
Vitamin D and Hypoxia: Points of Interplay in Cancer



Abstract
:Simple Summary
Abstract
1. Introduction
2. Vitamin D Synthesis and Metabolism
2.1. Canonical Vitamin D Metabolic Pathway
2.2. Noncanonical Vitamin D Metabolic Pathway
2.3. Hormonal Regulation of the Canonical Vitamin D Metabolic Pathway
3. Transcriptional Regulation of Target Gene Expression by Vitamin D
3.1. Genomic Response to Calcitriol
3.2. Nongenomic Response to Calcitriol
4. Vitamin D Crosstalk with HIF Signaling
4.1. Transcriptional Regulation of HIF-α and Calcitriol
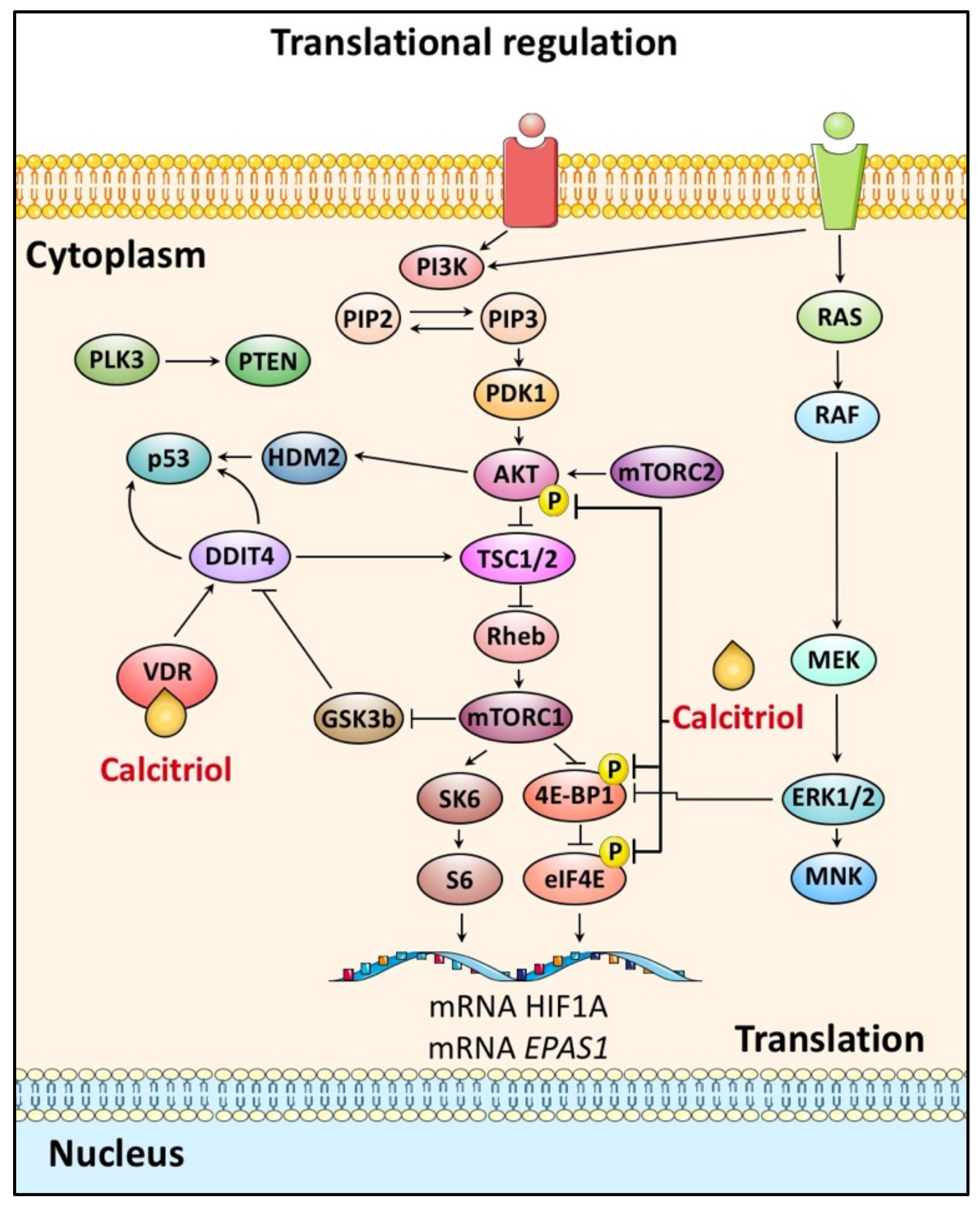
4.2. HIF-α Translation and Calcitriol
4.3. HIF-α Post-Translational Phosphorylation and Calcitriol
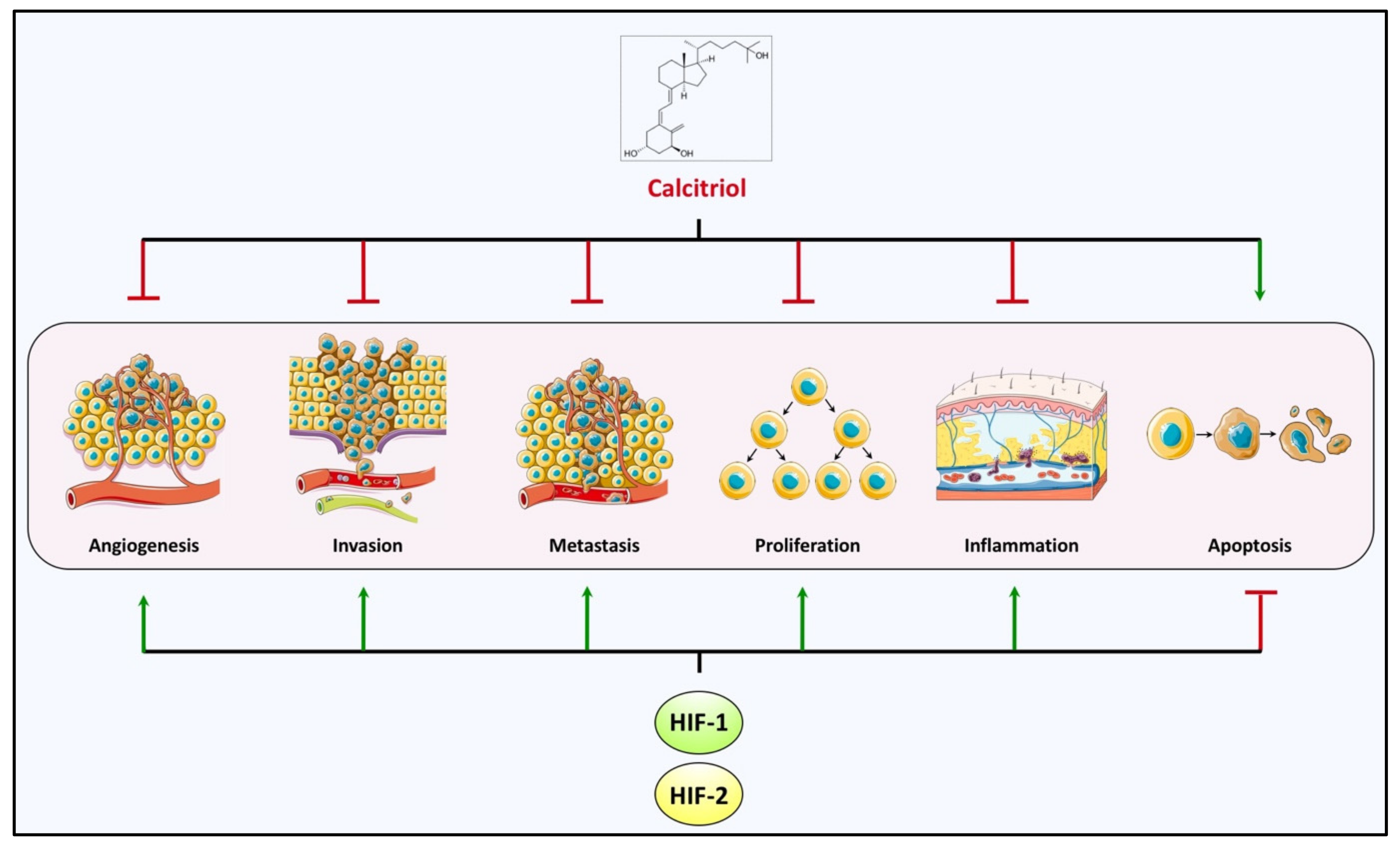
5. Cancer–Hypoxia–Vitamin D
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bouillon, R.; Carmeliet, G.; Verlinden, L.; van Etten, E.; Verstuyf, A.; Luderer, H.F.; Lieben, L.; Mathieu, C.; DeMay, M. Vitamin D and Human Health: Lessons from Vitamin D Receptor Null Mice. Endocr. Rev. 2008, 29, 726–776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Naughton, D.P. Vitamin D in health and disease: Current perspectives. Nutr. J. 2010, 9, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikle, D. Nonclassic Actions of Vitamin D. J. Clin. Endocrinol. Metab. 2009, 94, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouillon, R.; Eelen, G.; Verlinden, L.; Mathieu, C.; Carmeliet, G.; Verstuyf, A. Vitamin D and cancer. J. Steroid Biochem. Mol. Biol. 2006, 102, 156–162. [Google Scholar] [CrossRef]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef]
- Masuda, S.; Jones, G. Promise of vitamin D analogues in the treatment of hyperproliferative conditions. Mol. Cancer Ther. 2006, 5, 797–808. [Google Scholar] [CrossRef] [Green Version]
- Bikle, D.D. Extraskeletal actions of vitamin D. Ann. N. Y. Acad. Sci. 2016, 1376, 29–52. [Google Scholar] [CrossRef] [Green Version]
- Feldman, D.; Krishnan, A.V.; Swami, S.; Giovannucci, E.; Feldman, B.J. The role of vitamin D in reducing cancer risk and progression. Nat. Cancer 2014, 14, 342–357. [Google Scholar] [CrossRef]
- Holick, M.F. Sunlight and vitamin D for bone health and prevention of autoimmune diseases, cancers, and cardiovascular disease. Am. J. Clin. Nutr. 2004, 80, 1678S–1688S. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Chen, W.; Li, D.; Yin, X.; Zhang, X.; Olsen, N.; Zheng, S.G. Vitamin D and Chronic Diseases. Aging Dis. 2017, 8, 346–353. [Google Scholar] [CrossRef] [Green Version]
- Garland, C.; Garland, F.C.; Shaw, E.; Comstock, G.W.; Helsing, K.J.; Gorham, E.D. Serum 25-Hydroxyvitamin D and Colon Cancer: Eight-Year Prospective Study. Lancet 1989, 334, 1176–1178. [Google Scholar] [CrossRef]
- Engel, P.; Fagherazzi, G.; Boutten, A.; Dupré, T.; Mesrine, S.; Boutron-Ruault, M.-C.; Clavel-Chapelon, F. Serum 25(OH) Vitamin D and Risk of Breast Cancer: A Nested Case-Control Study from the French E3N Cohort. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2341–2350. [Google Scholar] [CrossRef] [Green Version]
- Ahonen, M.H.; Tenkanen, L.; Teppo, L.; Hakama, M.; Tuohimaa, P. Prostate cancer risk and prediagnostic serum 25-hydroxyvitamin D levels (Finland). Cancer Causes Control 2000, 11, 847–852. [Google Scholar] [CrossRef]
- Tretli, S.; Hernes, E.; Berg, J.P.; Hestvik, U.E.; Robsahm, T.E. Association between serum 25(OH)D and death from prostate cancer. Br. J. Cancer 2009, 100, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Giovannucci, E.; Liu, Y.; Rimm, E.B.; Hollis, B.W.; Fuchs, C.S.; Stampfer, M.J.; Willett, W.C. Prospective Study of Predictors of Vitamin D Status and Cancer Incidence and Mortality in Men. J. Natl. Cancer Inst. 2006, 98, 451–459. [Google Scholar] [CrossRef] [Green Version]
- Fleet, J.C.; Desmet, M.; Johnson, R.; Li, Y. Vitamin D and cancer: A review of molecular mechanisms. Biochem. J. 2011, 441, 61–76. [Google Scholar] [CrossRef] [Green Version]
- Giammanco, M.; Di Majo, D.; La Guardia, M.; Aiello, S.; Crescimannno, M.; Flandina, C.; Tumminello, F.M.; Leto, G. Vitamin D in cancer chemoprevention. Pharm. Biol. 2015, 53, 1399–1434. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Trump, D.L.; Johnson, C.S. Vitamin D in combination cancer treatment. J. Cancer 2010, 1, 101–107. [Google Scholar] [CrossRef] [Green Version]
- Mehta, R.G.; Peng, X.; Alimirah, F.; Murillo, G.; Mehta, R. Vitamin D and breast cancer: Emerging concepts. Cancer Lett. 2013, 334, 95–100. [Google Scholar] [CrossRef]
- Angeli-Terzidou, A.E.; Gkotinakou, I.-M.; Pazaitou-Panayiotou, K.; Tsakalof, A. Inhibition of calcitriol inactivating enzyme CYP24A1 gene expression by flavonoids in hepatocellular carcinoma cells under normoxia and hypoxia. Arch. Biochem. Biophys. 2021, 704, 108889. [Google Scholar] [CrossRef]
- Semenza, G.L. The Genomics and Genetics of Oxygen Homeostasis. Annu. Rev. Genom. Hum. Genet. 2020, 21, 183–204. [Google Scholar] [CrossRef] [Green Version]
- Mylonis, I.; Simos, G.; Paraskeva, E. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 2019, 8, 214. [Google Scholar] [CrossRef] [Green Version]
- Schito, L.; Semenza, G.L. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016, 2, 758–770. [Google Scholar] [CrossRef] [Green Version]
- Triantafyllou, A.; Mylonis, I.; Simos, G.; Bonanou, S.; Tsakalof, A. Flavonoids induce HIF-1α but impair its nuclear accumulation and activity. Free Radic. Biol. Med. 2008, 44, 657–670. [Google Scholar] [CrossRef]
- Mylonis, I.; Chachami, G.; Simos, G. Specific Inhibition of HIF Activity: Can Peptides Lead the Way? Cancers 2021, 13, 410. [Google Scholar] [CrossRef]
- Zhong, J.-C.; Li, X.-B.; Lyu, W.-Y.; Ye, W.-C.; Zhang, D.-M. Natural products as potent inhibitors of hypoxia-inducible factor-1α in cancer therapy. Chin. J. Nat. Med. 2020, 18, 696–703. [Google Scholar] [CrossRef]
- Choudhry, H.; Harris, A.L. Advances in Hypoxia-Inducible Factor Biology. Cell Metab. 2018, 27, 281–298. [Google Scholar] [CrossRef]
- Pinto, J.T.; Cooper, A.J.L. From Cholesterogenesis to Steroidogenesis: Role of Riboflavin and Flavoenzymes in the Biosynthesis of Vitamin D. Adv. Nutr. Int. Rev. J. 2014, 5, 144–163. [Google Scholar] [CrossRef] [Green Version]
- Heaney, R.P. Vitamin D in Health and Disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1535–1541. [Google Scholar] [CrossRef] [Green Version]
- Jäpelt, R.B.; Jakobsen, J. Vitamin D in plants: A review of occurrence, analysis, and biosynthesis. Front. Plant Sci. 2013, 4, 136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.; Prosser, D.E.; Kaufmann, M. Cytochrome P450-mediated metabolism of vitamin D. J. Lipid Res. 2014, 55, 1331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster, I. Cytochromes P450 are essential players in the vitamin D signaling system. Biochim. Biophys. Acta 2011, 1814, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D. Vitamin D Metabolism, Mechanism of Action, and Clinical Applications. Chem. Biol. 2014, 21, 319–329. [Google Scholar] [CrossRef] [Green Version]
