
FOXC1 Binds Enhancers and Promotes Cisplatin Resistance in Bladder Cancer

 ,
,
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. SP Assays
2.3. AcceSssIble Assays
2.4. RNA Sequencing (RNA-seq)
2.5. Chromatin Immunoprecipitation Coupled with Sequencing (ChIP-seq)
2.6. Motif Analysis
2.7. FOXC1 Knockout
2.8. Western Blotting
2.9. Real-Time qPCR (RT-qPCR)
2.10. Flow Cytometry Analysis of FOXC1 after Cisplatin Treatment
2.11. Cell Viability Assays with Cisplatin Treatment
2.12. Statistical Analysis and Graphical Packages
3. Results
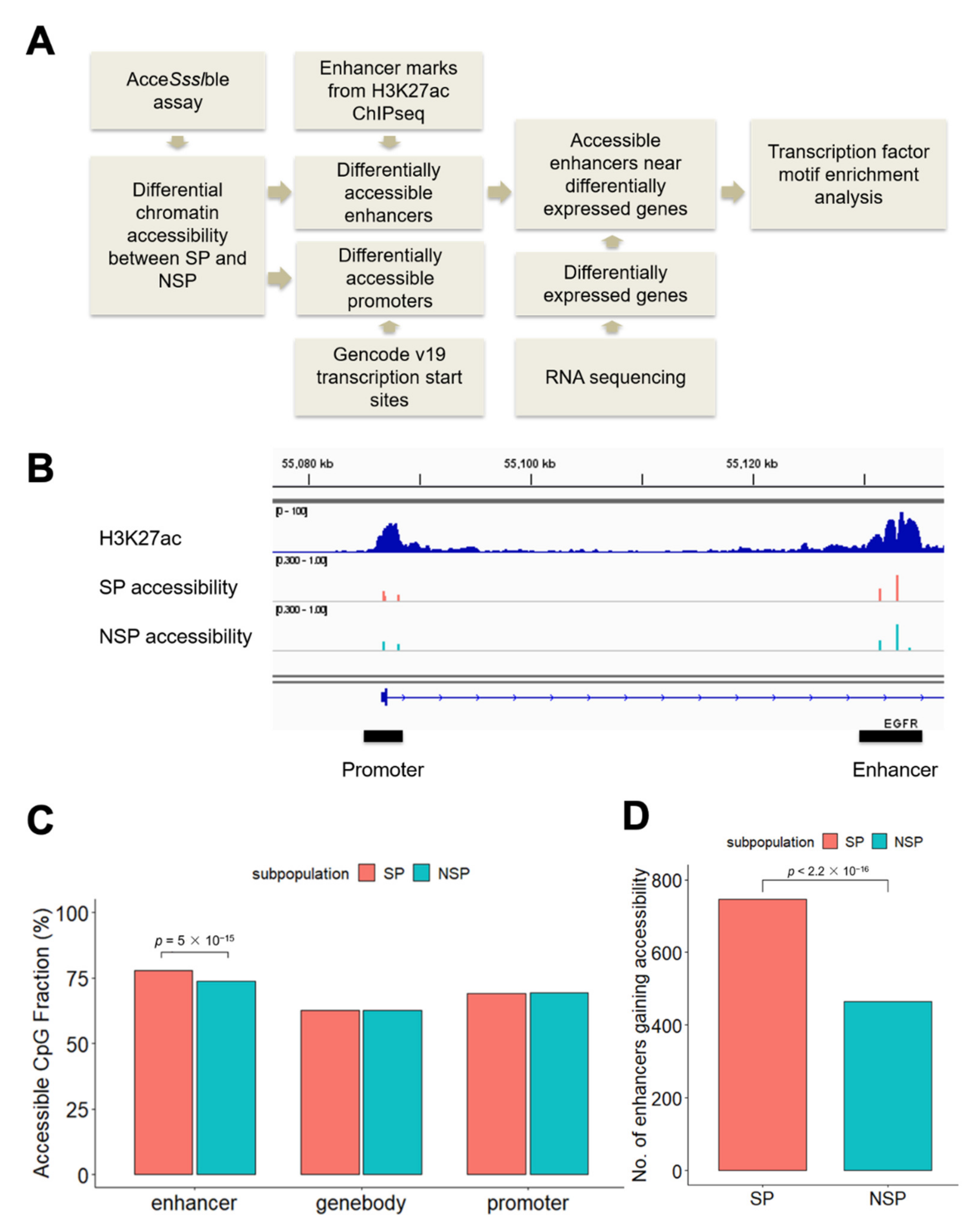
3.1. Differential Accessibility at Regulatory Elements between SP and NSP Cells
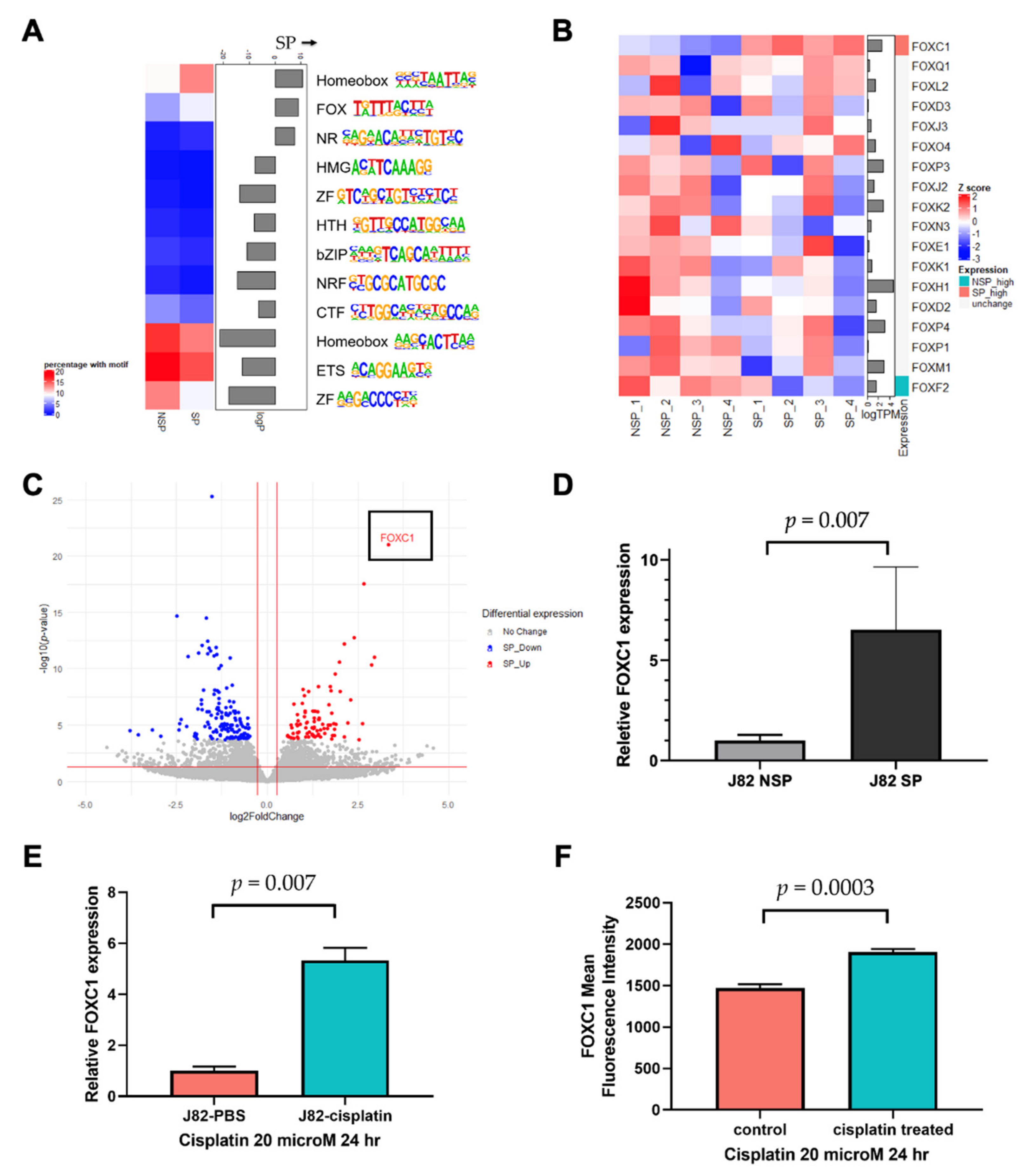
3.2. Linking FOXC1 to Increased Enhancer Activity in SP Cells
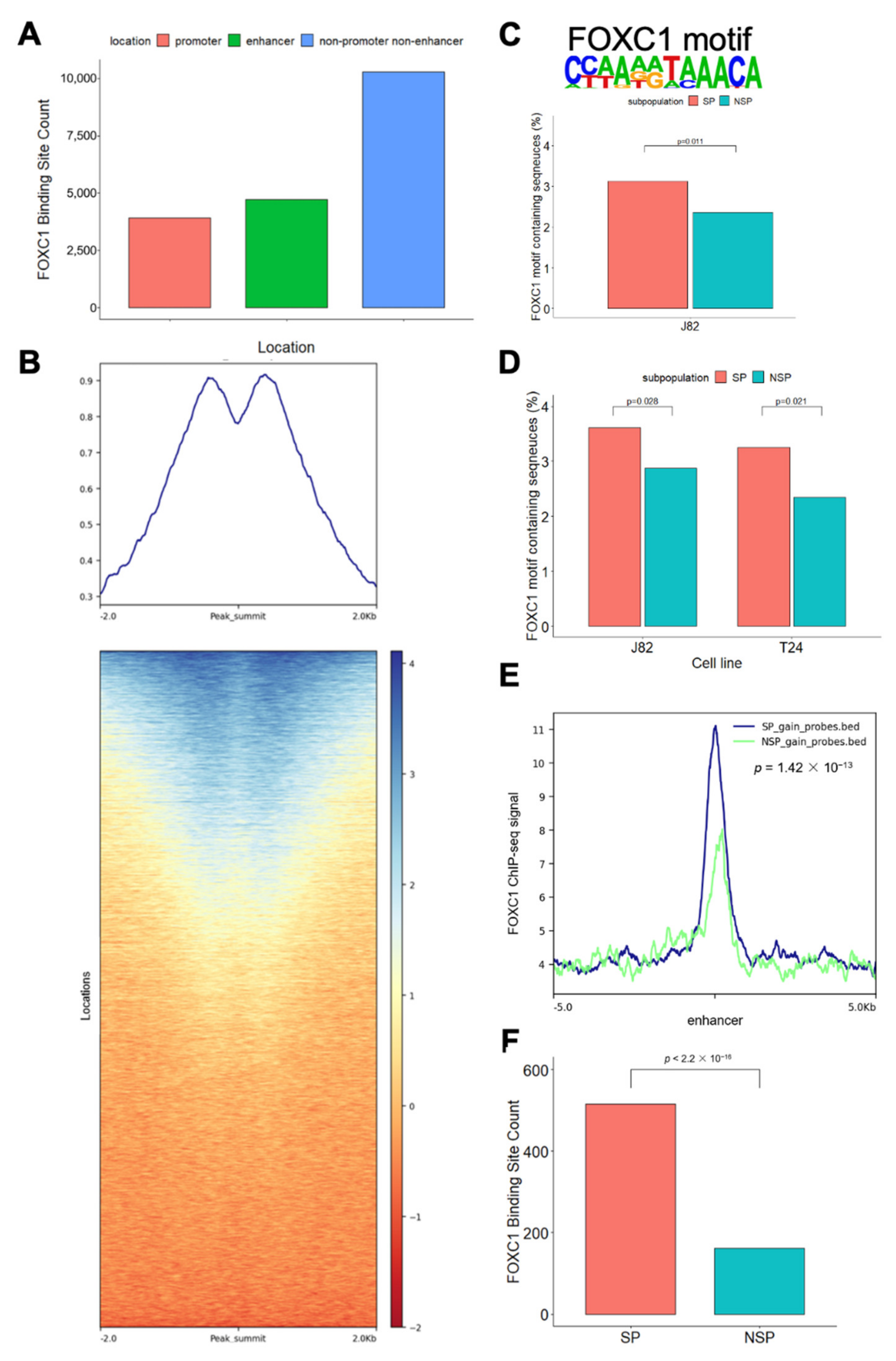
3.3. FOXC1 Binding Sites Are Significantly More Accessible in SP Cells
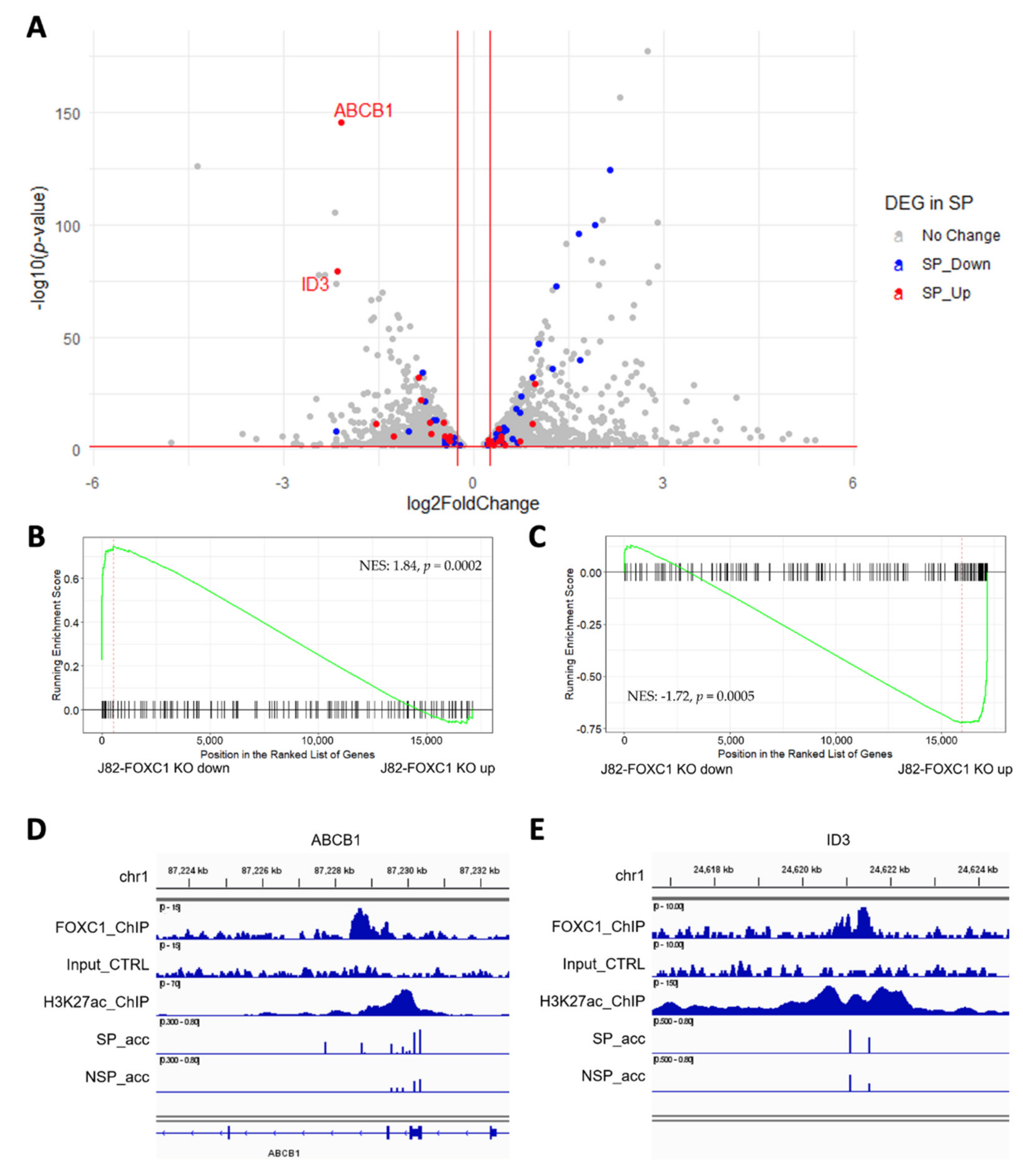
3.4. FOXC1 Controls Genes Regulating Drug Resistance and Cancer Stemness
3.5. FOXC1 Regulates the Transition to the SP Phenotype and Cisplatin Resistance in Bladder Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Tran, L.; Xiao, J.F.; Agarwal, N.; Duex, J.E.; Theodorescu, D. Advances in bladder cancer biology and therapy. Nat. Rev. Cancer 2021, 21, 104–121. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, S.; Paskeh, M.D.A.; Hashemi, F.; Zabolian, A.; Hashemi, M.; Entezari, M.; Tabari, T.; Ashrafizadeh, M.; Raee, P.; Aghamiri, S.; et al. Long non-coding RNAs as new players in bladder cancer: Lessons from pre-clinical and clinical studies. Life Sci. 2022, 288, 119948. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Xu, T.; Goldkorn, A. Cancer cells cyclically lose and regain drug-resistant highly tumorigenic features characteristic of a cancer stem-like phenotype. Mol. Cancer Ther. 2011, 10, 938–948. [Google Scholar] [CrossRef] [Green Version]
- He, K.; Xu, T.; Xu, Y.; Ring, A.; Kahn, M.; Goldkorn, A. Cancer cells acquire a drug resistant, highly tumorigenic, cancer stem-like phenotype through modulation of the PI3K/Akt/β-catenin/CBP pathway. Int. J. Cancer 2014, 134, 43–54. [Google Scholar] [CrossRef]
- Xu, T.; Li, H.T.; Wei, J.; Li, M.; Hsieh, T.C.; Lu, Y.T.; Lakshminarasimhan, R.; Xu, R.; Hodara, E.; Morrison, G.; et al. Epigenetic plasticity potentiates a rapid cyclical shift to and from an aggressive cancer phenotype. Int. J. Cancer 2020, 146, 3065–3076. [Google Scholar] [CrossRef]
- Roesch, A.; Vultur, A.; Bogeski, I.; Wang, H.; Zimmermann, K.M.; Speicher, D.; Korbel, C.; Laschke, M.W.; Gimotty, P.A.; Philipp, S.E.; et al. Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1B(high) cells. Cancer Cell 2013, 23, 811–825. [Google Scholar] [CrossRef] [Green Version]
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Goktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e813. [Google Scholar] [CrossRef] [Green Version]
- Herz, H.M. Enhancer deregulation in cancer and other diseases. Bioessays 2016, 38, 1003–1015. [Google Scholar] [CrossRef]
