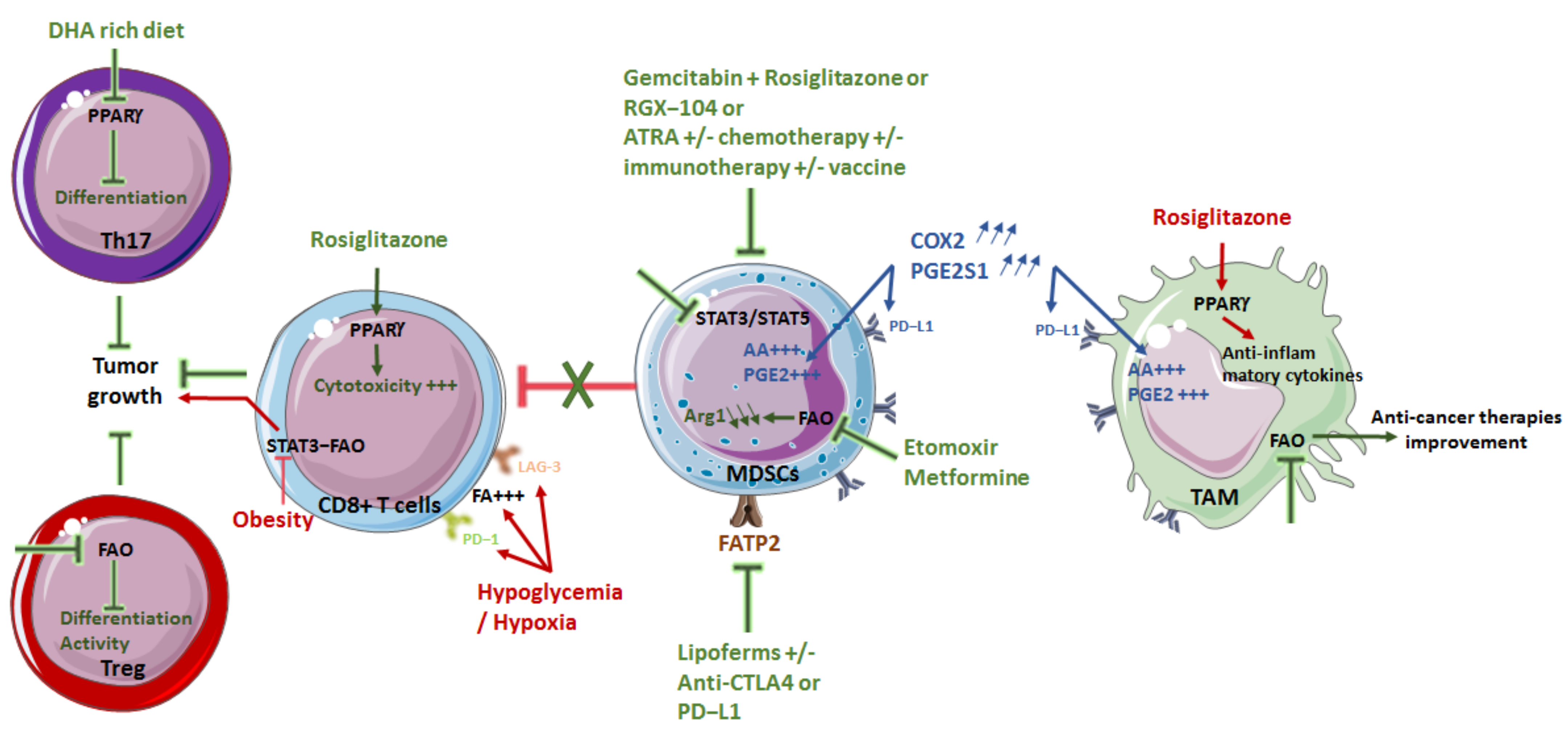
4. Targeting Lipid Metabolism as a Therapeutic Strategy
4.1. Limiting Macrophage Pro-Tumor Effect
4.2. Diminishing MDSC Immunosuppression
4.3. Modulating T Cells
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and Lymphocytes Prevent Primary Tumour Development and Shape Tumour Immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, Immunity and Cancer Immunoediting. Nat. Reviews. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Fendt, S.M.; Frezza, C.; Erez, A. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov 2020, 10, 1797–1807. [Google Scholar] [CrossRef] [PubMed]
- Hensley, C.T.; Faubert, B.; Yuan, Q.; Lev-Cohain, N.; Jin, E.; Kim, J.; Jiang, L.; Ko, B.; Skelton, R.; Loudat, L.; et al. Metabolic Heterogeneity in Human Lung Tumors. Cell 2016, 164, 681–694. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, S.; Glauben, R. Fatty Acid Metabolism in Myeloid-Derived Suppressor Cells and Tumor-Associated Macrophages: Key Factor in Cancer Immune Evasion. Cancers 2022, 14, 250. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Oyang, L.; Lin, J.; Tan, S.; Han, Y.; Wu, N.; Yi, P.; Tang, L.; Pan, Q.; Rao, S.; et al. The Cancer Metabolic Reprogramming and Immune Response. Mol. Cancer 2021, 20, 28. [Google Scholar] [CrossRef]
- Najafi, M.; Hashemi Goradel, N.; Farhood, B.; Salehi, E.; Nashtaei, M.S.; Khanlarkhani, N.; Khezri, Z.; Majidpoor, J.; Abouzaripour, M.; Habibi, M.; et al. Macrophage Polarity in Cancer: A Review. J. Cell. Biochem. 2019, 120, 2756–2765. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.V.; Connors, T.J.; Farber, D.L. Human T Cell Development, Localization, and Function throughout Life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-Derived Suppressor Cells as Regulators of the Immune System. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Greten, T.F.; Manns, M.P.; Korangy, F. Myeloid Derived Suppressor Cells in Human Diseases. Int. Immunopharmacol. 2011, 11, 802–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, D.; Adeshakin, A.O.; Xu, M.; Afolabi, L.O.; Zhang, G.; Chen, Y.H.; Wan, X. Lipid Metabolic Pathways Confer the Immunosuppressive Function of Myeloid-Derived Suppressor Cells in Tumor. Front. Immunol. 2019, 10, 1399. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K. Metabolic Reprogramming of Immune Cells in Cancer Progression. Immunity 2015, 43, 435–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remmerie, A.; Scott, C.L. Macrophages and Lipid Metabolism. Cell Immunol. 2018, 330, 27–42. [Google Scholar] [CrossRef]
- Ramel, E.; Lillo, S.; Daher, B.; Fioleau, M.; Daubon, T.; Saleh, M. The Metabolic Control of Myeloid Cells in the Tumor Microenvironment. Cells 2021, 10, 2960. [Google Scholar] [CrossRef]
- Odegaard, J.I.; Chawla, A. Alternative Macrophage Activation and Metabolism. Annu. Rev. Pathol. 2011, 6, 275–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varol, C.; Mildner, A.; Jung, S. Macrophages: Development and Tissue Specialization. Annu. Rev. Immunol. 2015, 33, 643–675. [Google Scholar] [CrossRef]
- Biswas, S.K.; Allavena, P.; Mantovani, A. Tumor-Associated Macrophages: Functional Diversity, Clinical Significance, and Open Questions. Semin. Immunopathol. 2013, 35, 585–600. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-Associated Macrophages: An Accomplice in Solid Tumor Progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, V.; Pradhan, P.; Braud, L.; Fuchs, H.R.; Gueler, F.; Motterlini, R.; Foresti, R.; Immenschuh, S. Human and Murine Macrophages Exhibit Differential Metabolic Responses to Lipopolysaccharide—A Divergent Role for Glycolysis. Redox Biol. 2019, 22, 101147. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.K.; Mantovani, A. Orchestration of Metabolism by Macrophages. Cell Metab. 2012, 15, 432–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Liu, R.; Yu, Q.; Dong, L.; Bi, Y.; Liu, G. Metabolic Reprogramming of Macrophages during Infections and Cancer. Cancer Lett. 2019, 452, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-Intrinsic Lysosomal Lipolysis Is Essential for Alternative Activation of Macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kume, N.; Moriwaki, H.; Kataoka, H.; Minami, M.; Murase, T.; Sawamura, T.; Masaki, T.; Kita, T. Inducible Expression of LOX-1, a Novel Receptor for Oxidized LDL, in Macrophages and Vascular Smooth Muscle Cells. Ann. N. Y. Acad. Sci. 2000, 902, 323–327. [Google Scholar] [CrossRef]
