
Genetic Clonality as the Hallmark Driving Evolution of Non-Small Cell Lung Cancer


Abstract
Simple Summary
Abstract
1. Introduction
2. Clonality of Genetic Alterations
3. The Cell of Origin Theory
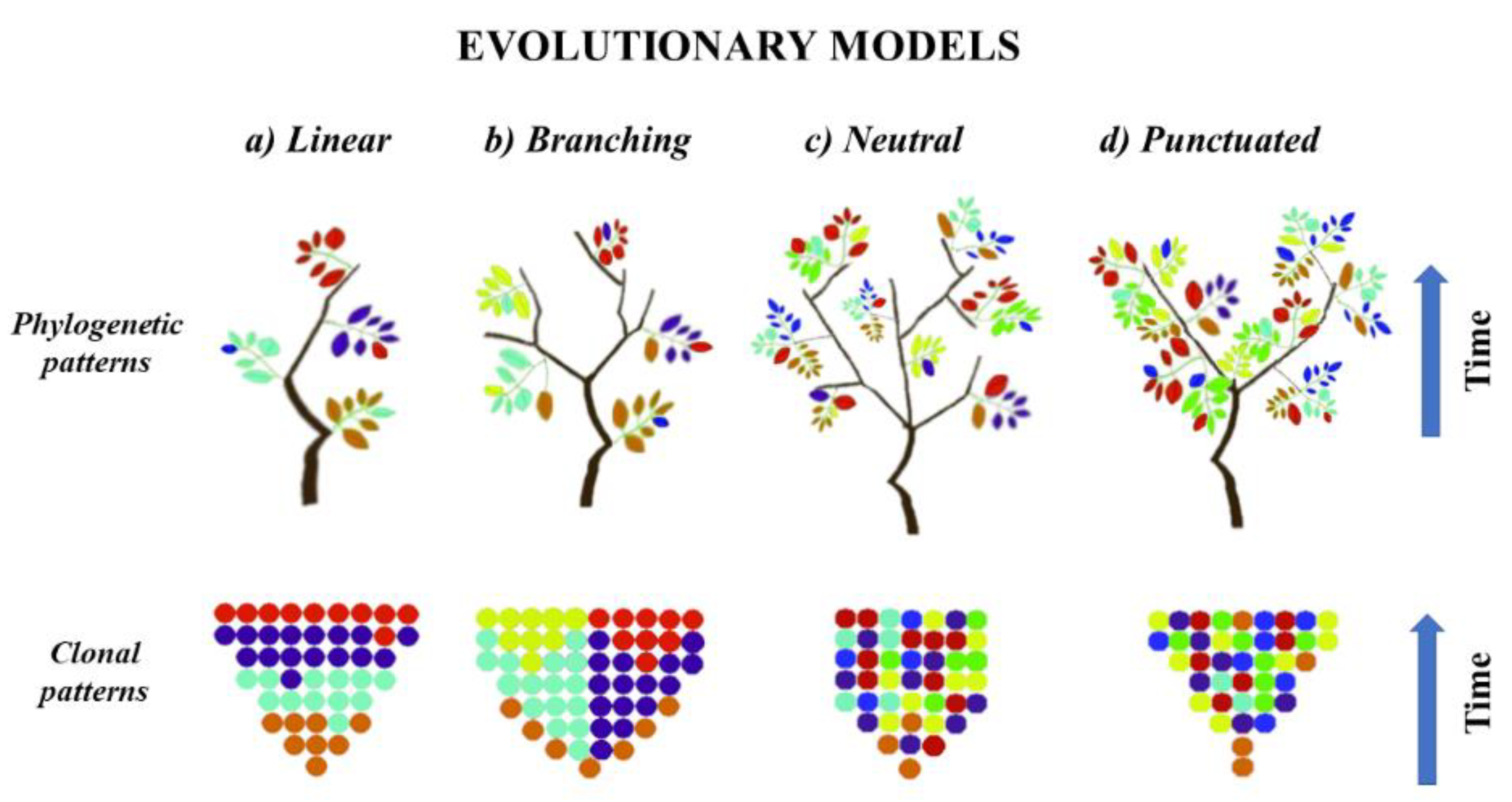
4. General Tumor Evolution Theory
5. Clinical Implication of Driver Alterations
6. Metastatic NSCLC Cells
7. Clonality of Metastases
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vendramin, R.; Litchfield, K.; Swanton, C. Cancer evolution: Darwin and beyond. EMBO J. 2021, 40, e108389. [Google Scholar] [CrossRef] [PubMed]
- Swanton, C. Intratumor Heterogeneity: Evolution through Space and Time. Cancer Res. 2012, 72, 4875–4882. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra54. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Gerold, J.M.; Nowak, M.A. Quantifying Clonal and Subclonal Passenger Mutations in Cancer Evolution. PLoS Comput. Biol. 2016, 12, e1004731. [Google Scholar] [CrossRef]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.; Gao, R.; Navin, N. Tumor evolution: Linear, branching, neutral or punctuated? Biochim. Biophys. Acta 2017, 1867, 151–161. [Google Scholar] [CrossRef]
- Caswell, D.R.; Swanton, C. The role of tumour heterogeneity and clonal cooperativity in metastasis, immune evasion and clinical outcome. BMC Med. 2017, 15, 133. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Biological and Therapeutic Impact of Intratumor Heterogeneity in Cancer Evolution. Cancer Cell 2015, 27, 15–26. [Google Scholar] [CrossRef]
- McGranahan, N.; Swanton, C. Clonal Heterogeneity and Tumor Evolution: Past, Present, and the Future. Cell 2017, 168, 613–628. [Google Scholar] [CrossRef]
- Turajlic, S.; Swanton, C. Metastasis as an evolutionary process. Science 2016, 352, 169–175. [Google Scholar] [CrossRef]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet. 2020, 21, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Neelakantan, D.; Drasin, D.J.; Ford, H.L. Intratumoral heterogeneity: Clonal cooperation in epithelial-to-mesenchymal transition and metastasis. Cell Adhes. Migr. 2014, 9, 265–276. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Birkbak, N.J.; McGranahan, N. Cancer Genome Evolutionary Trajectories in Metastasis. Cancer Cell 2020, 37, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.A. Parallel progression of primary tumours and metastases. Nat. Cancer 2009, 9, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Heyde, A.; Reiter, J.G.; Naxerova, K.; Nowak, M.A. Consecutive seeding and transfer of genetic diversity in metastasis. Proc. Natl. Acad. Sci. USA 2019, 116, 14129–14137. [Google Scholar] [CrossRef] [PubMed]
- Karreman, M.A.; Winkler, F. The mechanics of metastatic seeding. Nat. Cell Biol. 2018, 20, 860–862. [Google Scholar] [CrossRef]
- Tamura, T.; Kurishima, K.; Nakazawa, K.; Kagohashi, K.; Ishikawa, H.; Satoh, H.; Hizawa, N. Specific organ metastases and survival in metastatic non-small-cell lung cancer. Mol. Clin. Oncol. 2014, 3, 217–221. [Google Scholar] [CrossRef]
- Li, J.; Zhu, H.; Sun, L.; Xu, W.; Wang, X. Prognostic value of site-specific metastases in lung cancer: A population based study. J. Cancer 2019, 10, 3079–3086. [Google Scholar] [CrossRef]
- Nones, K.; Patch, A.-M. The Impact of Next Generation Sequencing in Cancer Research. Cancers 2020, 12, 2928. [Google Scholar] [CrossRef]
- Mardis, E.R. The Impact of Next-Generation Sequencing on Cancer Genomics: From Discovery to Clinic. Cold Spring Harb. Perspect. Med. 2019, 9, a036269. [Google Scholar] [CrossRef] [PubMed]
- Andor, N.; Graham, T.A.; Jansen, M.; Xia, L.C.; Aktipis, C.A.; Petritsch, C.; Ji, H.P.; Maley, C.C. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nat. Med. 2016, 22, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Werner, B.; Barnes, C.P.; Graham, T.A.; Sottoriva, A. Identification of neutral tumor evolution across cancer types. Nat. Genet. 2016, 48, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Beerenwinkel, N.; Schwarz, R.; Gerstung, M.; Markowetz, F. Cancer Evolution: Mathematical Models and Computational Inference. Syst. Biol. 2015, 64, e1–e25. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef]
- Goto, T.; Hirotsu, Y.; Mochizuki, H.; Nakagomi, T.; Shikata, D.; Yokoyama, Y.; Oyama, T.; Amemiya, K.; Okimoto, K.; Omata, M. Mutational analysis of multiple lung cancers: Discrimination between primary and metastatic lung cancers by genomic profile. Oncotarget 2017, 8, 31133–31143. [Google Scholar] [CrossRef]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- Yuan, M.; Huang, L.-L.; Chen, J.-H.; Wu, J.; Xu, Q. The emerging treatment landscape of targeted therapy in non-small-cell lung cancer. Signal Transduct. Target. Ther. 2019, 4, 61. [Google Scholar] [CrossRef]