- Liau, B.B.; Sievers, C.; Donohue, L.K.; Gillespie, S.M.; Flavahan, W.A.; Miller, T.E.; Venteicher, A.S.; Hebert, C.H.; Carey, C.D.; Rodig, S.J.; et al. Adaptive Chromatin Remodeling Drives Glioblastoma Stem Cell Plasticity and Drug Tolerance. Cell Stem Cell 2017, 20, 233–246.e7. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.C.; Fennell, K.A.; Chan, Y.C.; Rambow, F.; Yeung, M.M.; Vassiliadis, D.; Lara, L.; Yeh, P.; Martelotto, L.G.; Rogiers, A.; et al. Targeting enhancer switching overcomes non-genetic drug resistance in acute myeloid leukaemia. Nat. Commun. 2019, 10, 2723. [Google Scholar] [CrossRef] [Green Version]
- Becket, E.; Chopra, S.; Duymich, C.E.; Lin, J.J.; You, J.S.; Pandiyan, K.; Nichols, P.W.; Siegmund, K.D.; Charlet, J.; Weisenberger, D.J.; et al. Identification of DNA Methylation-Independent Epigenetic Events Underlying Clear Cell Renal Cell Carcinoma. Cancer Res. 2016, 76, 1954–1964. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Zhang, L.; Li, H.; Hinoue, T.; Zhou, W.; Ohtani, H.; El-Khoueiry, A.; Daniels, J.; O’Connell, C.; Dorff, T.B.; et al. Integrative Epigenetic Analysis Reveals Therapeutic Targets to the DNA Methyltransferase Inhibitor Guadecitabine (SGI-110) in Hepatocellular Carcinoma. Hepatology 2018, 68, 1412–1428. [Google Scholar] [CrossRef] [Green Version]
- Pandiyan, K.; You, J.S.; Yang, X.; Dai, C.; Zhou, X.J.; Baylin, S.B.; Jones, P.A.; Liang, G. Functional DNA demethylation is accompanied by chromatin accessibility. Nucleic Acids Res. 2013, 41, 3973–3985. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Frankish, A.; Diekhans, M.; Ferreira, A.M.; Johnson, R.; Jungreis, I.; Loveland, J.; Mudge, J.M.; Sisu, C.; Wright, J.; Armstrong, J.; et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019, 47, D766–D773. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Pattison, J.M.; Posternak, V.; Cole, M.D. Transcription Factor KLF5 Binds a Cyclin E1 Polymorphic Intronic Enhancer to Confer Increased Bladder Cancer Risk. Mol. Cancer Res. 2016, 14, 1078–1086. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. 2013. Available online: https://ui.adsabs.harvard.edu/abs/2013arXiv1303.3997L (accessed on 20 October 2020).
- Zhang, Y.; Liu, T.; Meyer, C.A.; Eeckhoute, J.; Johnson, D.S.; Bernstein, B.E.; Nusbaum, C.; Myers, R.M.; Brown, M.; Li, W.; et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008, 9, R137. [Google Scholar] [CrossRef] [Green Version]
- Blattler, A.; Yao, L.; Witt, H.; Guo, Y.; Nicolet, C.M.; Berman, B.P.; Farnham, P.J. Global loss of DNA methylation uncovers intronic enhancers in genes showing expression changes. Genome Biol. 2014, 15, 469. [Google Scholar] [CrossRef]
- Ramirez, F.; Ryan, D.P.; Gruning, B.; Bhardwaj, V.; Kilpert, F.; Richter, A.S.; Heyne, S.; Dundar, F.; Manke, T. deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016, 44, W160–W165. [Google Scholar] [CrossRef]
- Consortium, E.P.; Moore, J.E.; Purcaro, M.J.; Pratt, H.E.; Epstein, C.B.; Shoresh, N.; Adrian, J.; Kawli, T.; Davis, C.A.; Dobin, A.; et al. Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 2020, 583, 699–710. [Google Scholar] [CrossRef]
- Rhie, S.K.; Perez, A.A.; Lay, F.D.; Schreiner, S.; Shi, J.; Polin, J.; Farnham, P.J. A high-resolution 3D epigenomic map reveals insights into the creation of the prostate cancer transcriptome. Nat. Commun. 2019, 10, 4154. [Google Scholar] [CrossRef] [Green Version]
