
Cell Type-Specific Roles of STAT3 Signaling in the Pathogenesis and Progression of K-ras Mutant Lung Adenocarcinoma



Abstract
:Simple Summary
Abstract
1. Introduction
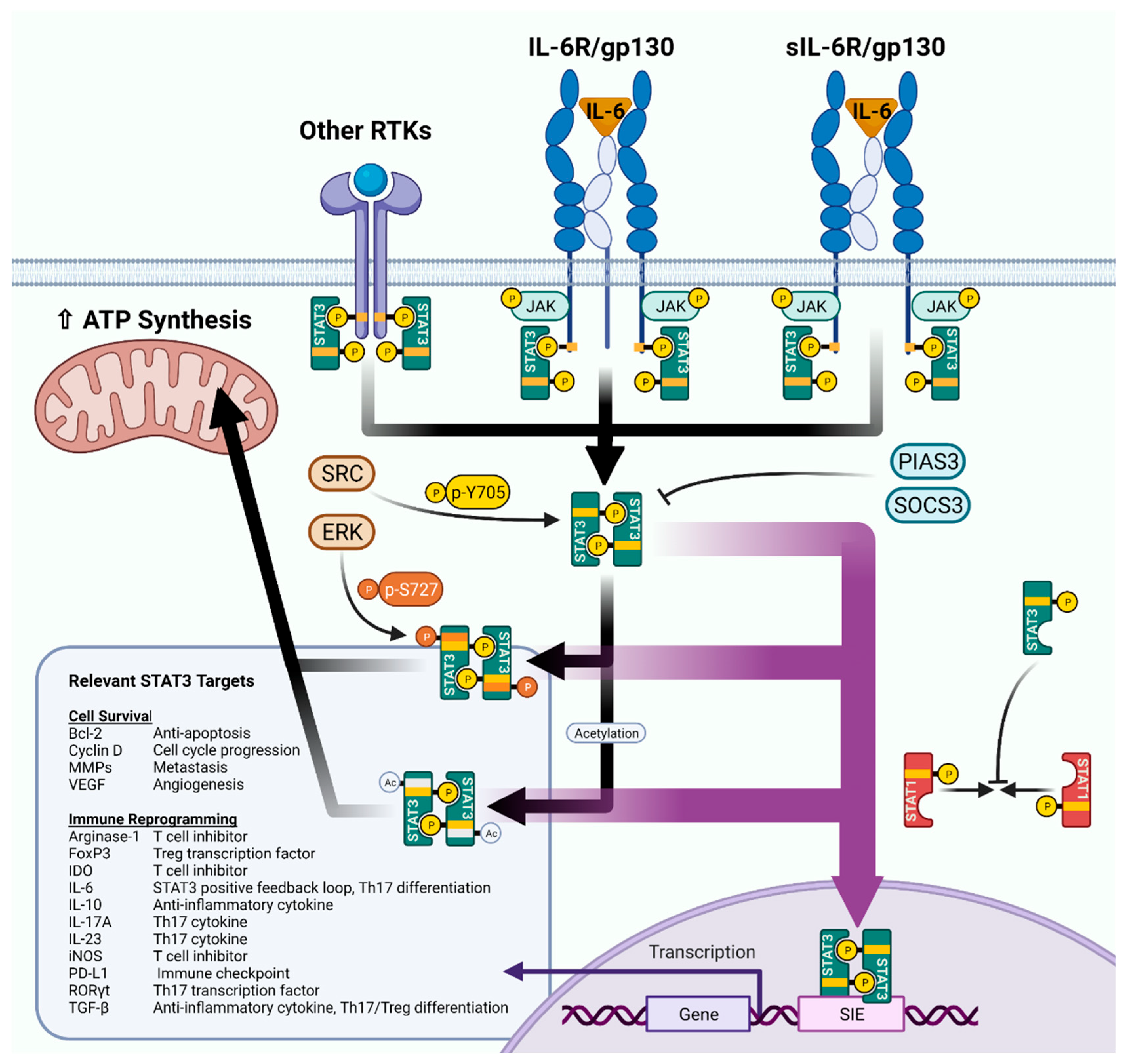
2. STAT3 Structure and Signaling: The Basics
3. Studying STAT3 by Cell Type in KM-LUAD Models
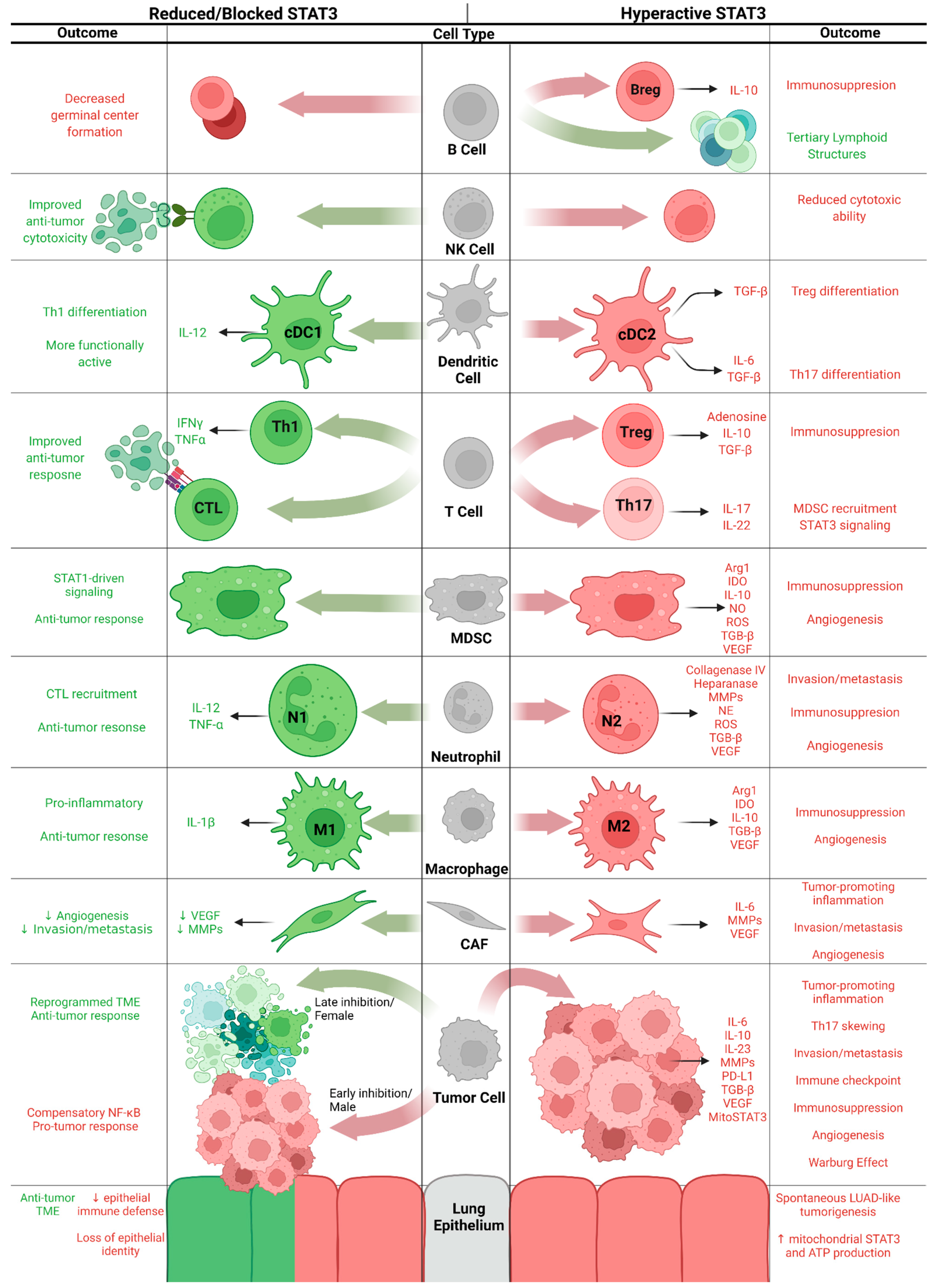
4. Cell-Specific Functions of STAT3 in KM-LUAD
4.1. Lung Epithelium/Tumor Cells
4.2. Cancer-Associated Fibroblasts
4.3. Macrophages
4.4. Neutrophils
4.5. Myeloid-Derived Suppressor Cells
4.6. T Cells
4.7. Dendritic Cells
4.8. Natural Killer Cells
4.9. B Cells
5. STAT3 Inhibition
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Goldstraw, P.; Ball, D.; Jett, J.R.; Le Chevalier, T.; Lim, E.; Nicholson, A.G.; Shepherd, F.A. Non-small-cell lung cancer. Lancet 2011, 378, 1727–1740. [Google Scholar] [CrossRef]
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical relevance of KRAS in human cancers. BioMed Res. Int. 2010, 2010, 150960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayekar, M.K.; Bivona, T.G. Current landscape of targeted therapy in lung cancer. Clin. Pharmacol. Ther. 2017, 102, 757–764. [Google Scholar] [CrossRef]
- Ji, H.; Houghton, A.; Mariani, T.; Perera, S.; Kim, C.; Padera, R.; Tonon, G.; McNamara, K.; Marconcini, L.; Hezel, A. K-ras activation generates an inflammatory response in lung tumors. Oncogene 2006, 25, 2105–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moghaddam, S.J.; Li, H.; Cho, S.-N.; Dishop, M.K.; Wistuba, I.I.; Ji, L.; Kurie, J.M.; Dickey, B.F.; De Mayo, F.J. Promotion of lung carcinogenesis by chronic obstructive pulmonary disease—Like airway inflammation in a K-ras—Induced mouse model. Am. J. Respir. Cell Mol. Biol. 2009, 40, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Jiang, G.; Zhang, P.; Fan, J. Programmed cell death and its role in inflammation. Mil. Med. Res. 2015, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Busch, S.E.; Hanke, M.L.; Kargl, J.; Metz, H.E.; MacPherson, D.; Houghton, A.M. Lung Cancer Subtypes Generate Unique Immune Responses. J. Immunol. 2016, 197, 4493–4503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, B.M.; Pine, S.R.; Chaturvedi, A.K.; Caporaso, N.; Harris, C.C. A combined prognostic serum interleukin-8 and interleukin-6 classifier for stage 1 lung cancer in the prostate, lung, colorectal, and ovarian cancer screening trial. J. Thorac. Oncol. 2014, 9, 1494–1503. [Google Scholar] [CrossRef] [Green Version]
- Barrera, L.; Montes-Servín, E.; Barrera, A.; Ramírez-Tirado, L.A.; Salinas-Parra, F.; Bañales-Méndez, J.L.; Sandoval-Ríos, M.; Arrieta, Ó. Cytokine profile determined by data-mining analysis set into clusters of non-small-cell lung cancer patients according to prognosis. Ann. Oncol. 2015, 26, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Brooks, G.D.; McLeod, L.; Alhayyani, S.; Miller, A.; Russell, P.A.; Ferlin, W.; Rose-John, S.; Ruwanpura, S.; Jenkins, B.J. IL6 Trans-signaling Promotes KRAS-Driven Lung Carcinogenesis. Cancer Res. 2016, 76, 866–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haura, E.B.; Livingston, S.; Coppola, D. Autocrine interleukin-6/interleukin-6 receptor stimulation in non-small-cell lung cancer. Clin. Lung Cancer 2006, 7, 273–275. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Caetano, M.S.; Zhang, H.; Cumpian, A.M.; Gong, L.; Unver, N.; Ostrin, E.J.; Daliri, S.; Chang, S.H.; Ochoa, C.E.; Hanash, S.; et al. IL6 Blockade Reprograms the Lung Tumor Microenvironment to Limit the Development and Progression of K-ras-Mutant Lung Cancer. Cancer Res. 2016, 76, 3189–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; Wang, J.; Jiang, N.; Pan, H.; Li, D. Correlation between p-STAT3 overexpression and prognosis in lung cancer: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0182282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, M. Autocrine IL-6 signaling: A key event in tumorigenesis? Cancer Cell 2008, 13, 7–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpenter, R.L.; Lo, H.W. STAT3 Target Genes Relevant to Human Cancers. Cancers 2014, 6, 897–925. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutta, P.; Sabri, N.; Li, J.; Li, W.X. Role of STAT3 in lung cancer. JAKSTAT 2014, 3, e999503. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, P.C.; Behrmann, I.; Haan, S.; Hermanns, H.M.; Müller-Newen, G.; Schaper, F. Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 2003, 374 Pt 1, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Delgoffe, G.M.; Vignali, D.A. STAT heterodimers in immunity: A mixed message or a unique signal? JAKSTAT 2013, 2, e23060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Investig. 2011, 121, 3375–3383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.J.; Zhao, Y.; Chait, B.T.; Lathem, W.W.; Ritzi, M.; Knippers, R.; Darnell, J.E. Ser727-dependent recruitment of MCM5 by Stat1alpha in IFN-gamma-induced transcriptional activation. EMBO J. 1998, 17, 6963–6971. [Google Scholar] [CrossRef] [Green Version]
- Tesoriere, A.; Dinarello, A.; Argenton, F. The Roles of Post-Translational Modifications in STAT3 Biological Activities and Functions. Biomedicines 2021, 9, 956. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, M.; Li, J.; Xiao, M.; Chin, Y.E.; Cheng, J.; Yeh, E.T.; Yang, J.; Yi, J. SUMOylation and SENP3 regulate STAT3 activation in head and neck cancer. Oncogene 2016, 35, 5826–5838. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to IL-6 and activates transcription by binding to NFkappaB. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krebs, D.L.; Hilton, D.J. SOCS proteins: Negative regulators of cytokine signaling. Stem Cells 2001, 19, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Shuai, K.; Liu, B. Regulation of gene-activation pathways by PIAS proteins in the immune system. Nat. Rev. Immunol. 2005, 5, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wormald, S.; Zhang, J.G.; Krebs, D.L.; Mielke, L.A.; Silver, J.; Alexander, W.S.; Speed, T.P.; Nicola, N.A.; Hilton, D.J. The comparative roles of suppressor of cytokine signaling-1 and -3 in the inhibition and desensitization of cytokine signaling. J. Biol. Chem. 2006, 281, 11135–11143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Looyenga, B.D.; Hutchings, D.; Cherni, I.; Kingsley, C.; Weiss, G.J.; Mackeigan, J.P. STAT3 is activated by JAK2 independent of key oncogenic driver mutations in non-small cell lung carcinoma. PLoS ONE 2012, 7, e30820. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Song, H.; Huang, C.; Yao, E.; Gacayan, R.; Xu, S.M.; Chuang, P.T. Alveolar type II cells possess the capability of initiating lung tumor development. PLoS ONE 2012, 7, e53817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tichelaar, J.W.; Lu, W.; Whitsett, J.A. Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J. Biol. Chem. 2000, 275, 11858–11864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, G.H.; Wellen, S.L.; Klimstra, D.; Lenczowski, J.M.; Tichelaar, J.W.; Lizak, M.J.; Whitsett, J.A.; Koretsky, A.; Varmus, H.E. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001, 15, 3249–3262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drosten, M.; Guerra, C.; Barbacid, M. Genetically Engineered Mouse Models of K-Ras-Driven Lung and Pancreatic Tumors: Validation of Therapeutic Targets. Cold Spring Harb. Perspect. Med. 2018, 8, a031542. [Google Scholar] [CrossRef] [PubMed]
- Anghelina, D.; Lam, E.; Falck-Pedersen, E. Diminished Innate Antiviral Response to Adenovirus Vectors in cGAS/STING-Deficient Mice Minimally Impacts Adaptive Immunity. J. Virol. 2016, 90, 5915–5927. [Google Scholar] [CrossRef] [Green Version]
- Tanner, L.; Single, A.B. Animal Models Reflecting Chronic Obstructive Pulmonary Disease and Related Respiratory Disorders: Translating Pre-Clinical Data into Clinical Relevance. J. Innate Immun. 2020, 12, 203–225. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.C.; Takano, Y. NNK-Induced Lung Tumors: A Review of Animal Model. J. Oncol. 2011, 2011, 635379. [Google Scholar] [CrossRef] [Green Version]