- Masuda, S.; Byford, V.; Arabian, A.; Sakai, Y.; Demay, M.B.; St-Arnaud, R.; Jones, G. Altered Pharmacokinetics of 1α,25-Dihydroxyvitamin D3and 25-Hydroxyvitamin D3in the Blood and Tissues of the 25-Hydroxyvitamin D-24-Hydroxylase (Cyp24a1) Null Mouse. Endocrinology 2005, 146, 825–834. [Google Scholar] [CrossRef] [Green Version]
- St-Arnaud, R.; Arabian, A.; Travers, R.; Barletta, F.; Raval-Pandya, M.; Chapin, K.; Depovere, J.; Mathieu, C.; Christakos, S.; DeMay, M.B.; et al. Deficient Mineralization of Intramembranous Bone in Vitamin D-24-Hydroxylase-Ablated Mice Is Due to Elevated 1,25-Dihydroxyvitamin D and Not to the Absence of 24,25-Dihydroxyvitamin D*. Endocrinology 2000, 141, 2658–2666. [Google Scholar] [CrossRef]
- Slominski, A.; Semak, I.; Zjawiony, J.; Wortsman, J.; Li, W.; Szczesniewski, A.; Tuckey, R.C. The cytochrome P450scc system opens an alternate pathway of vitamin D3 metabolism. FEBS J. 2005, 272, 4080–4090. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.; Zmijewski, M.; Semak, I.; Zbytek, B.; Pisarchik, A.; Li, W.; Zjawiony, J.; Tuckey, R. Cytochromes P450 and Skin Cancer: Role of Local Endocrine Pathways. Anti Cancer Agents Med. Chem. 2014, 14, 77–96. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.T.; Brozyna, A.; Skobowiat, C.; Zmijewski, M.; Kim, T.-K.; Janjetovic, Z.; Oak, A.S.; Jóźwicki, W.; Jetten, A.; Mason, R.; et al. On the role of classical and novel forms of vitamin D in melanoma progression and management. J. Steroid Biochem. Mol. Biol. 2018, 177, 159–170. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.T.; Brożyna, A.A.; Zmijewski, M.A.; Janjetovic, Z.; Kim, T.-K.; Slominski, R.M.; Tuckey, R.C.; Mason, R.S.; Jetten, A.M.; Guroji, P.; et al. The Role of Classical and Novel Forms of Vitamin D in the Pathogenesis and Progression of Nonmelanoma Skin Cancers. Adv. Exp. Med. Biol. 2020, 1268, 257–283. [Google Scholar] [CrossRef]
- Tongkao-On, W.; Carter, S.; Reeve, V.E.; Dixon, K.M.; Gordon-Thomson, C.; Halliday, G.M.; Tuckey, R.C.; Mason, R.S. CYP11A1 in skin: An alternative route to photoprotection by vitamin D compounds. J. Steroid Biochem. Mol. Biol. 2015, 148, 72–78. [Google Scholar] [CrossRef]
- Jones, G.; Prosser, D.E.; Kaufmann, M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): Its important role in the degradation of vitamin D. Arch. Biochem. Biophys. 2012, 523, 9–18. [Google Scholar] [CrossRef]
- Zierold, C.; Darwish, H.M.; DeLuca, H.F. Two vitamin D response elements function in the rat 1,25-dihydroxyvitamin D 24-hydroxylase promoter. J. Biol. Chem. 1995, 270, 1675–1678. [Google Scholar] [CrossRef] [Green Version]
- Meyer, M.B.; Goetsch, P.D.; Pike, J.W. A Downstream Intergenic Cluster of Regulatory Enhancers Contributes to the Induction of CYP24A1 Expression by 1α,25-Dihydroxyvitamin D3. J. Biol. Chem. 2010, 285, 15599–15610. [Google Scholar] [CrossRef] [Green Version]
- Khundmiri, S.J.; Murray, R.D.; Lederer, E. PTH and Vitamin D. Compr. Physiol. 2016, 6, 561–601. [Google Scholar]
- Rost, C.R.; Bikle, D.D.; Kaplan, R.A. In Vitro Stimulation of 25-Hydroxycholecalciferol lα- Hydroxylation by Parathyroid Hormone in Chick Kidney Slices: Evidence for a Role for Adenosine 3′,5′- Monophosphate *. Endocrinology 1981, 108, 1002–1006. [Google Scholar] [CrossRef]
- Zierold, C.; Mings, J.A.; DeLuca, H.F. Parathyroid hormone regulates 25-hydroxyvitamin D3-24-hydroxylase mRNA by altering its stability. Proc. Natl. Acad. Sci. USA 2001, 98, 13572–13576. [Google Scholar] [CrossRef] [Green Version]
- Perwad, F.; Zhang, M.Y.H.; Tenenhouse, H.S.; Portale, A.A. Fibroblast growth factor 23 impairs phosphorus and vitamin D metabolism in vivo and suppresses 25-hydroxyvitamin D-1α-hydroxylase expression in vitro. Am. J. Physiol. Physiol. 2007, 293, F1577–F1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haussler, M.R.; Jurutka, P.W.; Mizwicki, M.; Norman, A.W. Vitamin D receptor (VDR)-mediated actions of 1α,25(OH)2vitamin D3: Genomic and non-genomic mechanisms. Best. Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 543–559. [Google Scholar] [CrossRef] [PubMed]
- Haussler, M.R.; Whitfield, G.K.; Kaneko, I.; Haussler, C.A.; Hsieh, D.; Hsieh, J.-C.; Jurutka, P.W. Molecular Mechanisms of Vitamin D Action. Calcif. Tissue Int. 2013, 92, 77–98. [Google Scholar] [CrossRef]
- Pike, J.W.; Meyer, M.B. Fundamentals of vitamin D hormone-regulated gene expression. J. Steroid Biochem. Mol. Biol. 2013, 144, 5–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, A.; Hernandez, E.D.; Reina-Campos, M.; Castilla, E.A.; Subramaniam, S.; Raghunandan, S.; Roberts, L.R.; Kisseleva, T.; Karin, M.; Diaz-Meco, M.T.; et al. p62/SQSTM1 by Binding to Vitamin D Receptor Inhibits Hepatic Stellate Cell Activity, Fibrosis, and Liver Cancer. Cancer Cell 2016, 30, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Haberle, V.; Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell Biol. 2018, 19, 621–637. [Google Scholar] [CrossRef]
- Carlberg, C.; Muñoz, A. An update on vitamin D signaling and cancer. Semin. Cancer Biol. 2020, 79, 217–230. [Google Scholar] [CrossRef]
- Seuter, S.; Neme, A.; Carlberg, C. Epigenome-wide effects of vitamin D and their impact on the transcriptome of human monocytes involve CTCF. Nucleic Acids Res. 2016, 44, 4090–4104. [Google Scholar] [CrossRef] [Green Version]
- Pereira, F.; Barbáchano, A.; Silva, J.; Bonilla, F.; Campbell, M.J.; Muñoz, A.; Larriba, M.J. KDM6B/JMJD3 histone deme-thylase is induced by vitamin D and modulates its effects in colon cancer cells. Hum. Mol. Genet. 2011, 20, 4655–4665. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Yoshihara, E.; He, N.; Hah, N.; Fan, W.; Pinto, A.F.; Huddy, T.; Wang, Y.; Ross, B.; Estepa, G.; et al. Vitamin D Switches BAF Complexes to Protect β Cells. Cell 2018, 173, 1135–1149.e15. [Google Scholar] [CrossRef] [Green Version]
- Chichiarelli, S.; Altieri, F.; Paglia, G.; Rubini, E.; Minacori, M.; Eufemi, M. ERp57/PDIA3: New insight. Cell. Mol. Biol. Lett. 2022, 27, 12. [Google Scholar] [CrossRef]
- Kranz, P.; Neumann, F.; Wolf, A.; Classen, F.; Pompsch, M.; Ocklenburg, T.; Baumann, J.; Janke, K.; Baumann, M.; Goepelt, K.; et al. PDI is an essential redox-sensitive activator of PERK during the unfolded protein response (UPR). Cell Death Dis. 2017, 8, e2986. [Google Scholar] [CrossRef] [Green Version]
- Ivanova, I.G.; Park, C.V.; Yemm, A.I.; Kenneth, N.S. PERK/eIF2α signaling inhibits HIF-induced gene expression during the unfolded protein response via YB1-dependent regulation of HIF1α translation. Nucleic Acids Res. 2018, 46, 3878–3890. [Google Scholar] [CrossRef] [Green Version]
- Doroudi, M.; Schwartz, Z.; Boyan, B.D. Membrane-mediated actions of 1,25-dihydroxy vitamin D3: A review of the roles of phospholipase A2 activating protein and Ca2+/calmodulin-dependent protein kinase II. J. Steroid Biochem. Mol. Biol. 2015, 147, 81–84. [Google Scholar] [CrossRef] [Green Version]
- Nemere, I.; Safford, S.E.; Rohe, B.; DeSouza, M.M.; Farach-Carson, M.C. Identification and characterization of 1,25D3-membrane-associated rapid response, steroid (1,25D3-MARRS) binding protein. J. Steroid Biochem. Mol. Biol. 2004, 89, 281–285. [Google Scholar] [CrossRef]