- Rhie, S.K.; Schreiner, S.; Witt, H.; Armoskus, C.; Lay, F.D.; Camarena, A.; Spitsyna, V.N.; Guo, Y.; Berman, B.P.; Evgrafov, O.V.; et al. Using 3D epigenomic maps of primary olfactory neuronal cells from living individuals to understand gene regulation. Sci. Adv. 2018, 4, eaav8550. [Google Scholar] [CrossRef] [Green Version]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, B.; Zhou, B.; Qu, Y.; Gao, B.; Xu, Y.; Chung, S.; Tanaka, H.; Yang, W.; Giuliano, A.E.; Cui, X. FOXC1-induced non-canonical WNT5A-MMP7 signaling regulates invasiveness in triple-negative breast cancer. Oncogene 2018, 37, 1399–1408. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Qu, Y.; Jin, Y.; Yu, Y.; Deng, N.; Wawrowsky, K.; Zhang, X.; Li, N.; Bose, S.; Wang, Q.; et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015, 13, 1046–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.G.; Sikic, B.I. Molecular pathways: Regulation and therapeutic implications of multidrug resistance. Clin. Cancer Res. 2012, 18, 1863–1869. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.J.; Li, C.F.; Chu, Y.Y.; Wang, Y.H.; Hour, T.C.; Yen, C.J.; Chang, W.C.; Wang, J.M. Inhibition of the EGFR/STAT3/CEBPD Axis Reverses Cisplatin Cross-resistance with Paclitaxel in the Urothelial Carcinoma of the Urinary Bladder. Clin. Cancer Res. 2017, 23, 503–513. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, C.A.; Kreso, A.; Ryan, P.; Hermans, K.G.; Gibson, L.; Wang, Y.; Tsatsanis, A.; Gallinger, S.; Dick, J.E. ID1 and ID3 regulate the self-renewal capacity of human colon cancer-initiating cells through p21. Cancer Cell 2012, 21, 777–792. [Google Scholar] [CrossRef] [Green Version]