- Shaurova, T.; Zhang, L.; Goodrich, D.W.; Hershberger, P.A. Understanding Lineage Plasticity as a Path to Targeted Therapy Failure in EGFR-Mutant Non-small Cell Lung Cancer. Front. Genet. 2020, 11, 281. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Govindan, R.; Ding, L.; Griffith, M.; Subramanian, J.; Dees, N.D.; Kanchi, K.L.; Maher, C.A.; Fulton, R.; Fulton, L.; Wallis, J.; et al. Genomic Landscape of Non-Small Cell Lung Cancer in Smokers and Never-Smokers. Cell 2012, 150, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Avci, N.; Hayar, M.; Altmisdortoglu, O.; Tanriverdi, O.; Deligonul, A.; Ordu, C.; Evrensel, T. Smoking habits are an independent prognostic factor in patients with lung cancer. Clin. Respir. J. 2015, 11, 579–584. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.D.; Alexandrov, A.; Kim, J.; Wala, J.; Berger, A.H.; Pedamallu, C.S.; Shukla, S.A.; Guo, G.; Brooks, A.N.; Murray, B.A.; et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat. Genet. 2016, 48, 607–616. [Google Scholar] [CrossRef]
- Imielinski, M.; Berger, A.H.; Hammerman, P.S.; Hernandez, B.; Pugh, T.J.; Hodis, E.; Cho, J.; Suh, J.; Capelletti, M.; Sivachenko, A.; et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell 2012, 150, 1107–1120. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas (TCGA) Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef]
- Shi, J.; Hua, X.; Zhu, B.; Ravichandran, S.; Wang, M.; Nguyen, C.; Brodie, S.A.; Palleschi, A.; Alloisio, M.; Pariscenti, G.; et al. Somatic Genomics and Clinical Features of Lung Adenocarcinoma: A Retrospective Study. PLOS Med. 2016, 13, e1002162. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- Watkins, T.B.K.; Lim, E.L.; Petkovic, M.; Elizalde, S.; Birkbak, N.J.; Wilson, G.A.; Moore, D.A.; Grönroos, E.; Rowan, A.; Dewhurst, S.M.; et al. Pervasive chromosomal instability and karyotype order in tumour evolution. Nature 2020, 587, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Devarakonda, S.; Govindan, R. TRAC(ERx)-ing lung cancer evolution. Ann. Oncol. 2017, 28, 1690–1692. [Google Scholar] [CrossRef] [PubMed]
- Vincenten, J.P.L.; Van Essen, H.F.; Lissenberg-Witte, B.I.; Bulkmans, N.W.J.; Krijgsman, O.; Sie, D.; Eijk, P.P.; Smit, E.F.; Ylstra, B.; Thunnissen, E. Clonality analysis of pulmonary tumors by genome-wide copy number profiling. PLoS ONE 2019, 14, e0223827. [Google Scholar] [CrossRef]
- Zhang, J.; Fujimoto, J.; Zhang, J.; Wedge, D.C.; Song, X.; Zhang, J.; Seth, S.; Chow, C.-W.; Cao, Y.; Gumbs, C.; et al. Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 2014, 346, 256–259. [Google Scholar] [CrossRef]
- Saber, A.; Hiltermann, T.N.; Kok, K.; Terpstra, M.M.; De Lange, K.; Timens, W.; Groen, H.J.; Berg, A.V.D. Mutation patterns in small cell and non-small cell lung cancer patients suggest a different level of heterogeneity between primary and metastatic tumors. Carcinogenesis 2017, 38, 144–151. [Google Scholar] [CrossRef]
- Hashida, S.; Soh, J.; Toyooka, S.; Tanaka, T.; Furukawa, M.; Shien, K.; Yamamoto, H.; Asano, H.; Tsukuda, K.; Hagiwara, K.; et al. Presence of the minor EGFR T790M mutation is associated with drug-sensitive EGFR mutations in lung adenocarcinoma patients. Oncol. Rep. 2014, 32, 145–152. [Google Scholar] [CrossRef]
- Inukai, M.; Toyooka, S.; Ito, S.; Asano, H.; Ichihara, S.; Soh, J.; Suehisa, H.; Ouchida, M.; Aoe, K.; Aoe, M.; et al. Presence of Epidermal Growth Factor Receptor Gene T790M Mutation as a Minor Clone in Non–Small Cell Lung Cancer. Cancer Res. 2006, 66, 7854–7858. [Google Scholar] [CrossRef]
- Majeed, U.; Manochakian, R.; Zhao, Y.; Lou, Y. Targeted therapy in advanced non-small cell lung cancer: Current advances and future trends. J. Hematol. Oncol. 2021, 14, 108. [Google Scholar] [CrossRef]
- Senosain, M.F.; Massion, P.P. Intratumor Heterogeneity in Early Lung Adenocarcinoma. Front Oncol. 2020, 10, 349. [Google Scholar] [CrossRef]
- Wei, Q.; Ye, Z.; Zhong, X.; Li, L.; Wang, C.; Myers, R.E.; Palazzo, J.P.; Fortuna, D.; Yan, A.; Waldman, S.A.; et al. Multiregion whole-exome sequencing of matched primary and metastatic tumors revealed genomic heterogeneity and suggested polyclonal seeding in colorectal cancer metastasis. Ann. Oncol. 2017, 28, 2135–2141. [Google Scholar] [CrossRef]