- Kortylewski, M.; Kujawski, M.; Wang, T.; Wei, S.; Zhang, S.; Pilon-Thomas, S.; Niu, G.; Kay, H.; Mulé, J.; Kerr, W.G.; et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat. Med. 2005, 11, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Maeda, Y.; Davé, V.; Whitsett, J.A. Transcriptional control of lung morphogenesis. Physiol. Rev. 2007, 87, 219–244. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, S.; Shi, J.; Martin, B.L.; Crawford, H.C.; Petrenko, O.; Reich, N.C. STAT3 is a master regulator of epithelial identity and KRAS-driven tumorigenesis. Genes Dev. 2018, 32, 1175–1187. [Google Scholar] [CrossRef] [Green Version]
- Ware, H.H.; Kulkarni, V.V.; Wang, Y.; Pantaleón García, J.; Leiva Juarez, M.; Kirkpatrick, C.T.; Wali, S.; Syed, S.; Kontoyiannis, A.D.; Sikkema, W.K.A.; et al. Inducible lung epithelial resistance requires multisource reactive oxygen species generation to protect against bacterial infections. PLoS ONE 2019, 14, e0208216. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V. Pulmonary Innate Immune Response Determines the Outcome of Inflammation During Pneumonia and Sepsis-Associated Acute Lung Injury. Front. Immunol. 2020, 11, 1722. [Google Scholar] [CrossRef]
- Hanna, J.M.; Onaitis, M.W. Cell of origin of lung cancer. J. Carcinog. 2013, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. The cytokine network in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2009, 41, 631–638. [Google Scholar] [CrossRef] [PubMed]
- Decker, T.; Kovarik, P. Serine phosphorylation of STATs. Oncogene 2000, 19, 2628–2637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alhayyani, S.; McLeod, L.; West, A.C.; Balic, J.J.; Hodges, C.; Yu, L.; Smith, J.A.; Prodanovic, Z.; Bozinovski, S.; Kumar, B.; et al. Oncogenic dependency on STAT3 serine phosphorylation in KRAS mutant lung cancer. Oncogene 2022, 41, 809–823. [Google Scholar] [CrossRef]
- Karni, R.; Jove, R.; Levitzki, A. Inhibition of pp60c-Src reduces Bcl-XL expression and reverses the transformed phenotype of cells overexpressing EGF and HER-2 receptors. Oncogene 1999, 18, 4654–4662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, M.; Suzui, M.; Yasumatu, R.; Nakashima, T.; Kuratomi, Y.; Azuma, K.; Tomita, K.; Komiyama, S.; Weinstein, I.B. Constitutive activation of signal transducers and activators of transcription 3 correlates with cyclin D1 overexpression and may provide a novel prognostic marker in head and neck squamous cell carcinoma. Cancer Res. 2002, 62, 3351–3355. [Google Scholar] [PubMed]
- Marzec, M.; Zhang, Q.; Goradia, A.; Raghunath, P.N.; Liu, X.; Paessler, M.; Wang, H.Y.; Wysocka, M.; Cheng, M.; Ruggeri, B.A.; et al. Oncogenic kinase NPM/ALK induces through STAT3 expression of immunosuppressive protein CD274 (PD-L1, B7-H1). Proc. Natl. Acad. Sci. USA 2008, 105, 20852–20857. [Google Scholar] [CrossRef] [Green Version]
- Garama, D.J.; Harris, T.J.; White, C.L.; Rossello, F.J.; Abdul-Hay, M.; Gough, D.J.; Levy, D.E. A Synthetic Lethal Interaction between Glutathione Synthesis and Mitochondrial Reactive Oxygen Species Provides a Tumor-Specific Vulnerability Dependent on STAT3. Mol. Cell. Biol. 2015, 35, 3646–3656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 14133–14138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, Y.; Osaki, T.; Naka, T.; Iwahori, K.; Furukawa, M.; Nagatomo, I.; Kijima, T.; Kumagai, T.; Yoshida, M.; Tachibana, I.; et al. Overexpression of PIAS3 suppresses cell growth and restores the drug sensitivity of human lung cancer cells in association with PI3-K/Akt inactivation. Neoplasia 2006, 8, 817–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Du, H.; Qin, Y.; Roberts, J.; Cummings, O.W.; Yan, C. Activation of the signal transducers and activators of the transcription 3 pathway in alveolar epithelial cells induces inflammation and adenocarcinomas in mouse lung. Cancer Res. 2007, 67, 8494–8503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ihara, S.; Kida, H.; Arase, H.; Tripathi, L.P.; Chen, Y.A.; Kimura, T.; Yoshida, M.; Kashiwa, Y.; Hirata, H.; Fukamizu, R.; et al. Inhibitory roles of signal transducer and activator of transcription 3 in antitumor immunity during carcinogen-induced lung tumorigenesis. Cancer Res. 2012, 72, 2990–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabner, B.; Schramek, D.; Mueller, K.M.; Moll, H.P.; Svinka, J.; Hoffmann, T.; Bauer, E.; Blaas, L.; Hruschka, N.; Zboray, K.; et al. Disruption of STAT3 signalling promotes KRAS-induced lung tumorigenesis. Nat. Commun. 