- Su, P.; Wang, Q.; Bi, E.; Ma, X.; Liu, L.; Yang, M.; Qian, J.; Yi, Q. Enhanced Lipid Accumulation and Metabolism Are Required for the Differentiation and Activation of Tumor-Associated Macrophages. Cancer Res. 2020, 80, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Mosca, M.; Polentarutti, N.; Mangano, G.; Apicella, C.; Doni, A.; Mancini, F.; De Bortoli, M.; Coletta, I.; Polenzani, L.; Santoni, G.; et al. Regulation of the Microsomal Prostaglandin E Synthase-1 in Polarized Mononuclear Phagocytes and Its Constitutive Expression in Neutrophils. J. Leukoc. Biol. 2007, 82, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.; Reilly, S.M.; Karabacak, V.; Gangl, M.R.; Fitzgerald, K.; Hatano, B.; Lee, C.H. Adipocyte-derived Th2 Cytokines and Myeloid PPARdelta Regulate Macrophage Polarization and Insulin Sensitivity. Cell Metab 2008, 7, 485–495. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 Activation of Kupffer Cells by PPARdelta Ameliorates Obesity-Induced Insulin Resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, E.L.; Pearce, E.J. Metabolic Pathways in Immune Cell Activation and Quiescence. Immunity 2013, 38, 633–643. [Google Scholar] [CrossRef] [Green Version]
- Thomas, G.D.; Ruckerl, D.; Maskrey, B.H.; Whitfield, P.D.; Blaxter, M.L.; Allen, J.E. The Biology of Nematode- and IL4Ralpha-Dependent Murine Macrophage Polarization in Vivo as Defined by RNA-Seq and Targeted Lipidomics. Blood 2012, 120, e93–e104. [Google Scholar] [CrossRef] [Green Version]
- Porta, C.; Marino, A.; Consonni, F.M.; Bleve, A.; Mola, S.; Storto, M.; Riboldi, E.; Sica, A. Metabolic Influence on the Differentiation of Suppressive Myeloid Cells in Cancer. Carcinogenesis 2018, 39, 1095–1104. [Google Scholar] [CrossRef] [Green Version]
- Spann, N.J.; Garmire, L.X.; McDonald, J.G.; Myers, D.S.; Milne, S.B.; Shibata, N.; Reichart, D.; Fox, J.N.; Shaked, I.; Heudobler, D.; et al. Regulated Accumulation of Desmosterol Integrates Macrophage Lipid Metabolism and Inflammatory Responses. Cell 2012, 151, 138–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Wynn, T.A. Obstacles and Opportunities for Understanding Macrophage Polarization. J. Leukoc. Biol. 2011, 89, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.I.; Celis, E.; Lennox, B.; et al. Lipid Accumulation and Dendritic Cell Dysfunction in Cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, R.; Tyurin, V.A.; Veglia, F.; Condamine, T.; Amoscato, A.; Mohammadyani, D.; Johnson, J.J.; Zhang, L.M.; Klein-Seetharaman, J.; Celis, E.; et al. Oxidized Lipids Block Antigen Cross-Presentation by Dendritic Cells in Cancer. J. Immunol. 2014, 192, 2920–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeyda, M.; Saemann, M.D.; Stuhlmeier, K.M.; Mascher, D.G.; Nowotny, P.N.; Zlabinger, G.J.; Waldhausl, W.; Stulnig, T.M. Polyunsaturated Fatty Acids Block Dendritic Cell Activation and Function Independently of NF-kappaB Activation. J. Biol. Chem. 2005, 280, 14293–14301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weatherill, A.R.; Lee, J.Y.; Zhao, L.; Lemay, D.G.; Youn, H.S.; Hwang, D.H. Saturated and Polyunsaturated Fatty Acids Reciprocally Modulate Dendritic Cell Functions Mediated through TLR4. J. Immunol. 2005, 174, 5390–5397. [Google Scholar] [CrossRef] [PubMed]
- Aliberti, J.; Hieny, S.; Reis e Sousa, C.; Serhan, C.N.; Sher, A. Lipoxin-Mediated Inhibition of IL-12 Production by DCs: A Mechanism for Regulation of Microbial Immunity. Nat. Immunol. 2002, 3, 76–82. [Google Scholar] [CrossRef]