- Sachindra; Larribere, L.; Novak, D.; Wu, H.; Huser, L.; Granados, K.; Orouji, E.; Utikal, J. New role of ID3 in melanoma adaptive drug-resistance. Oncotarget 2017, 8, 110166–110175. [Google Scholar] [CrossRef] [Green Version]
- Marine, J.C.; Dawson, S.J.; Dawson, M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 2020, 20, 743–756. [Google Scholar] [CrossRef]
- Cao, S.; Wang, Z.; Gao, X.; He, W.; Cai, Y.; Chen, H.; Xu, R. FOXC1 induces cancer stem cell-like properties through upregulation of beta-catenin in NSCLC. J. Exp. Clin. Cancer Res. 2018, 37, 220. [Google Scholar] [CrossRef] [Green Version]
- Sabapathi, N.; Sabarimurugan, S.; Madurantakam Royam, M.; Kumarasamy, C.; Xu, X.; Xu, G.; Jayaraj, R. Prognostic Significance of FOXC1 in Various Cancers: A Systematic Review and Meta-Analysis. Mol. Diagn. Ther. 2019, 23, 695–706. [Google Scholar] [CrossRef]
- Tkocz, D.; Crawford, N.T.; Buckley, N.E.; Berry, F.B.; Kennedy, R.D.; Gorski, J.J.; Harkin, D.P.; Mullan, P.B. BRCA1 and GATA3 corepress FOXC1 to inhibit the pathogenesis of basal-like breast cancers. Oncogene 2012, 31, 3667–3678. [Google Scholar] [CrossRef] [Green Version]
- Tang, L.; Jin, J.; Xu, K.; Wang, X.; Tang, J.; Guan, X. SOX9 interacts with FOXC1 to activate MYC and regulate CDK7 inhibitor sensitivity in triple-negative breast cancer. Oncogenesis 2020, 9, 47. [Google Scholar] [CrossRef]
- Zhang, P.; Long, Q.; Zeng, S.; Wen, M.; Lu, Q. FOXC1-induced LINC01123 acts as a mediator in triple negative breast cancer. Cancer Cell Int. 2020, 20, 199. [Google Scholar] [CrossRef]
