
HuR Plays a Role in Double-Strand Break Repair in Pancreatic Cancer Cells and Regulates Functional BRCA1-Associated-Ring-Domain-1(BARD1) Isoforms

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Drugs
2.3. Plasmid and siRNA Transfections
2.4. RT-qPCR and mRNA Expression Analysis
2.5. Analysis of BARD1 mRNA Stability in HuR-Silenced Cells by Actinomycin D
2.6. Ribonucleo-Protein Immunoprecipitation (RNP-IP) Assay
2.7. Microarray and Bioinformatics Analysis (GEO Accession # GSE166951)
2.8. Gene Set Enrichment Analysis of siHuR vs. siSCR RNA-seq Database (GEO Accession # GSE167525)
2.9. Comet Assay
2.10. pDR-GFP Assay to Assess HRR
2.11. Cell Survival and Colony Formation
2.12. Immunoblot Analysis
2.13. Immunofluorescence
2.14. Cell Cycle Analysis
2.15. BARD1 mRNA Expression and Survival Analysis
2.16. Statistical Analysis
3. Results
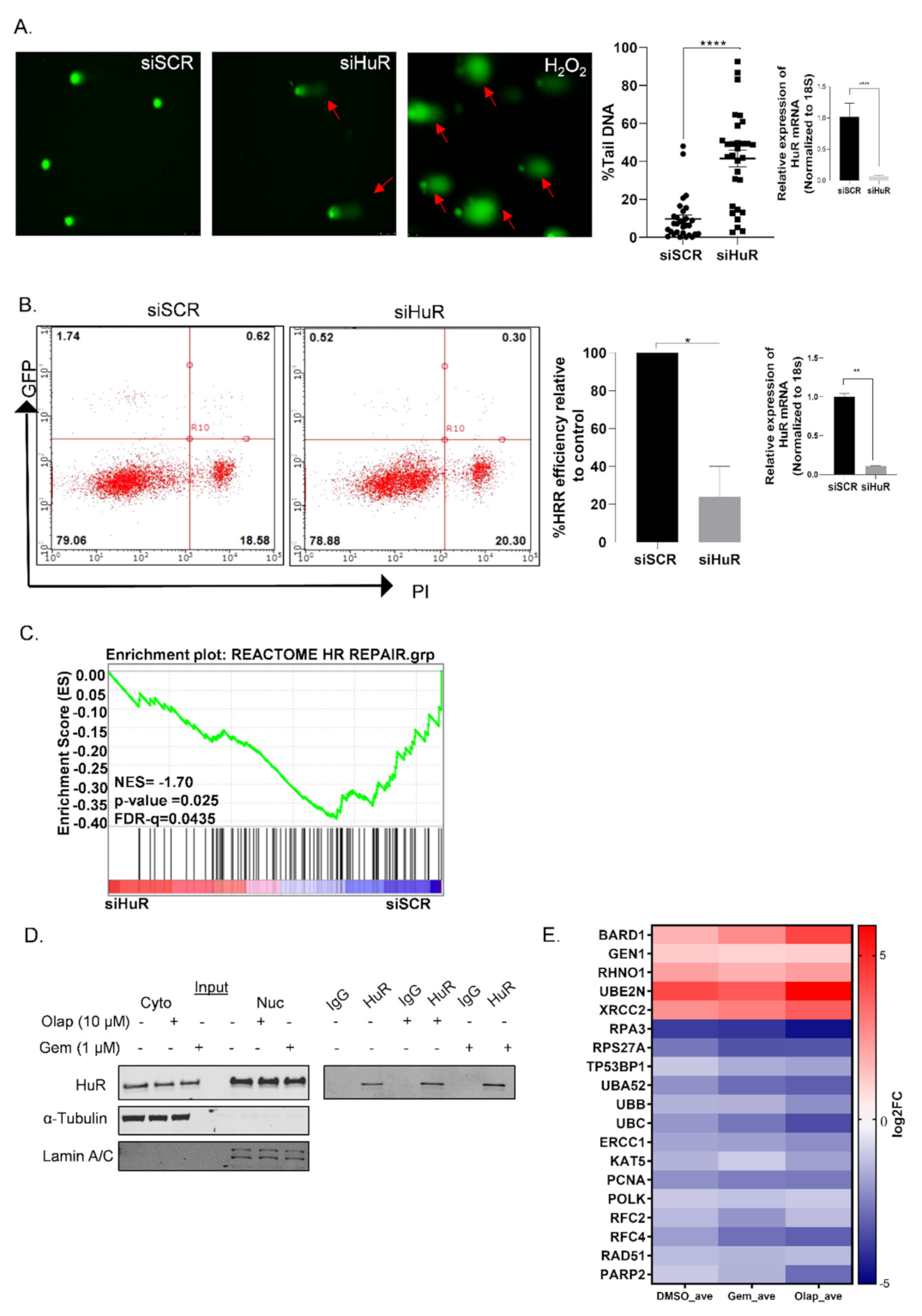
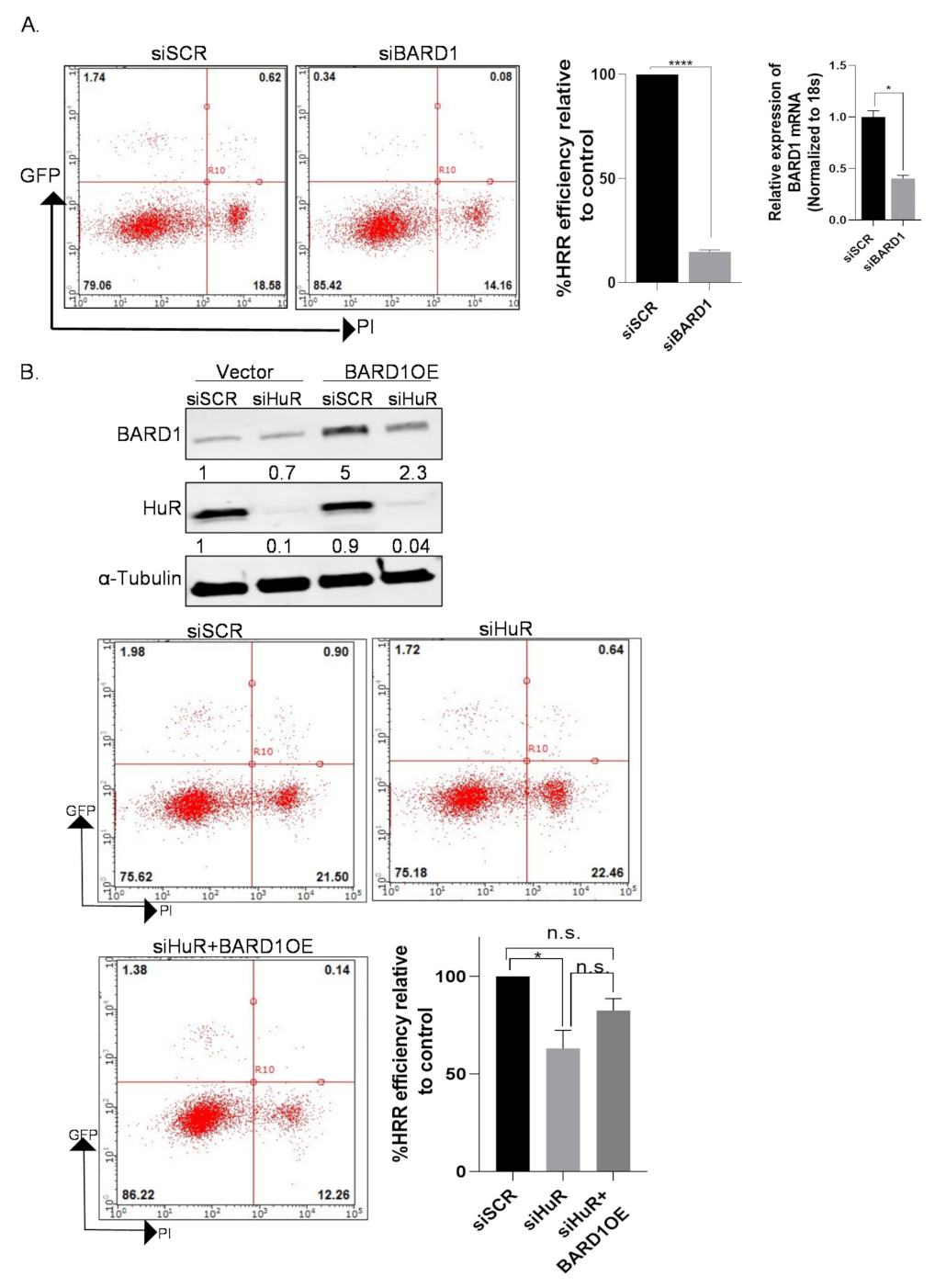
3.1. HuR Regulates Homologous Recombination Repair in PDAC Cells
3.2. A Subset of DDR Genes Are Targets of HuR: BARD1 Is a Novel HuR Target
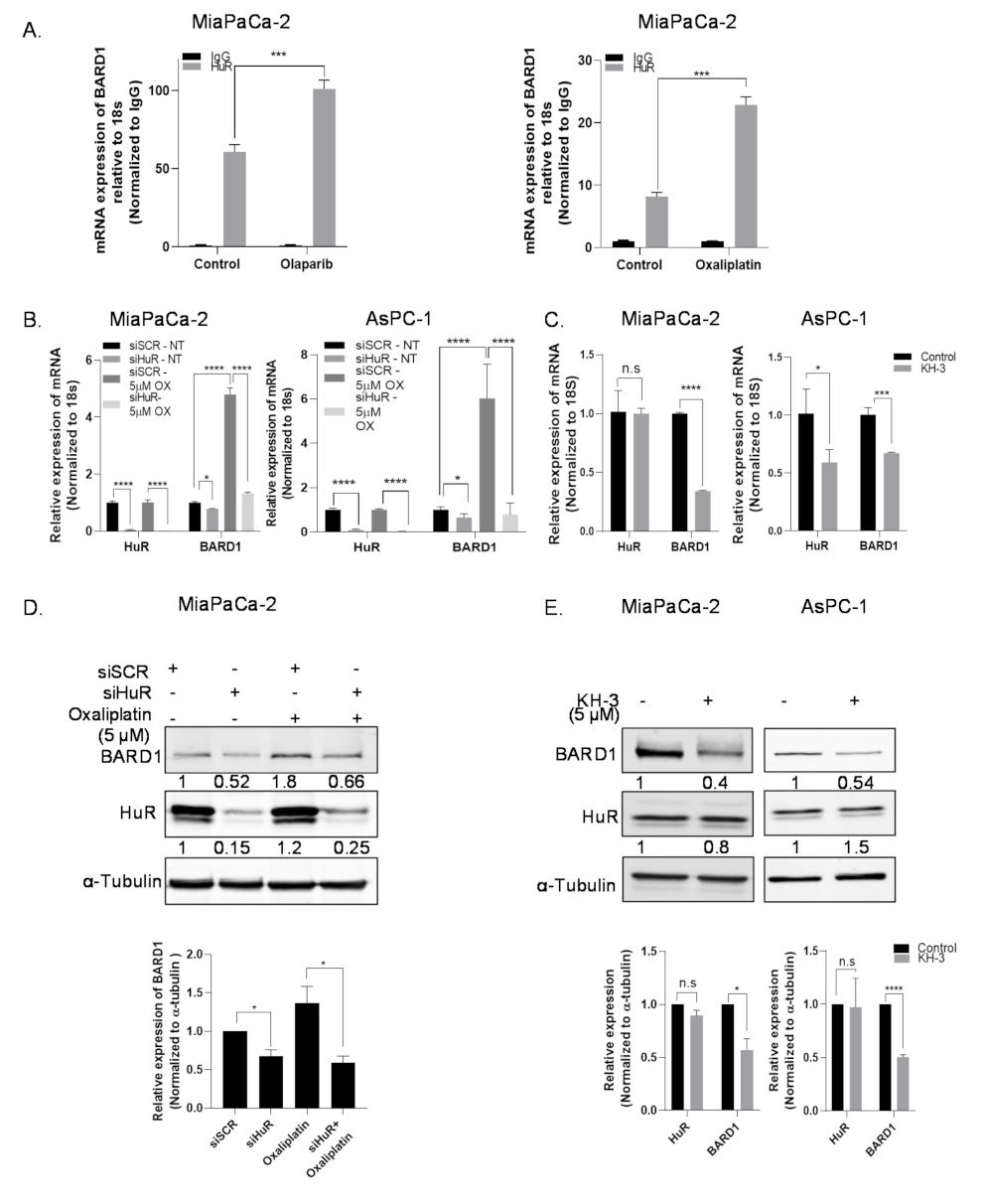
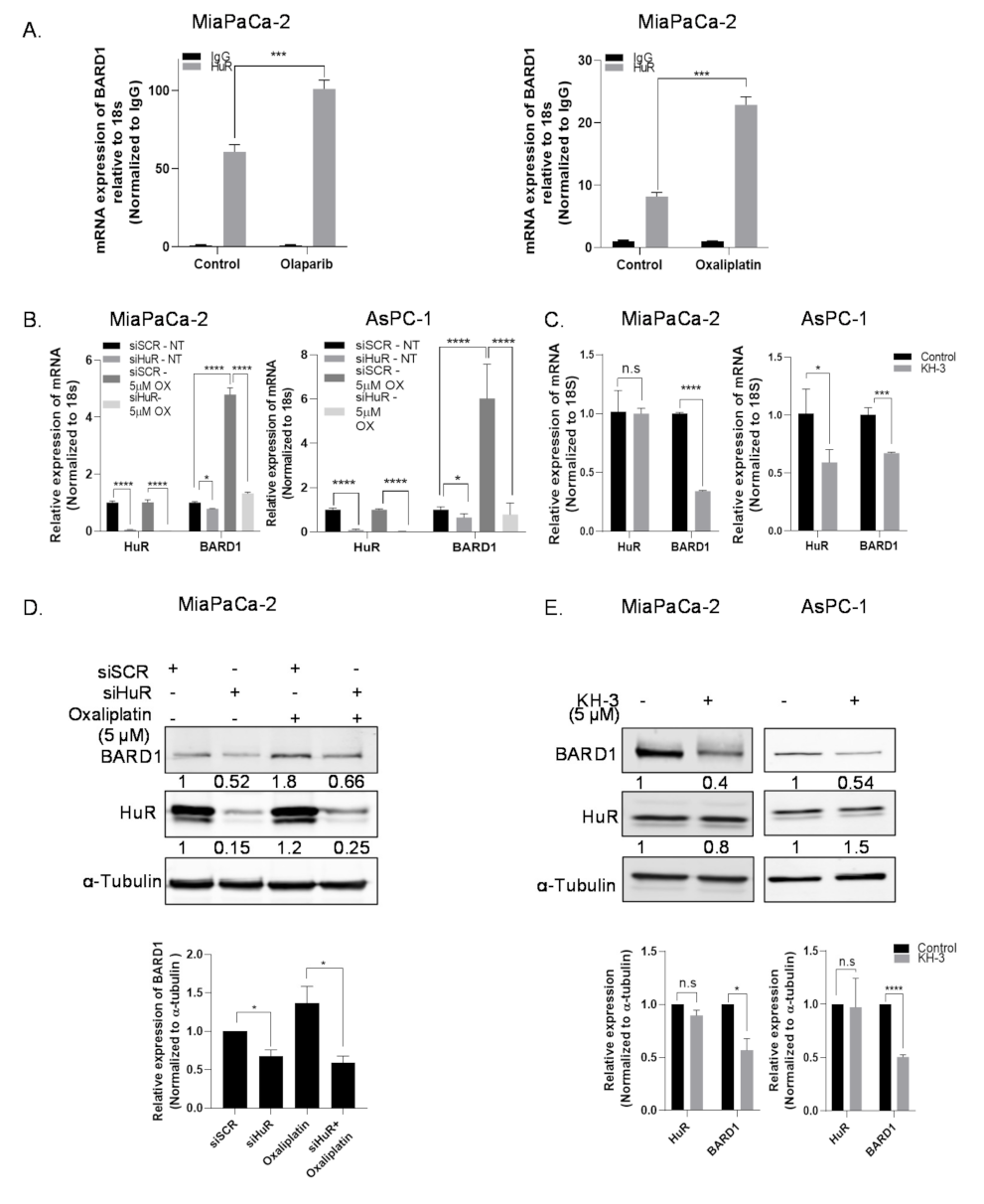
3.3. HuR Regulates mRNA Expression of BARD1
3.4. HuR Impacts Total Protein Expression of BARD1
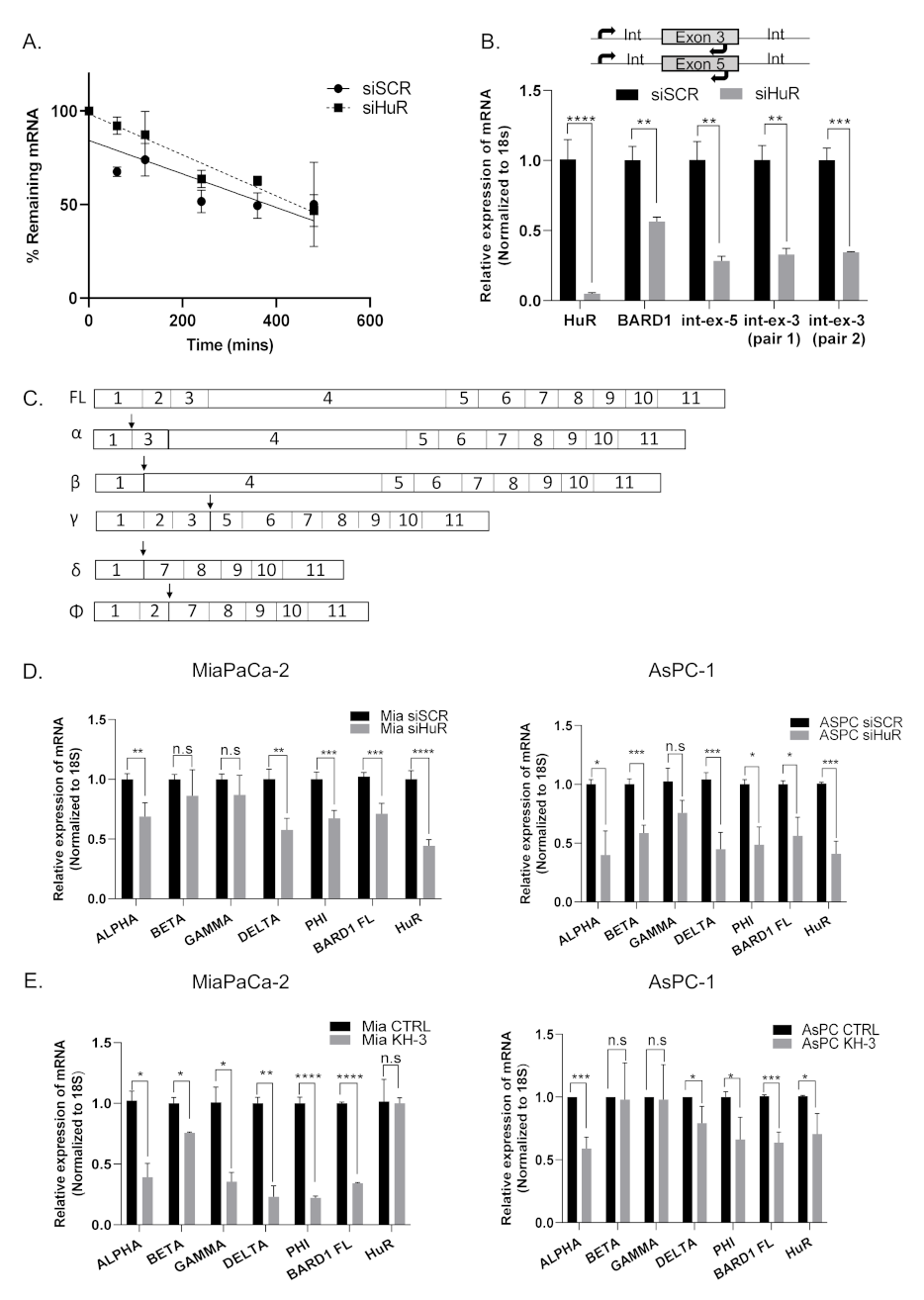
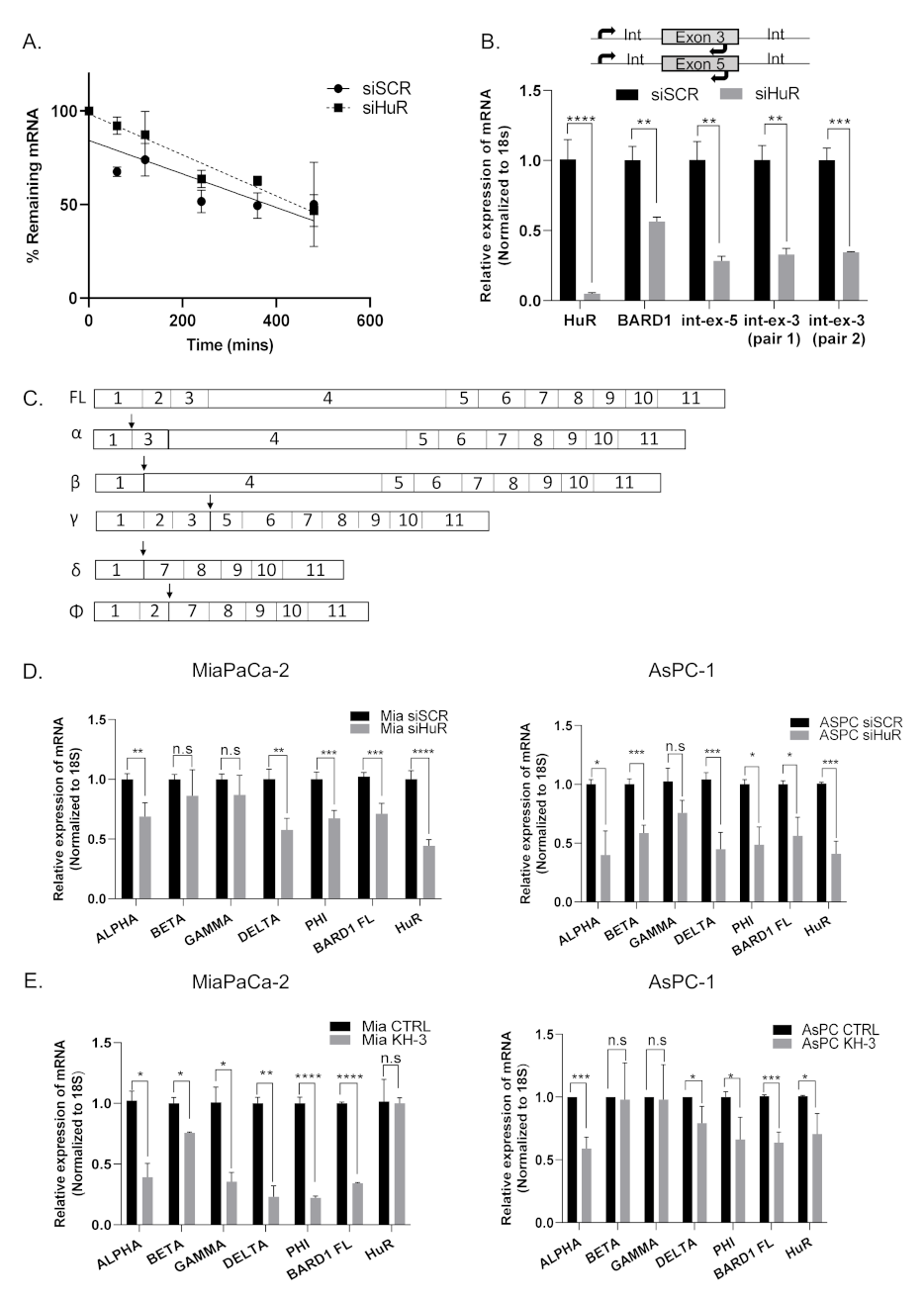
3.5. HuR Does Not Directly Stabilize BARD1 mRNA
3.6. HuR Regulates BARD1 in a Non-Canonical Manner
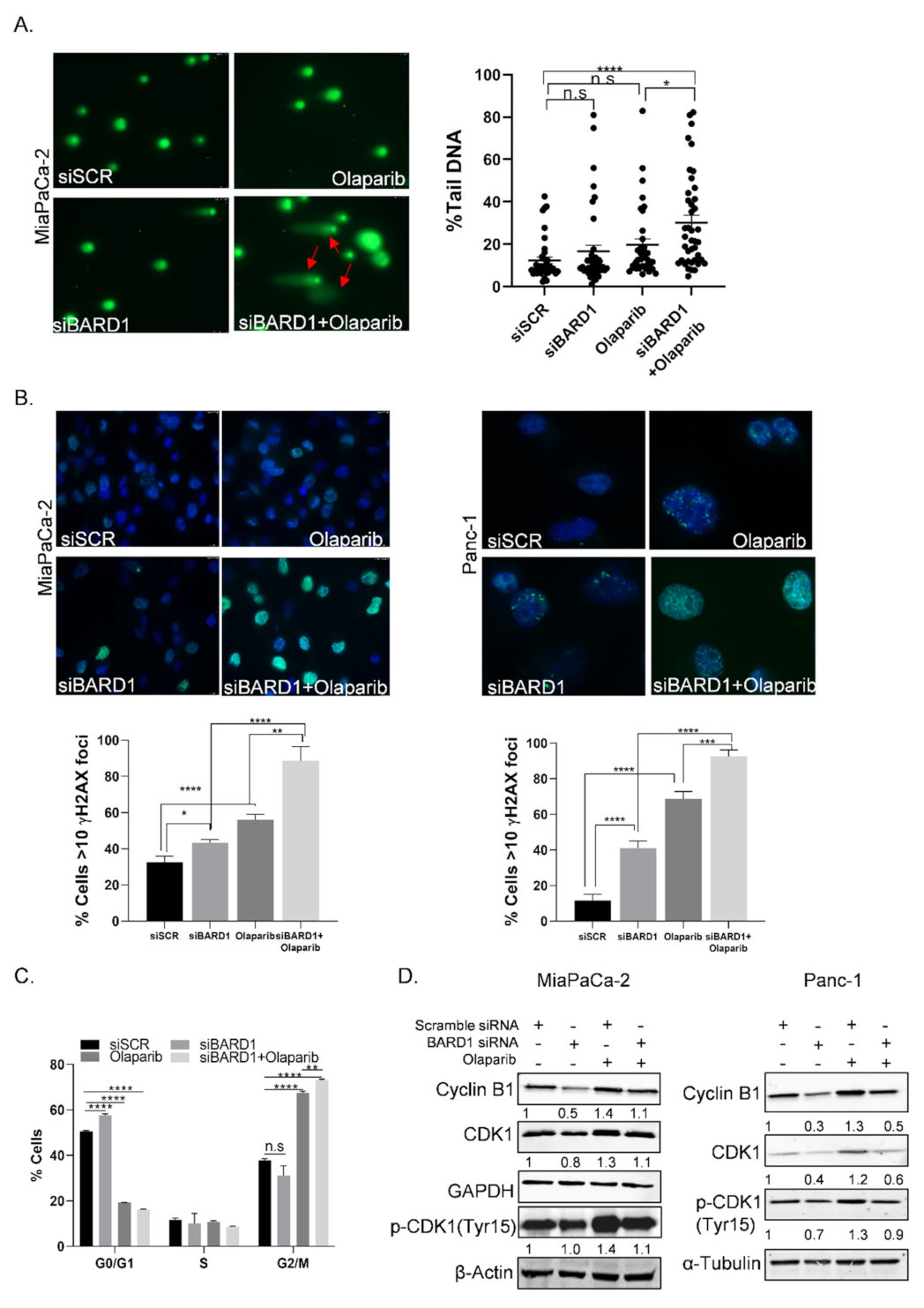
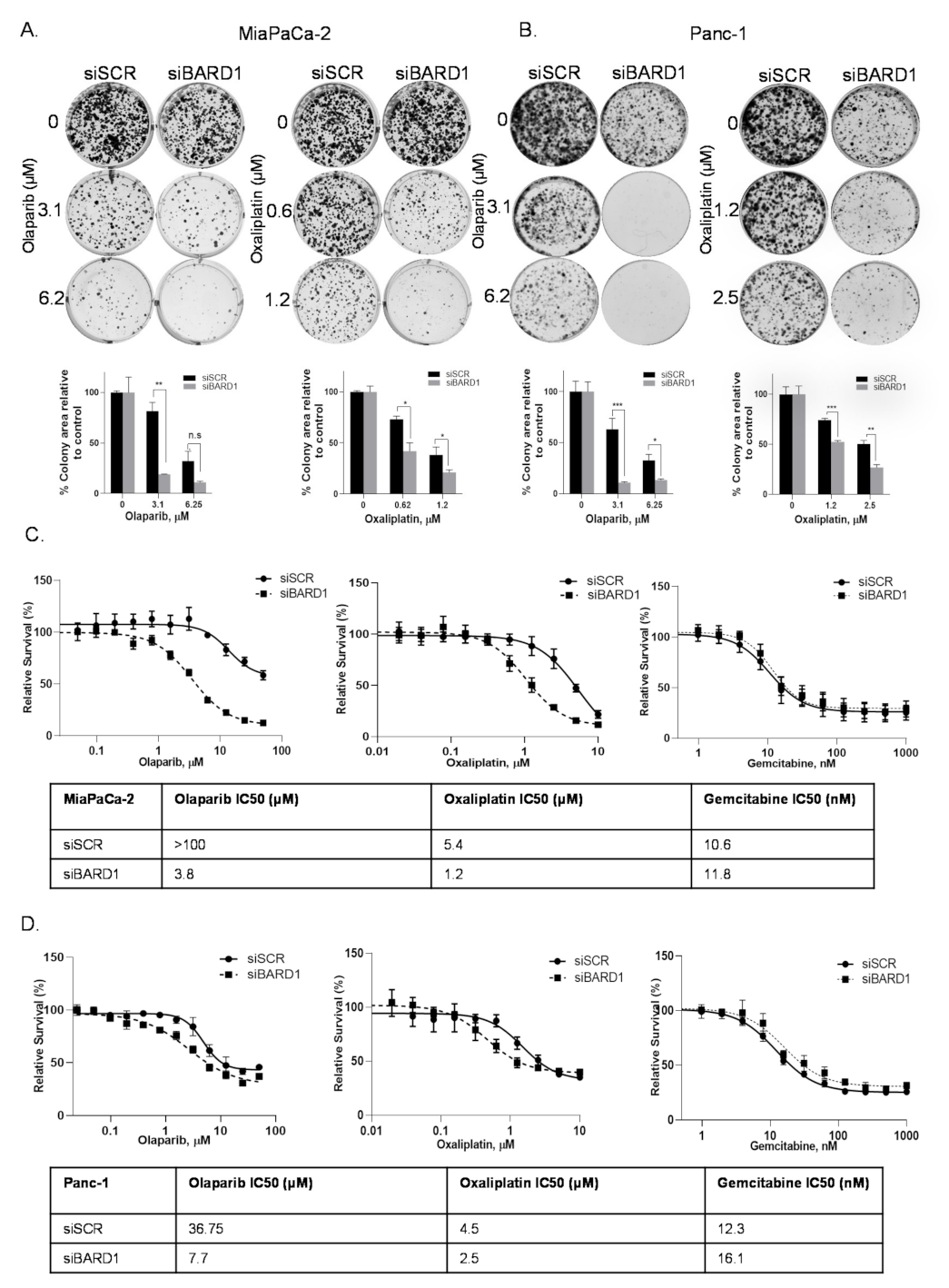
3.7. BARD1 Supports PDAC Growth and Modulates Drug Responses
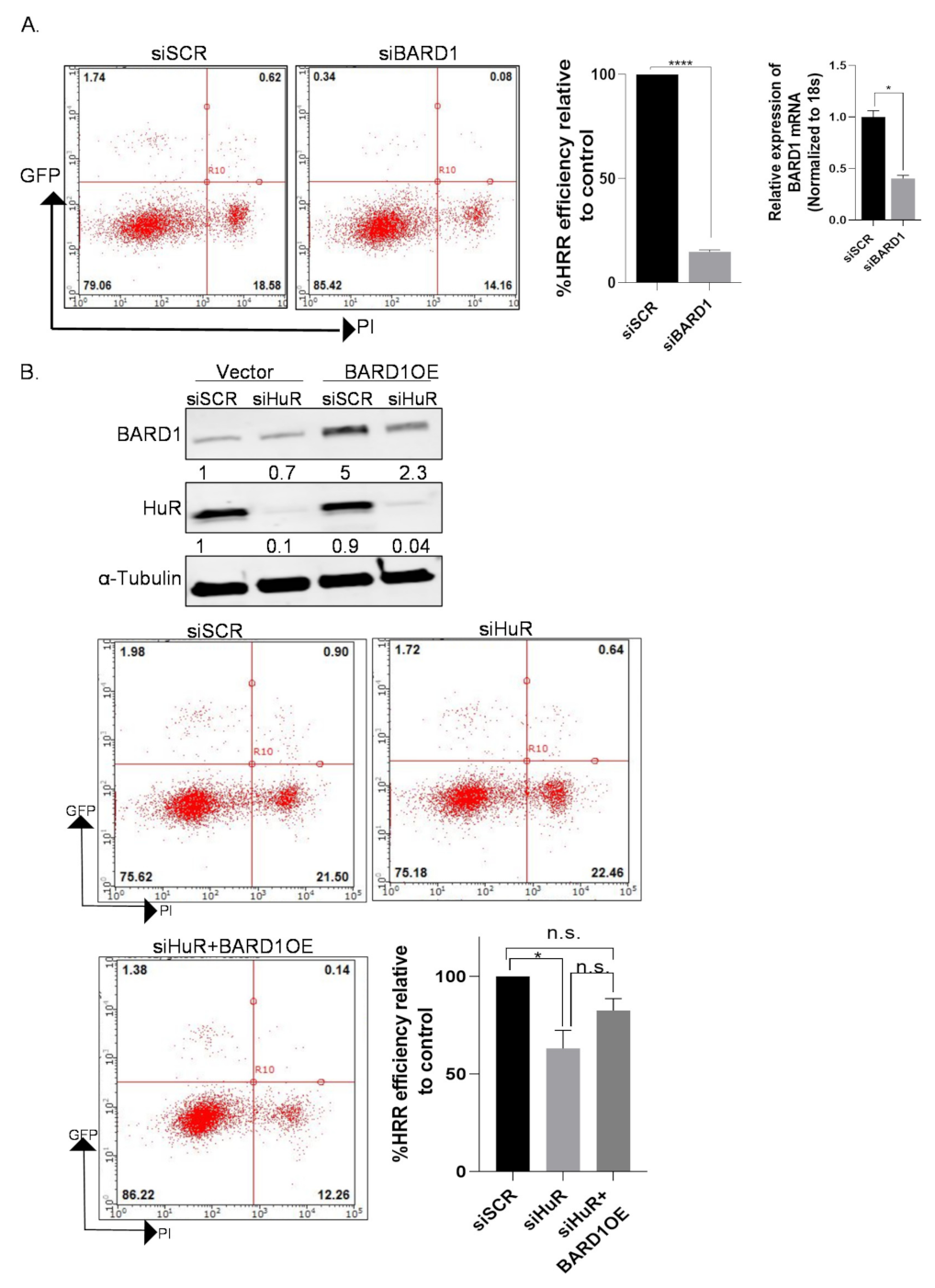
3.8. Knockdown of BARD1 Increases Drug-Induced DNA Damage
3.9. Overexpression of BARD1 in HuR Silenced Cells Partially Rescues HRR Phenotype
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Satyananda, V.; Gupta, R.; Hari, D.M.; Yeh, J.; Chen, K.T. Advances in translational research and clinical care in pancreatic cancer: Where are we headed? Gastroenterol. Res. Pract. 2019, 2019, 7690528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheahan, A.V.; Biankin, A.V.; Parish, C.R.; Khachigian, L.M. Targeted therapies in the management of locally advanced and metastatic pancreatic cancer: A systematic review. Oncotarget 2018, 9, 21613–21627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjoquist, K.M.; Chin, V.T.; Chantrill, L.A.; O’Connor, C.; Hemmings, C.; Chang, D.K.; Chou, A.; Pajic, M.; Johns, A.L.; Nagrial, A.M.; et al. Personalising pancreas cancer treatment: When tissue is the issue. World J. Gastroenterol. 