- Testa, U.; Castelli, G.; Pelosi, E. Lung Cancers: Molecular Characterization, Clonal Heterogeneity and Evolution, and Cancer Stem Cells. Cancers 2018, 10, 248. [Google Scholar] [CrossRef] [PubMed]
- Marjanovic, N.D.; Weinberg, R.A.; Chaffer, C.L. Cell Plasticity and Heterogeneity in Cancer. Clin. Chem. 2013, 59, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Baccelli, I.; Trumpp, A. The evolving concept of cancer and metastasis stem cells. J. Cell Biol. 2012, 198, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Nolte, S.M.; Venugopal, C.; McFarlane, N.; Morozova, O.; Hallett, R.M.; O’Farrell, E.; Manoranjan, B.; Murty, N.K.; Klurfan, P.; Kachur, E.; et al. A Cancer Stem Cell Model for Studying Brain Metastases from Primary Lung Cancer. JNCI J. Natl. Cancer Inst. 2013, 105, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Walcher, L.; Kistenmacher, A.-K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.-R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells—Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Ferone, G.; Lee, M.C.; Sage, J.; Berns, A. Cells of origin of lung cancers: Lessons from mouse studies. Genes Dev. 2020, 34, 1017–1032. [Google Scholar] [CrossRef]
- Sanchez-Danes, A.; Blanpain, C. Deciphering the cells of origin of squamous cell carcinomas. Nat. Cancer 2018, 18, 549–561. [Google Scholar] [CrossRef]
- Kreso, A.; Dick, J.E. Evolution of the Cancer Stem Cell Model. Cell Stem Cell 2014, 14, 275–291. [Google Scholar] [CrossRef]
- Eramo, A.; Lotti, F.; Sette, G.; Pilozzi, E.; Biffoni, M.; Di Virgilio, A.; Conticello, C.; Ruco, L.; Peschle, C.; De Maria, R. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell Death Differ. 2008, 15, 504–514. [Google Scholar] [CrossRef]
- Bertolini, G.; Roz, L.; Perego, P.; Tortoreto, M.; Fontanella, E.; Gatti, L.; Pratesi, G.; Fabbri, A.; Andriani, F.; Tinelli, S.; et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc. Natl. Acad. Sci. USA 2009, 106, 16281–16286. [Google Scholar] [CrossRef]
- Liu, Y.-P.; Yang, C.-J.; Huang, M.-S.; Yeh, C.-T.; Wu, A.T.; Lee, Y.-C.; Lai, T.-C.; Lee, C.-H.; Hsiao, Y.-W.; Lu, J.; et al. Cisplatin Selects for Multidrug-Resistant CD133+Cells in Lung Adenocarcinoma by Activating Notch Signaling. Cancer Res. 2013, 73, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.P.; Spinola, M.; Dodge, M.; Raso, M.G.; Behrens, C.; Gao, B.; Schuster, K.; Shao, C.; Larsen, J.; Sullivan, L.A.; et al. Aldehyde Dehydrogenase Activity Selects for Lung Adenocarcinoma Stem Cells Dependent on Notch Signaling. Cancer Res. 2010, 70, 9937–9948. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Yu, L.; Wang, D.; Zhou, L.; Cheng, Z.; Chai, D.; Ma, L.; Tao, Y. Aberrant expression of CD133 in non-small cell lung cancer and its relationship to vasculogenic mimicry. BMC Cancer 2012, 12, 535. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Lu, L.; Zander, D.S.; Sreerama, L.; Coco, D.; Moreb, J.S. ALDH1A1 and ALDH3A1 expression in lung cancers: Correlation with histologic type and potential precursors. Lung Cancer 2008, 59, 340–349. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, K.D.; Proost, N.; Brouns, I.; Adriaensen, D.; Song, J.-Y.; Berns, A. Cell of Origin of Small Cell Lung Cancer: Inactivation of Trp53 and Rb1 in Distinct Cell Types of Adult Mouse Lung. Cancer Cell 2011, 19, 754–764. [Google Scholar] [CrossRef]
- Hardavella, G.; George, R.; Sethi, T. Lung cancer stem cells—characteristics, phenotype. Transl. Lung Cancer Res. 2016, 5, 272–279. [Google Scholar] [CrossRef]
- Fukui, T.; Shaykhiev, R.; Agosto-Perez, F.; Mezey, J.G.; Downey, R.J.; Travis, W.D.; Crystal, R.G. Lung adenocarcinoma subtypes based on expression of human airway basal cell genes. Eur. Respir. J. 2013, 42, 1332–1344. [Google Scholar] [CrossRef]
- Kim, C.F.B.; Jackson, E.L.; Woolfenden, A.E.; Lawrence, S.; Babar, I.; Vogel, S.; Crowley, D.; Bronson, R.T.; Jacks, T. Identification of Bronchioalveolar Stem Cells in Normal Lung and Lung Cancer. Cell 2005, 121, 823–835. [Google Scholar] [CrossRef]
- Salnikov, A.V.; Gladkich, J.; Moldenhauer, G.; Volm, M.; Herr, I. CD133 is indicative for a resistance phenotype but does not represent a prognostic marker for survival of non-small cell lung cancer patients. Int. J. Cancer 2010, 126, 950–958. [Google Scholar] [CrossRef]
- Hassan, K.A.; Wang, L.; Korkaya, H.; Chen, G.; Maillard, I.; Etherton-Beer, C.; Kalemkerian, G.P.; Wicha, M.S. Notch Pathway Activity Identifies Cells with Cancer Stem Cell–like Properties and Correlates with Worse Survival in Lung Adenocarcinoma. Clin. Cancer Res. 2013, 19, 1972–1980. [Google Scholar] [CrossRef]