- Shamshiev, A.T.; Ampenberger, F.; Ernst, B.; Rohrer, L.; Marsland, B.J.; Kopf, M. Dyslipidemia Inhibits Toll-like Receptor-Induced Activation of CD8alpha-Negative Dendritic Cells and Protective Th1 Type Immunity. J. Exp. Med. 2007, 204, 441–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, J.K.; Mamotte, C.D.; Patel, P.; Yeoh, T.L.; Jackaman, C.; Nelson, D.J. Mesothelioma Tumor Cells Modulate Dendritic Cell Lipid Content, Phenotype and Function. PLoS ONE 2015, 10, e0123563. [Google Scholar] [CrossRef] [Green Version]
- Jiang, L.; Fang, X.; Wang, H.; Li, D.; Wang, X. Ovarian Cancer-Intrinsic Fatty Acid Synthase Prevents Anti-tumor Immunity by Disrupting Tumor-Infiltrating Dendritic Cells. Front. Immunol. 2018, 9, 2927. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Gabrilovich, D.I. Dendritic Cells in Cancer: The Role Revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S.; et al. Paracrine Wnt5a-beta-Catenin Signaling Triggers a Metabolic Program that Drives Dendritic Cell Tolerization. Immunity 2018, 48, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubillos-Ruiz, J.R.; Silberman, P.C.; Rutkowski, M.R.; Chopra, S.; Perales-Puchalt, A.; Song, M.; Zhang, S.; Bettigole, S.E.; Gupta, D.; Holcomb, K.; et al. ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 2015, 161, 1527–1538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- den Brok, M.H.; Raaijmakers, T.K.; Collado-Camps, E.; Adema, G.J. Lipid Droplets as Immune Modulators in Myeloid Cells. Trends Immunol. 2018, 39, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Al-Khami, A.A.; Rodriguez, P.C.; Ochoa, A.C. Metabolic Reprogramming of Myeloid-Derived Suppressor Cells (MDSC) in Cancer. Oncoimmunology 2016, 5, e1200771. [Google Scholar] [CrossRef] [Green Version]
- Al-Khami, A.A.; Zheng, L.; Del Valle, L.; Hossain, F.; Wyczechowska, D.; Zabaleta, J.; Sanchez, M.D.; Dean, M.J.; Rodriguez, P.C.; Ochoa, A.C. Exogenous Lipid Uptake Induces Metabolic and Functional Reprogramming of Tumor-Associated Myeloid-Derived Suppressor Cells. Oncoimmunology 2017, 6, e1344804. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty Acid Transport Protein 2 Reprograms Neutrophils in Cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef]
- Hossain, F.; Al-Khami, A.A.; Wyczechowska, D.; Hernandez, C.; Zheng, L.; Reiss, K.; Valle, L.D.; Trillo-Tinoco, J.; Maj, T.; Zou, W.; et al. Inhibition of Fatty Acid Oxidation Modulates Immunosuppressive Functions of Myeloid-Derived Suppressor Cells and Enhances Cancer Therapies. Cancer Immunol. Res. 2015, 3, 1236–1247. [Google Scholar] [CrossRef] [Green Version]
- Cao, M.; Huang, W.; Chen, Y.; Li, G.; Liu, N.; Wu, Y.; Wang, G.; Li, Q.; Kong, D.; Xue, T.; et al. Chronic Restraint Stress Promotes the Mobilization and Recruitment of Myeloid-Derived Suppressor Cells through Beta-Adrenergic-Activated CXCL5-CXCR2-Erk Signaling Cascades. Int. J. Cancer 2021, 149, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated Regulation of Myeloid Cells by Tumours. Nat. Reviews. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trikha, P.; Carson, W.E., 3rd. Signaling Pathways Involved in MDSC Regulation. Biochim. Biophys. Acta 2014, 1846, 55–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donkor, M.K.; Lahue, E.; Hoke, T.A.; Shafer, L.R.; Coskun, U.; Solheim, J.C.; Gulen, D.; Bishay, J.; Talmadge, J.E. Mammary Tumor Heterogeneity in the Expansion of Myeloid-Derived Suppressor Cells. Int. Immunopharmacol. 2009, 9, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Dominguez, G.A.; Youn, J.I.; Kossenkov, A.V.; Mony, S.; Alicea-Torres, K.; Tcyganov, E.; Hashimoto, A.; Nefedova, Y.; Lin, C.; et al. Lectin-Type Oxidized LDL Receptor-1 Distinguishes Population of Human Polymorphonuclear Myeloid-Derived Suppressor Cells in Cancer Patients. Sci. Immunol. 2016, 1, aaf8943. [Google Scholar] [CrossRef] [Green Version]
