
Autophagy, Oxidative Stress and Cancer Development


Abstract
:Simple Summary
Abstract
1. Introduction
2. The Link between Endoplasmic Reticulum (ER) Stress, Oxidative Stress, Autophagy and Tumor Initiation
2.1. ER Stress and Autophagy
2.2. Implication of Fasting and Calorie Restriction in Autophagy
2.3. Oxidative Stress and Autophagy
2.4. Tumor Necrosis Factor (TNF)-Induced Necroptosis and Autophagy
3. The Role of Autophagy in Cancer
3.1. Autophagy as a Novel Therapeutic Target in Cancer
3.2. Autophagy Biomarkers in Cancer
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [Green Version]
- White, E.; Mehnert, J.M.; Chan, C.S. Autophagy, Metabolism, and Cancer. Clin. Cancer Res. 2015, 21, 5037–5046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabinowitz, J.D.; White, E. Autophagy and Metabolism. Science 2010, 330, 1344–1348. [Google Scholar] [CrossRef] [Green Version]
- Onorati, A.V.; Dyczynski, M.; Ojha, R.; Amaravadi, R.K. Targeting Autophagy in Cancer. Cancer 2018, 124, 3307–3318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network Organization of the Human Autophagy System. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin Is an Autophagy Receptor for Damaged Mitochondria in Parkin-Mediated Mitophagy That Is Disrupted by an ALS-Linked Mutation. Proc. Natl. Acad. Sci. USA 2014, 111, e4439–e4448. [Google Scholar] [CrossRef] [Green Version]
- Cheng, M.; Qian, L.; Shen, G.; Bian, G.; Xu, T.; Xu, W.; Shen, G.; Hu, S. Microbiota Modulate Tumoral Immune Surveillance in Lung through a ΓδT17 Immune Cell-Dependent Mechanism. Cancer Res. 2014, 74, 4030–4041. [Google Scholar] [CrossRef] [Green Version]
- Sarparanta, J.; García-Macia, M.; Singh, R. Autophagy and Mitochondria in Obesity and Type 2 Diabetes. Curr. Diabetes Rev. 2017, 13. [Google Scholar] [CrossRef]
- Amaravadi, R.K.; Lippincott-Schwartz, J.; Yin, X.M.; Weiss, W.A.; Takebe, N.; Timmer, W.; DiPaola, R.S.; Lotze, M.T.; White, E. Principles and Current Strategies for Targeting Autophagy for Cancer Treatment. Clin. Cancer Res. 2011, 17, 654–666. [Google Scholar] [CrossRef] [Green Version]
- Kaur, J.; Debnath, J. Autophagy at the Crossroads of Catabolism and Anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The Machinery of Macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filomeni, G.; de Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide Metabolism in Mammalian Organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [CrossRef]
- Wagner, B.A.; Venkataraman, S.; Buettner, G.R. The Rate of Oxygen Utilization by Cells. Free Radic. Biol. Med. 2011, 51, 700–712. [Google Scholar] [CrossRef] [Green Version]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Disulfide Relays and Phosphorylative Cascades: Partners in Redox-Mediated Signaling Pathways. Cell Death Differ. 2005, 12, 1555–1563. [Google Scholar] [CrossRef] [Green Version]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Cell Signalling and the Glutathione Redox System. Biochem. Pharmacol. 2002, 64, 1057–1064. [Google Scholar] [CrossRef]
- Bindoli, A.; Fukuto, J.M.; Forman, H.J. Thiol Chemistry in Peroxidase Catalysis and Redox Signaling. Antioxid. Redox Signal. 2008, 10, 1549–1564. [Google Scholar] [CrossRef]
- Filomeni, G.; Desideri, E.; Cardaci, S.; Rotilio, G.; Ciriolo, M.R. Under the ROS…thiol Network Is the Principal Suspect for Autophagy Commitment. Autophagy 2010, 6, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.J.; Lei, Y.H.; Yao, N.; Wang, C.R.; Hu, N.; Ye, W.C.; Zhang, D.M.; Chen, Z.S. Autophagy and Multidrug Resistance in Cancer. Chin. J. Cancer 2017, 36, 52. [Google Scholar] [CrossRef]