2014, 20, 7849–7863. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Pishvaian, M.J.; Bender, R.J.; Halverson, D.; Rahib, L.; Hendifar, A.E.; Mikhail, S.; Chung, V.; Picozzi, V.J.; Sohal, D.; Blais, E.M.; et al. Molecular profiling of pancreatic cancer patients: Initial results from the Know Your Tumor initiative. Clin. Cancer Res. 2018, 24, 5018–5027. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Pishvaian, M.J.; Wang, H.; Parenti, S.; He, A.R.; Hwang, J.J.; Ley, L.; Difebo, H.; Smaglo, B.G.; Kim, S.S.; Weinberg, B.A.; et al. Final report of a phase I/II study of veliparib (Vel) in combination with 5-FU and oxaliplatin (FOLFOX) in patients (pts) with metastatic pancreatic cancer (mPDAC). J. Clin. Oncol. 2019, 37 (Suppl. 15), 4015. [Google Scholar] [CrossRef]
- Blanco, F.F.; Jimbo, M.; Wulfkuhle, J.; Gallagher, I.; Deng, J.; Enyenihi, L.; Meisner-Kober, N.; Londin, E.; Rigoutsos, I.; Sawicki, J.A.; et al. The mRNA-binding protein HuR promotes hypoxia-induced chemoresistance through posttranscriptional regulation of the proto-oncogene PIM1 in pancreatic cancer cells. Oncogene 2016, 35, 2529–2541. [Google Scholar] [CrossRef] [PubMed]
- Jimbo, M.; Blanco, F.F.; Huang, Y.H.; Telonis, A.G.; Screnci, B.A.; Cosma, G.L.; Alexeev, V.; Gonye, G.E.; Yeo, C.J.; Sawicki, J.A.; et al. Targeting the mRNA-binding protein HuR impairs malignant characteristics of pancreatic ductal adenocarcinoma cells. Oncotarget 2015, 6, 27312–27331. [Google Scholar] [CrossRef] [Green Version]
- Brody, J.R.; Dixon, D.A. Complex HuR function in pancreatic cancer cells. Wiley Interdiscip. Rev. RNA 2018, 9, e1469. [Google Scholar] [CrossRef] [PubMed]
- Schultz, C.W.; Preet, R.; Dhir, T.; Dixon, D.A.; Brody, J.R. Understanding and targeting the disease-related RNA binding protein human antigen R (HuR). Wiley Interdiscip. Rev. RNA 2020, 11, e1581. [Google Scholar] [CrossRef] [PubMed]
- Richards, N.G.; Rittenhouse, D.W.; Freydin, B.; Cozzitorto, J.A.; Grenda, D.; Rui, H.; Gonye, G.; Kennedy, E.P.; Yeo, C.J.; Brody, J.R.; et al. HuR status is a powerful marker for prognosis and response to gemcitabine-based chemotherapy for resected pancreatic ductal adenocarcinoma patients. Ann. Surg. 2010, 252, 499–506. [Google Scholar] [CrossRef]