- Allison, M.W.; Robert, S.; Thao, T.; Michael, E.; Benjamin, K.; Meckling, K.A. Differential effects of the 1,25D3-MARRS receptor (ERp57/PDIA3) on murine mammary gland development depend on the vitamin D3 dose. Steroids 2020, 158, 108621. [Google Scholar]
- Axanova, L.S.; Chen, Y.Q.; McCoy, T.; Sui, G.; Cramer, S.D. 1,25-dihydroxyvitamin D3 and PI3K/AKT inhibitors synergistically inhibit growth and induce senescence in prostate cancer cells. Prostate 2010, 70, 1658–1671. [Google Scholar] [CrossRef] [Green Version]
- Nemere, I.; Garbi, N.; Winger, Q. The 1,25D3-MARRS receptor/PDIA3/ERp57 and lifespan. J. Cell. Biochem. 2015, 116, 380–385. [Google Scholar] [CrossRef]
- Buitrago, C.; Pardo, V.G.; Boland, R. Role of VDR in 1α,25-dihydroxyvitamin D3-dependent non-genomic activation of MAPKs, Src and Akt in skeletal muscle cells. J. Steroid Biochem. Mol. Biol. 2013, 136, 125–130. [Google Scholar] [CrossRef]
- Zhao, G.; Simpson, R.U. Membrane localization, Caveolin-3 association and rapid actions of vitamin D receptor in cardiac myocytes. Steroids 2010, 75, 555–559. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Doroudi, M.; Cheung, J.; Grozier, A.L.; Schwartz, Z.; Boyan, B.D. Plasma membrane Pdia3 and VDR interact to elicit rapid responses to 1α,25(OH)2D3. Cell. Signal. 2013, 25, 2362–2373. [Google Scholar] [CrossRef]
- Ema, M.; Taya, S.; Yokotani, N.; Sogawa, K.; Matsuda, Y.; Fujii-Kuriyama, Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 4273–4278. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.Z.; Moran, S.M.; Hogenesch, J.B.; Wartman, L.; Bradfield, C.A. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Exp. 1998, 7, 205–213. [Google Scholar]
- Semenza, G.L.; Wang, G.L. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol. Cell. Biol. 1992, 12, 5447–5454. [Google Scholar] [CrossRef]
- Arany, Z.; Huang, L.E.; Eckner, R.; Bhattacharya, S.; Jiang, C.; Goldberg, M.A.; Bunn, H.F.; Livingston, D.M. An essential role for p300/CBP in the cellular response to hypoxia. Proc. Natl. Acad. Sci. USA 1996, 93, 12969–12973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.N.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, L.C.; Simon, M.C. Hypoxia-Inducible Factors in Cancer. Cancer Res. 2022, 82, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.E.; Bagnall, J.; Mason, D.; Levy, R.; Fernig, D.G.; See, V. Differential sub-nuclear distribution of hypoxia-inducible factors (HIF)-1 and -2 alpha impacts on their stability and mobility. Open Biol. 2016, 6, 160195. [Google Scholar] [CrossRef] [Green Version]
- Menrad, H.; Werno, C.; Schmid, T.; Copanaki, E.; Deller, T.; Dehne, N.; Brüne, B. Roles of hypoxia-inducible factor-1alpha (HIF-1alpha) versus HIF-2alpha in the survival of hepatocellular tumor spheroids. Hepatology 2010, 51, 2183–2192. [Google Scholar] [CrossRef]
- Smythies, J.A.; Sun, M.; Masson, N.; Salama, R.; Simpson, P.D.; Murray, E.; Neumann, V.; Cockman, M.A.-O.; Choudhry, H.; Ratcliffe, P.A.-O.X.; et al. Inherent DNA-binding specificities of the HIF-1α and HIF-2α transcription factors in chromatin. EMBO Rep. 2019, 20, e46401. [Google Scholar] [CrossRef]
- Albanese, A.; Daly, L.; Mennerich, D.; Kietzmann, T.; Sée, V. The Role of Hypoxia-Inducible Factor Post-Translational Modifications in Regulating Its Localisation, Stability, and Activity. Int. J. Mol. Sci. 2020, 22, 268. [Google Scholar] [CrossRef]
- Battello, N.; Zimmer, A.D.; Goebel, C.; Dong, X.; Behrmann, I.; Haan, C.; Hiller, K.; Wegner, A. The role of HIF-1 in oncostatin M-dependent metabolic reprogramming of hepatic cells. Cancer Metab. 2016, 4, 3. [Google Scholar] [CrossRef] [Green Version]
- D’Ignazio, L.; Bandarra, D.; Rocha, S. NF-κB and HIF crosstalk in immune responses. FEBS J. 2016, 283, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Obacz, J.; Pastorekova, S.; Vojtesek, B.; Hrstka, R. Cross-talk between HIF and p53 as mediators of molecular responses to physiological and genotoxic stresses. Mol. Cancer 2013, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Taylor, C.T. Interdependent roles for hypoxia inducible factor and nuclear factor-κB in hypoxic inflammation. J. Physiol. 2008, 586, 4055–4059. [Google Scholar] [CrossRef]
- Papadakis, A.I.; Paraskeva, E.; Peidis, P.; Muaddi, H.; Li, S.; Raptis, L.; Pantopoulos, K.; Simos, G.; Koromilas, A.E. eIF2α Kinase PKR Modulates the Hypoxic Response by Stat3-Dependent Transcriptional Suppression of HIF-1α. Cancer Res. 2010, 70, 7820–7829. [Google Scholar] [CrossRef] [Green Version]
- Al Taleb, Z.; Petry, A.; Chi, T.F.; Mennerich, D.; Görlach, A.; Dimova, E.; Kietzmann, T. Differential transcriptional regulation of hypoxia-inducible factor-1α by arsenite under normoxia and hypoxia: Involvement of Nrf2. Klin. Wochenschr. 2016, 94, 1153–1166. [Google Scholar] [CrossRef] [Green Version]
- Lacher, S.E.; Levings, D.; Freeman, S.; Slattery, M. Identification of a functional antioxidant response element at the HIF1A locus. Redox Biol. 2018, 19, 401–411. [Google Scholar] [CrossRef]
- Frede, S.; Stockmann, C.; Freitag, P.; Fandrey, J. Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappa, B. Biochem. J. 2006, 396, 517–527. [Google Scholar] [CrossRef]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [Green Version]
- Van Uden, P.; Kenneth, N.S.; Rocha, S. Regulation of hypoxia-inducible factor-1α by NF-κB. Biochem. J. 2008, 412, 477–484. [Google Scholar] [CrossRef] [Green Version]
- BelAiba, R.S.; Bonello, S.; Zähringer, C.; Schmidt, S.; Hess, J.; Kietzmann, T.; Görlach, A. Hypoxia Up-Regulates Hypoxia-Inducible Factor-1α Transcription by Involving Phosphatidylinositol 3-Kinase and Nuclear Factor κB in Pulmonary Artery Smooth Muscle Cells. Mol. Biol. Cell 2007, 18, 4691–4697. [Google Scholar] [CrossRef] [Green Version]
- Tsapournioti, S.; Mylonis, I.; Hatziefthimiou, A.; Ioannou, M.G.; Stamatiou, R.; Koukoulis, G.K.; Simos, G.; Molyvdas, P.-A.; Paraskeva, E. TNFα induces expression of HIF-1α mRNA and protein but inhibits hypoxic stimulation of HIF-1 transcriptional activity in airway smooth muscle cells. J. Cell Physiol. 2013, 228, 1745–1753. [Google Scholar] [CrossRef]
- Takeda, K.; Ho, V.C.; Takeda, H.; Duan, L.-J.; Nagy, A.; Fong, G.-H. Placental but Not Heart Defects Are Associated with Elevated Hypoxia-Inducible Factor α Levels in Mice Lacking Prolyl Hydroxylase Domain Protein 2. Mol. Cell. Biol. 2006, 26, 8336–8346. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Duan, B.; Zhao, X.; Chen, Y.; Sun, S.; Deng, W.; Zhang, Y.; Du, J.; Chen, Y.; Gu, L. MBD3 mediates epigenetic regulation on EPAS1 promoter in cancer. Tumor Biol. 2016, 37, 13455–13467. [Google Scholar] [CrossRef]
- Mohlin, S.; Hamidian, A.; Von Stedingk, K.; Bridges, E.; Wigerup, C.; Bexell, D.; Påhlman, S. PI3K–mTORC2 but not PI3K-mTORC1 Regulates Transcription of HIF2A/EPAS1 and Vascularization in Neuroblastoma. Cancer Res. 2015, 75, 4617–4628. [Google Scholar] [CrossRef] [Green Version]
