
Apelin Resistance Contributes to Muscle Loss during Cancer Cachexia in Mice

 , , , , , , , ,
, , , , , , , ,  and
and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Adeno-Associated Vectors, Plasmids, and Drugs
2.3. Protein Degradation Assay
2.4. Diameter Measurements
2.5. Luciferase-Based Assays
2.6. Apelin Peptide Synthesis
2.7. RNA Isolation from Cultured Cells or Muscles and Reverse Transcription
2.8. Quantitative Real-Time Polymerase Chain Reaction (PCR)
2.9. Protein Extraction and Western Blot
2.10. Enzyme-Linked Immunosorbent Assay (ELISA)
2.11. MALDI-TOF Analysis
2.12. In Vivo Animal Models
2.13. Muscle Sample Processing and Fiber Size Measurements
2.14. Patients Data Analysis
2.15. Sulforhodamine B (SRB) Assay
2.16. Statistical Analysis
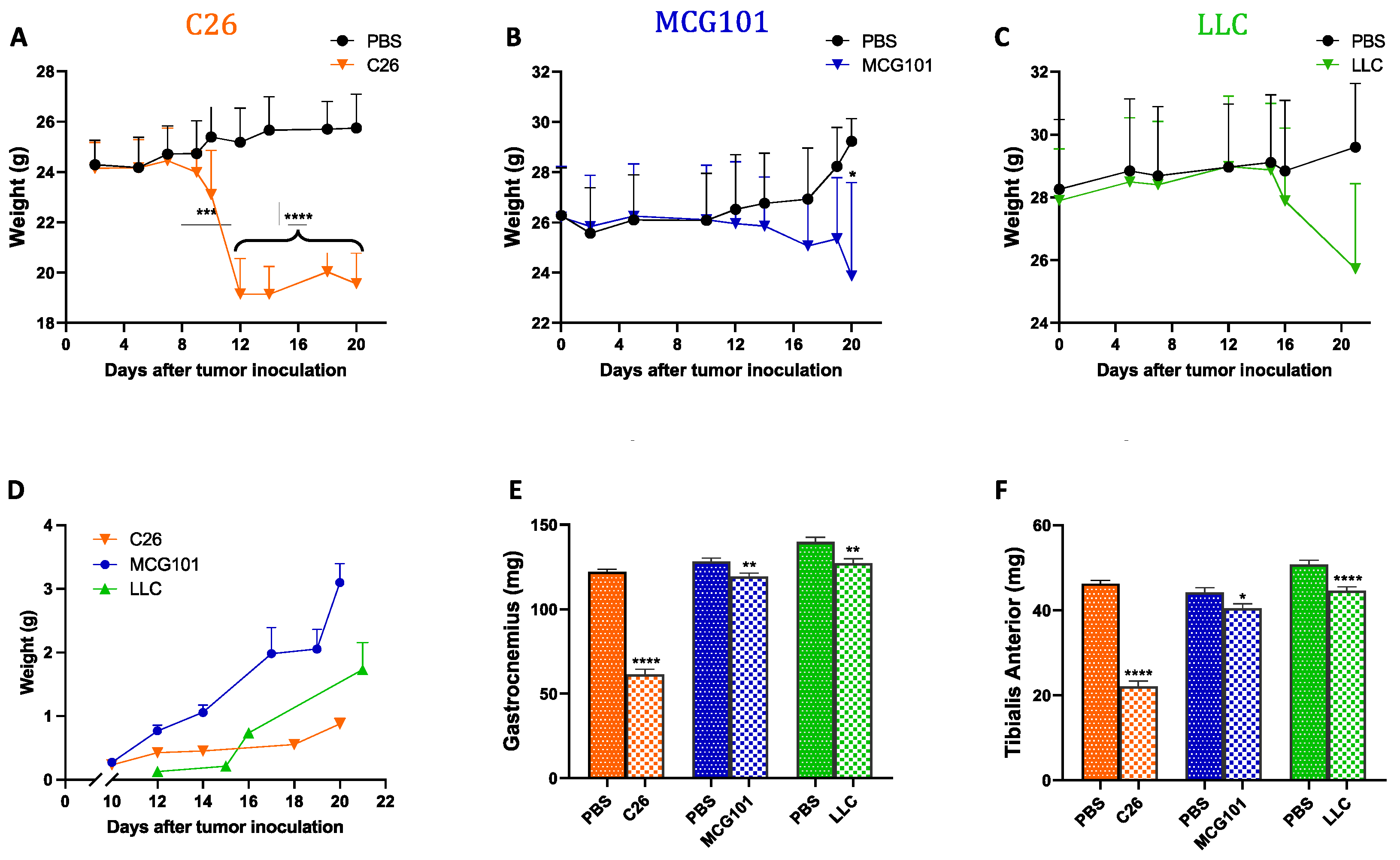
3. Results
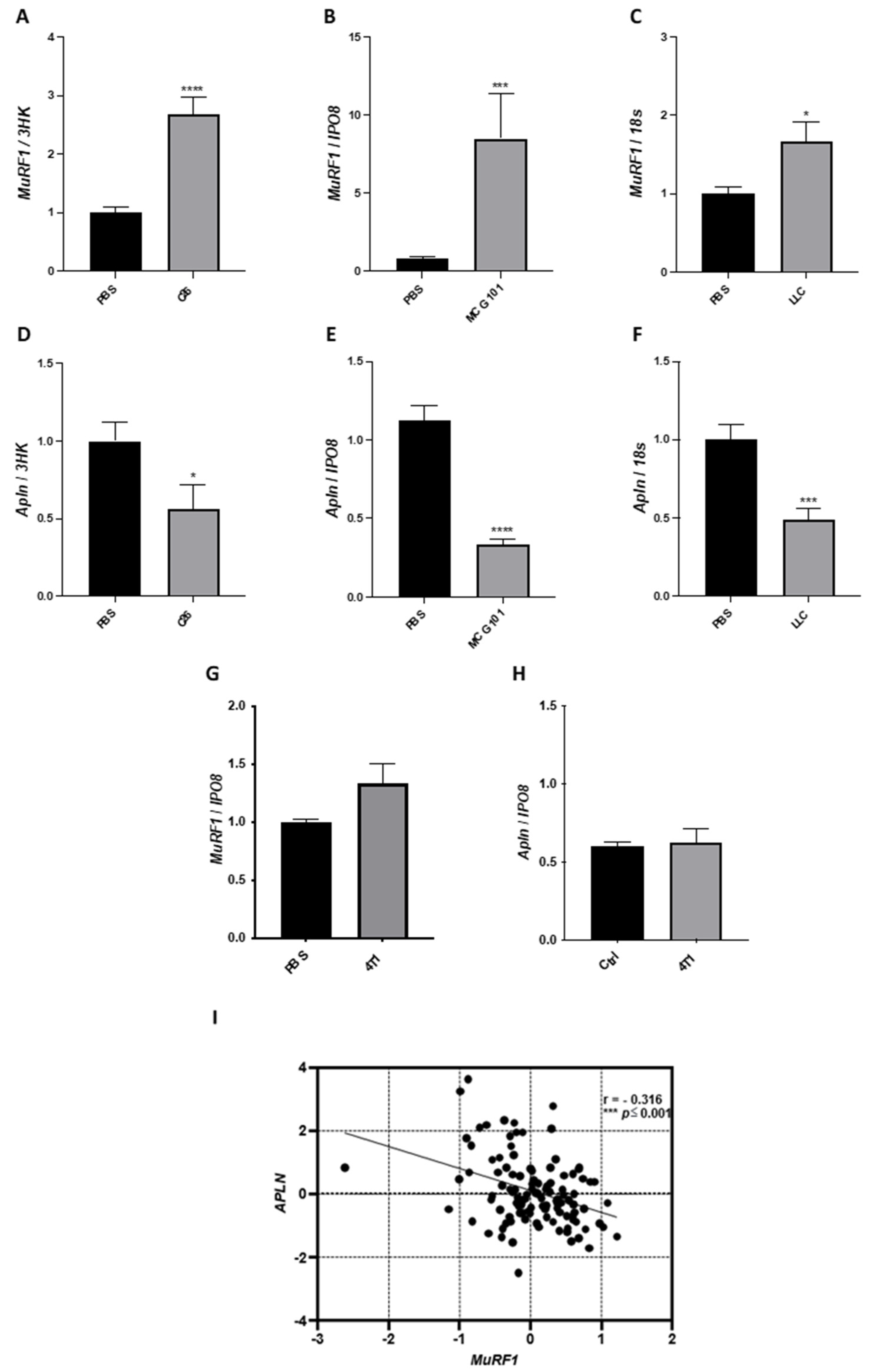
3.1. The Expression of Apelin Is Low in Cachectic Muscles of Rodents and Patients with Cancer
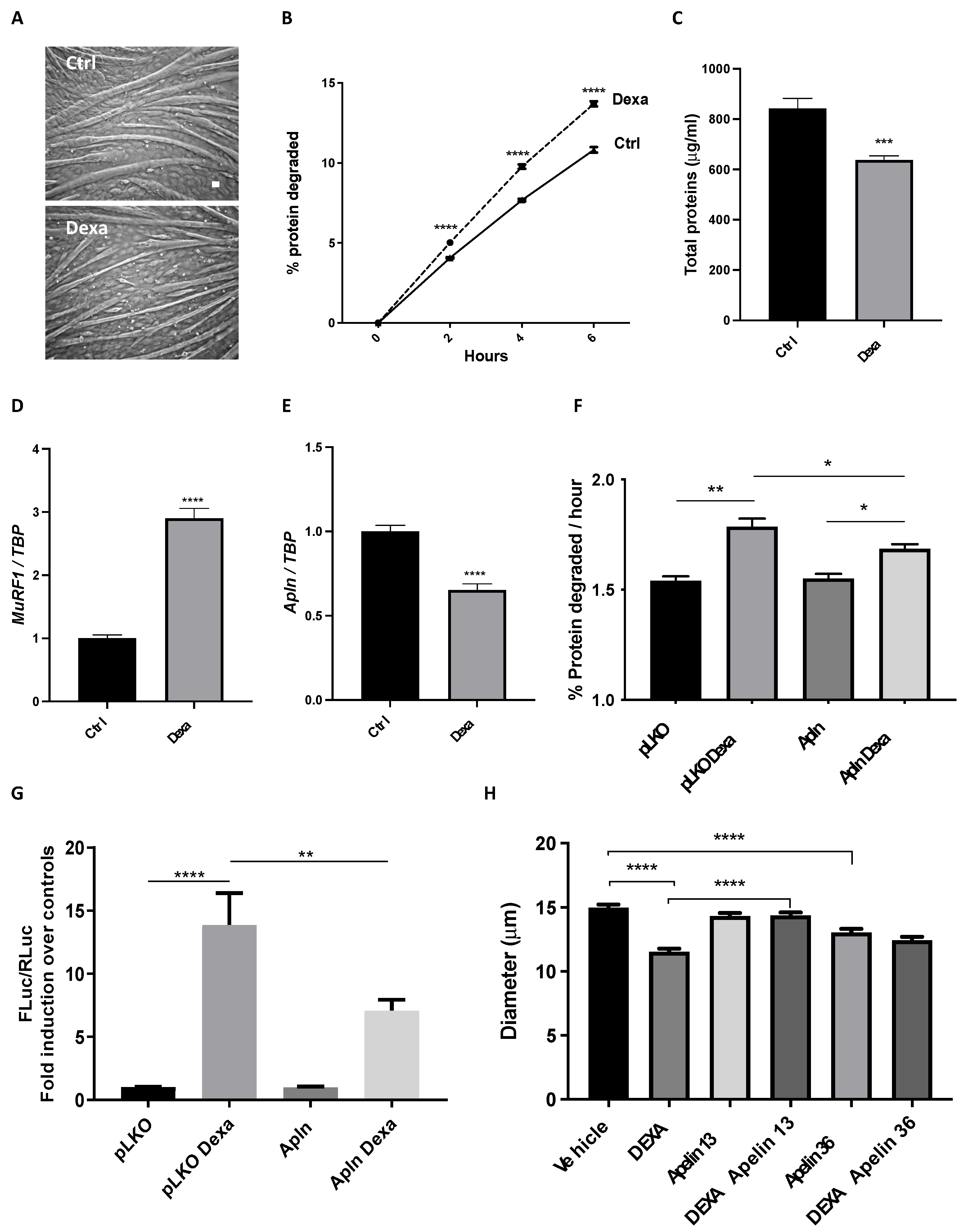
3.2. Apelin Exerts Anti-Catabolic Action on Atrophying Myotubes
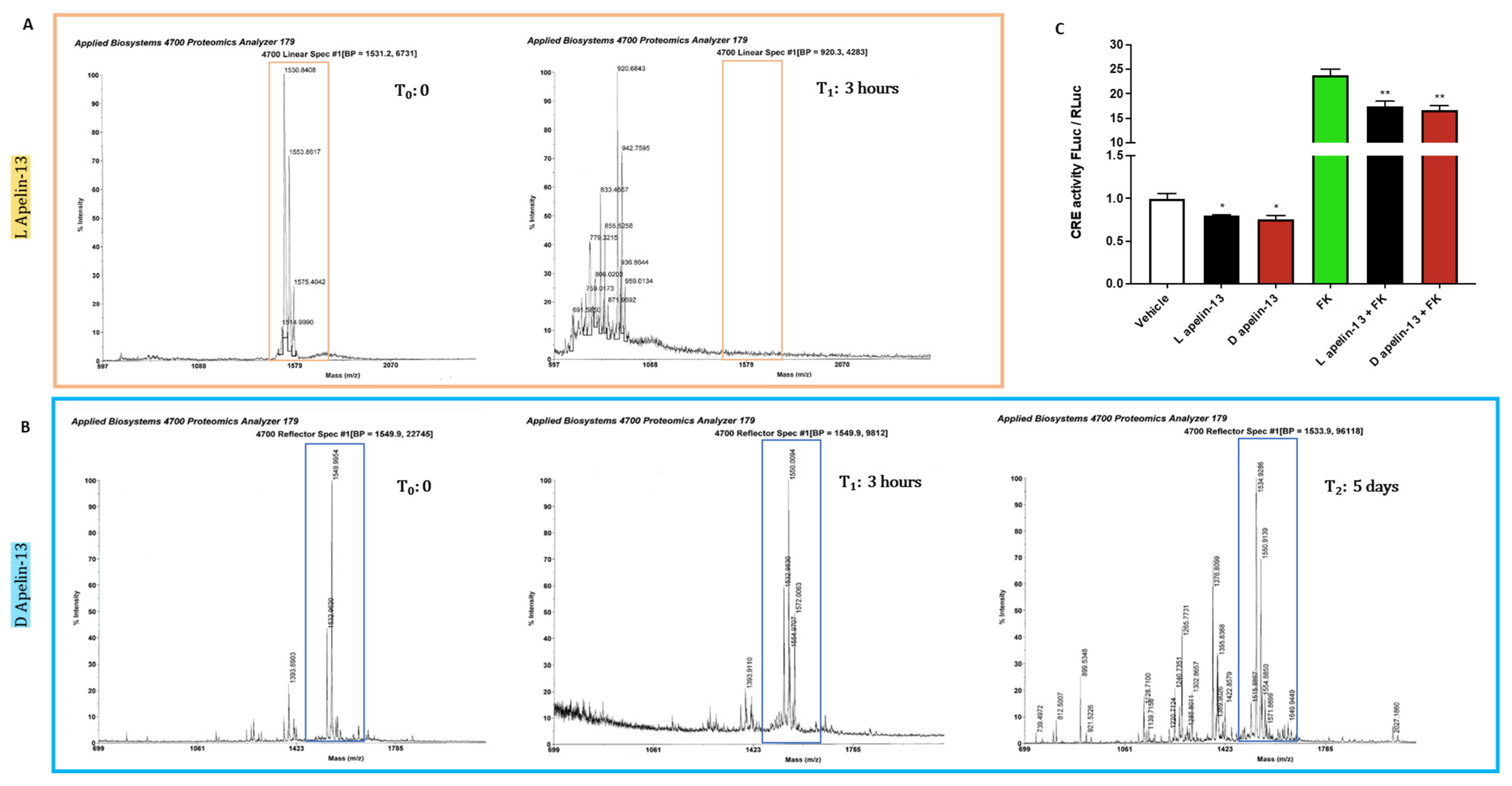
3.3. Apelin 13 Carrying All the Amino Acids in D-Isomers Has Much Longer Half-Life In Vivo
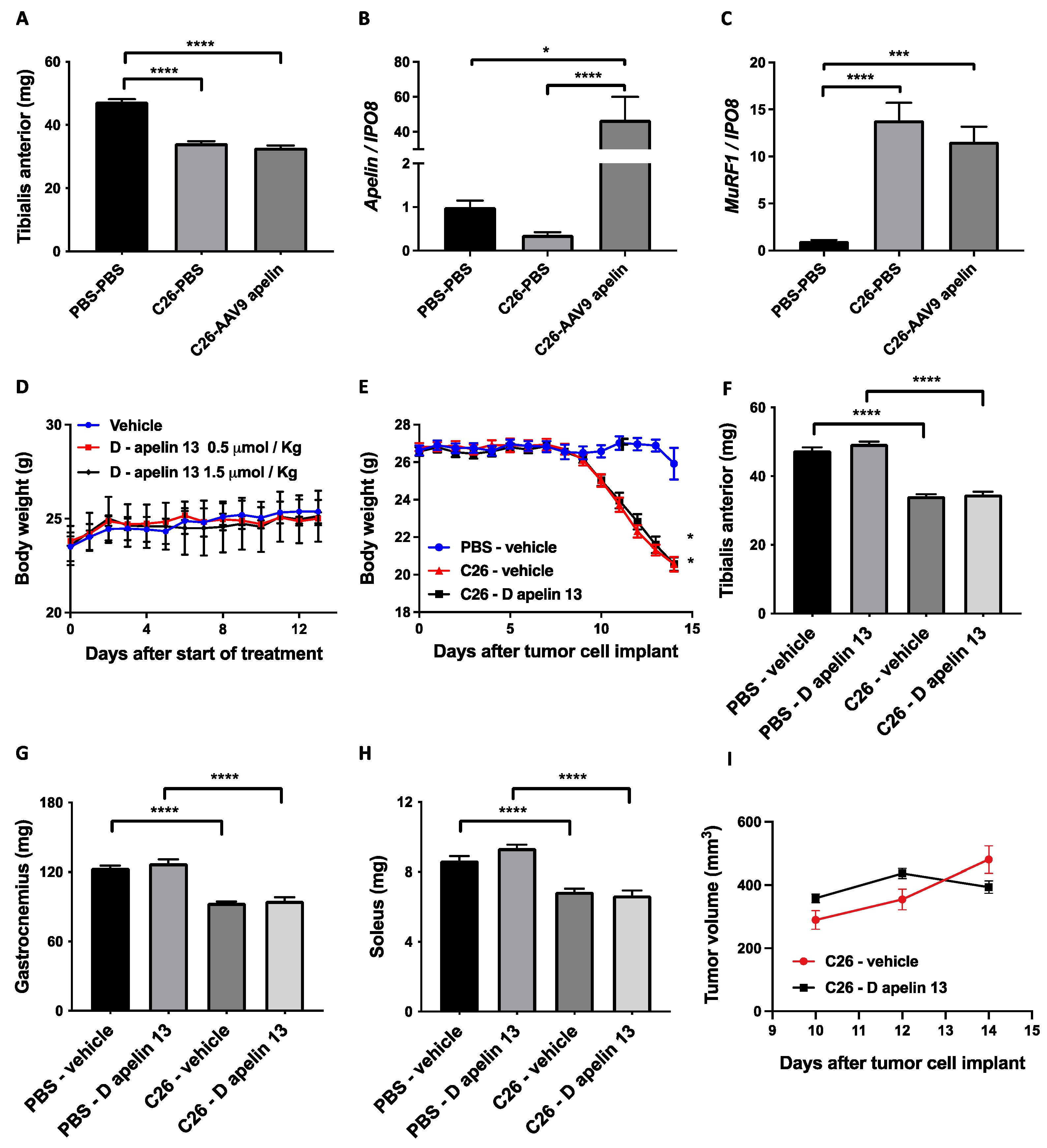
3.4. Preproapelin or Apelin Given to Cancer-Bearing Mice Does Not Affect Muscle Size at All
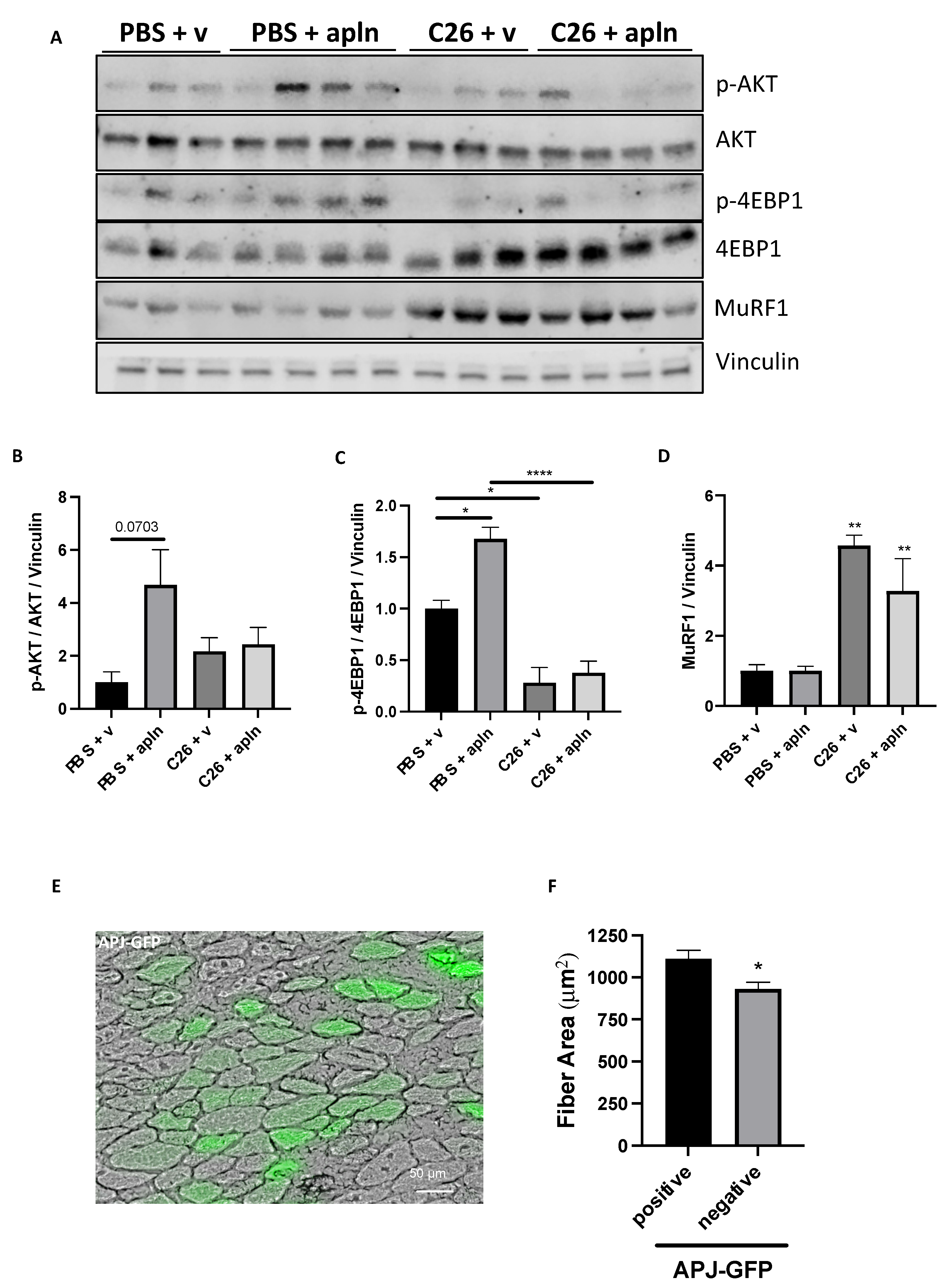
3.5. Muscles from Mice with Cancer Cachexia Exhibit Undesirable Apelin Resistance
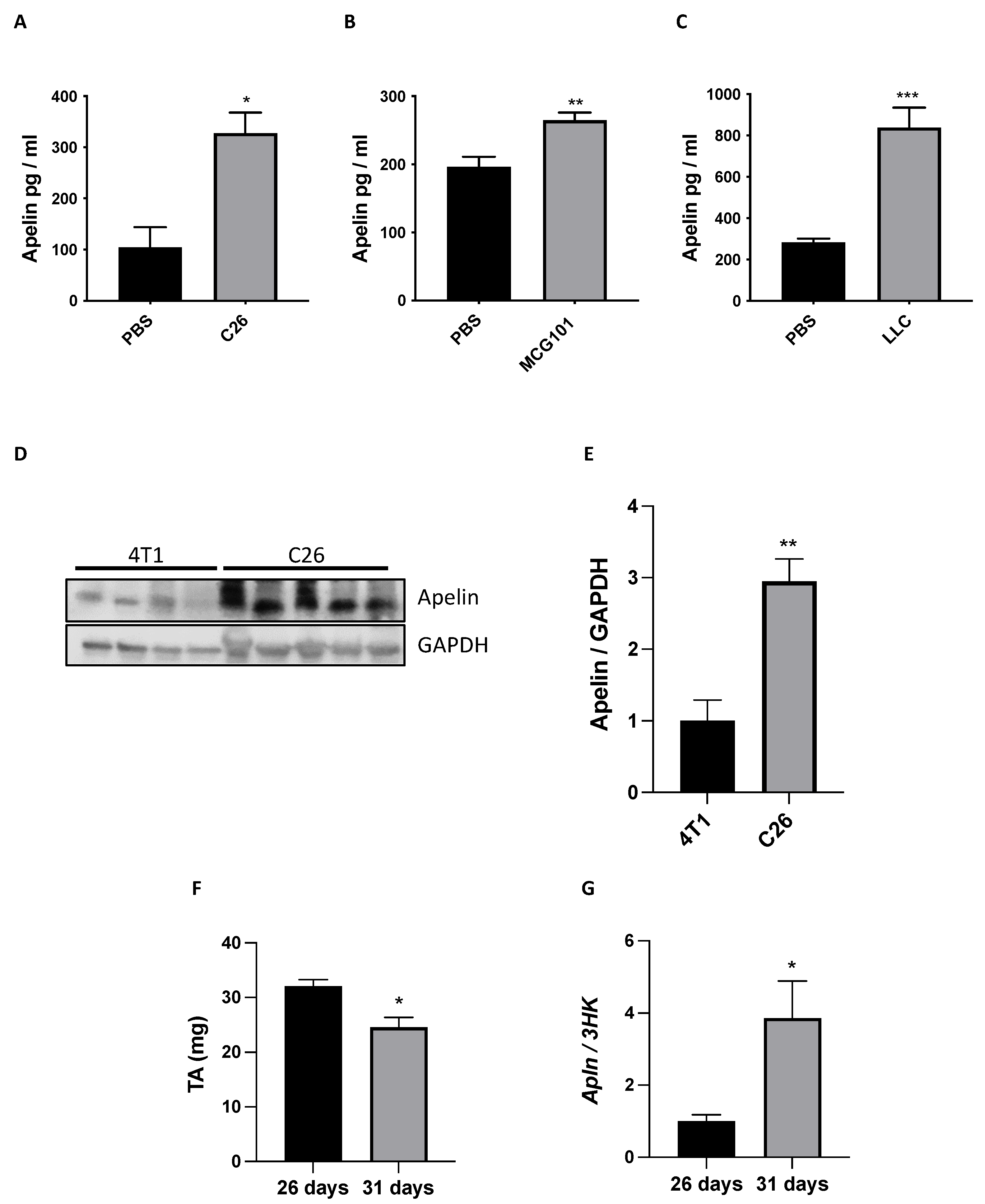
3.6. Plasma Apelin Is Increased in Cachectic Cancer-Bearing Mice
4. Discussion
4.1. Apelin Is Regulated in Opposite Ways in Muscle and Plasma during Cancer Cachexia
4.2. How Apelin and Glucocorticoids May Be Linked
4.3. Insulin Resistance and Apelin Resistance in Cancer Cachexia?
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Piccirillo, R.; Demontis, F.; Perrimon, N.; Goldberg, A.L. Mechanisms of muscle growth and atrophy in mammals and drosophila. Dev. Dyn. 2014, 243, 201–215. [Google Scholar] [CrossRef] [Green Version]
- Argiles, J.M.; Busquets, S.; Stemmler, B.; Lopez-Soriano, F.J. Cancer cachexia: Understanding the molecular basis. Nat. Rev. Cancer 2014, 14, 754–762. [Google Scholar] [CrossRef]
- Pretto, F.; Ghilardi, C.; Moschetta, M.; Bassi, A.; Rovida, A.; Scarlato, V.; Talamini, L.; Fiordaliso, F.; Bisighini, C.; Damia, G.; et al. Sunitinib prevents cachexia and prolongs survival of mice bearing renal cancer by restraining STAT3 and MuRF-1 activation in muscle. Oncotarget 2015, 6, 3043–3054. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.L.; Lu, J.; Song, Y.; Kwak, K.S.; Jiao, Q.; Rosenfeld, R.; Chen, Q.; Boone, T.; Simonet, W.S.; et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 2010, 142, 531–543. [Google Scholar] [CrossRef] [Green Version]