2015, 6, 6285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Qu, Z.; Yan, S.; Sun, F.; Whitsett, J.A.; Shapiro, S.D.; Xiao, G. Differential roles of STAT3 in the initiation and growth of lung cancer. Oncogene 2015, 34, 3804–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caetano, M.S.; Hassane, M.; Van, H.T.; Bugarin, E.; Cumpian, A.M.; McDowell, C.L.; Cavazos, C.G.; Zhang, H.; Deng, S.; Diao, L. Sex specific function of epithelial STAT3 signaling in pathogenesis of K-ras mutant lung cancer. Nat. Commun. 2018, 9, 4589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, S.; Ramos-Castaneda, M.; Velasco, W.V.; Clowers, M.J.; Gutierrez, B.A.; Noble, O.; Dong, Y.; Zarghooni, M.; Alvarado, L.; Caetano, M.S.; et al. Interplay between Estrogen and Stat3/NF-κB Driven Immunomodulation in Lung Cancer. Carcinogenesis 2020, 41, 1529–1542. [Google Scholar] [CrossRef] [PubMed]
- Thomas, L.; Doyle, L.A.; Edelman, M.J. Lung cancer in women: Emerging differences in epidemiology, biology, and therapy. Chest 2005, 128, 370–381. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Zhang, L.; Zhang, H.; Peng, Q.; Montgrain, P.R.; Wang, Y.; Song, Y.; Li, J.; Li, W.X. Unphosphorylated STAT3 in heterochromatin formation and tumor suppression in lung cancer. BMC Cancer 2020, 20, 145. [Google Scholar] [CrossRef] [PubMed]
- Augsten, M. Cancer-associated fibroblasts as another polarized cell type of the tumor microenvironment. Front. Oncol. 2014, 4, 62. [Google Scholar] [CrossRef]
- Wu, F.; Yang, J.; Liu, J.; Wang, Y.; Mu, J.; Zeng, Q.; Deng, S.; Zhou, H. Signaling pathways in cancer-associated fibroblasts and targeted therapy for cancer. Signal Transduct. Target. Ther. 2021, 6, 218. [Google Scholar] [CrossRef]
- Wang, L.; Cao, L.; Wang, H.; Liu, B.; Zhang, Q.; Meng, Z.; Wu, X.; Zhou, Q.; Xu, K. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget 2017, 8, 76116–76128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, L.; Huang, G.; Wang, R.; Pan, Y.; He, Z.; Chu, X.; Song, H.; Chen, L. Cancer-associated fibroblasts treated with cisplatin facilitates chemoresistance of lung adenocarcinoma through IL-11/IL-11R/STAT3 signaling pathway. Sci. Rep. 2016, 6, 38408. [Google Scholar] [CrossRef] [PubMed]
- Ardain, A.; Marakalala, M.J.; Leslie, A. Tissue-resident innate immunity in the lung. Immunology 2020, 159, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.M.; Pikor, L.A.; Kung, S.H.; Hamilton, M.J.; Lam, S.; Lam, W.L.; Bennewith, K.L. Macrophages, Inflammation, and Lung Cancer. Am. J. Respir. Crit. Care Med. 2016, 193, 116–130. [Google Scholar] [CrossRef] [PubMed]
- Rébé, C.; Végran, F.; Berger, H.; Ghiringhelli, F. STAT3 activation: A key factor in tumor immunoescape. JAKSTAT 2013, 2, e23010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kortylewski, M.; Kujawski, M.; Herrmann, A.; Yang, C.; Wang, L.; Liu, Y.; Salcedo, R.; Yu, H. Toll-like receptor 9 activation of signal transducer and activator of transcription 3 constrains its agonist-based immunotherapy. Cancer Res. 2009, 69, 2497–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Qu, Z.; Sun, F.; Han, L.; Li, L.; Yan, S.; Stabile, L.P.; Chen, L.F.; Siegfried, J.M.; Xiao, G. Myeloid STAT3 Promotes Lung Tumorigenesis by Transforming Tumor Immunosurveillance into Tumor-Promoting Inflammation. Cancer Immunol. Res. 2017, 5, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Hiwatashi, K.; Tamiya, T.; Hasegawa, E.; Fukaya, T.; Hashimoto, M.; Kakoi, K.; Kashiwagi, I.; Kimura, A.; Inoue, N.; Morita, R.; et al. Suppression of SOCS3 in macrophages prevents cancer metastasis by modifying macrophage phase and MCP2/CCL8 induction. Cancer Lett. 2011, 308, 172–180. [Google Scholar] [CrossRef]
