
Next Generation Plasma Proteomics Identifies High-Precision Biomarker Candidates for Ovarian Cancer

 ,
,  , , , , ,
, , , , ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Protein Measurements
2.3. Statistical Analysis
3. Results
3.1. Protein Measurements
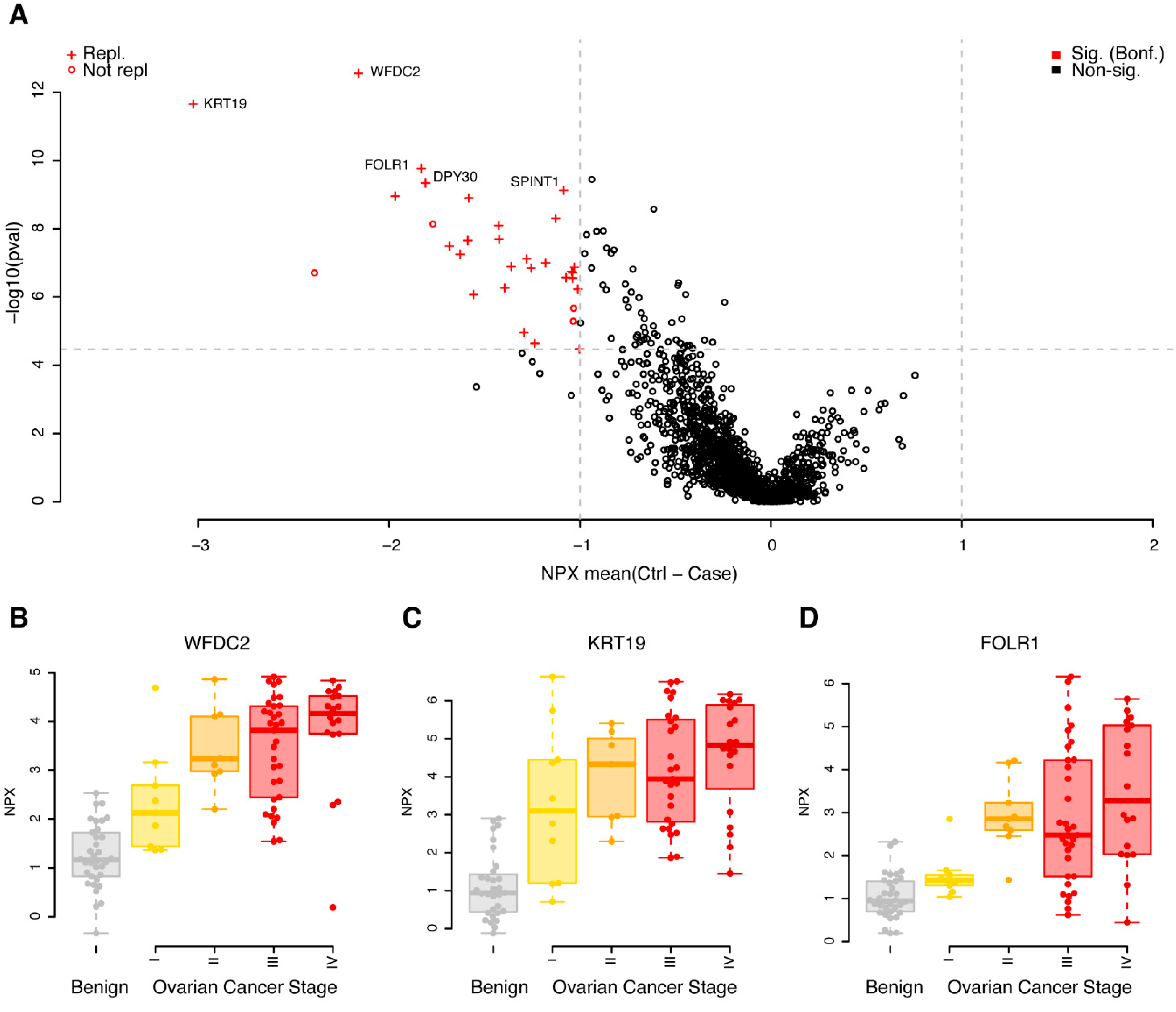
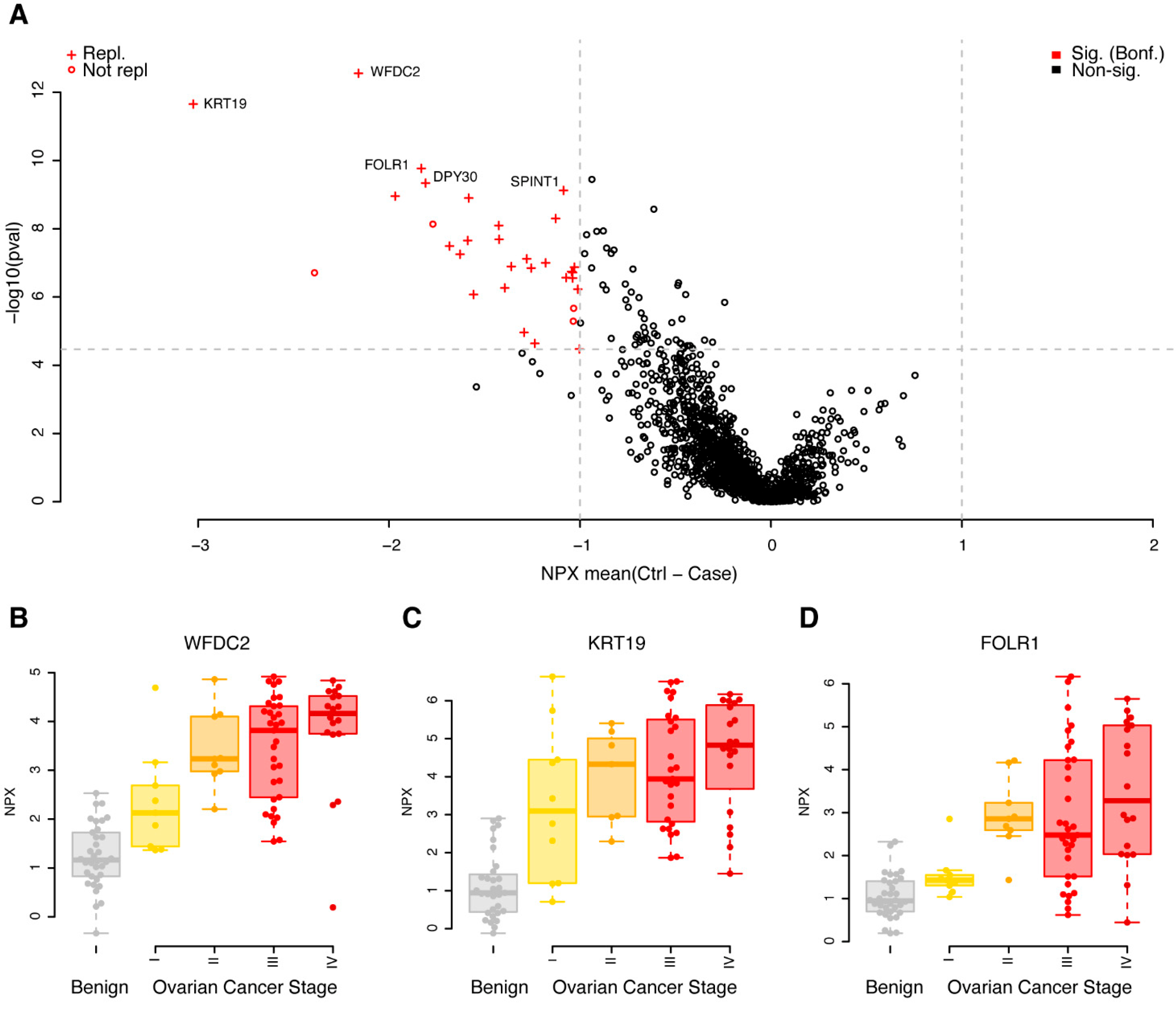
3.2. Replicated Single Valued Biomarkers for Ovarian Cancer
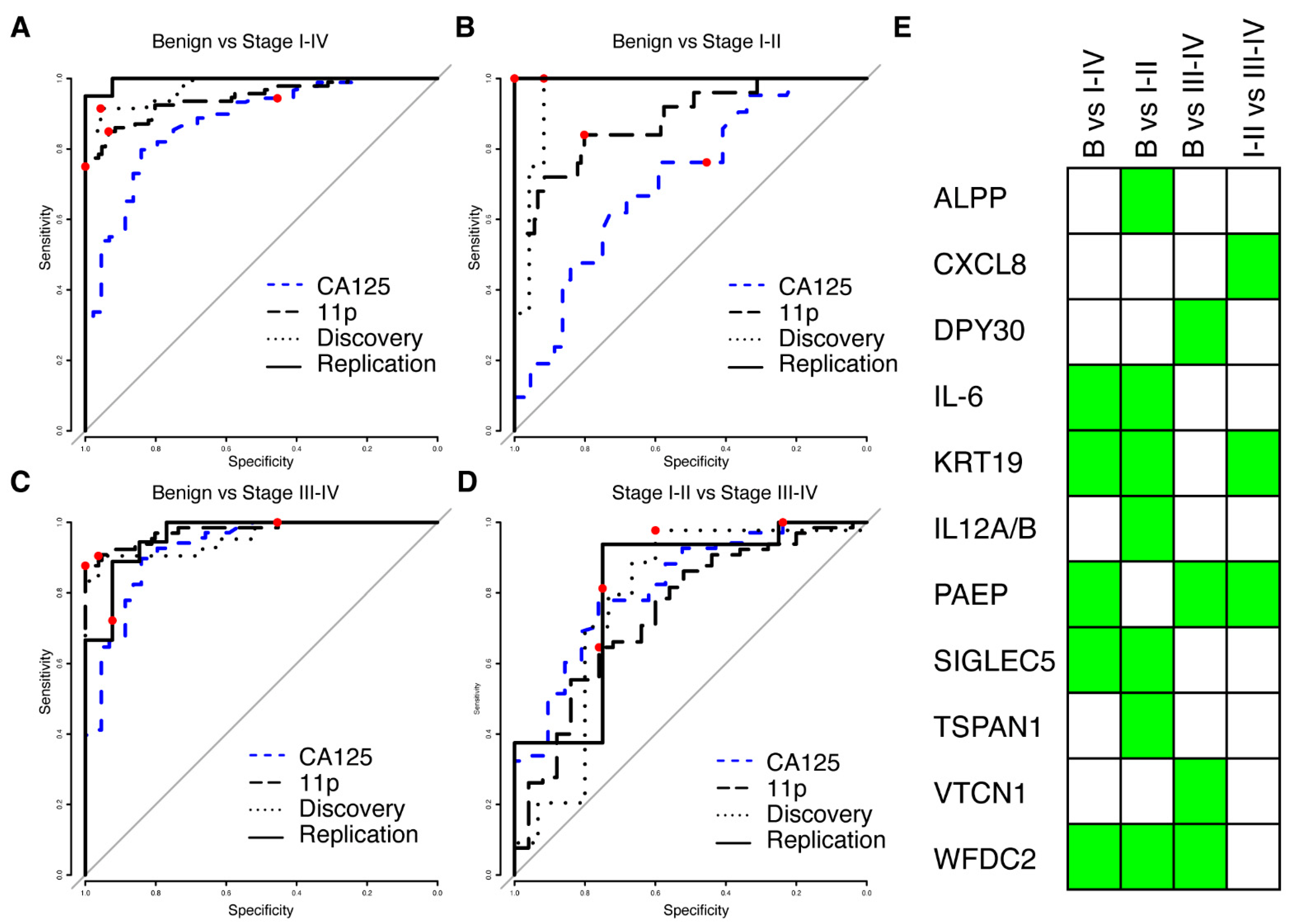
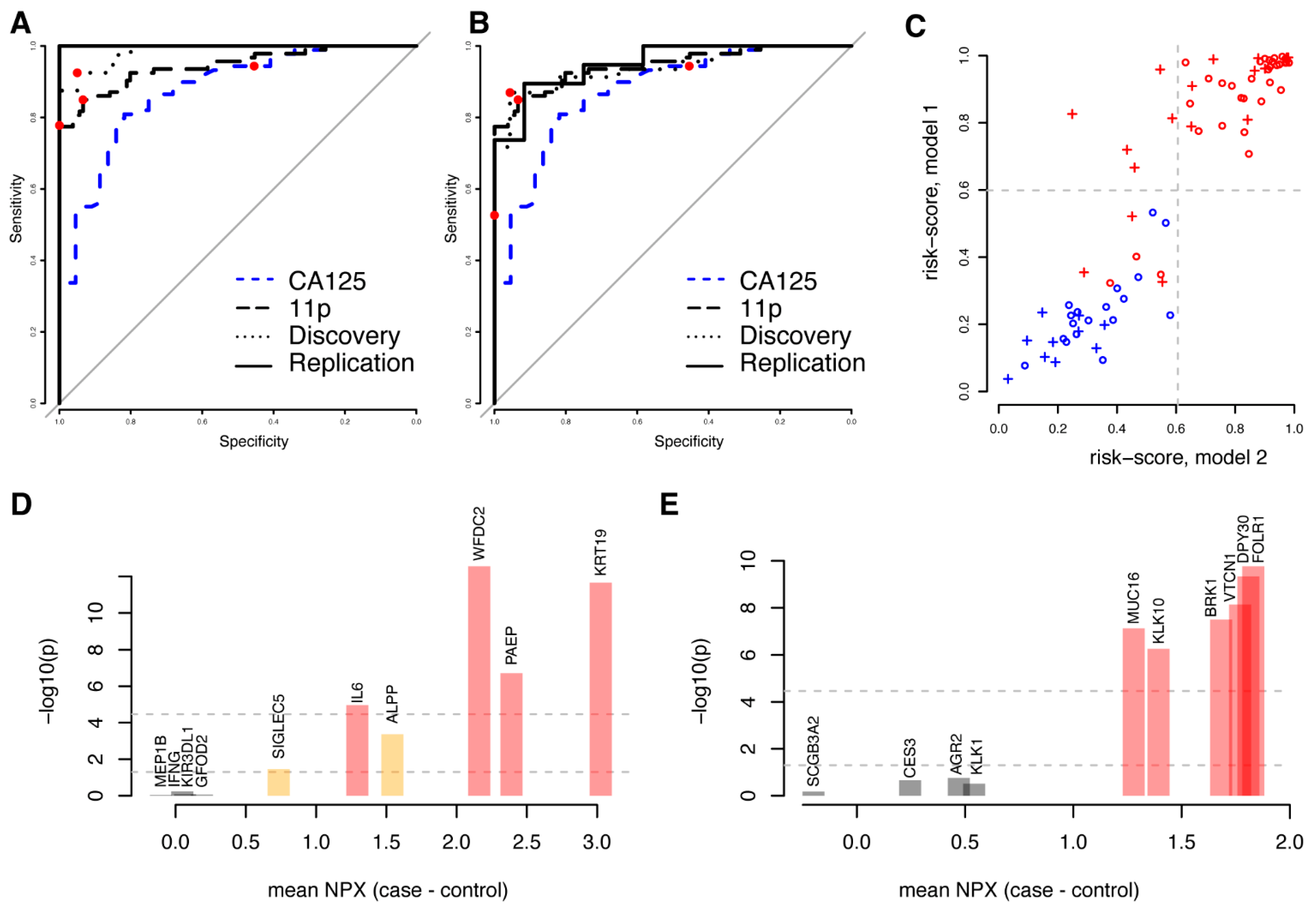
3.3. New Multivariate Models Outperform Previous Biomarkers
3.4. High Performance Models Are Not Unique
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian Cancer Statistics, 2018. CA A Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, I.J.; Menon, U.; Ryan, A.; Gentry-Maharaj, A.; Burnell, M.; Kalsi, J.K.; Amso, N.N.; Apostolidou, S.; Benjamin, E.; Cruickshank, D.; et al. Ovarian Cancer Screening and Mortality in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A Randomised Controlled Trial. Lancet 2016, 387, 945–956. [Google Scholar] [CrossRef] [Green Version]