- Figueroa-Clarevega, A.; Bilder, D. Malignant drosophila tumors interrupt insulin signaling to induce cachexia-like wasting. Dev. Cell 2015, 33, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Kwon, Y.; Song, W.; Droujinine, I.A.; Hu, Y.; Asara, J.M.; Perrimon, N. Systemic organ wasting induced by localized expression of the secreted Insulin/IGF antagonist ImpL2. Dev. Cell 2015, 33, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Asp, M.L.; Tian, M.; Wendel, A.A.; Belury, M.A. Evidence for the contribution of insulin resistance to the development of cachexia in tumor-bearing mice. Int. J. Cancer 2010, 126, 756–763. [Google Scholar] [CrossRef]
- Honors, M.A.; Kinzig, K.P. The role of insulin resistance in the development of muscle wasting during cancer cachexia. J. Cachexia Sarcopenia Muscle 2012, 3, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Lecker, S.H.; Jagoe, R.T.; Gilbert, A.; Gomes, M.; Baracos, V.; Bailey, J.; Price, S.R.; Mitch, W.E.; Goldberg, A.L. Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J. 2004, 18, 39–51. [Google Scholar] [CrossRef]
- Martinelli, G.B.; Olivari, D.; Re Cecconi, A.D.; Talamini, L.; Ottoboni, L.; Lecker, S.H.; Stretch, C.; Baracos, V.E.; Bathe, O.F.; Resovi, A.; et al. Activation of the SDF1/CXCR4 pathway retards muscle atrophy during cancer cachexia. Oncogene 2016, 48, 6212–6222. [Google Scholar] [CrossRef]
- Indrakusuma, I.; Sell, H.; Eckel, J. Novel mediators of adipose tissue and muscle crosstalk. Curr. Obes. Rep. 2015, 4, 411–417. [Google Scholar] [CrossRef]
- Brailoiu, G.; Dun, S.L.; Yang, J.; Ohsawa, M.; Chang, J.K.; Dun, N.J. Apelin-immunoreactivity in the rat hypothalamus and pituitary. Neurosci. Lett. 2002, 327, 193–197. [Google Scholar] [CrossRef]
- Dray, C.; Knauf, C.; Daviaud, D.; Waget, A.; Boucher, J.; Buleon, M.; Cani, P.D.; Attane, C.; Guigne, C.; Carpene, C.; et al. Apelin stimulates glucose utilization in normal and obese insulin-resistant mice. Cell Metab. 2008, 8, 437–445. [Google Scholar] [CrossRef]
- Yue, P.; Jin, H.; Aillaud, M.; Deng, A.C.; Azuma, J.; Asagami, T.; Kundu, R.K.; Reaven, G.M.; Quertermous, T.; Tsao, P.S. Apelin is necessary for the maintenance of insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E59–E67. [Google Scholar] [CrossRef] [Green Version]
- Tatemoto, K.; Hosoya, M.; Habata, Y.; Fujii, R.; Kakegawa, T.; Zou, M.X.; Kawamata, Y.; Fukusumi, S.; Hinuma, S.; Kitada, C.; et al. Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor. Biochem. Biophys. Res. Commun. 1998, 251, 471–476. [Google Scholar] [CrossRef]
- Hosoya, M.; Kawamata, Y.; Fukusumi, S.; Fujii, R.; Habata, Y.; Hinuma, S.; Kitada, C.; Honda, S.; Kurokawa, T.; Onda, H.; et al. Molecular and functional characteristics of APJ. tissue distribution of MRNA and interaction with the endogenous ligand apelin. J. Biol. Chem. 2000, 275, 21061–21067. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Li, H.; Chen, L. Targeting drugs to APJ receptor: The prospect of treatment of hypertension and other cardiovascular diseases. Curr. Drug Targets 2015, 16, 148–155. [Google Scholar] [CrossRef]