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125, 5591–5596. [Google Scholar] [CrossRef] [PubMed]
- Albini, A.; Bruno, A.; Gallo, C.; Pajardi, G.E.; Noonan, U.M.; Dallaglio, K. Cancer stem cells and the tumor microenvironment: Interplay in tumor heterogeneity. Connect. Tissue Res. 2015, 56, 414–425. [Google Scholar] [CrossRef] [PubMed]
- MacDonagh, L.; Gray, S.; Breen, E.; Cuffe, S.; Finn, S.; O’Byrne, K.J.; Barr, M.P. Lung cancer stem cells: The root of resistance. Cancer Lett. 2016, 372, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.; Mlecnik, B.; Vasaturo, A.; Bindea, G.; Fredriksen, T.; Lafontaine, L.; Buttard, B.; Morgand, E.; Bruni, D.; Jouret-Mourin, A.; et al. Evolution of Metastases in Space and Time under Immune Selection. Cell 2018, 175, 751–765.e16. [Google Scholar] [CrossRef] [PubMed]
- Ben-David, U.; Beroukhim, R.; Golub, T.R. Genomic evolution of cancer models: Perils and opportunities. Nat. Cancer 2019, 19, 97–109. [Google Scholar] [CrossRef]
- Reiter, J.G.; Makohon-Moore, A.P.; Gerold, J.M.; Heyde, A.; Attiyeh, M.A.; Kohutek, Z.A.; Tokheim, C.J.; Brown, A.; DeBlasio, R.M.; Niyazov, J.; et al. Minimal functional driver gene heterogeneity among untreated metastases. Science 2018, 361, 1033–1037. [Google Scholar] [CrossRef]
- Marusyk, A.; Tabassum, D.P.; Altrock, P.; Almendro, V.; Michor, F.; Polyak, K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014, 514, 54–58. [Google Scholar] [CrossRef]
- Xie, S.; Wu, Z.; Qi, Y.; Wu, B.; Zhu, X. The metastasizing mechanisms of lung cancer: Recent advances and therapeutic challenges. Biomed. Pharmacother. 2021, 138, 111450. [Google Scholar] [CrossRef]
- Nguyen, L.; Vanner, R.; Dirks, P.B.; Eaves, C.J. Cancer stem cells: An evolving concept. Nat. Cancer 2012, 12, 133–143. [Google Scholar] [CrossRef]
- Plaks, V.; Kong, N.; Werb, Z. The Cancer Stem Cell Niche: How Essential Is the Niche in Regulating Stemness of Tumor Cells? Cell Stem Cell 2015, 16, 225–238. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Jordan, E.J.; Kim, H.R.; Arcila, M.E.; Barron, D.; Chakravarty, D.; Gao, J.; Chang, M.T.; Ni, A.; Kundra, R.; Jonsson, P.; et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017, 7, 596–609. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Forjaz, G.; Mooradian, M.J.; Meza, R.; Kong, C.Y.; Cronin, K.A.; Mariotto, A.B.; Lowy, D.R.; Feuer, E.J. The Effect of Advances in Lung-Cancer Treatment on Population Mortality. N. Engl. J. Med. 2020, 383, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Lewis, W.E.; Hong, L.; Mott, F.E.; Simon, G.; Wu, C.C.; Rinsurongkawong, W.; Lee, J.J.; Lam, V.K.; Heymach, J.V.; Zhang, J.; et al. Efficacy of Targeted Inhibitors in Metastatic Lung Squamous Cell Carcinoma with EGFR or ALK Alterations. JTO Clin. Res. Rep. 2021, 2, 100237. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.E.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Nathanson, T.; Rizvi, H.; Creelan, B.C.; Sanchez-Vega, F.; Ahuja, A.; Ni, A.; Novik, J.B.; Mangarin, L.M.; Abu-Akeel, M.; et al. Genomic Features of Response to Combination Immunotherapy in Patients with Advanced Non-Small-Cell Lung Cancer. Cancer Cell 2018, 33, 843–852. [Google Scholar] [CrossRef]
- Rizvi, H.; Sanchez-Vega, F.; La, K.; Chatila, W.; Jonsson, P.; Halpenny, D.; Plodkowski, A.; Long, N.; Sauter, J.L.; Rekhtman, N.; et al. Molecular Determinants of Response to Anti–Programmed Cell Death (PD)-1 and Anti–Programmed Death-Ligand 1 (PD-L1) Blockade in Patients with Non–Small-Cell Lung Cancer Profiled with Targeted Next-Generation Sequencing. J. Clin. Oncol. 2018, 36, 633–641. [Google Scholar] [CrossRef]
- Abbosh, C.; Birkbak, N.J.; Wilson, G.A.; Jamal-Hanjani, M.; Constantin, T.; Salari, R.; Le Quesne, J.; Moore, D.A.; Veeriah, S.; Rosenthal, R.; et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017, 545, 446–451. [Google Scholar] [CrossRef]
- Vavalà, T.; Monica, V.; Iacono, M.L.; Mele, T.; Busso, S.; Righi, L.; Papotti, M.; Scagliotti, G.V.; Novello, S. Precision medicine in age-specific non-small-cell-lung-cancer patients: Integrating biomolecular results into clinical practice—A new approach to improve personalized translational research. Lung Cancer 2017, 107, 84–90. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [PubMed]
- NIH. Clinical Trials.gov. Available online: https://clinicaltrials.gov/ (accessed on 13 February 2022).