- Chand, S.N.; Zarei, M.; Schiewer, M.J.; Kamath, A.R.; Romeo, C.; Lal, S.; Cozzitorto, J.A.; Nevler, A.; Scolaro, L.; Londin, E.; et al. Posttranscriptional regulation of PARG mRNA by HuR facilitates DNA repair and resistance to PARP inhibitors. Cancer Res. 2017, 77, 5011–5025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, S.; Burkhart, R.A.; Beeharry, N.; Bhattacharjee, V.; Londin, E.R.; Cozzitorto, J.A.; Romeo, C.; Jimbo, M.; Norris, Z.A.; Yeo, C.J.; et al. HuR posttranscriptionally regulates WEE1: Implications for the DNA damage response in pancreatic cancer cells. Cancer Res. 2014, 74, 1128–1140. [Google Scholar] [CrossRef] [Green Version]
- Andrade, D.; Mehta, M.; Griffith, J.; Oh, S.; Corbin, J.; Babu, A.; De, S.; Chen, A.; Zhao, Y.D.; Husain, S.; et al. HuR reduces radiation-induced DNA damage by enhancing expression of ARID1A. Cancers 2019, 11, 2014. [Google Scholar] [CrossRef] [Green Version]
- Mazan-Mamczarz, K.; Galban, S.; Lopez de Silanes, I.; Martindale, J.L.; Atasoy, U.; Keene, J.D.; Gorospe, M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. USA 2003, 100, 8354–8359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Furneaux, H.; Cheng, H.; Caldwell, M.C.; Hutter, D.; Liu, Y.; Holbrook, N.; Gorospe, M. HuR regulates p21 mRNA stabilization by UV light. Mol. Cell. Biol. 2000, 20, 760–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Pierce, A.J.; Jasin, M. Measuring recombination proficiency in mouse embryonic stem cells. Methods Mol. Biol. 2005, 291, 373–384. [Google Scholar] [CrossRef] [PubMed]
- Pierce, A.J.; Johnson, R.D.; Thompson, L.H.; Jasin, M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999, 13, 2633–2638. [Google Scholar] [CrossRef] [Green Version]
- Grammatikakis, I.; Abdelmohsen, K.; Gorospe, M. Posttranslational control of HuR function. Wiley Interdiscip. Rev. RNA 2017, 8, e1372. [Google Scholar] [CrossRef] [Green Version]
- Nowotarski, S.L.; Shantz, L.M. Cytoplasmic accumulation of the RNA-binding protein HuR stabilizes the ornithine decarboxylase transcript in a murine nonmelanoma skin cancer model. J. Biol. Chem. 2010, 285, 31885–31894. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Watanabe, S.; Sagara, Y.; Mitsunari, K.; Matsuo, T.; Ohba, K.; Sakai, H. High expression of HuR in cytoplasm, but not nuclei, is associated with malignant aggressiveness and prognosis in bladder cancer. PLoS ONE 2013, 