- Wysocka, M.B.; Pietraszek-Gremplewicz, K.; Nowak, D. The role of apelin in cardiovascular diseases, obesity and cancer. Front. Physiol. 2018, 9, 557. [Google Scholar] [CrossRef]
- Masri, B.; Knibiehler, B.; Audigier, Y. Apelin signalling: A promising pathway from cloning to pharmacology. Cell Signal. 2005, 17, 415–426. [Google Scholar] [CrossRef]
- Vinel, C.; Lukjanenko, L.; Batut, A.; Deleruyelle, S.; Pradère, J.-P.; Le Gonidec, S.; Dortignac, A.; Geoffre, N.; Pereira, O.; Karaz, S.; et al. The exerkine apelin reverses age-associated sarcopenia. Nat. Med. 2018, 24, 1360–1371. [Google Scholar] [CrossRef]
- Lee, D.K.; Cheng, R.; Nguyen, T.; Fan, T.; Kariyawasam, A.P.; Liu, Y.; Osmond, D.H.; George, S.R.; O’Dowd, B.F. Characterization of apelin, the ligand for the APJ receptor. J. Neurochem. 2000, 74, 34–41. [Google Scholar] [CrossRef]
- Reed, A.B.; Lanman, B.A.; Holder, J.R.; Yang, B.H.; Ma, J.; Humphreys, S.C.; Wang, Z.; Chan, J.C.Y.; Miranda, L.P.; Swaminath, G.; et al. Half-Life extension of Peptidic APJ agonists by N-Terminal lipid conjugation. Bioorg. Med. Chem. Lett. 2020, 30, 127499. [Google Scholar] [CrossRef]
- Jiang, Y.; Yan, M.; Wang, C.; Wang, Q.; Chen, X.; Zhang, R.; Wan, L.; Ji, B.; Dong, B.; Wang, H.; et al. The effects of apelin and elabela ligands on apelin receptor distinct signaling profiles. Front. Pharmacol. 2021, 12, 630548. [Google Scholar] [CrossRef]
- Zhen, E.Y.; Higgs, R.E.; Gutierrez, J.A. Pyroglutamyl Apelin-13 identified as the major apelin isoform in human plasma. Anal. Biochem. 2013, 442, 1–9. [Google Scholar] [CrossRef]
- Resovi, A.; Borsotti, P.; Ceruti, T.; Passoni, A.; Zucchetti, M.; Berndt, A.; Riser, B.L.; Taraboletti, G.; Belotti, D. CCN-Based therapeutic peptides modify pancreatic ductal adenocarcinoma microenvironment and decrease tumor growth in combination with chemotherapy. Cells 2020, 9, 952. [Google Scholar] [CrossRef]
- Kawahara, H.; Naito, H.; Takara, K.; Wakabayashi, T.; Kidoya, H.; Takakura, N. Tumor endothelial cell-specific drug delivery system using apelin-conjugated liposomes. PLoS ONE 2013, 8, e65499. [Google Scholar] [CrossRef] [Green Version]
- Kidoya, H.; Naito, H.; Takakura, N. Apelin Induces enlarged and nonleaky blood vessels for functional recovery from ischemia. Blood 2010, 115, 3166–3174. [Google Scholar] [CrossRef] [Green Version]
- Arsic, N.; Zacchigna, S.; Zentilin, L.; Ramirez-Correa, G.; Pattarini, L.; Salvi, A.; Sinagra, G.; Giacca, M. Vascular endothelial growth factor stimulates skeletal muscle regeneration in vivo. Mol. Ther. 2004, 10, 844–854. [Google Scholar] [CrossRef]
- Ayuso, E.; Mingozzi, F.; Montane, J.; Leon, X.; Anguela, X.M.; Haurigot, V.; Edmonson, S.A.; Africa, L.; Zhou, S.; High, K.A.; et al. High AAV vector purity results in serotype- and tissue-independent enhancement of transduction efficiency. Gene Ther. 2010, 17, 503–510. [Google Scholar] [CrossRef] [Green Version]