- Altorki, N.K.; Markowitz, G.J.; Gao, D.; Port, J.L.; Saxena, A.; Stiles, B.; McGraw, T.; Mittal, V. The lung microenvironment: An important regulator of tumour growth and metastasis. Nat. Rev. Cancer 2019, 19, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Arai, E.; Takahashi, Y.; Totsuka, H.; Chiku, S.; Taniguchi, H.; Katai, H.; Sakamoto, H.; Yoshida, T.; Kanai, Y. Cooperative participation of epigenomic and genomic alterations in the clinicopathological diversity of gastric adenocarcinomas: Significance of cell adhesion and epithelial–mesenchymal transition-related signaling pathways. Carcinogenesis 2020, 41, 1473–1484. [Google Scholar] [CrossRef]
- LaBarge, M.A. The Difficulty of Targeting Cancer Stem Cell Niches. Clin. Cancer Res. 2010, 16, 3121–3129. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Parsana, P.; Amend, S.R.; Hernandez, J.; Pienta, K.J.; Battle, A. Identifying global expression patterns and key regulators in epithelial to mesenchymal transition through multi-study integration. BMC Cancer 2017, 17, 447. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Tsai, E.B.; Pomykala, K.; Ruchalski, K.; Genshaft, S.; Abtin, F.; Gutierrez, A.; Kim, H.J.; Li, A.; Adame, C.; Jalalian, A.; et al. Feasibility and Safety of Intrathoracic Biopsy and Repeat Biopsy for Evaluation of Programmed Cell Death Ligand–1 Expression for Immunotherapy in Non–Small Cell Lung Cancer. Radiology 2018, 287, 326–332. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Cieślik, M.; Hoang, S.A.; Baranova, N.; Chodaparambil, S.; Kumar, M.; Allison, D.F.; Xu, X.; Wamsley, J.J.; Gray, L.; Jones, D.R.; et al. Epigenetic coordination of signaling pathways during the epithelial-mesenchymal transition. Epigenet. Chromatin 2013, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Gordian, E.; Welsh, E.A.; Gimbrone, N.; Siegel, E.M.; Shibata, D.; Creelan, B.C.; Cress, W.D.; Eschrich, S.A.; Haura, E.B.; Muñoz-Antonia, T. Transforming growth factor β-induced epithelial-to-mesenchymal signature predicts metastasis-free survival in non-small cell lung cancer. Oncotarget 2019, 10, 810–824. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Januario, T.; Eberhard, D.A.; Cavet, G.; Zhu, W.; Fu, L.; Pham, T.Q.; Soriano, R.; Stinson, J.; Seshagiri, S.; et al. Epithelial versus Mesenchymal Phenotype Determines In vitro Sensitivity and Predicts Clinical Activity of Erlotinib in Lung Cancer Patients. Clin. Cancer Res. 2005, 11, 8686–8698. [Google Scholar] [CrossRef] [PubMed]
- Byers, L.A.; Diao, L.; Wang, J.; Saintigny, P.; Girard, L.; Peyton, M.; Shen, L.; Fan, Y.; Giri, U.; Tumula, P.K.; et al. An Epithelial–Mesenchymal Transition Gene Signature Predicts Resistance to EGFR and PI3K Inhibitors and Identifies Axl as a Therapeutic Target for Overcoming EGFR Inhibitor Resistance. Clin. Cancer Res. 2012, 19, 279–290. [Google Scholar] [CrossRef]
- Liang, S.-Q.; Marti, T.M.; Dorn, P.B.; Froment, L.; Hall, S.R.R.; Berezowska, S.; Kocher, G.J.; Schmid, R.A.; Peng, R. Blocking the epithelial-to-mesenchymal transition pathway abrogates resistance to anti-folate chemotherapy in lung cancer. Cell Death Dis. 2015, 6, e1824. [Google Scholar] [CrossRef]
- Chae, Y.K.; Chang, S.; Ko, T.; Anker, J.; Agte, S.; Iams, W.; Choi, W.M.; Lee, K.; Cruz, M. Epithelial-mesenchymal transition (EMT) signature is inversely associated with T-cell infiltration in non-small cell lung cancer (NSCLC). Sci. Rep. 2018, 8, 2918. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Nagathihalli, N.S.; Massion, P.P.; Gonzalez, A.L.; Lu, P.; Datta, P.K. Smoking Induces Epithelial-to-Mesenchymal Transition in Non–Small Cell Lung Cancer through HDAC-Mediated Downregulation of E-Cadherin. Mol. Cancer Ther. 2012, 11, 2362–2372. [Google Scholar] [CrossRef]
- Vu, T.; Jin, L.; Datta, P.K. Effect of Cigarette Smoking on Epithelial to Mesenchymal Transition (EMT) in Lung Cancer. J. Clin. Med. 2016, 5, 44. [Google Scholar] [CrossRef]