- Sridhar, S.; Botbol, Y.; MacIan, F.; Cuervo, A.M. Autophagy and Disease: Always Two Sides to a Problem. J. Pathol. 2012, 226, 255–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchser, W.J.; Laskow, T.C.; Pavlik, P.J.; Lin, H.M.; Lotze, M.T. Cell-Mediated Autophagy Promotes Cancer Cell Survival. Cancer Res. 2012, 72, 2970–2979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide Is the Major Reactive Oxygen Species Regulating Autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Shvets, E.; Elazar, Z. Oxidation as a Post-Translational Modification That Regulates Autophagy. Autophagy 2007, 3, 371–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levonen, A.L.; Hill, B.G.; Kansanen, E.; Zhang, J.; Darley-Usmar, V.M. Redox Regulation of Antioxidants, Autophagy, and the Response to Stress: Implications for Electrophile Therapeutics. Free Radic. Biol. Med. 2014, 71, 196–207. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.L.; Loeb, L.A. The Contribution of Endogenous Sources of DNA Damage to the Multiple Mutations in Cancer. Mutat. Res. 2001, 477, 7–21. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative Stress, Inflammation, and Cancer: How Are They Linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storz, P. Reactive Oxygen Species in Tumor Progression. Front. Biosci. A J. Virtual Libr. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [Green Version]
- Kocaturk, N.M.; Gozuacik, D. Crosstalk Between Mammalian Autophagy and the Ubiquitin-Proteasome System. Front. Cell Dev. Biol. 2018, 6, 128. [Google Scholar] [CrossRef]
- Lin, Y.; Jiang, M.; Chen, W.; Zhao, T.; Wei, Y. Cancer and ER Stress: Mutual Crosstalk between Autophagy, Oxidative Stress and Inflammatory Response. Biomed. Pharmacother. Biomed. Pharmacother. 2019, 118, 109249. [Google Scholar] [CrossRef]
- Kleizen, B.; Braakman, I. Protein Folding and Quality Control in the Endoplasmic Reticulum. Curr. Opin. Cell Biol. 2004, 16, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J. The Endoplasmic Reticulum: A Multifunctional Signaling Organelle. Cell Calcium 2002, 32, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Voeltz, G.K.; Rolls, M.M.; Rapoport, T.A. Structural Organization of the Endoplasmic Reticulum. EMBO Rep. 2002, 3, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; LaFerla, F.M.; Chan, S.L.; Leissring, M.A.; Shepel, P.N.; Geiger, J.D. Calcium Signaling in the ER: Its Role in Neuronal Plasticity and Neurodegenerative Disorders. Trends Neurosci. 2000, 23, 222–229. [Google Scholar] [CrossRef]
- Fagone, P.; Jackowski, S. Membrane Phospholipid Synthesis and Endoplasmic Reticulum Function. J. Lipid Res. 2009, 50 Suppl. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Kaufman, R.J. The Impact of the Unfolded Protein Response on Human Disease. J. Cell Biol. 2012, 197, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Grandi, A.; Santi, A.; Campagnoli, S.; Parri, M.; de Camilli, E.; Song, C.; Jin, B.; Lacombe, A.; Castori-Eppenberger, S.; Sarmientos, P.; et al. ERMP1, a Novel Potential Oncogene Involved in UPR and Oxidative Stress Defense, Is Highly Expressed in Human Cancer. Oncotarget 2016, 7, 63596–63610. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.; Emi, M.; Tanabe, K.; Murakami, S. Role of the Unfolded Protein Response in Cell Death. Apoptosis Int. J. Program. Cell Death 2006, 11, 5–13. [Google Scholar] [CrossRef]