8, e59095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebedeva, S.; Jens, M.; Theil, K.; Schwanhausser, B.; Selbach, M.; Landthaler, M.; Rajewsky, N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol. Cell 2011, 43, 340–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakheet, T.; Hitti, E.; Al-Saif, M.; Moghrabi, W.N.; Khabar, K.S.A. The AU-rich element landscape across human transcriptome reveals a large proportion in introns and regulation by ELAVL1/HuR. Biochim. Biophys. Acta Gene Regul. Mech. 2018, 1861, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Tarsounas, M.; Sung, P. The antitumorigenic roles of BRCA1-BARD1 in DNA repair and replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Daza-Martin, M.; Densham, R.M.; Morris, J.R. BRCA1-BARD1: The importance of being in shape. Mol. Cell. Oncol. 2019, 6, e1656500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irminger-Finger, I.; Leung, W.C. BRCA1-dependent and independent functions of BARD1. Int. J. Biochem. Cell Biol. 2002, 34, 582–587. [Google Scholar] [CrossRef]
- McCarthy, E.E.; Celebi, J.T.; Baer, R.; Ludwig, T. Loss of Bard1, the heterodimeric partner of the Brca1 tumor suppressor, results in early embryonic lethality and chromosomal instability. Mol. Cell. Biol. 2003, 23, 5056–5063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irminger-Finger, I.; Ratajska, M.; Pilyugin, M. New concepts on BARD1: Regulator of BRCA pathways and beyond. Int. J. Biochem. Cell Biol. 2016, 72, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Ryser, S.; Dizin, E.; Pils, D.; Krainer, M.; Jefford, C.E.; Bertoni, F.; Zeillinger, R.; Irminger-Finger, I. Oncogenic BARD1 isoforms expressed in gynecological cancers. Cancer Res. 2007, 67, 11876–11885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sporn, J.C.; Hothorn, T.; Jung, B. BARD1 expression predicts outcome in colon cancer. Clin. Cancer Res. 2011, 17, 5451–5462. [Google Scholar] [CrossRef] [Green Version]
- Feki, A.; Jefford, C.E.; Durand, P.; Harb, J.; Lucas, H.; Krause, K.H.; Irminger-Finger, I. BARD1 expression during spermatogenesis is associated with apoptosis and hormonally regulated. Biol. Reprod. 2004, 71, 1614–1624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cimmino, F.; Avitabile, M.; Lasorsa, V.A.; Pezone, L.; Cardinale, A.; Montella, A.; Cantalupo, S.; Iolascon, A.; Capasso, M. Functional characterization of full-length BARD1 strengthens its role as a tumor suppressor in neuroblastoma. J. Cancer 2020, 11, 1495–1504. [Google Scholar] [CrossRef] [Green Version]