- Vasaikar, S.V.; Deshmukh, A.P.; Hollander, P.D.; Addanki, S.; Kuburich, N.A.; Kudaravalli, S.; Joseph, R.; Chang, J.T.; Soundararajan, R.; Mani, S.A. EMTome: A resource for pan-cancer analysis of epithelial-mesenchymal transition genes and signatures. Br. J. Cancer 2021, 124, 259–269. [Google Scholar] [CrossRef]
- Ingangi, V.; Minopoli, M.; Ragone, C.; Motti, M.L.; Carriero, M.V.; Ingangi, V.; Minopoli, M.; Ragone, C.; Motti, M.L.; Carriero, M.V. Role of Microenvironment on the Fate of Disseminating Cancer Stem Cells. Front. Oncol. 2019, 9, 82. [Google Scholar] [CrossRef] [PubMed]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.; Assen, F.P.; Leithner, A.; Abe, J.; Schachner, H.; Asfour, G.; Bago-Horvath, Z.; Stein, J.V.; Uhrin, P.; Sixt, M.; et al. Lymph node blood vessels provide exit routes for metastatic tumor cell dissemination in mice. Science 2018, 359, 1408–1411. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Li, Z.; Ma, Z.; Curtis, C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat. Genet. 2020, 52, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Fu, R.; Liang, Y.; Lin, J.; Qiu, Z.; Wu, Y.; Zhong, W. Genomic Evolution of Lung Cancer Metastasis: Current Status and Perspectives. Cancer Commun. 2021, 41, 1252–1256. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-C.; Reuben, A.; Hu, X.; McGranahan, N.; Chen, R.; Jalali, A.; Negrao, M.V.; Hubert, S.M.; Tang, C.; Wu, C.-C.; et al. Multiomics profiling of primary lung cancers and distant metastases reveals immunosuppression as a common characteristic of tumor cells with metastatic plasticity. Genome Biol. 2020, 21, 271. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; Van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef]
- Zhao, Z.-M.; Zhao, B.; Bai, Y.; Iamarino, A.; Gaffney, S.G.; Schlessinger, J.; Lifton, R.P.; Rimm, D.L.; Townsend, J.P. Early and multiple origins of metastatic lineages within primary tumors. Proc. Natl. Acad. Sci. USA 2016, 113, 2140–2145. [Google Scholar] [CrossRef]
- Chen, X.; Bu, Q.; Yan, X.; Li, Y.; Yu, Q.; Zheng, H.; Zhao, L.; Zeng, Y.; Lu, L.; Lan, D.; et al. Genomic Mutations of Primary and Metastatic Lung Adenocarcinoma in Chinese Patients. J. Oncol. 2020, 2020, 6615575. [Google Scholar] [CrossRef]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; De Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; Van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef]
- Rud, A.K.; Boye, K.; Fodstad, Ø.; Juell, S.; Jørgensen, L.H.; Solberg, S.; Helland, Å.; Brustugun, O.T.; Mælandsmo, G.M. Detection of disseminated tumor cells in lymph nodes from patients with early stage non-small cell lung cancer. Diagn. Pathol. 2016, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Tezel, C.; Dogruyol, T.; Alpay, L.; Akyıl, M.; Evman, S.; Metin, S.; Baysungur, V.; Yalcinkaya, I. Prognostic Importance of the Lymph Node Factor in Surgically Resected Non-Small Cell Lung Cancer. Thorac. Cardiovasc. Surg. 2018, 68, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Okamoto, T.; Fukuyama, S.; Maehara, Y. Therapeutic strategy for postoperative recurrence in patients with non-small cell lung cancer. World J. Clin. Oncol. 2014, 5, 1048–1054. [Google Scholar] [CrossRef]
- Pari, A.A.A.; Singhal, M.; Augustin, H.G. Emerging paradigms in metastasis research. J. Exp. Med. 2021, 218, e20190218. [Google Scholar] [CrossRef]
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative clinical genomics of metastatic cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Nwabo, K.A.H.; Takam, K.P.; Tagne, S.R.; Vecchio, L.; Seke, E.P.F.; Muller, J.-M.; Bassi, G.; Lukong, E.; Kumar, G.R.; Mbo, A.J.; et al. Developmental pathways associated with cancer metastasis: Notch, Wnt, and Hedgehog. Cancer Biol. Med. 2017, 14, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Shih, D.J.H.; Nayyar, N.; Bihun, I.; Dagogo-Jack, I.; Gill, C.M.; Aquilanti, E.; Bertalan, M.; Kaplan, A.; D’Andrea, M.R.; Chukwueke, U.; et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat. Genet. 2020, 52, 371–377. [Google Scholar] [CrossRef]