- Menon, U.; Gentry-Maharaj, A.; Burnell, M.; Singh, N.; Ryan, A.; Karpinskyj, C.; Carlino, G.; Taylor, J.; Massingham, S.K.; Raikou, M.; et al. Ovarian Cancer Population Screening and Mortality after Long-Term Follow-up in the UK Collaborative Trial of Ovarian Cancer Screening (UKCTOCS): A Randomised Controlled Trial. Lancet 2021, 397, 2182–2193. [Google Scholar] [CrossRef]
- Bast, R.C.; Han, C.Y.; Lu, Z.; Lu, K.H. Next Steps in the Early Detection of Ovarian Cancer. Commun. Med. 2021, 1, 36. [Google Scholar] [CrossRef] [PubMed]
- Lycke, M.; Kristjansdottir, B.; Sundfeldt, K. A Multicenter Clinical Trial Validating the Performance of HE4, CA125, Risk of Ovarian Malignancy Algorithm and Risk of Malignancy Index. Gynecol. Oncol. 2018, 151, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C.; Klug, T.L.; John, E.S.; Jenison, E.; Niloff, J.M.; Lazarus, H.; Berkowitz, R.S.; Leavitt, T.; Griffiths, C.T.; Parker, L.; et al. A Radioimmunoassay Using a Monoclonal Antibody to Monitor the Course of Epithelial Ovarian Cancer. N. Engl. J. Med. 1983, 309, 883–887. [Google Scholar] [CrossRef]