- Gong, L.; Cumpian, A.M.; Caetano, M.S.; Ochoa, C.E.; De la Garza, M.M.; Lapid, D.J.; Mirabolfathinejad, S.G.; Dickey, B.F.; Zhou, Q.; Moghaddam, S.J. Promoting effect of neutrophils on lung tumorigenesis is mediated by CXCR2 and neutrophil elastase. Mol. Cancer 2013, 12, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kargl, J.; Busch, S.E.; Yang, G.H.; Kim, K.H.; Hanke, M.L.; Metz, H.E.; Hubbard, J.J.; Lee, S.M.; Madtes, D.K.; McIntosh, M.W.; et al. Neutrophils dominate the immune cell composition in non-small cell lung cancer. Nat. Commun. 2017, 8, 14381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaul, M.E.; Fridlender, Z.G. Neutrophils as active regulators of the immune system in the tumor microenvironment. J. Leukoc. Biol. 2017, 102, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef] [Green Version]
- Acuff, H.B.; Carter, K.J.; Fingleton, B.; Gorden, D.L.; Matrisian, L.M. Matrix metalloproteinase-9 from bone marrow-derived cells contributes to survival but not growth of tumor cells in the lung microenvironment. Cancer Res. 2006, 66, 259–266. [Google Scholar] [CrossRef] [Green Version]
- Peranzoni, E.; Zilio, S.; Marigo, I.; Dolcetti, L.; Zanovello, P.; Mandruzzato, S.; Bronte, V. Myeloid-derived suppressor cell heterogeneity and subset definition. Curr. Opin. Immunol. 2010, 22, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Welch, D.R.; Schissel, D.J.; Howrey, R.P.; Aeed, P.A. Tumor-elicited polymorphonuclear cells, in contrast to “normal” circulating polymorphonuclear cells, stimulate invasive and metastatic potentials of rat mammary adenocarcinoma cells. Proc. Natl. Acad. Sci. USA 1989, 86, 5859–5863. [Google Scholar] [CrossRef] [Green Version]
- Houghton, A.M.; Rzymkiewicz, D.M.; Ji, H.; Gregory, A.D.; Egea, E.E.; Metz, H.E.; Stolz, D.B.; Land, S.R.; Marconcini, L.A.; Kliment, C.R.; et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat. Med. 2010, 16, 219–223. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I. The Dawn of Myeloid-Derived Suppressor Cells: Identification of Arginase I as the Mechanism of Immune Suppression. Cancer Res. 2021, 81, 3953–3955. [Google Scholar] [CrossRef] [PubMed]
- Karin, N. The Development and Homing of Myeloid-Derived Suppressor Cells: From a Two-Stage Model to a Multistep Narrative. Front. Immunol. 2020, 11, 557586. [Google Scholar] [CrossRef]
- Nefedova, Y.; Nagaraj, S.; Rosenbauer, A.; Muro-Cacho, C.; Sebti, S.M.; Gabrilovich, D.I. Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res. 2005, 65, 9525–9535. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I. Myeloid-Derived Suppressor Cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Kujawski, M.; Kortylewski, M.; Lee, H.; Herrmann, A.; Kay, H.; Yu, H. Stat3 mediates myeloid cell-dependent tumor angiogenesis in mice. J. Clin. Investig. 2008, 118, 3367–3377. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Kusmartsev, S.; Nefedova, Y.; Yoder, D.; Gabrilovich, D.I. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J. Immunol. 2004, 172, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Wölfle, S.J.; Strebovsky, J.; Bartz, H.; Sähr, A.; Arnold, C.; Kaiser, C.; Dalpke, A.H.; Heeg, K. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur. J. Immunol. 2011, 41, 413–424. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor necrosis factor signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef] [Green Version]
- Golubovskaya, V.; Wu, L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers 2016, 8, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capone, A.; Volpe, E. Transcriptional Regulators of T Helper 17 Cell Differentiation in Health and Autoimmune Diseases. Front. Immunol. 2020, 11, 348. [Google Scholar] [CrossRef]