- Pilyugin, M.; Andre, P.A.; Ratajska, M.; Kuzniacka, A.; Limon, J.; Tournier, B.B.; Colas, J.; Laurent, G.; Irminger-Finger, I. Antagonizing functions of BARD1 and its alternatively spliced variant BARD1delta in telomere stability. Oncotarget 2017, 8, 9339–9353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Cohen, M.; Wu, J.; Sow, M.H.; Nikolic, B.; Bischof, P.; Irminger-Finger, I. Identification of BARD1 splice-isoforms involved in human trophoblast invasion. Int. J. Biochem. Cell Biol. 2007, 39, 1659–1672. [Google Scholar] [CrossRef] [PubMed]
- Bosse, K.R.; Diskin, S.J.; Cole, K.A.; Wood, A.C.; Schnepp, R.W.; Norris, G.; Nguyen, L.B.; Jagannathan, J.; Laquaglia, M.; Winter, C.; et al. Common variation at BARD1 results in the expression of an oncogenic isoform that influences neuroblastoma susceptibility and oncogenicity. Cancer Res. 2012, 72, 2068–2078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Q.; Pilyugin, M.; Kuester, D.; Leoni, V.P.; Li, L.; Casula, G.; Zorcolo, L.; Schneider-Stock, R.; Atzori, L.; Irminger-Finger, I. Expression of oncogenic BARD1 isoforms affects colon cancer progression and correlates with clinical outcome. Br. J. Cancer 2012, 107, 675–683. [Google Scholar] [CrossRef] [Green Version]
- Dhir, T.; Schultz, C.W.; Jain, A.; Brown, S.Z.; Haber, A.; Goetz, A.; Xi, C.; Su, G.H.; Xu, L.; Posey, J., 3rd; et al. Abemaciclib is effective against pancreatic cancer cells and synergizes with HuR and YAP1 inhibition. Mol. Cancer Res. 2019, 17, 2029–2041. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Agostini, L.C.; McCarthy, G.A.; Chand, S.N.; Ramirez, A.; Nevler, A.; Cozzitorto, J.; Schultz, C.W.; Lowder, C.Y.; Smith, K.M.; et al. Poly (ADP) ribose glycohydrolase can be effectively targeted in pancreatic cancer. Cancer Res. 2019, 79, 4491–4502. [Google Scholar] [CrossRef]
- Jain, A.; Brown, S.Z.; Thomsett, H.L.; Londin, E.; Brody, J.R. Evaluation of post-transcriptional gene regulation in pancreatic cancer cells: Studying RNA binding [roteins and their mRNA targets. Methods Mol. Biol. 2019, 1882, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef] [PubMed]