- Sölétormos, G.; Duffy, M.J.; Othman Abu Hassan, S.; Verheijen, R.H.M.; Tholander, B.; Bast, R.C.; Gaarenstroom, K.N.; Sturgeon, C.M.; Bonfrer, J.M.; Petersen, P.H.; et al. Clinical Use of Cancer Biomarkers in Epithelial Ovarian Cancer: Updated Guidelines from the European Group on Tumor Markers. In Proceedings of the International Journal of Gynecological Cancer; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2016; Volume 26, pp. 43–51. [Google Scholar]
- Bulska-Będkowska, W.; Chelmecka, E.; Owczarek, A.J.; Mizia-Stec, K.; Witek, A.; Szybalska, A.; Grodzicki, T.; Olszanecka-Glinianowicz, M.; Chudek, J. CA125 as a Marker of Heart Failure in the Older Women: Population-Based Analysis. J. Clin. Med. 2019, 8, 607. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Zhou, Y.X.; He, C.; Ai, J.Y.; Lan, G.L.; Xiong, H.F.; He, W.H.; Xia, L.; Zhu, Y.; Lu, N.H. Elevated CA125 Levels Are Associated with Adverse Clinical Outcomes in Acute Pancreatitis: A Propensity Score-Matched Study. Pancreatol. Off. J. Int. Assoc. Pancreatol. (IAP) 2020, 20, 789–794. [Google Scholar] [CrossRef]
- Lycke, M.; Ulfenborg, B.; Kristjansdottir, B.; Sundfeldt, K. Increased Diagnostic Accuracy of Adnexal Tumors with A Combination of Established Algorithms and Biomarkers. J. Clin. Med. 2020, 9, 299. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.G.; McMeekin, D.S.; Brown, A.K.; DiSilvestro, P.; Miller, M.C.; Allard, W.J.; Gajewski, W.; Kurman, R.; Bast, R.C., Jr.; Skates, S.J. A Novel Multiple Marker Bioassay Utilizing HE4 and CA125 for the Prediction of Ovarian Cancer in Patients with a Pelvic Mass. Gynecol. Oncol. 2009, 112, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Cui, R.; Wang, Y.; Li, Y.; Li, Y. Clinical Value of ROMA Index in Diagnosis of Ovarian Cancer: Meta-Analysis. Cancer Manag. Res. 2019, 11, 2545. [Google Scholar] [CrossRef] [Green Version]