- Nicos, M.; Harbers, L.; Patrucco, E.; Kramer-Drauberg, M.; Zhang, X.; Voena, C.; Kowalczyk, A.; Bozyk, A.; Peksa, R.; Jarosz, B.; et al. Comparative genomic profiling identifies targetable brain metastasis drivers in non-small cell lung cancer. Res. Square 2022. [Google Scholar] [CrossRef]
- Kim, N.; Kim, H.K.; Lee, K.; Hong, Y.; Cho, J.H.; Choi, J.W.; Lee, J.-I.; Suh, Y.-L.; Ku, B.M.; Eum, H.H.; et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020, 11, 2285. [Google Scholar] [CrossRef]
- Zhang, L.; Mao, S.; Yao, M.; Chao, N.; Yang, Y.; Ni, Y.; Song, T.; Liu, Z.; Yang, Y.; Li, W. Spatial Transcriptome Sequencing Revealed Spatial Trajectory in the Non-Small Cell Lung Carcinoma. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rozenberg, J.M.; Filkov, G.I.; Trofimenko, A.V.; Karpulevich, E.A.; Parshin, V.D.; Royuk, V.V.; Sekacheva, M.I.; Durymanov, M.O. Biomedical Applications of Non-Small Cell Lung Cancer Spheroids. Front. Oncol. 2021, 11, 791069. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, H.; Ding, Q.; Xing, Y.; Xu, Z.; Lu, C.; Luo, D.; Xu, L.; Xia, W.; Zhou, C.; et al. Establishment of patient-derived tumor spheroids for non-small cell lung cancer. PLoS ONE 2018, 13, e0194016. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Type of Driver Alterations | Gene | Clonality Type | |
|---|---|---|---|
| Mutational level | SNVs, substitutions, small indels, genes rearrangements/fusions | TP53 | clonal/subclonal |
| KRAS | clonal/subclonal | ||
| EGFR | clonal | ||
| KEAP1 | clonal | ||
| KMT2C | clonal | ||
| KMT2D | clonal | ||
| NF1 | subclonal | ||
| PIK3CA | subclonal | ||
| COL5A2 | clonal | ||
| ATM | subclonal | ||
| ARID1A | clonal | ||
| BRAF | clonal | ||
| NOTCH1 | clonal/subclonal | ||
| ALK | clonal | ||
| PTEN | clonal | ||
| MET | clonal | ||
| CNAs level | Amplifications | TRIO | subclonal |
| SOX | clonal | ||
| PIK3CA | clonal | ||
| TERT | clonal/subclonal | ||
| NKX2-1 | subclonal | ||
| TP63 | clonal | ||
| MUC1 | subclonal | ||
| MYC | subclonal | ||
| RICTOR | subclonal | ||
| FGFR1 | clonal | ||
| CCND1 | subclonal | ||
| CDK4 | subclonal | ||
| Deletions | CDKN2A | subclonal | |
| CDKN2B | subclonal | ||
| DMRTA1 | subclonal | ||
| STC1 | subclonal | ||
| NSCLC Subtype | Driver Gene | % TCGA Frequency | Targeted Drug | Status (Registered/Open Trial) |
|---|---|---|---|---|
| non-smoking related (LUAD) | EGFR | 16.6% | erlotinib, gefitinib, afatinib, dacomitinib, osimertinib | registered |
| KEAP1 | 15.2% | telaglenastat | NCT04265534 | |
| sotorasib | NCT05054725 | |||
| STK11 | 13.2% | talazoparib | NCT04265534 | |
| sotorasib | NCT04933695 | |||
| BRAF | 5.6% | dabrafenib + trametinib | registered | |
| encorafenib + binimetinib | NCT03915951 | |||
| ALK-EML4 (fusion) | 2.9/2.5% | crizotinib, alectinib, ceritinib, brigatinib, lorlatinib | registered | |
| ROS1 (fusion) | 1.3% | crizotinib, entrectinib | registered | |
| repotrectinib | NCT03093116 | |||
| MET | 3.6% | crizotinib | breakthrough therapy NCT04084717 | |
| tepotinib | registered NCT04739358 NCT04647838 | |||
| capmatinib | registered NCT04427072 NCT03693339 | |||
| capmatinib + spartalizumab | NCT04323436 | |||
| cabozantinib | NCT03911193 | |||
| ningetinib | NCT04992858 | |||
| RET (fusion) | 1.5% | cabozantinib | NCT01639508 NCT04131543 | |
| selpercatinib | registered NCT04194944 NCT04268550 | |||
| entrectinib | NCT04302025 | |||
| pralsetinib | registered NCT03037385 NCT04222972 | |||
| anlotinib | NCT04073537 | |||
| Smoking related (LUAD + LUSC) | TP53 | 60.1% | ALRN-6924 | NCT04022876 |
| tedopi | NCT04884282 | |||
| KRAS | 24% | binimetinib + palbociclib | NCT03170206 | |
| sotorasib | registered NCT05118854 | |||
| JDQ443 | NCT05132075 | |||
| adagrasib | breakthrough therapy NCT04685135 | |||
| sotorasib + RMC-4630 | NCT05054725 | |||
| PIK3CA | 8.3% | serabelisib + canagliflozin | NCT04073680 | |
| TPST-1495 | NCT04344795 | |||
| BRCA1/2 | 7.5% | rucaparib | NCT03845296 | |
| olaparib + cediranib + durvalumab | NCT02484404 | |||
| niraparib + pembrolizumab | NCT04475939 | |||
| CDKN2A | 7.3% | erlotinib + trastuzumab | NCT04591431 | |
| RB1 | 5.6% | IBI188 + GM-CSF | NCT04861948 | |