- Re Cecconi, A.D.; Forti, M.; Chiappa, M.; Zhu, Z.; Zingman, L.V.; Cervo, L.; Beltrame, L.; Marchini, S.; Piccirillo, R. Musclin, A Myokine induced by aerobic exercise, retards muscle atrophy during cancer cachexia in mice. Cancers 2019, 11, 1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, 0034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Resovi, A.; Bani, M.R.; Porcu, L.; Anastasia, A.; Minoli, L.; Allavena, P.; Cappello, P.; Novelli, F.; Scarpa, A.; Morandi, E.; et al. Soluble stroma-related biomarkers of pancreatic cancer. EMBO Mol. Med. 2018, 10, e8741. [Google Scholar] [CrossRef]
- Michaelis, K.A.; Zhu, X.; Burfeind, K.G.; Krasnow, S.M.; Levasseur, P.R.; Morgan, T.K.; Marks, D.L. Establishment and characterization of a novel murine model of pancreatic cancer cachexia. J. Cachex-Sarcopenia Muscle 2017, 8, 824–838. [Google Scholar] [CrossRef] [PubMed]
- Bedard, N.; Jammoul, S.; Moore, T.; Wykes, L.; Hallauer, P.L.; Hastings, K.E.; Stretch, C.; Baracos, V.; Chevalier, S.; Plourde, M.; et al. Inactivation of the ubiquitin-specific protease 19 deubiquitinating enzyme protects against muscle wasting. FASEB J. 2015, 29, 3889–3898. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Latres, E.; Baumhueter, S.; Lai, V.K.-M.; Nunez, L.; Clarke, B.A.; Poueymirou, W.T.; Panaro, F.J.; Na, E.; Dharmarajan, K.; et al. Identification of Ubiquitin Ligases Required for Skeletal Muscle Atrophy. Science 2001, 294, 1704–1708. [Google Scholar] [CrossRef]
- Sartori, R.; Schirwis, E.; Blaauw, B.; Bortolanza, S.; Zhao, J.; Enzo, E.; Stantzou, A.; Mouisel, E.; Toniolo, L.; Ferry, A.; et al. BMP signaling controls muscle mass. Nat. Genet. 2013, 45, 1309–1318. [Google Scholar] [CrossRef]
- Biferali, B.; Proietti, D.; Mozzetta, C.; Madaro, L. Fibro-Adipogenic progenitors cross-talk in skeletal muscle: The social network. Front. Physiol. 2019, 10, 1074. [Google Scholar] [CrossRef]
- Latres, E.; Amini, A.R.; Amini, A.A.; Griffiths, J.; Martin, F.J.; Wei, Y.; Lin, H.C.; Yancopoulos, G.D.; Glass, D.J. Insulin-like growth Factor-1 (IGF-1) inversely regulates atrophy-induced genes via the phosphatidylinositol 3-Kinase/Akt/Mammalian target of rapamycin (PI3K/Akt/MTOR) pathway. J. Biol. Chem. 2005, 280, 2737–2744. [Google Scholar] [CrossRef] [Green Version]
- Muto, J.; Shirabe, K.; Yoshizumi, T.; Ikegami, T.; Aishima, S.; Ishigami, K.; Yonemitsu, Y.; Ikeda, T.; Soejima, Y.; Maehara, Y. The Apelin-APJ system induces tumor Arteriogenesis in hepatocellular carcinoma. Anticancer Res. 2014, 34, 5313–5320. [Google Scholar]