- Russell, M.R.; Graham, C.; D’Amato, A.; Gentry-Maharaj, A.; Ryan, A.; Kalsi, J.K.; Whetton, A.D.; Menon, U.; Jacobs, I.; Graham, R.L.J. Diagnosis of Epithelial Ovarian Cancer Using a Combined Protein Biomarker Panel. Br. J. Cancer 2019, 121, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Enroth, S.; Berggrund, M.; Lycke, M.; Broberg, J.; Lundberg, M.; Assarsson, E.; Olovsson, M.; Stålberg, K.; Sundfeldt, K.; Gyllensten, U. High Throughput Proteomics Identifies a High-Accuracy 11 Plasma Protein Biomarker Signature for Ovarian Cancer. Commun. Biol. 2019, 2, 221. [Google Scholar] [CrossRef] [Green Version]
- Glimelius, B.; Melin, B.; Enblad, G.; Alafuzoff, I.; Beskow, A.; Ahlström, H.; Bill-Axelson, A.; Birgisson, H.; Björ, O.; Edqvist, P.H.; et al. U-CAN: A Prospective Longitudinal Collection of Biomaterials and Clinical Information from Adult Cancer Patients in Sweden. Acta Oncol. 2018, 57, 187–194. [Google Scholar] [CrossRef] [Green Version]
- Region Västra Götaland. Gothia Forum För Klinisk Forskning: Biobank Väst. Available online: https://www.gothiaforum.com/web/en (accessed on 6 December 2021).