- Harris, T.J.; Grosso, J.F.; Yen, H.R.; Xin, H.; Kortylewski, M.; Albesiano, E.; Hipkiss, E.L.; Getnet, D.; Goldberg, M.V.; Maris, C.H.; et al. Cutting edge: An in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J. Immunol. 2007, 179, 4313–4317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Yao, Z.; Painter, S.L.; Fanslow, W.C.; Ulrich, D.; Macduff, B.M.; Spriggs, M.K.; Armitage, R.J. Human IL-17: A novel cytokine derived from T cells. J. Immunol. 1995, 155, 5483–5486. [Google Scholar] [PubMed]
- Tobin, R.P.; Jordan, K.R.; Kapoor, P.; Spongberg, E.; Davis, D.; Vorwald, V.M.; Couts, K.L.; Gao, D.; Smith, D.E.; Borgers, J.S.W.; et al. IL-6 and IL-8 Are Linked With Myeloid-Derived Suppressor Cell Accumulation and Correlate With Poor Clinical Outcomes in Melanoma Patients. Front. Oncol. 2019, 9, 1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbay, E.A.; Koyama, S.; Liu, Y.; Dries, R.; Bufe, L.E.; Silkes, M.; Alam, M.M.; Magee, D.M.; Jones, R.; Jinushi, M.; et al. Interleukin-17A Promotes Lung Tumor Progression through Neutrophil Attraction to Tumor Sites and Mediating Resistance to PD-1 Blockade. J. Thorac. Oncol. 2017, 12, 1268–1279. [Google Scholar] [CrossRef] [Green Version]
- Gorczynski, R.M. IL-17 Signaling in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1240, 47–58. [Google Scholar] [CrossRef]
- Deng, S.; Clowers, M.J.; Velasco, W.V.; Ramos-Castaneda, M.; Moghaddam, S.J. Understanding the Complexity of the Tumor Microenvironment in K-ras Mutant Lung Cancer: Finding an Alternative Path to Prevention and Treatment. Front. Oncol. 2019, 9, 1556. [Google Scholar] [CrossRef]
- Chang, S.H.; Mirabolfathinejad, S.G.; Katta, H.; Cumpian, A.M.; Gong, L.; Caetano, M.S.; Moghaddam, S.J.; Dong, C. T helper 17 cells play a critical pathogenic role in lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 5664–5669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalmin, F.; Mignot, G.; Bruchard, M.; Chevriaux, A.; Végran, F.; Hichami, A.; Ladoire, S.; Derangère, V.; Vincent, J.; Masson, D.; et al. Stat3 and Gfi-1 transcription factors control Th17 cell immunosuppressive activity via the regulation of ectonucleotidase expression. Immunity 2012, 36, 362–373. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, H.; Hashimoto, M.; Minagawa, S.; Takasaka, N.; Ma, R.; Moermans, C.; Ito, S.; Araya, J.; Budelsky, A.; Goodsell, A.; et al. Role of IL-17A in murine models of COPD airway disease. Am. J. Physiol. Lung Cell Mol. Physiol. 2017, 312, L122–L130. [Google Scholar] [CrossRef] [PubMed]
- Carrega, P.; Loiacono, F.; Di Carlo, E.; Scaramuccia, A.; Mora, M.; Conte, R.; Benelli, R.; Spaggiari, G.M.; Cantoni, C.; Campana, S.; et al. NCR+ILC3 concentrate in human lung cancer and associate with intratumoral lymphoid structures. Nat. Commun. 2015, 6, 8280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asadzadeh, Z.; Mohammadi, H.; Safarzadeh, E.; Hemmatzadeh, M.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Azizi, G.; Baradaran, B. The paradox of Th17 cell functions in tumor immunity. Cell. Immunol. 2017, 322, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, G.F.; Fouser, L.A.; Artis, D. Border patrol: Regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 2011, 12, 383–390. [Google Scholar] [CrossRef]
- Tufman, A.; Huber, R.M.; Völk, S.; Aigner, F.; Edelmann, M.; Gamarra, F.; Kiefl, R.; Kahnert, K.; Tian, F.; Boulesteix, A.L.; et al. Interleukin-22 is elevated in lavage from patients with lung cancer and other pulmonary diseases. BMC Cancer 2016, 16, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khosravi, N.; Caetano, M.S.; Cumpian, A.M.; Unver, N.; De la Garza Ramos, C.; Noble, O.; Daliri, S.; Hernandez, B.J.; Gutierrez, B.A.; Evans, S.E.; et al. IL22 Promotes Kras Mutant Lung Cancer by Induction of a Pro-Tumor Immune Response and Protection of Stemness Properties. Cancer Immunol. Res. 2018, 6, 788–797. [Google Scholar] [CrossRef] [Green Version]