- Chai, E.; Zhang, L.; Li, C. LOX-1+ PMN-MDSC Enhances Immune Suppression Which Promotes Glioblastoma Multiforme Progression. Cancer Manag. Res. 2019, 11, 7307–7315. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Weidinger, C.; Schmidt, F.; Keye, J.; Friedrich, M.; Yerinde, C.; Willimsky, G.; Qin, Z.; Siegmund, B.; Glauben, R. Oleate but Not Stearate Induces the Regulatory Phenotype of Myeloid Suppressor Cells. Sci. Rep. 2017, 7, 7498. [Google Scholar] [CrossRef]
- Zhao, T.; Du, H.; Blum, J.S.; Yan, C. Critical Role of PPARgamma in Myeloid-Derived Suppressor Cell-Stimulated Cancer Cell Proliferation and Metastasis. Oncotarget 2016, 7, 1529–1543. [Google Scholar] [CrossRef]
- Park, J.; Lee, S.E.; Hur, J.; Hong, E.B.; Choi, J.I.; Yang, J.M.; Kim, J.Y.; Kim, Y.C.; Cho, H.J.; Peters, J.M.; et al. M-CSF from Cancer Cells Induces Fatty Acid Synthase and PPARbeta/delta Activation in Tumor Myeloid Cells, Leading to Tumor Progression. Cell Rep. 2015, 10, 1614–1625. [Google Scholar] [CrossRef] [Green Version]
- Waisman, A.; Lukas, D.; Clausen, B.E.; Yogev, N. Dendritic Cells as Gatekeepers of Tolerance. Semin. Immunopathol. 2017, 39, 153–163. [Google Scholar] [CrossRef]
- Nefedova, Y.; Fishman, M.; Sherman, S.; Wang, X.; Beg, A.A.; Gabrilovich, D.I. Mechanism of all-trans retinoic acid effect on tumor-associated myeloid-derived suppressor cells. Cancer Res 2007, 67, 11021–11028. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting Edge: Distinct Glycolytic and Lipid Oxidative Metabolic Programs Are Essential for Effector and Regulatory CD4+ T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Lochner, M.; Berod, L.; Sparwasser, T. Fatty Acid Metabolism in the Regulation of T Cell Function. Trends Immunol. 2015, 36, 81–91. [Google Scholar] [CrossRef]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bahre, H.; et al. De Novo Fatty Acid Synthesis Controls the Fate between Regulatory T and T Helper 17 Cells. Nat. Med. 2014, 20, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Macintyre, A.N.; Gerriets, V.A.; Nichols, A.G.; Michalek, R.D.; Rudolph, M.C.; Deoliveira, D.; Anderson, S.M.; Abel, E.D.; Chen, B.J.; Hale, L.P.; et al. The Glucose Transporter Glut1 is Selectively Essential for CD4 T Cell Activation and Effector Function. Cell Metab. 2014, 20, 61–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linke, M.; Fritsch, S.D.; Sukhbaatar, N.; Hengstschlager, M.; Weichhart, T. mTORC1 and mTORC2 as Regulators of Cell Metabolism in Immunity. FEBS Lett. 2017, 591, 3089–3103. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.H.; Ortiz, D.; Academia, E.C.; Anies, A.C.; Liao, C.Y.; Kennedy, B.K. Rapamycin-Mediated mTORC2 Inhibition Is Determined by the Relative Expression of FK506-Binding Proteins. Aging Cell 2015, 14, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a Metabolic Gene Regulatory Network Downstream of mTOR Complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Z.; Zhang, W. Role of mTOR in Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2018, 19, 2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronova, S.; Wedaman, K.; Aronov, P.A.; Fontes, K.; Ramos, K.; Hammock, B.D.; Powers, T. Regulation of Ceramide Biosynthesis by TOR Complex 2. Cell Metab. 2008, 7, 148–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidani, Y.; Elsaesser, H.; Hock, M.B.; Vergnes, L.; Williams, K.J.; Argus, J.P.; Marbois, B.N.; Komisopoulou, E.; Wilson, E.B.; Osborne, T.F.; et al. Sterol Regulatory Element-Binding Proteins Are Essential for the Metabolic Programming of Effector T Cells and Adaptive Immunity. Nat. Immunol. 2013, 14, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angela, M.; Endo, Y.; Asou, H.K.; Yamamoto, T.; Tumes, D.J.; Tokuyama, H.; Yokote, K.; Nakayama, T. Fatty Acid Metabolic Reprogramming via mTOR-Mediated Inductions of PPARgamma Directs Early Activation of T Cells. Nat. Commun. 2016, 7, 13683. [Google Scholar] [CrossRef]