- Assarsson, E.; Lundberg, M.; Holmquist, G.; Björkesten, J.; Thorsen, S.B.; Ekman, D.; Eriksson, A.; Dickens, E.R.; Ohlsson, S.; Edfeldt, G.; et al. Homogenous 96-Plex PEA Immunoassay Exhibiting High Sensitivity, Specificity, and Excellent Scalability. PLoS ONE 2014, 9, e95192. [Google Scholar] [CrossRef] [Green Version]
- Wik, L.; Nordberg, N.; Broberg, J.; Björkesten, J.; Assarsson, E.; Henriksson, S.; Grundberg, I.; Pettersson, E.; Westerberg, C.; Liljeroth, E.; et al. Proximity Extension Assay in Combination with Next Generation Sequencing for High-Throughput Proteome-Wide Analysis. Mol. Cell. Proteom. 2021, 20, 100168. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; ISBN 3-900051-07-0. [Google Scholar]
- Eklund, A.; Trimble, J. The Bee Swarm Plot, an Alternative to Stripchart. 2021. Available online: https://cran.r-project.org/web/packages/beeswarm/beeswarm.pdf (accessed on 6 December 2021).
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Sander, C.; Stuart, J.M.; Chang, K.; Creighton, C.J.; et al. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1. [Google Scholar] [CrossRef] [Green Version]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. PROC: An Open-Source Package for R and S+ to Analyze and Compare ROC Curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- Wang, Y.; Li, L.; Douville, C.; Cohen, J.D.; Yen, T.T.; Kinde, I.; Sundfelt, K.; Kjær, S.K.; Hruban, R.H.; Shih, I.M.; et al. Evaluation of Liquid from the Papanicolaou Test and Other Liquid Biopsies for the Detection of Endometrial and Ovarian Cancers. Sci. Transl. Med. 2018, 10, eaap8793. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and Localization of Surgically Resectable Cancers with a Multi-Analyte Blood Test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef] [Green Version]
- Skates, S.J.; Greene, M.H.; Buys, S.S.; Mai, P.L.; Brown, P.; Piedmonte, M.; Rodriguez, G.; Schorge, J.O.; Sherman, M.; Daly, M.B.; et al. Early Detection of Ovarian Cancer Using the Risk of Ovarian Cancer Algorithm with Frequent CA125 Testing in Women at Increased Familial Risk—Combined Results from Two Screening Trials. Clin. Cancer Res. 2017, 23, 3628–3637. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, A.N.; Fraser, L.S.M.; Philpott, S.; Manchanda, R.; Burnell, M.; Badman, P.; Hadwin, R.; Rizzuto, I.; Benjamin, E.; Singh, N.; et al. Evidence of Stage Shift in Women Diagnosed with Ovarian Cancer during Phase II of the United Kingdom Familial Ovarian Cancer Screening Study. J. Clin. Oncol. 2017, 35, 1411–1420. [Google Scholar] [CrossRef]
- Gao, A.; Zhang, L.; Chen, X.; Chen, Y.; Xu, Z.; Liu, Y.; Zhu, W. Effect of VTCN1 on Progression and Metastasis of Ovarian Carcinoma in Vitro and Vivo. Biomed. Pharmacother. Biomed. Pharmacother. 2015, 73, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Scholz, C.; Heublein, S.; Lenhard, M.; Friese, K.; Mayr, D.; Jeschke, U. Glycodelin A Is a Prognostic Marker to Predict Poor Outcome in Advanced Stage Ovarian Cancer Patients. BMC Res. Notes 2012, 5, 551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kampan, N.C.; Madondo, M.T.; Reynolds, J.; Hallo, J.; McNally, O.M.; Jobling, T.W.; Stephens, A.N.; Quinn, M.A.; Plebanski, M. Pre-Operative Sera Interleukin-6 in the Diagnosis of High-Grade Serous Ovarian Cancer. Sci. Rep. 2020, 10, 2213. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, S.; Li, A.; Zhang, A.; Zhang, S.; Chen, L. DPY30 Is Required for the Enhanced Proliferation, Motility and Epithelial-Mesenchymal Transition of Epithelial Ovarian Cancer Cells. Int. J. Mol. Med. 2018, 42, 3065–3072. [Google Scholar] [CrossRef]