- Zheng, S.G.; Wang, J.; Wang, P.; Gray, J.D.; Horwitz, D.A. IL-2 is essential for TGF-beta to convert naive CD4+CD25− cells to CD25+Foxp3+ regulatory T cells and for expansion of these cells. J. Immunol. 2007, 178, 2018–2027. [Google Scholar] [CrossRef] [Green Version]
- Arce-Sillas, A.; Álvarez-Luquín, D.D.; Tamaya-Domínguez, B.; Gomez-Fuentes, S.; Trejo-García, A.; Melo-Salas, M.; Cárdenas, G.; Rodríguez-Ramírez, J.; Adalid-Peralta, L. Regulatory T Cells: Molecular Actions on Effector Cells in Immune Regulation. J. Immunol. Res. 2016, 2016, 1720827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, B.; Liu, Y.; Jiang, S.J. Prognostic value of tumor-infiltrating FoxP3+ regulatory T cells in cancers: A systematic review and meta-analysis. Sci. Rep. 2015, 5, 15179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallandre, J.R.; Brillard, E.; Créhange, G.; Radlovic, A.; Remy-Martin, J.P.; Saas, P.; Rohrlich, P.S.; Pivot, X.; Ling, X.; Tiberghien, P.; et al. Role of STAT3 in CD4+CD25+FOXP3+ regulatory lymphocyte generation: Implications in graft-versus-host disease and antitumor immunity. J. Immunol. 2007, 179, 7593–7604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowley, M.; Inaba, K.; Steinman, R.M. Dendritic cells are the principal cells in mouse spleen bearing immunogenic fragments of foreign proteins. J. Exp. Med. 1990, 172, 383–386. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef]
- Laouar, Y.; Welte, T.; Fu, X.Y.; Flavell, R.A. STAT3 is required for Flt3L-dependent dendritic cell differentiation. Immunity 2003, 19, 903–912. [Google Scholar] [CrossRef] [Green Version]
- Park, S.J.; Nakagawa, T.; Kitamura, H.; Atsumi, T.; Kamon, H.; Sawa, S.; Kamimura, D.; Ueda, N.; Iwakura, Y.; Ishihara, K.; et al. IL-6 regulates in vivo dendritic cell differentiation through STAT3 activation. J. Immunol. 2004, 173, 3844–3854. [Google Scholar] [CrossRef] [Green Version]
- Iwata-Kajihara, T.; Sumimoto, H.; Kawamura, N.; Ueda, R.; Takahashi, T.; Mizuguchi, H.; Miyagishi, M.; Takeda, K.; Kawakami, Y. Enhanced cancer immunotherapy using STAT3-depleted dendritic cells with high Th1-inducing ability and resistance to cancer cell-derived inhibitory factors. J. Immunol. 2011, 187, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chin, Y.E.; Weisiger, E.; Malter, C.; Tawara, I.; Toubai, T.; Gatza, E.; Mascagni, P.; Dinarello, C.A.; Reddy, P. Cutting edge: Negative regulation of dendritic cells through acetylation of the nonhistone protein STAT-3. J. Immunol. 2009, 182, 5899–5903. [Google Scholar] [CrossRef] [PubMed]
- Marcus, A.; Gowen, B.G.; Thompson, T.W.; Iannello, A.; Ardolino, M.; Deng, W.; Wang, L.; Shifrin, N.; Raulet, D.H. Recognition of tumors by the innate immune system and natural killer cells. Adv. Immunol. 2014, 122, 91–128. [Google Scholar] [CrossRef] [Green Version]
- Morvan, M.G.; Lanier, L.L. NK cells and cancer: You can teach innate cells new tricks. Nat. Rev. Cancer 2016, 16, 7–19. [Google Scholar] [CrossRef]
- Gotthardt, D.; Putz, E.M.; Straka, E.; Kudweis, P.; Biaggio, M.; Poli, V.; Strobl, B.; Müller, M.; Sexl, V. Loss of STAT3 in murine NK cells enhances NK cell-dependent tumor surveillance. Blood 2014, 124, 2370–2379. [Google Scholar] [CrossRef]
- Zhou, X.; Liu, S.; Liu, J.; Zhang, Z.; Mao, X.; Zhou, H. MicroRNA-130a enhances the killing ability of natural killer cells against non-small cell lung cancer cells by targeting signal transducers and activators of transcription 3. Biochem. Biophys. Res. Commun. 2020, 523, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.X.; Yu, C.R.; Dambuza, I.M.; Mahdi, R.M.; Dolinska, M.B.; Sergeev, Y.V.; Wingfield, P.T.; Kim, S.H.; Egwuagu, C.E. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat. Med. 2014, 20, 633–641. [Google Scholar] [CrossRef]