- Yang, K.; Shrestha, S.; Zeng, H.; Karmaus, P.W.; Neale, G.; Vogel, P.; Guertin, D.A.; Lamb, R.F.; Chi, H. T Cell Exit from Quiescence and Differentiation into Th2 Cells Depend on Raptor-mTORC1-Mediated Metabolic Reprogramming. Immunity 2013, 39, 1043–1056. [Google Scholar] [CrossRef] [Green Version]
- Zelba, H.; Weide, B.; Martens, A.; Derhovanessian, E.; Bailur, J.K.; Kyzirakos, C.; Pflugfelder, A.; Eigentler, T.K.; Di Giacomo, A.M.; Maio, M.; et al. Circulating CD4+ T cells that produce IL4 or IL17 when stimulated by melan-A but not by NY-ESO-1 have negative impacts on survival of patients with stage IV melanoma. Clin. Cancer Res. 2014, 20, 4390–4399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihaylova, M.M.; Shaw, R.J. The AMPK Signalling Pathway Coordinates Cell Growth, Autophagy and Metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T Cell Lineage Differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cluxton, D.; Petrasca, A.; Moran, B.; Fletcher, J.M. Differential Regulation of Human Treg and Th17 Cells by Fatty Acid Synthesis and Glycolysis. Front. Immunol. 2019, 10, 115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopf, H.; de la Rosa, G.M.; Howard, O.M.; Chen, X. Rapamycin Inhibits Differentiation of Th17 Cells and Promotes Generation of FoxP3+ T Regulatory Cells. Int. Immunopharmacol. 2007, 7, 1819–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gualdoni, G.A.; Mayer, K.A.; Goschl, L.; Boucheron, N.; Ellmeier, W.; Zlabinger, G.J. The AMP Analog AICAR Modulates the Treg/Th17 Axis through Enhancement of Fatty Acid Oxidation. FASEB J. 2016, 30, 3800–3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Jin, H.; Xu, Y.; Shan, J. Rapamycin Modulate Treg/Th17 Balance via Regulating Metabolic Pathways: A Study in Mice. Transpl. Proc. 2019, 51, 2136–2140. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y.; Ito, S.; Ogiyama, Y. Suspected Anemia Caused by Maternal Anti-Jra Antibodies: A Case Report. Biomark. Res. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, Y.H.; Carreiro, A.L.; Buhman, K.K. Dgat1 and Dgat2 Regulate Enterocyte Triacylglycerol Distribution and Alter Proteins Associated with Cytoplasmic Lipid Droplets in Response to Dietary Fat. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 600–614. [Google Scholar] [CrossRef] [PubMed]
- Graham, K.L.; Werner, B.J.; Moyer, K.M.; Patton, A.K.; Krois, C.R.; Yoo, H.S.; Tverskoy, M.; LaJevic, M.; Napoli, J.L.; Sobel, R.A.; et al. DGAT1 Inhibits Retinol-Dependent Regulatory T Cell Formation and Mediates Autoimmune Encephalomyelitis. Proc. Natl. Acad. Sci. USA 2019, 116, 3126–3135. [Google Scholar] [CrossRef] [Green Version]
- Howie, D.; Cobbold, S.P.; Adams, E.; Ten Bokum, A.; Necula, A.S.; Zhang, W.; Huang, H.; Roberts, D.J.; Thomas, B.; Hester, S.S.; et al. Foxp3 Drives Oxidative Phosphorylation and Protection from Lipotoxicity. JCI Insight 2017, 2, e89160. [Google Scholar] [CrossRef] [Green Version]
- van der Windt, G.J.; Pearce, E.L. Metabolic Switching and Fuel Choice during T-Cell Differentiation and Memory Development. Immunol. Rev. 2012, 249, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Angelin, A.; Gil-de-Gomez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J., 3rd; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charbonnier, L.M.; Cui, Y.; Stephen-Victor, E.; Harb, H.; Lopez, D.; Bleesing, J.J.; Garcia-Lloret, M.I.; Chen, K.; Ozen, A.; Carmeliet, P.; et al. Functional Reprogramming of Regulatory T Cells in the Absence of Foxp3. Nat. Immunol. 2019, 20, 1208–1219. [Google Scholar] [CrossRef] [PubMed]