- Uddin, M.M.; Gaire, B.; Vancurova, I. Interleukin-8 Induces Proliferation of Ovarian Cancer Cells in 3D Spheroids. Methods Mol. Biol. 2020, 2108, 117–124. [Google Scholar] [CrossRef]
- Kristjánsdóttir, B.; Partheen, K.; Fung, E.T.; Yip, C.; Levan, K.; Sundfeldt, K. Early Inflammatory Response in Epithelial Ovarian Tumor Cyst Fluids. Cancer Med. 2014, 3, 1302–1312. [Google Scholar] [CrossRef]
- Shin, H.Y.; Yang, W.; Chay, D.B.; Lee, E.J.; Chung, J.Y.; Kim, H.S.; Kim, J.H. Tetraspanin 1 Promotes Endometriosis Leading to Ovarian Clear Cell Carcinoma. Mol. Oncol. 2021, 15, 987–1004. [Google Scholar] [CrossRef]
- Wang, P.; Magdolen, V.; Seidl, C.; Dorn, J.; Drecoll, E.; Kotzsch, M.; Yang, F.; Schmitt, M.; Schilling, O.; Rockstroh, A.; et al. Kallikrein-Related Peptidases 4, 5, 6 and 7 Regulate Tumour-Associated Factors in Serous Ovarian Cancer. Br. J. Cancer 2018, 119, 823–831. [Google Scholar] [CrossRef] [Green Version]
- Orsaria, M.; Londero, A.P.; Marzinotto, S.; Loreto, C.D.; Marchesoni, D.; Mariuzzi, L. Placental Type Alkaline Phosphatase Tissue Expression in Ovarian Serous Carcinoma. Cancer Biomark. 2016, 17, 479–486. [Google Scholar] [CrossRef] [Green Version]
- Uppendahl, L.D.; Felices, M.; Bendzick, L.; Ryan, C.; Kodal, B.; Hinderlie, P.; Boylan, K.L.M.; Skubitz, A.P.N.; Miller, J.S.; Geller, M.A. Cytokine-Induced Memory-like Natural Killer Cells Have Enhanced Function, Proliferation, and in Vivo Expansion against Ovarian Cancer Cells. Gynecol. Oncol. 2019, 153, 149–157. [Google Scholar] [CrossRef]
- Montalbán-hernández, K.; Cantero-cid, R.; Lozano-rodríguez, R.; Pascual-iglesias, A.; Avendaño-ortiz, J.; Casalvilla-dueñas, J.C.; Pérez, G.C.B.; Guevara, J.; Marcano, C.; Barragán, C.; et al. Soluble SIGLEC5: A New Prognosis Marker in Colorectal Cancer Patients. Cancers 2021, 13, 3896. [Google Scholar] [CrossRef]
- Broberg, K.; Svensson, J.; Grahn, K.; Assarsson, E.; Åberg, M.; Selander, J.; Enroth, S. Evaluation of 92 Cardiovascular Proteins in Dried Blood Spots Collected under Field-Conditions: Off-the-Shelf Affinity-Based Multiplexed Assays Work Well, Allowing for Simplified Sample Collection. BioEssays 2021, 43, 2000299. [Google Scholar] [CrossRef]
- Björkesten, J.; Enroth, S.; Shen, Q.; Wik, L.; Hougaard, D.M.; Cohen, A.S.; Sörensen, L.; Giedraitis, V.; Ingelsson, M.; Larsson, A.; et al. Stability of Proteins in Dried Blood Spot Biobanks. Mol. Cell. Proteom. 2017, 16, 1286–1296. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
| Cohort | All | Benign | Ovarian Cancer | ||||
|---|---|---|---|---|---|---|---|
| I | II | III | IV | ||||
| No. of samples | Discovery | 111 | 37 | 10 | 9 | 35 | 20 |
| Replication | 37 | 14 | 4 | 0 | 10 | 9 | |
| Age at diag. a | Discovery | 60.1 (13.2) | 56.2 (15.1) | 62.0 (12.2) | 66.4 (6.2) | 60.6 (12.5) | 62.4 (12.4) |