- Higuchi, K.; Masaki, T.; Gotoh, K.; Chiba, S.; Katsuragi, I.; Tanaka, K.; Kakuma, T.; Yoshimatsu, H. Apelin, an APJ receptor ligand, regulates body adiposity and favors the messenger ribonucleic acid expression of uncoupling proteins in mice. Endocrinology 2007, 148, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Evans, B.J.; King, A.T.; Katsifis, A.; Matesic, L.; Jamie, J.F. Methods to enhance the metabolic stability of peptide-based PET radiopharmaceuticals. Molecules 2020, 25, 2314. [Google Scholar] [CrossRef] [PubMed]
- Deterre, P.; Paupardin-Tritsch, D.; Bockaert, J.; Gerschenfeld, H.M. CAMP-Mediated decrease in K+ conductance evoked by serotonin and dopamine in the same neuron: A biochemical and physiological single-cell study. Proc. Natl. Acad. Sci. USA 1982, 79, 7934–7938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masri, B.; Morin, N.; Pedebernade, L.; Knibiehler, B.; Audigier, Y. The apelin receptor is coupled to Gi1 or Gi2 protein and is differentially desensitized by apelin fragments. J. Biol. Chem. 2006, 281, 18317–18326. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Kim, K.-M. Multifactorial regulation of G protein-coupled receptor endocytosis. Biomol. Ther. 2017, 25, 26–43. [Google Scholar] [CrossRef] [Green Version]
- Moo, E.V.; van Senten, J.R.; Bräuner-Osborne, H.; Møller, T.C. Arrestin-Dependent and -independent internalization of G protein-coupled receptors: Methods, mechanisms, and implications on cell signaling. Mol. Pharmacol. 2021, 99, 242–255. [Google Scholar] [CrossRef]
- Mueller, T.C.; Burmeister, M.A.; Bachmann, J.; Martignoni, M.E. Cachexia and pancreatic cancer: Are there treatment options? World J. Gastroenterol. 2014, 20, 9361–9373. [Google Scholar] [CrossRef]
- Rupert, J.E.; Narasimhan, A.; Jengelley, D.H.; Jiang, Y.; Liu, J.; Au, E.; Silverman, L.M.; Sandusky, G.; Bonetto, A.; Cao, S.; et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J. Exp. Med. 2021, 218, 20190450. [Google Scholar] [CrossRef]
- Olson, B.; Zhu, X.; Norgard, M.A.; Levasseur, P.R.; Butler, J.T.; Buenafe, A.; Burfeind, K.G.; Michaelis, K.A.; Pelz, K.R.; Mendez, H.; et al. Lipocalin 2 mediates appetite suppression during pancreatic cancer cachexia. Nat. Commun. 2021, 12, 2057. [Google Scholar] [CrossRef]
- Mannelli, M.; Gamberi, T.; Magherini, F.; Fiaschi, T. The adipokines in cancer cachexia. Int. J. Mol. Sci. 2020, 21, 4860. [Google Scholar] [CrossRef]
- Minetti, G.C.; Feige, J.N.; Rosenstiel, A.; Bombard, F.; Meier, V.; Werner, A.; Bassilana, F.; Sailer, A.W.; Kahle, P.; Lambert, C.; et al. Gαi2 signaling promotes skeletal muscle hypertrophy, myoblast differentiation, and muscle regeneration. Sci. Signal. 2011, 4, ra80. [Google Scholar] [CrossRef] [PubMed]