- Moniz, S.; Bandarra, D.; Biddlestone, J.; Campbell, K.; Komander, D.; Bremm, A.; Rocha, S. Cezanne regulates E2F1-dependent HIF2α expression. J. Cell Sci. 2015, 128, 3082–3093. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, M.S.; Eisinger-Mathason, T.S.K.; Sadri, N.; Ochocki, J.D.; Gade, T.P.F.; Amin, R.K.; Simon, M.C. Epigenetic re-expression of HIF-2α suppresses soft tissue sarcoma growth. Nat. Commun. 2016, 7, 10539. [Google Scholar] [CrossRef] [Green Version]
- Garcia, R.; Bowman, T.L.; Niu, G.; Yu, H.; Minton, S.; Muro-Cacho, C.A.; Cox, C.E.; Falcone, R.; Fairclough, R.; Parsons, S.; et al. Constitutive activation of Stat3 by the Src and JAK tyrosine kinases participates in growth regulation of human breast carcinoma cells. Oncogene 2001, 20, 2499–2513. [Google Scholar] [CrossRef] [Green Version]
- So, J.Y.; Smolarek, A.K.; Salerno, D.M.; Maehr, H.; Uskokovic, M.; Liu, F.; Suh, N. Targeting CD44-STAT3 Signaling by Gemini Vitamin D Analog Leads to Inhibition of Invasion in Basal-Like Breast Cancer. PLoS ONE 2013, 8, e54020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.R.; Lee, J.H.; Park, M.S.; Hwang, J.E.; Shim, H.J.; Cho, S.H.; Chung, I.-J.; Bae, W.K. Suppressive Effect of 19-nor-1α-25-Dihydroxyvitamin D2 on Gastric Cancer Cells and Peritoneal Metastasis Model. J. Korean Med Sci. 2012, 27, 1037–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, A.V.; Feldman, D. Molecular pathways mediating the anti-inflammatory effects of calcitriol: Implications for prostate cancer chemoprevention and treatment. Endocr. Relat. Cancer 2010, 17, R19–R38. [Google Scholar] [CrossRef] [Green Version]
- Stio, M.; Martinesi, M.; Bruni, S.; Treves, C.; Mathieu, C.; Verstuyf, A.; d’Albasio, G.; Bagnoli, S.; Bonanomi, A.G. The Vitamin D analogue TX 527 blocks NF-kappaB activation in peripheral blood mononuclear cells of patients with Crohn’s disease. J. Steroid Biochem. Mol. Biol. 2007, 103, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Kong, J.; Duan, Y.; Szeto, F.L.; Liao, A.; Madara, J.L.; Li, Y.C. Increased NF-kappaB activity in fibroblasts lacking the vitamin D receptor. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E315–E322. [Google Scholar] [CrossRef]
- Yu, X.P.; Bellido, T.; Manolagas, S.C. Down-regulation of NF-kappa B protein levels in activated human lymphocytes by 1,25-dihydroxyvitamin D3. Proc. Natl. Acad. Sci. USA 1995, 92, 10990–10994. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Zhang, J.; Ge, X.; Du, J.; Deb, D.K.; Li, Y.C. Vitamin D receptor inhibits nuclear factor κB activation by interacting with IκB kinase β protein. J. Biol. Chem. 2013, 288, 19450–19458. [Google Scholar] [CrossRef] [Green Version]
- Riis, J.L.; Johansen, C.; Gesser, B.; Møller, K.; Larsen, C.G.; Kragballe, K.; Iversen, L. 1alpha,25(OH)(2)D(3) regulates NF-kappaB DNA binding activity in cultured normal human keratinocytes through an increase in IkappaBalpha expres-sion. Arch. Dermatol. Res. 2004, 296, 195–202. [Google Scholar] [CrossRef]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.-W. NF-kappaB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef]
- Li, F.; Zhang, J.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Kumar, A.P.; Ahn, K.S.; Sethi, G. NF-κB in cancer therapy. Arch. Toxicol. 2015, 89, 711–731. [Google Scholar] [CrossRef]
- Chung, I.; Han, G.; Seshadri, M.; Gillard, B.M.; Yu, W.-D.; Foster, B.A.; Trump, D.L.; Johnson, C.S. Role of Vitamin D Receptor in the Antiproliferative Effects of Calcitriol in Tumor-Derived Endothelial Cells and Tumor AngiogenesisIn vivo. Cancer Res. 2009, 69, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Zhang, J.; Xiang, J.; Li, Y.; Wu, D.; Xu, J. Calcitriol inhibits ROS-NLRP3-IL-1β signaling axis via activation of Nrf2-antioxidant signaling in hyperosmotic stress stimulated human corneal epithelial cells. Redox Biol. 2019, 21, 101093. [Google Scholar] [CrossRef]
- Bárdos, J.I.; Ashcroft, M. Negative and positive regulation of HIF-1: A complex network. Biochim. Biophys. Acta 2005, 1755, 107–120. [Google Scholar] [CrossRef]
- Akeno, N.; Robins, J.; Zhang, M.; Czyzyk-Krzeska, M.F.; Clemens, T.L. Induction of Vascular Endothelial Growth Factor by IGF-I in Osteoblast-Like Cells Is Mediated by the PI3K Signaling Pathway through the Hypoxia-Inducible Factor-2α. Endocrinology 2002, 143, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nayak, B.K.; Feliers, D.; Sudarshan, S.; Friedrichs, W.E.; Day, R.T.; New, D.D.; Fitzgerald, J.P.; Eid, A.; DeNapoli, T.; Parekh, D.J.; et al. Stabilization of HIF-2α through redox regulation of mTORC2 activation and initiation of mRNA translation. Oncogene 2013, 32, 3147–3155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toschi, A.; Lee, E.; Gadir, N.; Ohh, M.; Foster, D.A. Differential Dependence of Hypoxia-inducible Factors 1α and 2α on mTORC1 and mTORC2. J. Biol. Chem. 2008, 283, 34495–34499. [Google Scholar] [CrossRef] [Green Version]
- Proietti, S.; Cucina, A.; D’Anselmi, F.; Dinicola, S.; Pasqualato, A.; Lisi, E.; Bizzarri, M. Melatonin and vitamin D3 synergisti-cally down-regulate Akt and MDM2 leading to TGFβ-1-dependent growth inhibition of breast cancer cells. J. Pineal. Res. 2011, 50, 150–158. [Google Scholar]
- Gkotinakou, I.-M.; Kechagia, E.; Pazaitou-Panayiotou, K.; Mylonis, I.; Liakos, P.; Tsakalof, A. Calcitriol Suppresses HIF-1 and HIF-2 Transcriptional Activity by Reducing HIF-1/2α Protein Levels via a VDR-Independent Mechanism. Cells 2020, 9, 2440. [Google Scholar] [CrossRef]
- Abu El Maaty, M.A.; Wölfl, S. Vitamin D as a Novel Regulator of Tumor Metabolism: Insights on Potential Mechanisms and Implications for Anti-Cancer Therapy. Int. J. Mol. Sci. 2017, 18, 2184. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.-O.; Seo, S.-K.; Kim, Y.-S.; Woo, S.-H.; Lee, K.-H.; Yi, J.-Y.; Lee, S.-J.; Choe, T.-B.; Lee, J.-H.; An, S.; et al. TXNIP potentiates Redd1-induced mTOR suppression through stabilization of Redd1. Oncogene 2011, 30, 3792–3801. [Google Scholar] [CrossRef]
- Horak, P.; Crawford, A.R.; Vadysirisack, D.D.; Nash, Z.M.; DeYoung, M.P.; Sgroi, D.; Ellisen, L.W. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 4675–4680. [Google Scholar] [CrossRef] [Green Version]
- Flügel, D.; Görlach, A.; Michiels, C.; Kietzmann, T. Glycogen Synthase Kinase 3 Phosphorylates Hypoxia-Inducible Factor 1α and Mediates Its Destabilization in a VHL-Independent Manner. Mol. Cell. Biol. 2007, 27, 3253–3265. [Google Scholar] [CrossRef] [Green Version]
- Mottet, D.; Dumont, V.; Deccache, Y.; Demazy, C.; Ninane, N.; Raes, M.; Michiels, C. Regulation of hypoxia-inducible factor-1alpha protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3beta pathway in HepG2 cells. J. Biol. Chem. 2003, 278, 31277–31285. [Google Scholar] [CrossRef] [Green Version]
- Flügel, D.; Görlach, A.; Kietzmann, T. GSK-3β regulates cell growth, migration, and angiogenesis via Fbw7 and USP28-dependent degradation of HIF-1α. Blood 2012, 119, 1292–1301. [Google Scholar] [CrossRef] [Green Version]