- Oladipupo, F.O.; Yu, C.R.; Olumuyide, E.; Jittaysothorn, Y.; Choi, J.K.; Egwuagu, C.E. STAT3 deficiency in B cells exacerbates uveitis by promoting expansion of pathogenic lymphocytes and suppressing regulatory B cells (Bregs) and Tregs. Sci. Rep. 2020, 10, 16188. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Min, Z.; Zhang, D.; Wang, W.; Marincola, F.; Wang, X. Enhanced frequency and potential mechanism of B regulatory cells in patients with lung cancer. J. Transl. Med. 2014, 12, 304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Xin, H.; Zhang, W.; Yazaki, P.J.; Zhang, Z.; Le, K.; Li, W.; Lee, H.; Kwak, L.; Forman, S.; et al. CD5 Binds to Interleukin-6 and Induces a Feed-Forward Loop with the Transcription Factor STAT3 in B Cells to Promote Cancer. Immunity 2016, 44, 913–923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, C.; Chen, X.; Dascani, P.; Hu, X.; Bolli, R.; Zhang, H.G.; Mcleish, K.R.; Yan, J. STAT3 Signaling in B Cells Is Critical for Germinal Center Maintenance and Contributes to the Pathogenesis of Murine Models of Lupus. J. Immunol. 2016, 196, 4477–4486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, A.L.A.; Hirpara, J.L.; Pervaiz, S.; Eu, J.Q.; Sethi, G.; Goh, B.C. Do STAT3 inhibitors have potential in the future for cancer therapy? Expert Opin. Investig. Drugs 2017, 26, 883–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parakh, S.; Ernst, M.; Poh, A.R. Multicellular Effects of STAT3 in Non-small Cell Lung Cancer: Mechanistic Insights and Therapeutic Opportunities. Cancers 2021, 13, 6228. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, S.; Xu, L.; Lin, J.; Zhao, C.; Huang, X. Novel activators and small-molecule inhibitors of STAT3 in cancer. Cytokine Growth Factor Rev. 2019, 49, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Kasembeli, M.M.; Jiang, X.; Tweardy, B.J.; Tweardy, D.J. Chemical probes that competitively and selectively inhibit Stat3 activation. PLoS ONE 2009, 4, e4783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, K.M.; Bharadwaj, U.; Eckols, T.K.; Kolosov, M.; Kasembeli, M.M.; Fridley, C.; Siller, R.; Tweardy, D.J. Small-molecule targeting of signal transducer and activator of transcription (STAT) 3 to treat non-small cell lung cancer. Lung Cancer 2015, 90, 182–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayliss, T.J.; Smith, J.T.; Schuster, M.; Dragnev, K.H.; Rigas, J.R. A humanized anti-IL-6 antibody (ALD518) in non-small cell lung cancer. Expert Opin. Biol. Ther. 2011, 11, 1663–1668. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Approaches | Examples | Induction Method |
|---|---|---|
| Tissue-Specific Cre Recombinase | CCSPCre: lung epithelium | |
| SPCCre: whole lung rtTA-tetO: dox-inducible | Germline/AdCre | |
| LysMCre: myeloid CD4Cre: helper T cell CD19Cre: B cell NCR1Cre: NK cell | Germline/Adoptive Transfer | |
| Mutations | KrasLA2/+ KrasLSL-G12D/+ KrasLSL-G12V/+ ±p53lox/lox | Germline/AdCre |
| COPD-like Inflammation | NTHi LPS | Nebulization Intranasal |
| Cigarette Smoke Elastase | Smoke exposure Intratracheal | |
| Carcinogen | NNK Urethane | Intraperitoneal |
| Cigarette Smoke eCigarette | Smoke exposure Atomized nicotine | |
| Xenograft | Immunocompromised mouse | Orthotopic, subcutaneous, or intravenous |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Clowers, M.J.; Moghaddam, S.J. Cell Type-Specific Roles of STAT3 Signaling in the Pathogenesis and Progression of K-ras Mutant Lung Adenocarcinoma. Cancers 2022, 14, 1785. https://doi.org/10.3390/cancers14071785
Clowers MJ, Moghaddam SJ. Cell Type-Specific Roles of STAT3 Signaling in the Pathogenesis and Progression of K-ras Mutant Lung Adenocarcinoma. Cancers. 2022; 14(7):1785. https://doi.org/10.3390/cancers14071785
Chicago/Turabian StyleClowers, Michael J., and Seyed Javad Moghaddam. 2022. "Cell Type-Specific Roles of STAT3 Signaling in the Pathogenesis and Progression of K-ras Mutant Lung Adenocarcinoma" Cancers 14, no. 7: 1785. https://doi.org/10.3390/cancers14071785
APA StyleClowers, M. J., & Moghaddam, S. J. (2022). Cell Type-Specific Roles of STAT3 Signaling in the Pathogenesis and Progression of K-ras Mutant Lung Adenocarcinoma. Cancers, 14(7), 1785. https://doi.org/10.3390/cancers14071785