- Lokireddy, S.; Kukushkin, N.V.; Goldberg, A.L. CAMP-Induced phosphorylation of 26S proteasomes on Rpn6/PSMD11 enhances their activity and the degradation of misfolded proteins. Proc. Natl. Acad. Sci. USA 2015, 112, E7176–E7185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.; Ehrlich, L.; Venter, J.; O’Brien, A.; White, T.; Zhou, T.; Dang, T.; Meng, F.; Invernizzi, P.; Bernuzzi, F.; et al. Inhibition of the apelin/apelin receptor axis decreases cholangiocarcinoma growth. Cancer Lett. 2017, 386, 179–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uribesalgo, I.; Hoffmann, D.; Zhang, Y.; Kavirayani, A.; Lazovic, J.; Berta, J.; Novatchkova, M.; Pai, T.-P.; Wimmer, R.A.; László, V.; et al. Apelin inhibition prevents resistance and metastasis associated with anti-angiogenic therapy. EMBO Mol. Med. 2019, 11, e9266. [Google Scholar] [CrossRef]
- Diakowska, D.; Markocka-Mączka, K.; Szelachowski, P.; Grabowski, K. Serum Levels of resistin, adiponectin, and apelin in gastroesophageal cancer patients. Dis. Markers 2014, 2014, 619649. [Google Scholar] [CrossRef] [Green Version]
- Geiger, T.; Velic, A.; Macek, B.; Lundberg, E.; Kampf, C.; Nagaraj, N.; Uhlen, M.; Cox, J.; Mann, M. Initial quantitative proteomic map of 28 mouse tissues using the SILAC mouse. Mol. Cell. Proteom. 2013, 12, 1709–1722. [Google Scholar] [CrossRef] [Green Version]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [Green Version]
- Williams, E.G.; Wu, Y.; Jha, P.; Dubuis, S.; Blattmann, P.; Argmann, C.A.; Houten, S.M.; Amariuta, T.; Wolski, W.; Zamboni, N.; et al. Systems proteomics of liver mitochondria function. Science 2016, 352, aad0189. [Google Scholar] [CrossRef] [Green Version]
- Kawamoto, S.; Matsumoto, Y.; Mizuno, K.; Okubo, K.; Matsubara, K. Expression profiles of active genes in human and mouse livers. Gene 1996, 174, 151–158. [Google Scholar] [CrossRef]
- Pope, G.R.; Tilve, S.; McArdle, C.A.; Lolait, S.J.; O’Carroll, A.-M. Agonist-Induced Internalization and Desensitization of the Apelin Receptor. Mol. Cell. Endocrinol. 2016, 437, 108–119. [Google Scholar] [CrossRef] [Green Version]
- Besse-Patin, A.; Montastier, E.; Vinel, C.; Castan-Laurell, I.; Louche, K.; Dray, C.; Daviaud, D.; Mir, L.; Marques, M.A.; Thalamas, C.; et al. Effect of endurance training on skeletal muscle myokine expression in obese men: Identification of apelin as a novel myokine. Int. J. Obes. 2014, 38, 707–713. [Google Scholar] [CrossRef] [PubMed]
- Fujie, S.; Sato, K.; Miyamoto-Mikami, E.; Hasegawa, N.; Fujita, S.; Sanada, K.; Hamaoka, T.; Iemitsu, M. Reduction of arterial stiffness by exercise training is associated with increasing plasma apelin level in middle-aged and older adults. PLoS ONE 2014, 9, e93545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papangeli, I.; Kim, J.; Maier, I.; Park, S.; Lee, A.; Kang, Y.; Tanaka, K.; Khan, O.F.; Ju, H.; Kojima, Y.; et al. MicroRNA 139-5p coordinates APLNR-CXCR4 crosstalk during vascular maturation. Nat. Commun. 2016, 7, 11268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandri, M.; Lin, J.; Handschin, C.; Yang, W.; Arany, Z.P.; Lecker, S.H.; Goldberg, A.L.; Spiegelman, B.M. PGC-1alpha protects skeletal muscle from atrophy by suppressing FoxO3 action and atrophy-specific gene transcription. Proc. Natl. Acad. Sci. USA 2006, 103, 16260–16265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Eda, H.; Tanaka, T.; Udagawa, T.; Ishikawa, T.; Horii, I.; Ishitsuka, H.; Kataoka, T.; Taguchi, T. Experimental cancer cachexia induced by transplantable colon 26 adenocarcinoma in mice. Cancer Res. 1990, 50, 2290–2295. [Google Scholar]