- González-Sancho, J.M.; Larriba, M.J.; Muñoz, A. Wnt and Vitamin D at the Crossroads in Solid Cancer. Cancers 2020, 12, 3434. [Google Scholar] [CrossRef]
- Xu, D.; Yao, Y.; Lu, L.; Costa, M.; Dai, W. Plk3 Functions as an Essential Component of the Hypoxia Regulatory Pathway by Direct Phosphorylation of HIF-1α. J. Biol. Chem. 2010, 285, 38944–38950. [Google Scholar] [CrossRef] [Green Version]
- Bullen, J.W.; Tchernyshyov, I.; Holewinski, R.J.; DeVine, L.; Wu, F.; Venkatraman, V.; Kass, D.L.; Cole, R.N.; Van Eyk, J.; Semenza, G.L. Protein kinase A—Dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci. Signal. 2016, 9, ra56. [Google Scholar] [CrossRef] [Green Version]
- Hubbi, M.E.; Gilkes, D.M.; Hu, H.; Kshitiz; Ahmed, I.; Semenza, G.L. Cyclin-dependent kinases regulate lysosomal degradation of hypoxia-inducible factor 1α to promote cell-cycle progression. Proc. Natl. Acad. Sci. USA 2014, 111, E3325–E3334. [Google Scholar] [CrossRef] [Green Version]
- Warfel, N.A.; Dolloff, N.G.; Dicker, D.T.; Malysz, J.; El-Deiry, W.S. CDK1 stabilizes HIF-1α via direct phosphorylation of Ser668 to promote tumor growth. Cell Cycle 2013, 12, 3689–3701. [Google Scholar] [CrossRef] [Green Version]
- Peehl, D.M.; Shinghal, R.; Nonn, L.; Seto, E.; Krishnan, A.V.; Brooks, J.D.; Feldman, D. Molecular activity of 1,25-dihydroxyvitamin D3 in primary cultures of human prostatic epithelial cells revealed by cDNA microarray analysis. J. Steroid Biochem. Mol. Biol. 2004, 92, 131–141. [Google Scholar] [CrossRef]
- Swami, S.; Raghavachari, N.; Muller, U.R.; Bao, Y.P.; Feldman, D. Vitamin D Growth Inhibition of Breast Cancer Cells: Gene Expression Patterns Assessed by cDNA Microarray. Breast Cancer Res. Treat. 2003, 80, 49–62. [Google Scholar] [CrossRef]
- Gradin, K.; Takasaki, C.; Fujii-Kuriyama, Y.; Sogawa, K. The Transcriptional Activation Function of the HIF-like Factor Requires Phosphorylation at a Conserved Threonine. J. Biol. Chem. 2002, 277, 23508–23514. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, D.E.; McNeill, L.A.; McDonough, M.A.; Aplin, R.T.; Hewitson, K.S.; Pugh, C.W.; Ratcliffe, P.J.; Schofield, C.J. Disruption of dimerization and substrate phosphorylation inhibit factor inhibiting hypoxia-inducible factor (FIH) activity. Biochem. J. 2004, 383, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Yu, W.-D.; Ma, Y.; Chernov, M.; Trump, D.L.; Johnson, C.S. Inhibition of Protein Kinase CK2 ReducesCyp24a1Expression and Enhances 1,25-Dihydroxyvitamin D3Antitumor Activity in Human Prostate Cancer Cells. Cancer Res. 2013, 73, 2289–2297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pangou, E.; Befani, C.; Mylonis, I.; Samiotaki, M.; Panayotou, G.; Simos, G.; Liakos, P. HIF-2α phosphorylation by CK1delta promotes erythropoietin secretion in liver cancer cells under hypoxia. J. Cell Sci. 2016, 129, 4213–4226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalousi, A.; Mylonis, I.; Politou, A.S.; Chachami, G.; Paraskeva, E.; Simos, G. Casein kinase 1 regulates human hypoxia-inducible factor HIF-1. J. Cell Sci. 2010, 123, 2976–2986. [Google Scholar] [CrossRef] [Green Version]
- Kourti, M.; Ikonomou, G.; Giakoumakis, N.-N.; Rapsomaniki, M.A.; Landegren, U.; Siniossoglou, S.; Lygerou, Z.; Simos, G.; Mylonis, I. CK1delta restrains lipin-1 induction, lipid droplet formation and cell proliferation under hypoxia by reducing HIF-1α/ARNT complex formation. Cell. Signal. 2015, 27, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Gkotinakou, I.-M.; Befani, C.; Simos, G.; Liakos, P. ERK1/2 phosphorylates HIF-2α and regulates its activity by controlling its CRM1-dependent nuclear shuttling. J. Cell Sci. 2019, 132, jcs225698. [Google Scholar] [CrossRef] [Green Version]
- Koukoulas, K.; Giakountis, A.; Karagiota, A.; Samiotaki, M.; Panayotou, G.; Simos, G.; Mylonis, I. ERK signaling controls productive HIF-1 binding to chromatin and cancer cell adaptation to hypoxia through HIF-1α interaction with NPM1. Mol. Oncol. 2021, 15, 3468–3489. [Google Scholar] [CrossRef]
- Mylonis, I.; Chachami, G.; Samiotaki, M.; Panayotou, G.; Paraskeva, E.; Kalousi, A.; Georgatsou, E.; Bonanou, S.; Simos, G. Identification of MAPK Phosphorylation Sites and Their Role in the Localization and Activity of Hypoxia-inducible Factor-1α. J. Biol. Chem. 2006, 281, 33095–33106. [Google Scholar] [CrossRef] [Green Version]
- Mylonis, I.; Chachami, G.; Paraskeva, E.; Simos, G. Atypical CRM1-dependent Nuclear Export Signal Mediates Regulation of Hypoxia-inducible Factor-1α by MAPK. J. Biol. Chem. 2008, 283, 27620–27627. [Google Scholar] [CrossRef] [Green Version]
- Gkotinakou, I.M.; Befani, C.; Samiotaki, M.; Panayotou, G.; Liakos, P. Novel HIF-2α interaction with Reptin52 impairs HIF-2 transcriptional activity and EPO secretion. Biochem. Biophys. Res. Commun. 2021, 557, 143–150. [Google Scholar] [CrossRef]
- Zhang, A.; Huang, Z.; Tao, W.; Zhai, K.; Wu, Q.; Rich, J.N.; Zhou, W.; Bao, S. USP33 deubiquitinates and stabilizes HIF-2alpha to promote hypoxia response in glioma stem cells. EMBO J. 2022, 41, e109187. [Google Scholar] [CrossRef]
- Cordes, T.; Diesing, D.; Becker, S.; Diedrich, K.; Reichrath, J.; Friedrich, M. Modulation of MAPK ERK1 and ERK2 in VDR-positive and -negative Breast Cancer Cell Lines. Anticancer Res. 2006, 26, 2749–2754. [Google Scholar]
- Semenza, G.L. Heritable disorders of oxygen sensing. Am. J. Med Genet. Part A 2021, 185, 2576–2581. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene 2009, 29, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Semenza, G.L. Pharmacologic Targeting of Hypoxia-Inducible Factors. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 379–403. [Google Scholar] [CrossRef]
- Huang, C.-Y.; Weng, Y.-T.; Li, P.-C.; Hsieh, N.-T.; Li, C.-I.; Liu, H.-S.; Lee, M.-F. Calcitriol Suppresses Warburg Effect and Cell Growth in Human Colorectal Cancer Cells. Life 2021, 11, 963. [Google Scholar] [CrossRef]
- Pálmer, H.G.; González-Sancho, J.M.; Espada, J.; Berciano, M.T.; Puig, I.; Baulida, J.; Quintanilla, M.; Cano, A.; de Herreros, A.G.; Lafarga, M.; et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J. Cell Biol. 2001, 154, 369–387. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.; Islam, N.; Dakshanamurthy, S.; Rizvi, I.; Rao, M.; Herrell, R.; Zinser, G.; Valrance, M.; Aranda, A.; Moras, D.; et al. The Molecular Basis of Vitamin D Receptor and β-Catenin Crossregulation. Mol. Cell 2006, 21, 799–809. [Google Scholar] [CrossRef]
- Xu, H.; Posner, G.H.; Stevenson, M.; Campbell, F.C. Apc MIN modulation of vitamin D secosteroid growth control. Carcinogenesis 2010, 31, 1434–1441. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, O.; Peña, C.; García, J.M.; Jesús Larriba, M.; Ordóñez-Morán, P.; Navarro, D.; Barbáchano, A.; López de Silanes, I.; Ballestar, E.; Fraga, M.F.; et al. The Wnt antagonist DICKKOPF-1 gene is induced by 1,25-dihydroxyvitamin D3 associated to the differentiation of human colon cancer cells. Carcinogenesis 2007, 28, 1877–1884. [Google Scholar] [CrossRef]