- Haber, A.O.; Jain, A.; Mani, C.; Nevler, A.; Agostini, L.C.; Golan, T.; Palle, K.; Yeo, C.J.; Gmeiner, W.H.; Brody, J.R. AraC-FdUMP[10] (CF10) is a next generation fluoropyrimidine with potent antitumor activity in PDAC and synergy with PARG inhibition. Mol. Cancer Res. 2021, 19, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, C.D.; Simeone, D.M.; Binkley, C.; Arumugam, T.; Greenson, J.K.; Giordano, T.J.; Misek, D.E.; Hanash, S. Molecular profiling of pancreatic adenocarcinoma and chronic pancreatitis identifies multiple genes differentially regulated in pancreatic cancer. Cancer Res. 2003, 63, 2649–2657. [Google Scholar] [PubMed]
- Badea, L.; Herlea, V.; Dima, S.O.; Dumitrascu, T.; Popescu, I. Combined gene expression analysis of whole-tissue and microdissected pancreatic ductal adenocarcinoma identifies genes specifically overexpressed in tumor epithelia. Hepatogastroenterology 2008, 55, 2016–2027. [Google Scholar]
- Nagy, A.; Lanczky, A.; Menyhart, O.; Gyorffy, B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci. Rep. 2018, 8, 9227. [Google Scholar] [CrossRef]
- Gyori, B.M.; Venkatachalam, G.; Thiagarajan, P.S.; Hsu, D.; Clement, M.V. OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2014, 2, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, N.; Corcoran, D.L.; Nusbaum, J.D.; Reid, D.W.; Georgiev, S.; Hafner, M.; Ascano, M., Jr.; Tuschl, T.; Ohler, U.; Keene, J.D. Integrative regulatory mapping indicates that the RNA-binding protein HuR couples pre-mRNA processing and mRNA stability. Mol. Cell 2011, 43, 327–339. [Google Scholar] [CrossRef] [Green Version]
- Park, W.; Chen, J.; Chou, J.F.; Varghese, A.M.; Yu, K.H.; Wong, W.; Capanu, M.; Balachandran, V.; McIntyre, C.A.; El Dika, I.; et al. Genomic methods identify homologous recombination deficiency in pancreas adenocarcinoma and optimize treatment selection. Clin. Cancer Res. 2020, 26, 3239–3247. [Google Scholar] [CrossRef]
- Yadav, S.; Kasi, P.M.; Bamlet, W.R.; Ho, T.P.; Polley, E.C.; Hu, C.; Hart, S.N.; Rabe, K.G.; Boddicker, N.J.; Gnanaolivu, R.D.; et al. Effect of germline mutations in homologous recombination repair genes on overall survival of patients with pancreatic adenocarcinoma. Clin. Cancer Res. 2020, 26, 6505–6512. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Hart, S.N.; Bamlet, W.R.; Moore, R.M.; Nandakumar, K.; Eckloff, B.W.; Lee, Y.K.; Petersen, G.M.; McWilliams, R.R.; Couch, F.J. Prevalence of pathogenic mutations in cancer predisposition genes among pancreatic cancer patients. Cancer Epidemiol. Biomark. Prev. 2016, 25, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Gardashova, G.; Lan, L.; Han, S.; Zhong, C.; Marquez, R.T.; Wei, L.; Wood, S.; Roy, S.; Gowthaman, R.; et al. Targeting the interaction between RNA-binding protein HuR and FOXQ1 suppresses breast cancer invasion and metastasis. Commun. Biol. 2020, 3, 193. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.D.; Xu, H.; Baer, R. Ubiquitination and proteasomal degradation of the BRCA1 tumor suppressor is regulated during cell cycle progression. J. Biol. Chem. 2004, 279, 33909–33918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunus, J.M.; French, J.D.; Edwards, S.L.; Beveridge, D.J.; Hatchell, E.C.; Wagner, S.A.; Stein, S.R.; Davidson, A.; Simpson, K.J.; Francis, G.D.; et al. Posttranscriptional regulation of the breast cancer susceptibility gene BRCA1 by the RNA binding protein HuR. Cancer Res. 2008, 68, 9469–9478. [Google Scholar] [CrossRef] [Green Version]
- Izquierdo, J.M. Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J. Biol. Chem. 2008, 283, 19077–19084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, A.C.; Abdelmohsen, K.; Yoon, J.H.; Martindale, J.L.; Yang, X.; Curtis, J.; Mercken, E.M.; Chenette, D.M.; Zhang, Y.; Schneider, R.J.; et al. RNA-binding protein AUF1 promotes myogenesis by regulating MEF2C expression levels. Mol. Cell. Biol. 2014, 34, 3106–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nouaille, S.; Mondeil, S.; Finoux, A.L.; Moulis, C.; Girbal, L.; Cocaign-Bousquet, M. The stability of an mRNA is influenced by its concentration: A potential physical mechanism to regulate gene expression. Nucleic Acids Res. 2017, 45, 11711–11724. [Google Scholar] [CrossRef] [Green Version]
- Bernstein, J.A.; Lin, P.H.; Cohen, S.N.; Lin-Chao, S. Global analysis of Escherichia coli RNA degradosome function using DNA microarrays. Proc. Natl. Acad. Sci. USA 2004, 101, 2758–2763. [Google Scholar] [CrossRef] [Green Version]
- Joukov, V.; Chen, J.; Fox, E.A.; Green, J.B.; Livingston, D.M. Functional communication between endogenous BRCA1 and its partner, BARD1, during Xenopus laevis development. Proc. Natl. Acad. Sci. USA 2001, 98, 12078–12083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiba, N.; Parvin, J.D. Redistribution of BRCA1 among four different protein complexes following replication blockage. J. Biol. Chem. 2001, 276, 38549–38554. [Google Scholar] [CrossRef] [Green Version]