- Pacella, I.; Procaccini, C.; Focaccetti, C.; Miacci, S.; Timperi, E.; Faicchia, D.; Severa, M.; Rizzo, F.; Coccia, E.M.; Bonacina, F.; et al. Fatty Acid Metabolism Complements Glycolysis in the Selective Regulatory T Cell Expansion during Tumor Growth. Proc. Natl. Acad. Sci. USA 2018, 115, E6546–E6555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-Cell Memory by Modulating Fatty Acid Metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Lim, S.A.; Wei, J.; Nguyen, T.M.; Shi, H.; Su, W.; Palacios, G.; Dhungana, Y.; Chapman, N.M.; Long, L.; Saravia, J.; et al. Lipid Signalling Enforces Functional Specialization of Treg Cells in Tumours. Nature 2021, 591, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Kupper, T.S. Metabolic Reprogramming and Longevity of Tissue-Resident Memory T Cells. Front. Immunol. 2018, 9, 1347. [Google Scholar] [CrossRef] [Green Version]
- Almeida, L.; Lochner, M.; Berod, L.; Sparwasser, T. Metabolic Pathways in T Cell Activation and Lineage Differentiation. Semin. Immunol. 2016, 28, 514–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimeloe, S.; Mehling, M.; Frick, C.; Loeliger, J.; Bantug, G.R.; Sauder, U.; Fischer, M.; Belle, R.; Develioglu, L.; Tay, S.; et al. The Immune-Metabolic Basis of Effector Memory CD4+ T Cell Function under Hypoxic Conditions. J. Immunol. 2016, 196, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, M.C.; Lee, J.; Choi, Y. Tumor Necrosis Factor Receptor-Associated Factor 6 (TRAF6) Regulation of Development, Function, and Homeostasis of the Immune System. Immunol. Rev. 2015, 266, 72–92. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, D.; van der Windt, G.J.; Huang, S.C.; Curtis, J.D.; Chang, C.H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8(+) T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity 2014, 41, 75–88. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Chaudhary, O.; Rodriguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.M.; Williams, A.; Schulze, I.; et al. Uptake of Oxidized Lipids by the Scavenger Receptor CD36 Promotes Lipid Peroxidation and Dysfunction in CD8(+) T Cells in Tumors. Immunity 2021, 54, 1561–1577. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Phoon, Y.P.; Karlinsey, K.; Tian, Y.F.; Thapaliya, S.; Thongkum, A.; Qu, L.; Matz, A.J.; Cameron, M.; Cameron, C.; et al. A High OXPHOS CD8 T Cell Subset Is Predictive of Immunotherapy Resistance in Melanoma Patients. J. Exp. Med. 2022, 219, e20202084. [Google Scholar] [CrossRef]
- Iwama, Y.; Eguchi, M. Quantitative Evaluation of Leukemic Mitochondria with a Computer-Controlled Image Analyzer. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1986, 51, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.S.; Elizer, S.K.; Marshall, A.F.; Stocks, B.T.; Moore, D.J. Regulation of B Lymphocyte Responses to Toll-like Receptor Ligand Binding during Diabetes Prevention in Non-obese Diabetic (NOD) Mice. J. Diabetes 2016, 8, 120–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro-Maldonado, A.; Wang, R.; Nichols, A.G.; Kuraoka, M.; Milasta, S.; Sun, L.D.; Gavin, A.L.; Abel, E.D.; Kelsoe, G.; Green, D.R.; et al. Metabolic Reprogramming Is Required for Antibody Production That Is Suppressed in Anergic but Exaggerated in Chronically BAFF-Exposed B Cells. J. Immunol. 2014, 192, 3626–3636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Li, M.; Hou, T.; Gao, T.; Zhu, W.G.; Yang, Y. Sirtuins in Glucose and Lipid Metabolism. Oncotarget 2017, 8, 1845–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequeira, J.; Boily, G.; Bazinet, S.; Saliba, S.; He, X.; Jardine, K.; Kennedy, C.; Staines, W.; Rousseaux, C.; Mueller, R.; et al. sirt1-null Mice Develop an Autoimmune-like Condition. Exp. Cell Res. 2008, 314, 3069–3074. [Google Scholar] [CrossRef]