- Pendás-Franco, N.; García, J.M.; Peña, C.; Valle, N.; Pálmer, H.G.; Heinäniemi, M.; Carlberg, C.; Jiménez, B.; Bonilla, F.; Muñoz, A.; et al. DICKKOPF-4 is induced by TCF/beta-catenin and upregulated in human colon cancer, promotes tumour cell invasion and angiogenesis and is repressed by 1alpha,25-dihydroxyvitamin D3. Oncogene 2008, 27, 4467–4477. [Google Scholar] [CrossRef] [Green Version]
- Cordero, J.B.; Cozzolino, M.; Lu, Y.; Vidal, M.; Slatopolsky, E.; Stahl, P.D.; Barbieri, M.A.; Dusso, A. 1,25-Dihydroxyvitamin D Down-regulates Cell Membrane Growth- and Nuclear Growth-promoting Signals by the Epidermal Growth Factor Receptor. J. Biol. Chem. 2002, 277, 38965–38971. [Google Scholar] [CrossRef] [Green Version]
- McGaffin, K.R.; Chrysogelos, S.A. Identification and characterization of a response element in the EGFR promoter that mediates transcriptional repression by 1,25-dihydroxyvitamin D3 in breast cancer cells. J. Mol. Endocrinol. 2005, 35, 117–133. [Google Scholar] [CrossRef]
- Tong, W.M.; Hofer, H.; Ellinger, A.; Peterlik, M.; Cross, H.S. Mechanism of antimitogenic action of vitamin D in human colon carcinoma cells: Relevance for suppression of epidermal growth factor-stimulated cell growth. Oncol. Res. Featur. Preclin. Clin. Cancer Ther. 1999, 11, 77–84. [Google Scholar]
- Colston, K.W.; Perks, C.M.; Xie, S.P.; Holly, J.M. Growth inhibition of both MCF-7 and Hs578T human breast cancer cell lines by vitamin D analogues is associated with increased expression of insulin-like growth factor binding protein-3. J. Mol. Endocrinol. 1998, 20, 157–162. [Google Scholar] [CrossRef]
- Sprenger, C.C.; Peterson, A.; Lance, R.; Ware, J.L.; Drivdahl, R.H.; Plymate, S.R. Regulation of proliferation of prostate epithelial cells by 1,25-dihydroxyvitamin D3 is accompanied by an increase in insulin-like growth factor binding protein-3. J. Endocrinol. 2001, 170, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.S.; Burnstein, K.L. Vitamin D inhibits G1 to S progression in LNCaP prostate cancer cells through p27Kip1 stabilization and Cdk2 mislocalization to the cytoplasm. J. Biol. Chem. 2003, 278, 46862–46868. [Google Scholar] [CrossRef] [Green Version]
- Blutt, S.E.; McDonnell, T.J.; Polek, T.C.; Weigel, N.L. Calcitriol-induced apoptosis in LNCaP cells is blocked by overex-pression of Bcl-2. Endocrinology 2000, 141, 10–17. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.-D.; Schley, G.; Badiali, L.; Theres, H.; Scholz, H. 1,25-dihydroxyvitamin D3-induced apoptosis of retinoblastoma cells is associated with reciprocal changes of Bcl-2 and bax. Exp. Eye Res. 2003, 77, 1–9. [Google Scholar] [CrossRef]
- Reitsma, P.H.; Rothberg, P.G.; Astrin, S.M.; Trial, J.; Bar-Shavit, Z.; Hall, A.; Teitelbaum, S.; Kahn, A.J. Regulation of myc gene expression in HL-60 leukaemia cells by a vitamin D metabolite. Nature 1983, 306, 492–494. [Google Scholar] [CrossRef] [PubMed]
- Rohan, J.N.P.; Weigel, N.L. 1α,25-Dihydroxyvitamin D3 Reduces c-Myc Expression, Inhibiting Proliferation and Causing G1 Accumulation in C4-2 Prostate Cancer Cells. Endocrinology 2009, 150, 2046–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, J.; Krishnan, A.V.; Swami, S.; Nonn, L.; Peehl, D.M.; Feldman, D. Regulation of Prostaglandin Metabolism by Calcitriol Attenuates Growth Stimulation in Prostate Cancer Cells. Cancer Res. 2005, 65, 7917–7925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nonn, L.; Peng, L.; Feldman, D.; Peehl, D.M. Inhibition of p38 by Vitamin D Reduces Interleukin-6 Production in Normal Prostate Cells via Mitogen-Activated Protein Kinase Phosphatase 5: Implications for Prostate Cancer Prevention by Vitamin D. Cancer Res. 2006, 66, 4516–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harant, H.; Wolff, B.; Lindley, I.J. 1Alpha,25-dihydroxyvitamin D3 decreases DNA binding of nuclear factor-kappaB in human fibroblasts. FEBS Lett. 1998, 436, 329–334. [Google Scholar] [CrossRef] [Green Version]
- Iseki, K.; Tatsuta, M.; Uehara, H.; Iishi, H.; Yano, H.; Sakai, N.; Ishiguro, S. Inhibition of angiogenesis as a mechanism for inhibition by 1-hydroxyvitamin D3 and 1,25-dihydroxyvitamin D3 of colon carcinogenesis induced by azoxymethane in wistar rats. Int. J. Cancer 1999, 81, 730–733. [Google Scholar] [CrossRef]
- Merrigan, S.L.; Park, B.; Ali, Z.; Jensen, L.D.; Corson, T.W.; Kennedy, B.N. Calcitriol and non-calcemic vitamin D analogue, 22-oxacalcitriol, attenuate developmental and pathological choroidal vasculature angiogenesis ex vivo and in vivo. Oncotarget 2020, 11, 493–509. [Google Scholar] [CrossRef] [Green Version]
- Calvani, M.; Trisciuoglio, D.; Bergamaschi, C.; Shoemaker, R.H.; Melillo, G. Differential Involvement of Vascular Endothelial Growth Factor in the Survival of Hypoxic Colon Cancer Cells. Cancer Res. 2008, 68, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Görlach, A.; Diebold, I.; Schini-Kerth, V.B.; Berchner-Pfannschmidt, U.; Roth, U.; Brandes, R.P.; Kietzmann, T.; Busse, R. Thrombin activates the hypoxia-inducible factor-1 signaling pathway in vascular smooth muscle cells: Role of the p22(phox)-containing NADPH oxidase. Circ. Res. 2001, 89, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Kietzmann, T.; Mennerich, D.; Dimova, E.Y. Hypoxia-Inducible Factors (HIFs) and Phosphorylation: Impact on Stability, Localization, and Transactivity. Front. Cell Dev. Biol. 2016, 4, 11. [Google Scholar] [CrossRef]
- Richard, D.E.; Berra, E.; Pouysségur, J. Nonhypoxic Pathway Mediates the Induction of Hypoxia-inducible Factor 1α in Vascular Smooth Muscle Cells. J. Biol. Chem. 2000, 275, 26765–26771. [Google Scholar] [CrossRef]
- Stiehl, D.; Jelkmann, W.; Wenger, R.H.; Hellwig-Bürgel, T. Normoxic induction of the hypoxia-inducible factor 1α by insulin and interleukin-1β involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002, 512, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Liu, L.; Zhao, Y.; Zhang, J.; Wang, D.; Chen, J.; He, Y.; Wu, J.; Zhang, Z.; Liu, Z. Hypoxia Induces Genomic DNA Demethylation through the Activation of HIF-1α and Transcriptional Upregulation of MAT2A in Hepatoma Cells. Mol. Cancer Ther. 2011, 10, 1113–1123. [Google Scholar] [CrossRef] [Green Version]
- Minet, E.; Arnould, T.; Michel, G.; Roland, I.; Mottet, D.; Raes, M.; Remacle, J.; Michiels, C. ERK activation upon hypoxia: Involvement in HIF-1 activation. FEBS Lett. 2000, 468, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, A.V.; Feldman, D. Mechanisms of the Anti-Cancer and Anti-Inflammatory Actions of Vitamin D. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 311–336. [Google Scholar] [CrossRef] [Green Version]
- Schneikert, J.; Behrens, J. The canonical Wnt signalling pathway and its APC partner in colon cancer development. Gut 2007, 56, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Barbáchano, A.; Ordóñez-Morán, P.; García, J.M.; Sánchez, A.; Pereira, F.; Larriba, M.J.; Martínez, N.; Hernández, J.; Landolfi, S.; Bonilla, F.; et al. SPROUTY-2 and E-cadherin regulate reciprocally and dictate colon cancer cell tumourigenicity. Oncogene 2010, 29, 4800–4813. [Google Scholar] [CrossRef] [Green Version]