- Mazan-Mamczarz, K.; Hagner, P.R.; Dai, B.; Wood, W.H.; Zhang, Y.; Becker, K.G.; Liu, Z.; Gartenhaus, R.B. Identification of transformation-related pathways in a breast epithelial cell model using a ribonomics approach. Cancer Res. 2008, 68, 7730–7735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Yuan, S.; Chen, X.; Zhu, P.; Li, J.; Qin, L.; Liao, W. Up-regulation of BRCA1-associated RING Domain 1 Promotes Hepatocellular Carcinoma Progression by Targeting Akt Signaling. Sci. Rep. 2017, 7, 7649. [Google Scholar] [CrossRef]
- Pilyugin, M.; Irminger-Finger, I. Long non-coding RNA and microRNAs might act in regulating the expression of BARD1 mRNAs. Int. J. Biochem. Cell Biol. 2014, 54, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Vlastos, A.T.; Pelte, M.F.; Caligo, M.A.; Bianco, A.; Krause, K.H.; Laurent, G.J.; Irminger-Finger, I. Aberrant expression of BARD1 in breast and ovarian cancers with poor prognosis. Int. J. Cancer 2006, 118, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Steinfeld, J.B.; Liang, F.; Chen, X.; Maranon, D.G.; Jian Ma, C.; Kwon, Y.; Rao, T.; Wang, W.; Sheng, C.; et al. BRCA1-BARD1 promotes RAD51-mediated homologous DNA pairing. Nature 2017, 550, 360–365. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jain, A.; McCoy, M.; Coats, C.; Brown, S.Z.; Addya, S.; Pelz, C.; Sears, R.C.; Yeo, C.J.; Brody, J.R. HuR Plays a Role in Double-Strand Break Repair in Pancreatic Cancer Cells and Regulates Functional BRCA1-Associated-Ring-Domain-1(BARD1) Isoforms. Cancers 2022, 14, 1848. https://doi.org/10.3390/cancers14071848
Jain A, McCoy M, Coats C, Brown SZ, Addya S, Pelz C, Sears RC, Yeo CJ, Brody JR. HuR Plays a Role in Double-Strand Break Repair in Pancreatic Cancer Cells and Regulates Functional BRCA1-Associated-Ring-Domain-1(BARD1) Isoforms. Cancers. 2022; 14(7):1848. https://doi.org/10.3390/cancers14071848
Chicago/Turabian StyleJain, Aditi, Matthew McCoy, Carolyn Coats, Samantha Z. Brown, Sankar Addya, Carl Pelz, Rosalie C. Sears, Charles J. Yeo, and Jonathan R. Brody. 2022. "HuR Plays a Role in Double-Strand Break Repair in Pancreatic Cancer Cells and Regulates Functional BRCA1-Associated-Ring-Domain-1(BARD1) Isoforms" Cancers 14, no. 7: 1848. https://doi.org/10.3390/cancers14071848
APA StyleJain, A., McCoy, M., Coats, C., Brown, S. Z., Addya, S., Pelz, C., Sears, R. C., Yeo, C. J., & Brody, J. R. (2022). HuR Plays a Role in Double-Strand Break Repair in Pancreatic Cancer Cells and Regulates Functional BRCA1-Associated-Ring-Domain-1(BARD1) Isoforms. Cancers, 14(7), 1848. https://doi.org/10.3390/cancers14071848