| Replication | 57.4 (14.4) | 49.9 (14.9) | 70.4 (15.6) | 60.2 (6.8) | 60.3 (14.9) | ||
| Age diff p-value b | 0.21 | 0.11 | 0.36 | 0.52 | 0.83 | ||
| CA125 (U/mL) c | Discovery | 263 (358.8) | 41.5 (40) | 67 (65.2) | 240 (213.5) | 594 (551.5) | 1358 (1693.1) |
| Replication | 189 (255) | 28.5 (24.5) | 189 (235.7) | 640 (763.5) | 340 (315.8) | ||
| CA125 diff p-value b | 0.64 | 0.46 | 0.48 | 0.76 | 0.31 | ||
| AUC a | Sens b | Spec b | ||
|---|---|---|---|---|
| B vs. I–IV | Discovery | 0.98 (0.95–1.00) | 0.91 (0.83–0.98) | 0.96 (0.87–1.00) |
| Replication | 1.00 (0.99–1.00) | 0.75 (0.55–0.95) | 1.00 (1.00–1.00) | |
| p-value c | 0.17 | 0.11 | 1.00 | |
| B vs. I–II | Discovery | 0.96 (0.90–1.00) | 1.00 (1.00–1.00) | 0.92 (0.79–1.00) |
| Replication | 1.00 (1.00–1.00) | 1.00 (1.00–1.00) | 1.00 (1.00–1.00) | |
| p-value c | 0.22 | 1.00 | 0.53 | |
| B vs. III–IV | Discovery | 0.96 (0.92–1.00) | 0.90 (0.81–0.98) | 0.96 (0.89–1.00) |
| Replication | 0.96 (0.90–1.00) | 0.72 (0.50–0.89) | 0.92 (0.77–1.00) | |
| p-value c | 0.92 | 0.11 | 1.00 | |
| I–II vs. III–IV | Discovery | 0.78 (0.61–0.95) | 0.98 (0.93–1.00) | 0.60 (0.33–0.80) |
| Replication | 0.81 (0.51–1.00) | 0.81 (0.62–1.00) | 0.75 (0.25–1.00) | |
| p-value c | 0.85 | 0.054 | 1.00 |
| AUC a | Sens b | Spec b | ||
|---|---|---|---|---|
| Model 1 | Discovery | 0.98 (0.96–1.00) | 0.93 (0.82–1.00) | 0.95 (0.85–1.00) |
| Replication | 1.00 (1.00–1.00) | 0.78 (0.56–0.94) | 1.00 (1.00–1.00) | |
| p-value c | 0.16 | 0.19 | 1.00 | |
| Model 2 | Discovery | 0.94 (0.89–0.99) | 0.87 (0.76–0.96) | 0.96 (0.87–1.00) |
| Replication | 0.95 (0.89–1.00) | 0.53 (0.32–0.74) | 1.00 (1.00–1.00) | |
| p-value c | 0.78 | 0.0073 | 1.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gyllensten, U.; Hedlund-Lindberg, J.; Svensson, J.; Manninen, J.; Öst, T.; Ramsell, J.; Åslin, M.; Ivansson, E.; Lomnytska, M.; Lycke, M.; et al. Next Generation Plasma Proteomics Identifies High-Precision Biomarker Candidates for Ovarian Cancer. Cancers 2022, 14, 1757. https://doi.org/10.3390/cancers14071757
Gyllensten U, Hedlund-Lindberg J, Svensson J, Manninen J, Öst T, Ramsell J, Åslin M, Ivansson E, Lomnytska M, Lycke M, et al. Next Generation Plasma Proteomics Identifies High-Precision Biomarker Candidates for Ovarian Cancer. Cancers. 2022; 14(7):1757. https://doi.org/10.3390/cancers14071757
Chicago/Turabian StyleGyllensten, Ulf, Julia Hedlund-Lindberg, Johanna Svensson, Johanna Manninen, Torbjörn Öst, Jon Ramsell, Matilda Åslin, Emma Ivansson, Marta Lomnytska, Maria Lycke, and et al. 2022. "Next Generation Plasma Proteomics Identifies High-Precision Biomarker Candidates for Ovarian Cancer" Cancers 14, no. 7: 1757. https://doi.org/10.3390/cancers14071757
APA StyleGyllensten, U., Hedlund-Lindberg, J., Svensson, J., Manninen, J., Öst, T., Ramsell, J., Åslin, M., Ivansson, E., Lomnytska, M., Lycke, M., Axelsson, T., Liljedahl, U., Nordlund, J., Edqvist, P.-H., Sjöblom, T., Uhlén, M., Stålberg, K., Sundfeldt, K., Åberg, M., & Enroth, S. (2022). Next Generation Plasma Proteomics Identifies High-Precision Biomarker Candidates for Ovarian Cancer. Cancers, 14(7), 1757. https://doi.org/10.3390/cancers14071757