- Bhalla, S.; Gordon, L.I. Functional Characterization of NAD Dependent de-acetylases SIRT1 and SIRT2 in B-Cell Chronic Lymphocytic Leukemia (CLL). Cancer Biol. Ther. 2016, 17, 300–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyal, G.; Wong, K.; Nirschl, C.J.; Souders, N.; Neuberg, D.; Anandasabapathy, N.; Dranoff, G. PPARgamma Contributes to Immunity Induced by Cancer Cell Vaccines That Secrete GM-CSF. Cancer Immunol. Res. 2018, 6, 723–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prima, V.; Kaliberova, L.N.; Kaliberov, S.; Curiel, D.T.; Kusmartsev, S. COX2/mPGES1/PGE2 Pathway Regulates PD-L1 Expression in Tumor-Associated Macrophages and Myeloid-Derived Suppressor Cells. Proc. Natl. Acad. Sci. USA 2017, 114, 1117–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malandrino, M.I.; Fucho, R.; Weber, M.; Calderon-Dominguez, M.; Mir, J.F.; Valcarcel, L.; Escote, X.; Gomez-Serrano, M.; Peral, B.; Salvado, L.; et al. Enhanced Fatty Acid Oxidation in Adipocytes and Macrophages Reduces Lipid-Induced Triglyceride Accumulation and Inflammation. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E756–E769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Biase, S.; Lee, C.; Brandhorst, S.; Manes, B.; Buono, R.; Cheng, C.W.; Cacciottolo, M.; Martin-Montalvo, A.; de Cabo, R.; Wei, M.; et al. Fasting-Mimicking Diet Reduces HO-1 to Promote T Cell-Mediated Tumor Cytotoxicity. Cancer Cell 2016, 30, 136–146. [Google Scholar] [CrossRef] [Green Version]
- Mirza, N.; Fishman, M.; Fricke, I.; Dunn, M.; Neuger, A.M.; Frost, T.J.; Lush, R.M.; Antonia, S.; Gabrilovich, D.I. All-Trans-Retinoic Acid Improves Differentiation of Myeloid Cells and Immune Response in Cancer Patients. Cancer Res. 2006, 66, 9299–9307. [Google Scholar] [CrossRef] [Green Version]
- York, A.G.; Williams, K.J.; Argus, J.P.; Zhou, Q.D.; Brar, G.; Vergnes, L.; Gray, E.E.; Zhen, A.; Wu, N.C.; Yamada, D.H.; et al. Limiting Cholesterol Biosynthetic Flux Spontaneously Engages Type I IFN Signaling. Cell 2015, 163, 1716–1729. [Google Scholar] [CrossRef] [Green Version]
- Bunt, S.K.; Mohr, A.M.; Bailey, J.M.; Grandgenett, P.M.; Hollingsworth, M.A. Rosiglitazone and Gemcitabine in Combination Reduces Immune Suppression and Modulates T Cell Populations in Pancreatic Cancer. Cancer Immunol. Immunother. CII 2013, 62, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Adeshakin, A.O.; Liu, W.; Adeshakin, F.O.; Afolabi, L.O.; Zhang, M.; Zhang, G.; Wang, L.; Li, Z.; Lin, L.; Cao, Q.; et al. Regulation of ROS in Myeloid-Derived Suppressor Cells through Targeting Fatty Acid Transport Protein 2 Enhanced anti-PD-L1 Tumor Immunotherapy. Cell Immunol. 2021, 362, 104286. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-Associated Fibroblasts Neutralize the Anti-tumor Effect of CSF1 Receptor Blockade by Inducing PMN-MDSC Infiltration of Tumors. Cancer Cell 2017, 32, 654–668. [Google Scholar] [CrossRef] [Green Version]
- Divakaruni, A.S.; Hsieh, W.Y.; Minarrieta, L.; Duong, T.N.; Kim, K.K.O.; Desousa, B.R.; Andreyev, A.Y.; Bowman, C.E.; Caradonna, K.; Dranka, B.P.; et al. Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab. 2018, 28, 490–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Palmfeldt, J.; Gregersen, N.; Makhov, A.M.; Conway, J.F.; Wang, M.; McCalley, S.P.; Basu, S.; Alharbi, H.; St Croix, C.; et al. Mitochondrial Fatty Acid Oxidation and the Electron Transport Chain Comprise a Multifunctional Mitochondrial Protein Complex. J. Biol. Chem. 2019, 294, 12380–12391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.L.; Li, Q.; Yang, X.M.; Fang, F.; Li, J.; Wang, Y.H.; Yang, Q.; Zhu, L.; Nie, H.Z.; Zhang, X.L.; et al. SPON2 Promotes M1-like Macrophage Recruitment and Inhibits Hepatocellular Carcinoma Metastasis by Distinct Integrin-Rho GTPase-Hippo Pathways. Cancer Res. 2018, 78, 2305–2317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, P.; Yin, K.; Tang, X.; Tian, J.; Zhang, Y.; Ma, J.; Xu, H.; Xu, Q.; Wang, S. Metformin Inhibits the Function of Granulocytic Myeloid-Derived Suppressor Cells in Tumor-Bearing Mice. Biomed. Pharm. 2019, 120, 109458. [Google Scholar] [CrossRef]
- Huang, B.; Song, B.L.; Xu, C. Cholesterol Metabolism in Cancer: Mechanisms and Therapeutic Opportunities. Nat. Metab. 2020, 2, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavazoie, M.F.; Pollack, I.; Tanqueco, R.; Ostendorf, B.N.; Reis, B.S.; Gonsalves, F.C.; Kurth, I.; Andreu-Agullo, C.; Derbyshire, M.L.; Posada, J.; et al. LXR/ApoE Activation Restricts Innate Immune Suppression in Cancer. Cell 2018, 172, 825–840. [Google Scholar] [CrossRef] [Green Version]