- Goncalves, M.D.; Hwang, S.-K.; Pauli, C.; Murphy, C.J.; Cheng, Z.; Hopkins, B.D.; Wu, D.; Loughran, R.M.; Emerling, B.M.; Zhang, G.; et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc. Natl. Acad. Sci. USA 2018, 115, E743–E752. [Google Scholar] [CrossRef] [Green Version]
- Costelli, P.; Carbó, N.; Tessitore, L.; Bagby, G.J.; Lopez-Soriano, F.J.; Argilés, J.M.; Baccino, F.M. Tumor necrosis factor-alpha mediates changes in tissue protein turnover in a rat cancer cachexia model. J. Clin. Investig. 1993, 92, 2783–2789. [Google Scholar] [CrossRef] [Green Version]
- Tessitore, L.; Costelli, P.; Baccino, F.M. Pharmacological interference with tissue Hypercatabolism in Tumour-Bearing rats. Biochem. J. 1994, 299 Pt 1, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Cala, M.P.; Agulló-Ortuño, M.T.; Prieto-García, E.; González-Riano, C.; Parrilla-Rubio, L.; Barbas, C.; Díaz-García, C.V.; García, A.; Pernaut, C.; Adeva, J.; et al. Multiplatform plasma fingerprinting in cancer cachexia: A pilot observational and translational study. J. Cachexia Sarcopenia Muscle 2018, 9, 348–357. [Google Scholar] [CrossRef]
- Flint, T.R.; Janowitz, T.; Connell, C.M.; Roberts, E.W.; Denton, A.E.; Coll, A.P.; Jodrell, D.I.; Fearon, D.T. Tumor-Induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. 2016, 24, 672–684. [Google Scholar] [CrossRef]
- Knapp, M.L.; al-Sheibani, S.; Riches, P.G.; Hanham, I.W.; Phillips, R.H. Hormonal factors associated with weight loss in patients with advanced breast cancer. Ann. Clin. Biochem. 1991, 28 Pt 5, 480–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B.A.; Drujan, D.; Willis, M.S.; Murphy, L.O.; Corpina, R.A.; Burova, E.; Rakhilin, S.V.; Stitt, T.N.; Patterson, C.; Latres, E.; et al. The E3 ligase MuRF1 degrades myosin heavy chain protein in dexamethasone-treated skeletal muscle. Cell Metab. 2007, 6, 376–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eo, H.; Reed, C.H.; Valentine, R.J. Imoxin prevents dexamethasone-induced promotion of muscle-specific E3 ubiquitin ligases and stimulates anabolic signaling in C2C12 myotubes. Biomed. Pharmacother. 2020, 128, 110238. [Google Scholar] [CrossRef] [PubMed]
- Sacheck, J.M.; Ohtsuka, A.; McLary, S.C.; Goldberg, A.L. IGF-I Stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, Atrogin-1 and MuRF1. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E591–E601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.S.; Kong, I.D. Irisin prevents dexamethasone-induced atrophy in C2C12 Myotubes. Pflug. Arch. 2020, 472, 495–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.; Hou, X.; Tatemoto, K. Regulation of apelin MRNA expression by insulin and glucocorticoids in mouse 3T3-L1 adipocytes. Regul. Pept. 2005, 132, 27–32. [Google Scholar] [CrossRef]