- Zhang, Y.; Liao, Y.; Chen, C.; Sun, W.; Sun, X.; Liu, Y.; Xu, E.; Lai, M.; Zhang, H. p38-regulated FOXC1 stability is required for colorectal cancer metastasis. J. Pathol. 2020, 250, 217–230. [Google Scholar] [CrossRef]
- Melguizo, C.; Prados, J.; Luque, R.; Ortiz, R.; Rama, A.R.; Caba, O.; Rodriguez-Serrano, F.; Alvarez, P.J.; Aranega, A. Modulation of multidrug resistance gene expression in peripheral blood mononuclear cells of lung cancer patients and evaluation of their clinical significance. Cancer Chemother. Pharmacol. 2013, 71, 537–541. [Google Scholar] [CrossRef]
- Perk, J.; Iavarone, A.; Benezra, R. Id family of helix-loop-helix proteins in cancer. Nat. Rev. Cancer 2005, 5, 603–614. [Google Scholar] [CrossRef]
- Tseng, Y.J.; Huang, C.E.; Wen, C.N.; Lai, P.Y.; Wu, M.H.; Sun, Y.C.; Wang, H.Y.; Lu, J.J. Predicting breast cancer metastasis by using serum biomarkers and clinicopathological data with machine learning technologies. Int. J. Med. Inform. 2019, 128, 79–86. [Google Scholar] [CrossRef]
- Golson, M.L.; Kaestner, K.H. Fox transcription factors: From development to disease. Development 2016, 143, 4558–4570. [Google Scholar] [CrossRef] [Green Version]
- Iwafuchi-Doi, M.; Donahue, G.; Kakumanu, A.; Watts, J.A.; Mahony, S.; Pugh, B.F.; Lee, D.; Kaestner, K.H.; Zaret, K.S. The Pioneer Transcription Factor FoxA Maintains an Accessible Nucleosome Configuration at Enhancers for Tissue-Specific Gene Activation. Mol. Cell 2016, 62, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Gilding, L.N.; Somervaille, T.C.P. The Diverse Consequences of FOXC1 Deregulation in Cancer. Cancers 2019, 11, 184. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.; Yu, H.V.; Tseng, K.C.; Flath, M.; Fabian, P.; Segil, N.; Crump, J.G. Foxc1 establishes enhancer accessibility for craniofacial cartilage differentiation. eLife 2021, 10, e63595. [Google Scholar] [CrossRef]
- Huang, L.; Huang, Z.; Fan, Y.; He, L.; Ye, M.; Shi, K.; Ji, B.; Huang, J.; Wang, Y.; Li, Q. FOXC1 promotes proliferation and epithelial-mesenchymal transition in cervical carcinoma through the PI3K-AKT signal pathway. Am. J. Transl. Res. 2017, 9, 1297–1306. [Google Scholar]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.-T.; Xu, T.; Iqbal, M.; Hsieh, T.-C.; Luo, Z.; Liang, G.; Farnham, P.J.; Rhie, S.K.; Goldkorn, A. FOXC1 Binds Enhancers and Promotes Cisplatin Resistance in Bladder Cancer. Cancers 2022, 14, 1717. https://doi.org/10.3390/cancers14071717
Lu Y-T, Xu T, Iqbal M, Hsieh T-C, Luo Z, Liang G, Farnham PJ, Rhie SK, Goldkorn A. FOXC1 Binds Enhancers and Promotes Cisplatin Resistance in Bladder Cancer. Cancers. 2022; 14(7):1717. https://doi.org/10.3390/cancers14071717
Chicago/Turabian StyleLu, Yi-Tsung, Tong Xu, Maheen Iqbal, Tien-Chan Hsieh, Zhifei Luo, Gangning Liang, Peggy J. Farnham, Suhn K. Rhie, and Amir Goldkorn. 2022. "FOXC1 Binds Enhancers and Promotes Cisplatin Resistance in Bladder Cancer" Cancers 14, no. 7: 1717. https://doi.org/10.3390/cancers14071717
APA StyleLu, Y.-T., Xu, T., Iqbal, M., Hsieh, T.-C., Luo, Z., Liang, G., Farnham, P. J., Rhie, S. K., & Goldkorn, A. (2022). FOXC1 Binds Enhancers and Promotes Cisplatin Resistance in Bladder Cancer. Cancers, 14(7), 1717. https://doi.org/10.3390/cancers14071717