- Yan, D.; Yang, Q.; Shi, M.; Zhong, L.; Wu, C.; Meng, T.; Yin, H.; Zhou, J. Polyunsaturated Fatty Acids Promote the Expansion of Myeloid-Derived Suppressor Cells by Activating the JAK/STAT3 Pathway. Eur. J. Immunol. 2013, 43, 2943–2955. [Google Scholar] [CrossRef]
- Zhang, C.; Yue, C.; Herrmann, A.; Song, J.; Egelston, C.; Wang, T.; Zhang, Z.; Li, W.; Lee, H.; Aftabizadeh, M.; et al. STAT3 Activation-Induced Fatty Acid Oxidation in CD8(+) T Effector Cells Is Critical for Obesity-Promoted Breast Tumor Growth. Cell Metab. 2020, 31, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Myers, M.G., Jr.; Leibel, R.L.; Seeley, R.J.; Schwartz, M.W. Obesity and Leptin Resistance: Distinguishing Cause from Effect. Trends Endocrinol. Metab. 2010, 21, 643–651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Dela Cruz-Chuh, J.; Khojasteh, S.C.; Dragovich, P.S.; Pillow, T.H.; Zhang, D. Carfilzomib Is Not an Appropriate Payload of Antibody-Drug Conjugates Due to Rapid Inactivation by Lysosomal Enzymes. Drug Metab. Dispos. 2019, 47, 884–889. [Google Scholar] [CrossRef]
- Thurnher, M.; Gruenbacher, G. T Lymphocyte Regulation by Mevalonate Metabolism. Sci. Signal. 2015, 8, re4. [Google Scholar] [CrossRef] [Green Version]
- Perl, A. Activation of mTOR (Mechanistic Target of Rapamycin) in Rheumatic Diseases. Nat. Rev. Rheumatol. 2016, 12, 169–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Kurupati, R.; Liu, L.; Zhou, X.Y.; Zhang, G.; Hudaihed, A.; Filisio, F.; Giles-Davis, W.; Xu, X.; Karakousis, G.C.; et al. Enhancing CD8(+) T Cell Fatty Acid Catabolism within a Metabolically Challenging Tumor Microenvironment Increases the Efficacy of Melanoma Immunotherapy. Cancer Cell 2017, 32, 377–391. [Google Scholar] [CrossRef] [Green Version]
- Berger, H.; Vegran, F.; Chikh, M.; Gilardi, F.; Ladoire, S.; Bugaut, H.; Mignot, G.; Chalmin, F.; Bruchard, M.; Derangere, V.; et al. SOCS3 Transactivation by PPARgamma prevents IL-17-Driven Cancer Growth. Cancer Res. 2013, 73, 3578–3590. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Xu, J.; Zhao, J.; Yin, W.; Liu, D.; Chen, W.; Hou, S.X. Arf1-Mediated Lipid Metabolism Sustains Cancer Cells and Its Ablation Induces Anti-tumor Immune Responses in Mice. Nat. Commun. 2020, 11, 220. [Google Scholar] [CrossRef]
- Li, D.; Li, Y. The Interaction between Ferroptosis and Lipid Metabolism in Cancer. Signal. Transduct. Target. Ther. 2020, 5, 108. [Google Scholar] [CrossRef]
- Xue, C.C.; Li, M.H.; Zhao, Y.; Zhou, J.; Hu, Y.; Cai, K.Y.; Zhao, Y.; Yu, S.H.; Luo, Z. Tumor Microenvironment-Activatable Fe-Doxorubicin Preloaded Amorphous CaCO3 Nanoformulation Triggers Ferroptosis in Target Tumor Cells. Sci. Adv. 2020, 6, eaax1346. [Google Scholar] [CrossRef]
- Fu, S.; He, K.; Tian, C.; Sun, H.; Zhu, C.; Bai, S.; Liu, J.; Wu, Q.; Xie, D.; Yue, T.; et al. Impaired Lipid Biosynthesis Hinders Anti-tumor Efficacy of Intratumoral iNKT cells. Nat. Commun. 2020, 11, 438. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mabrouk, N.; Lecoeur, B.; Bettaieb, A.; Paul, C.; Végran, F. Impact of Lipid Metabolism on Antitumor Immune Response. Cancers 2022, 14, 1850. https://doi.org/10.3390/cancers14071850
Mabrouk N, Lecoeur B, Bettaieb A, Paul C, Végran F. Impact of Lipid Metabolism on Antitumor Immune Response. Cancers. 2022; 14(7):1850. https://doi.org/10.3390/cancers14071850
Chicago/Turabian StyleMabrouk, Nesrine, Baptiste Lecoeur, Ali Bettaieb, Catherine Paul, and Frédérique Végran. 2022. "Impact of Lipid Metabolism on Antitumor Immune Response" Cancers 14, no. 7: 1850. https://doi.org/10.3390/cancers14071850
APA StyleMabrouk, N., Lecoeur, B., Bettaieb, A., Paul, C., & Végran, F. (2022). Impact of Lipid Metabolism on Antitumor Immune Response. Cancers, 14(7), 1850. https://doi.org/10.3390/cancers14071850