- Shen, H.; Liu, T.; Fu, L.; Zhao, S.; Fan, B.; Cao, J.; Li, X. Identification of MicroRNAs involved in dexamethasone-induced muscle atrophy. Mol. Cell Biochem. 2013, 381, 105–113. [Google Scholar] [CrossRef]
- Dray, C.; Debard, C.; Jager, J.; Disse, E.; Daviaud, D.; Martin, P.; Attané, C.; Wanecq, E.; Guigné, C.; Bost, F.; et al. Apelin and APJ regulation in adipose tissue and skeletal muscle of type 2 Diabetic mice and humans. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1161–E1169. [Google Scholar] [CrossRef] [Green Version]
- Arcidiacono, B.; Iiritano, S.; Nocera, A.; Possidente, K.; Nevolo, M.T.; Ventura, V.; Foti, D.; Chiefari, E.; Brunetti, A. Insulin resistance and cancer risk: An overview of the pathogenetic mechanisms. Exp. Diabetes Res. 2012, 2012, 789174. [Google Scholar] [CrossRef] [Green Version]
- Aulino, P.; Berardi, E.; Cardillo, V.M.; Rizzuto, E.; Perniconi, B.; Ramina, C.; Padula, F.; Spugnini, E.P.; Baldi, A.; Faiola, F.; et al. Molecular, cellular and physiological characterization of the cancer cachexia-inducing C26 colon carcinoma in mouse. BMC Cancer 2010, 10, 363. [Google Scholar] [CrossRef]
- Toye, A.A.; Lippiat, J.; Proks, P.; Shimomura, K.; Bentley, L.; Hugill, A.; Mijat, V.; Goldsworthy, M.; Moir, L.; Haynes, A.; et al. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 2005, 48, 675–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Re Cecconi, A.D.; Barone, M.; Forti, M.; Lunardi, M.; Cagnotto, A.; Salmona, M.; Olivari, D.; Zentilin, L.; Resovi, A.; Persichitti, P.; et al. Apelin Resistance Contributes to Muscle Loss during Cancer Cachexia in Mice. Cancers 2022, 14, 1814. https://doi.org/10.3390/cancers14071814
Re Cecconi AD, Barone M, Forti M, Lunardi M, Cagnotto A, Salmona M, Olivari D, Zentilin L, Resovi A, Persichitti P, et al. Apelin Resistance Contributes to Muscle Loss during Cancer Cachexia in Mice. Cancers. 2022; 14(7):1814. https://doi.org/10.3390/cancers14071814
Chicago/Turabian StyleRe Cecconi, Andrea David, Mara Barone, Mara Forti, Martina Lunardi, Alfredo Cagnotto, Mario Salmona, Davide Olivari, Lorena Zentilin, Andrea Resovi, Perla Persichitti, and et al. 2022. "Apelin Resistance Contributes to Muscle Loss during Cancer Cachexia in Mice" Cancers 14, no. 7: 1814. https://doi.org/10.3390/cancers14071814
APA StyleRe Cecconi, A. D., Barone, M., Forti, M., Lunardi, M., Cagnotto, A., Salmona, M., Olivari, D., Zentilin, L., Resovi, A., Persichitti, P., Belotti, D., Palo, F., Takakura, N., Kidoya, H., & Piccirillo, R. (2022). Apelin Resistance Contributes to Muscle Loss during Cancer Cachexia in Mice. Cancers, 14(7), 1814. https://doi.org/10.3390/cancers14071814