- Cui, M.; Zhao, Y.; Hance, K.W.; Shao, A.; Wood, R.J.; Fleet, J.C. Effects of MAPK signaling on 1,25-dihydroxyvitamin D-mediated CYP24 gene expression in the enterocyte-like cell line, Caco-2. J. Cell. Physiol. 2009, 219, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Kovalenko, P.; Cui, M.; DeSmet, M.; Clinton, S.K.; Fleet, J.C. Constitutive activation of the mitogen-activated protein kinase pathway impairs vitamin D signaling in human prostate epithelial cells. J. Cell. Physiol. 2010, 224, 433–442. [Google Scholar] [CrossRef] [Green Version]
- Peng, L.; Malloy, P.J.; Feldman, D. Identification of a Functional Vitamin D Response Element in the Human Insulin-Like Growth Factor Binding Protein-3 Promoter. Mol. Endocrinol. 2004, 18, 1109–1119. [Google Scholar] [CrossRef] [Green Version]
- Boyle, B.J.; Zhao, X.Y.; Cohen, P.; Feldman, D. Insulin-like growth factor binding protein-3 mediates 1 al-pha,25-dihydroxyvitamin d(3) growth inhibition in the LNCaP prostate cancer cell line through p21/WAF1. J. Urol. 2001, 165, 1319–1324. [Google Scholar] [CrossRef]
- Blutt, S.E.; Allegretto, E.A.; Pike, J.W.; Weigel, N.L. 1,25-Dihydroxyvitamin D3 and 9-cis-Retinoic Acid Act Synergistically to Inhibit the Growth of LNCaP Prostate Cells and Cause Accumulation of Cells in G1*. Endocrinology 1997, 138, 1491–1497. [Google Scholar] [CrossRef]
- Liu, M.; Lee, M.H.; Cohen, M.; Bommakanti, M.; Freedman, L.P. Transcriptional activation of the Cdk inhibitor p21 by vitamin D3 leads to the induced differentiation of the myelomonocytic cell line U937. Genes Dev. 1996, 10, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Lin, R.; Nagai, Y.; Sladek, R.; Bastien, Y.; Ho, J.; Petrecca, K.; Sotiropoulou, G.; Diamandis, E.P.; Hudson, T.J.; White, J.H. Expression profiling in squamous carcinoma cells reveals pleiotropic effects of vitamin D3 analog EB1089 signaling on cell proliferation, differentiation, and immune system regulation. Mol. Endocrinol. 2002, 16, 1243–1256. [Google Scholar] [CrossRef]
- Lin, R.; White, J.H. The pleiotropic actions of vitamin D. BioEssays 2004, 26, 21–28. [Google Scholar] [CrossRef]
- Brahimi-Horn, M.C.; Ben-Hail, D.; Ilie, M.; Gounon, P.; Rouleau, M.; Hofman, V.; Doyen, J.; Mari, B.; Shoshan-Barmatz, V.; Hofman, P.; et al. Expression of a Truncated Active Form of VDAC1 in Lung Cancer Associates with Hypoxic Cell Survival and Correlates with Progression to Chemotherapy Resistance. Cancer Res. 2012, 72, 2140–2150. [Google Scholar] [CrossRef] [Green Version]
- Brahimi-Horn, M.C.; Giuliano, S.; Saland, E.; Lacas-Gervais, S.; Sheiko, T.; Pelletier, J.; Bourget, I.; Bost, F.; Feral, C.; Boulter, E.; et al. Knockout of Vdac1 activates hypoxia-inducible factor through reactive oxygen species generation and induces tumor growth by promoting metabolic reprogramming and inflammation. Cancer Metab. 2015, 3, 8. [Google Scholar] [CrossRef] [Green Version]
- Mylonis, I.; Kourti, M.; Samiotaki, M.; Panayotou, G.; Simos, G. Mortalin-mediated and ERK-controlled targeting of HIF-1α to mitochondria confers resistance to apoptosis under hypoxia. J. Cell Sci. 2017, 130, 466–479. [Google Scholar] [CrossRef] [Green Version]
- Allavena, P.; Garlanda, C.; Borrello, M.G.; Sica, A.; Mantovani, A. Pathways connecting inflammation and cancer. Curr. Opin. Genet. Dev. 2008, 18, 3–10. [Google Scholar] [CrossRef]
- Lucia, M.S.; Torkko, K.C. Inflammation as a target for prostate cancer chemoprevention: Pathological and laboratory rationale. J. Urol. 2004, 171, S30–S34. [Google Scholar]
- Eltzschig, H.K.; Bratton, D.L.; Colgan, S.P. Targeting hypoxia signalling for the treatment of ischaemic and inflammatory diseases. Nat. Rev. Drug Discov. 2014, 13, 852–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Rosmorduc, O.; Housset, C. Hypoxia: A Link between Fibrogenesis, Angiogenesis, and Carcinogenesis in Liver Disease. Semin. Liver Dis. 2010, 30, 258–270. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Selimovic, D.; Ghozlan, H.; Abdel-Kader, O. Hepatitis C virus core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology 2009, 49, 1469–1482. [Google Scholar] [CrossRef] [PubMed]
- Giaccia, A.; Siim, B.G.; Johnson, R. HIF-1 as a target for drug development. Nat. Rev. Drug Discov. 2003, 2, 803–811. [Google Scholar] [CrossRef]
- Rankin, E.B.; Giaccia, A.J. The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ. 2008, 15, 678–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Marschall, Z.; Cramer, T.; Höcker, M.; Finkenzeller, G.; Wiedenmann, B.; Rosewicz, S. Dual mechanism of vascular endothelial growth factor upregulation by hypoxia in human hepatocellular carcinoma. Gut 2001, 48, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Bougatef, F.; Quemener, C.; Kellouche, S.; Naïmi, B.; Podgorniak, M.-P.; Millot, G.; Gabison, E.E.; Calvo, F.; Dosquet, C.; Lebbé, C.; et al. EMMPRIN promotes angiogenesis through hypoxia-inducible factor-2α-mediated regulation of soluble VEGF isoforms and their receptor VEGFR-2. Blood 2009, 114, 5547–5556. [Google Scholar] [CrossRef] [Green Version]
- Imura, S.; Miyake, H.; Izumi, K.; Tashiro, S.; Uehara, H. Correlation of vascular endothelial cell proliferation with microvessel density and expression of vascular endothelial growth factor and basic fibroblast growth factor in hepatocellular carcinoma. J. Med. Investig. 2004, 51, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Simiantonaki, N.; Jayasinghe, C.; Michel-Schmidt, R.; Peters, K.; Hermanns, M.I.; Kirkpatrick, C.J. Hypoxia-induced epi-thelial VEGF-C/VEGFR-3 upregulation in carcinoma cell lines. Int. J. Oncol. 2008, 32, 585–592. [Google Scholar]
- Chiang, K.-C.; Chen, T.C. The anti-cancer actions of vitamin D. Anti Cancer Agents Med. Chem. 2013, 13, 126–139. [Google Scholar] [CrossRef]
- Ben-Shoshan, M.; Amir, S.; Dang, D.T.; Dang, L.H.; Weisman, Y.; Mabjeesh, N.J. 1α,25-dihydroxyvitamin D3 (Calcitriol) inhibits hypoxia-inducible factor-1/vascular endothelial growth factor pathway in human cancer cells. Mol. Cancer Ther. 2007, 6, 1433–1439. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, G.K.; Lenore, S.R.; Peter, W.J.; Heike, Z.; Anish, K.O.; Hope, T.L.D.; Carol, A.H.; Michael, A.G.; Michelle, L.T.; Carlos Encinas, D.; et al. Functionally relevant polymorphisms in the human nuclear vitamin D receptor gene. Mol. Cell. Endocrinol. 2001, 177, 145–159. [Google Scholar] [CrossRef]
- Uitterlinden, A.G.; Fang, Y.; van Meurs, J.B.; Pols, H.A.; van Leeuwen, J.P. Genetics and biology of vitamin D receptor polymorphisms. Gene 2004, 338, 143–156. [Google Scholar] [CrossRef] [Green Version]
- Raimondi, S.; Johansson, H.; Maisonneuve, P.; Gandini, S. Review and meta-analysis on vitamin D receptor polymorphisms and cancer risk. Carcinogenesis 2009, 30, 1170–1180. [Google Scholar] [CrossRef] [Green Version]