| NOTCH1 | 5% | tegavivint + osimertinib | NCT04780568 | |
| PTEN | 4.2% | elemene + 1st EGFR-TKIs generation | NCT04401059 |
ACTA1 | CDC42 | ELMO3 * | GRHL2 * | LTBP4 * | PARP1 | SH3YL1 * | TIMP1 |
| ACTN1 * | CDH1 *^” | EPB41L5 * | GSC | MAF * | PDGFB | SHANK3 * | TIMP2 * |
| ADGRF4 * | CDH2 ”^ | EPHA1 * | GSK3B | MAL2 * | PDLIM7 * | SHROOM3 * | TJP1 ” |
| AFAP1L2 * | CDH3 * | EPHB2 * | HNMT * | MAP1B | PEA15 * | SIP1 | TJP3 * |
| AHNAK | CDS1 * | EPN3 * | HRG | MAPK13 * | PIK3CD * | SKIL * | TMC4 * |
| AKT1 | CERCAM * | EPPK1 * | IGFBP4 | MAPRE2 * | PLAUR * | SMAD2 | TMC5 * |
| ANGPTL4 | CHST3 * | ERBB3 * | IGFBP5 | MBOAT2 * | PLEK2 * | SMAD3 | TMEFF1 |
| ANKLE2 * | CLDN12 | ESR1 | IL11 * | METRNL * | PLG | SMAD7 * | TMEM125 * |
| ANKRD22 * | CLDN23 | EVPL * | IL1RN | MITF | PMEPA1 * | SNAI1 *^” | TMEM132A |
| ANTXR2 * | CLDN3 | EXOC6 * | ILK | MLXIP * | PPARG * | SNAI2 ^” | TMEM30B * |
| AP1M2 * | CLDN4 * | EZH1 | INADL * | MMP10 | PPP1R13L * | SNAI3 | TMEM45B * |
| ARHGEF18 * | CLDN7 * | EZH2 | INHBA | MMP2 ”* | PPP1R18 * | SOX10 ” | TNFRSF21 * |
| ARHGEF40 * | CMTM3 * | F11R * | ITBG6 ” | MMP3 ” | PPPDE2 | SOX11 | TP53I3 * |
| AXL * | COL1A1 * | FGF1 | ITGA5 * | MMP9 ” | PRR5 * | SPARC | TPM1 * |
| BCL2 | COL1A2 | FGF5 | ITGAV | MPP7 * | PRSS22 * | SPINT2 * | TRIO * |
| BCL9 | COL3A1 | FGFBP1 | ITGB1 | MPZL2 * | PRSS8 * | SPP1 | TRMT10A * |
| BEAN1 * | COL5A1 | FGFR1 * | ITGB3 | MSN | PTK2 | SSH3 * | TSPAN13 |
| BICDL1 * | COL7A1 * | FHL1 | ITGB6 * | MST1R | PTP4A1 | ST14 * | TSPAN2 * |
| BIRC3 | CRB3 * | FLNA * | JAG1 | MTA3 | PTRF * | STAP2 * | TWIS1 ” |
| BMP1 * | CTGF | FN1 ” | JUNB * | MTAC2D1 * | PXDC1 * | STAT3 | TWIST1 |
| BMP7 | CTNNA1 * | FOXC2 | JUP * | MUC1 * | PXN | STEAP1 | VCAN * |
| BSPRY * | CTNNB1 | FOXC2 ” | KCTD11 * | MUC5AC * | PXN-AS1 * | TACSTD1 * | VIM *^” |
| C1ofr116 * | DACT1 | FRMD6 * | KLC3 * | MUC5B * | RAB25 * | TACSTD2 * | VPS13A |
| C1ofr171 * | DBN1 * | FXYD3 * | KLF7 * | NAV1 * | RAC1 | TBC1D30 * | WEE1 |
| C3orf21 * | DCN | FZD7 | KRT14 | NCOR2 * | RBM35A * | TCF3 | WNT11 |
| CALCR | DEPTOR * | GADD45A | KRT7 | NF1 * | RBPMS * | TCF4 | WNT2B |
| CALD1 * | DHFR | GADD45B * | KRTCAP3 * | NKAIN4 * | RGS2 | TFPI | WNT5A |
| CAMK2N1 | DLG1 | GALNT2 * | KTR19 * | NLK | RHOD * | TFPI2 | WNT5B |
| CARD6 * | DSC2 | GALNT3 * | LAMB3 | NODAL | S100A14 * | TGFB1 * | WNT7A * |
| CASP3 | DSP *” | GALNT5 * | LAMC2 * | NOTCH1 | SAMD4A * | TGFB1I1 * | WT1 |
| CAV2 | EEPD1 * | GNC ” | LIX1L * | NPPB | SCARB2 | TGFB2 | YWHAG |
| CCNB2 | EGF | GNG11 | LOXL2 | NRG1 | SCNN1A * | TGFB3 | ZEB1 *^ |
| CCND1 | EGFR | GPR110 * | LRRC54 * | NRP1 * | SCRIB | TGFBR1 * | ZEB2 ^ |
| CD36 | EHF * | GPR56 * | LTBP1 * | NUDT13 | SERINC2 * | THBS1 | ZFP36L1 * |
| CD47 | ELANE | GRHL1 * | LTBP3 * | OCLN | SERPINE1 * | THRB * |
| Metastatic Site | Alteration | Gene | % TCGA Frequency in Primary NSCLC |
|---|---|---|---|
| lymph nodes | SNVs | NPIPA1 | 0.3% |
| CNAs (amplifications) | NKX2/1 | 8% | |
| brain | SNVs | KMT2C | 10.4% |
| AHNAK2 | 10% | ||
| PDE4DIP | 7.5% | ||
| ANKRD36C | 4% | ||
| BAGE2 | 0.2% | ||
| CNAs (amplifications) | MYC | 7.4% | |
| RICTOR | 5.7% | ||
| DDR2 | 3.6% | ||
| NTRK1 | 3.5% | ||
| ERBB2 | 2.5% | ||
| CDK12 | 1.8% | ||
| MMP13 | 1% | ||
| YAP1 | 0.7% | ||
| CNAs (deletions) | CDKN2A | 15.3% | |
| CDKN2B | 15% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicoś, M.; Krawczyk, P. Genetic Clonality as the Hallmark Driving Evolution of Non-Small Cell Lung Cancer. Cancers 2022, 14, 1813. https://doi.org/10.3390/cancers14071813
Nicoś M, Krawczyk P. Genetic Clonality as the Hallmark Driving Evolution of Non-Small Cell Lung Cancer. Cancers. 2022; 14(7):1813. https://doi.org/10.3390/cancers14071813
Chicago/Turabian StyleNicoś, Marcin, and Paweł Krawczyk. 2022. "Genetic Clonality as the Hallmark Driving Evolution of Non-Small Cell Lung Cancer" Cancers 14, no. 7: 1813. https://doi.org/10.3390/cancers14071813
APA StyleNicoś, M., & Krawczyk, P. (2022). Genetic Clonality as the Hallmark Driving Evolution of Non-Small Cell Lung Cancer. Cancers, 14(7), 1813. https://doi.org/10.3390/cancers14071813