- Ben-Eltriki, M.; Deb, S.; Guns, E.S.T. Calcitriol in Combination Therapy for Prostate Cancer: Pharmacokinetic and Pharmacodynamic Interactions. J. Cancer 2016, 7, 391–407. [Google Scholar] [CrossRef] [Green Version]
- Hu, K.; Callen, D.F.; Li, J.; Zheng, H. Circulating Vitamin D and Overall Survival in Breast Cancer Patients: A Dose-Response Meta-Analysis of Cohort Studies. Integr. Cancer Ther. 2018, 17, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Trump, D.L. Calcitriol and cancer therapy: A missed opportunity. Bone Rep. 2018, 9, 110–119. [Google Scholar] [CrossRef]
- Cabrera-Cano, A.; Dávila-Borja, V.M.; Juárez-Méndez, S.; Marcial-Quino, J.; Gómez-Manzo, S.; Castillo-Rodríguez, R.A. Hypoxia as a modulator of cytochromes P450: Overexpression of the cytochromes CYP2S1 and CYP24A1 in human liver cancer cells in hypoxia. Cell Biochem. Funct. 2021, 39, 478–487. [Google Scholar] [CrossRef]
- Zhalehjoo, N.; Shakiba, Y.; Panjehpour, M. Gene expression profiles of CYP24A1 and CYP27B1 in malignant and normal breast tissues. Mol. Med. Rep. 2016, 15, 467–473. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Gu, Y.; Zhao, S.; Sun, J.; Groome, L.J.; Wang, Y. Expressions of vitamin D metabolic components VDBP, CYP2R1, CYP27B1, CYP24A1, and VDR in placentas from normal and preeclamptic pregnancies. Am. J. Physiol. Metab. 2012, 303, E928–E935. [Google Scholar] [CrossRef] [Green Version]
- Beer, T.M.; Myrthue, A. Calcitriol in the treatment of prostate cancer. Anticancer Res. 2006, 26, 2647–2652. [Google Scholar]
- Osborn, J.L.; Schwartz, G.G.; Smith, D.C.; Bahnson, R.; Day, R.; Trump, D.L. Phase II trial of oral 1,25-dihydroxyvitamin D (calcitriol) in hormone refractory prostate cancer. Urol. Oncol. Semin. Orig. Investig. 1995, 1, 195–198. [Google Scholar] [CrossRef]
- Beer, T.M.; Garzotto, M.; Katovic, N.M. High-Dose Calcitriol and Carboplatin in Metastatic Androgen-Independent Prostate Cancer. Am. J. Clin. Oncol. 2004, 27, 535–541. [Google Scholar] [CrossRef]
- Beer, T.M.; Eilers, K.M.; Garzotto, M.; Egorin, M.J.; Lowe, B.A.; Henner, W.D. Weekly High-Dose Calcitriol and Docetaxel in Metastatic Androgen-Independent Prostate Cancer. J. Clin. Oncol. 2003, 21, 123–128. [Google Scholar] [CrossRef]
- Trump, D.L.; Potter, D.M.; Muindi, J.; Brufsky, A.; Johnson, C.S. Phase II trial of high-dose, intermittent calcitriol (1,25 dihydroxyvitamin D3) and dexamethasone in androgen-independent prostate cancer. Cancer 2006, 106, 2136–2142. [Google Scholar] [CrossRef]
- Guan, X.; Ding, Y.; Qi, T. Safety and efficacy of high dose pulse calcitriol and docetaxel for androgen-independent prostate cancer. Med. Case Rep. Study Protoc. 2021, 2, e0151. [Google Scholar] [CrossRef]
- Sanghani, N.S.; Haase, V.H. Hypoxia-Inducible Factor Activators in Renal Anemia: Current Clinical Experience. Adv. Chronic Kidney Dis. 2019, 26, 253–266. [Google Scholar] [CrossRef]
- Chen, W.; Hill, H.; Christie, A.; Kim, M.S.; Holloman, E.; Pavia-Jimenez, A.; Homayoun, F.; Ma, Y.; Patel, N.; Yell, P.; et al. Targeting renal cell carcinoma with a HIF-2 antagonist. Nature 2016, 539, 112–117. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Du, X.; Rizzi, J.P.; Liberzon, E.; Chakraborty, A.A.; Gao, W.; Carvo, I.; Signoretti, S.; Bruick, R.K.; Josey, J.A.; et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 2016, 539, 107–111. [Google Scholar] [CrossRef] [Green Version]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I Dose-Escalation Trial of PT2385, a First-in-Class Hypoxia-Inducible Factor-2α Antagonist in Patients with Previously Treated Advanced Clear Cell Renal Cell Carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yang, N.; Yuan, M. Dietary and circulating vitamin D and risk of renal cell carcinoma: A meta-analysis of observational studies. Int. Braz. J. Urol 2021, 47, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Mylonis, I.; Lakka, A.; Tsakalof, A.; Simos, G. The dietary flavonoid kaempferol effectively inhibits HIF-1 activity and hepatoma cancer cell viability under hypoxic conditions. Biochem. Biophys. Res. Commun. 2010, 398, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.Y.; Ong, P.S.; Wang, L.; Goel, A.; Ding, L.; Wong, A.L.-A.; Ho, P.C.-L.; Sethi, G.; Xiang, X.; Goh, B.C. Celastrol in cancer therapy: Recent developments, challenges and prospects. Cancer Letters 2021, 521, 252–267. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Kwah, M.X.-Y.; Liu, C.; Ma, Z.; Shanmugam, M.K.; Ding, L.; Xiang, X.; Ho, P.C.-L.; Wang, L.; Ong, P.S.; et al. Resveratrol for cancer therapy: Challenges and future perspectives. Cancer Letters 2021, 515, 63–72. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Pathway/Process Affected by Calcitriol | References |
|---|---|---|
| HT29/SW480 cell lines and HT29 NOD/SCID mouse xenografts | Suppression of glycolysis and reduced tumor xenograft volume | [147] |
| SW480, SW620, SKBR-3, HEK 293, NCI-H28 cell lines | Inhibition of β-catenin–TCF mediated transcription | [148,149] |
| C57BL/6J Apc +/+ and Apc −/+ mice | Reduced β-catenin signaling and polyp number | [150] |
| SW480-ADH cells and SW480-ADH xenografts in immunodeficient mice | Induction of Wnt antagonist DKK-1 | [151] |
| SW480-ADH, HEK293Tcells | Reduction Wnt-activator DKK-4 | [152] |
| A431, NR6, HeLa, BT549, Caco-2 cells | EGFR targeting to early endosomes reduction of ERK1/2 activation | [153,154,155] |
| MCF-7, Hs578T, prostate epithelial, and immortalized prostate epithelial P153 cells | Inhibition of proliferation due to reduced IGF signaling | [156,157] |
| LNCaP and DU145 cells and prostate specific PTEN-knock out mouse | Inhibition of prostate cancer cell growth | [63] |
| LNCaP-FGC cell line | Cell cycle arrest and decreased cell proliferation due to CDK-2 downregulation | [158] |
| LNCaP and Y79 cell lines | Increased apoptosis; decreased Bcl-2 and Bcl-XL and increased Bax expression | [159,160] |
| HL-60, LNCaP, C4-2, and RWPE-1 cell lines | Decreased c-Myc expression and cell proliferation; promoted differentiation of HL-60 cells | [161,162] |
| LNCaP, PC-3, MRC-5 cell lines, and prostate adenocarcinoma samples | Inhibition of prostaglandin, IL-6, IL-8 and NF-κB signaling | [163,164,165] |
| Wistar rats, C57BL/6J mice, ex vivo mouse choroidal sprouting model, | Inhibition of angiogenesis | [166,167] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gkotinakou, I.-M.; Mylonis, I.; Tsakalof, A. Vitamin D and Hypoxia: Points of Interplay in Cancer. Cancers 2022, 14, 1791. https://doi.org/10.3390/cancers14071791
Gkotinakou I-M, Mylonis I, Tsakalof A. Vitamin D and Hypoxia: Points of Interplay in Cancer. Cancers. 2022; 14(7):1791. https://doi.org/10.3390/cancers14071791
Chicago/Turabian StyleGkotinakou, Ioanna-Maria, Ilias Mylonis, and Andreas Tsakalof. 2022. "Vitamin D and Hypoxia: Points of Interplay in Cancer" Cancers 14, no. 7: 1791. https://doi.org/10.3390/cancers14071791
APA StyleGkotinakou, I.-M., Mylonis, I., & Tsakalof, A. (2022). Vitamin D and Hypoxia: Points of Interplay in Cancer. Cancers, 14(7), 1791. https://doi.org/10.3390/cancers14071791