- Brown, J.M. Tumor Hypoxia in Cancer Therapy. Methods Enzymol. 2007, 435. [Google Scholar] [CrossRef]
- Jäger, R.; Bertrand, M.J.M.; Gorman, A.M.; Vandenabeele, P.; Samali, A. The Unfolded Protein Response at the Crossroads of Cellular Life and Death during Endoplasmic Reticulum Stress. Biol. Cell Auspices Eur. Cell Biol. Organ. 2012, 104, 259–270. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, N.R.; Rodvold, J.; Sepulveda, H.; Rossi, S.; Drew, A.F.; Zanetti, M. Transmission of Endoplasmic Reticulum Stress and Pro-Inflammation from Tumor Cells to Myeloid Cells. Proc. Natl. Acad. Sci. USA 2011, 108, 6561–6566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuda, M.; Kondoh, N.; Imazeki, N.; Tanaka, K.; Okada, T.; Mori, K.; Hada, A.; Arai, M.; Wakatsuki, T.; Matsubara, O.; et al. Activation of the ATF6, XBP1 and Grp78 Genes in Human Hepatocellular Carcinoma: A Possible Involvement of the ER Stress Pathway in Hepatocarcinogenesis. J. Hepatol. 2003, 38, 605–614. [Google Scholar] [CrossRef]
- Qi, Z.; Chen, L. Endoplasmic Reticulum Stress and Autophagy. Adv. Exp. Med. Biol. 2019, 1206, 167–177. [Google Scholar] [CrossRef]
- Kapoor, A.; Sanyal, A.J. Endoplasmic Reticulum Stress and the Unfolded Protein Response. Clin. Liver Dis. 2009, 13, 581–590. [Google Scholar] [CrossRef]
- Fujita, E.; Kouroku, Y.; Isoai, A.; Kumagai, H.; Misutani, A.; Matsuda, C.; Hayashi, Y.K.; Momoi, T. Two Endoplasmic Reticulum-Associated Degradation (ERAD) Systems for the Novel Variant of the Mutant Dysferlin: Ubiquitin/Proteasome ERAD(I) and Autophagy/Lysosome ERAD(II). Hum. Mol. Genet. 2007, 16, 618–629. [Google Scholar] [CrossRef]
- Kouroku, Y.; Fujita, E.; Tanida, I.; Ueno, T.; Isoai, A.; Kumagai, H.; Ogawa, S.; Kaufman, R.J.; Kominami, E.; Momoi, T. ER Stress (PERK/EIF2alpha Phosphorylation) Mediates the Polyglutamine-Induced LC3 Conversion, an Essential Step for Autophagy Formation. Cell Death Differ. 2007, 14, 230–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.Q.; Chen, Z.; Chen, L.X. Endoplasmic Reticulum Stress: A Novel Mechanism and Therapeutic Target for Cardiovascular Diseases. Acta Pharmacol. Sin. 2016, 37, 425–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallóczy, Z.; Jiang, W.; Virgin IV, H.W.; Leib, D.A.; Scheuner, D.; Kaufman, R.J.; Eskelinen, E.L.; Levine, B. Regulation of Starvation- and Virus-Induced Autophagy by the EIF2alpha Kinase Signaling Pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 190–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, M.; Hino, S.; Saito, A.; Morikawa, K.; Kondo, S.; Kanemoto, S.; Murakami, T.; Taniguchi, M.; Tanii, I.; Yoshinaga, K.; et al. Autophagy Is Activated for Cell Survival after Endoplasmic Reticulum Stress. Mol. Cell. Biol. 2006, 26, 9220–9231. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.C. Autophagy and Stem Cells: Self-Eating for Self-Renewal. Front. Cell Dev. Biol. 2020, 8, 138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponpuak, M.; Mandell, M.A.; Kimura, T.; Chauhan, S.; Cleyrat, C.; Deretic, V. Secretory Autophagy. Curr. Opin. Cell Biol. 2015, 35, 106–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Humeau, J.; Buqué, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with Chemotherapy in the Era of Immune Checkpoint Inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Kuma, A.; Hatano, M.; Matsui, M.; Yamamoto, A.; Nakaya, H.; Yoshimori, T.; Ohsumi, Y.; Tokuhisa, T.; Mizushima, N. The Role of Autophagy during the Early Neonatal Starvation Period. Nature 2004, 432, 1032–1036. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.L.; Russell, R.C.; Guan, K.L. Amino Acid Signalling Upstream of MTOR. Nat. Rev. Mol. Cell Biol. 2013, 14, 133–139. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition) 1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Kim, K.H.; Lee, M.S. Autophagy--a Key Player in Cellular and Body Metabolism. Nat. Rev. Endocrinol. 2014, 10, 322–337. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821.e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-San Pedro, J.M.; Sica, V.; Martins, I.; Pol, J.; Loos, F.; Maiuri, M.C.; Durand, S.; Bossut, N.; Aprahamian, F.; Anagnostopoulos, G.; et al. Acyl-CoA-Binding Protein Is a Lipogenic Factor That Triggers Food Intake and Obesity. Cell Metab. 2019, 30, 754–767. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in Major Human Diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef]
- Menikdiwela, K.R.; Ramalingam, L.; Rasha, F.; Wang, S.; Dufour, J.M.; Kalupahana, N.S.; Sunahara, K.K.S.; Martins, J.O.; Moustaid-Moussa, N. Autophagy in Metabolic Syndrome: Breaking the Wheel by Targeting the Renin-Angiotensin System. Cell Death Dis. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- He, C.; Bassik, M.C.; Moresi, V.; Sun, K.; Wei, Y.; Zou, Z.; An, Z.; Loh, J.; Fisher, J.; Sun, Q.; et al. Exercise-Induced BCL2-Regulated Autophagy Is Required for Muscle Glucose Homeostasis. Nature 2012, 481, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, Á.F.; Bárcena, C.; Martínez-García, G.G.; Tamargo-Gómez, I.; Suárez, M.F.; Pietrocola, F.; Castoldi, F.; Esteban, L.; Sierra-Filardi, E.; Boya, P.; et al. Autophagy Couteracts Weight Gain, Lipotoxicity and Pancreatic β-Cell Death upon Hypercaloric pro-Diabetic Regimens. Cell Death Dis. 2017, 8, e2970. [Google Scholar] [CrossRef]
- Lim, Y.M.; Lim, H.; Hur, K.Y.; Quan, W.; Lee, H.Y.; Cheon, H.; Ryu, D.; Koo, S.H.; Kim, H.L.; Kim, J.; et al. Systemic Autophagy Insufficiency Compromises Adaptation to Metabolic Stress and Facilitates Progression from Obesity to Diabetes. Nat. Commun. 2014, 5, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Settembre, C.; de Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Jürgen Klisch, T.; et al. TFEB Controls Cellular Lipid Metabolism through a Starvation-Induced Autoregulatory Loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romero, M.; Zorzano, A. Role of Autophagy in the Regulation of Adipose Tissue Biology. Cell Cycle 2019, 18, 1435–1445. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Davis, G.S. Review: Occupational and Environmental Lung Disease. Curr. Opin. Pulm. Med. 2002, 8, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, S.; Arias, E.; Kwon, H.; Lopez, N.M.; Athonvarangkul, D.; Sahu, S.; Schwartz, G.J.; Pessin, J.E.; Singh, R. Loss of Autophagy in Hypothalamic POMC Neurons Impairs Lipolysis. EMBO Rep. 2012, 13, 258–265. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Flanagan, C.H.; Smith, L.A.; McDonell, S.B.; Hursting, S.D. When Less May Be More: Calorie Restriction and Response to Cancer Therapy. BMC Med. 2017, 15, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Erustes, A.G.; Costa, A.J.; Nascimento, A.C.; Bincoletto, C.; Ureshino, R.P.; Pereira, G.J.S.; Smaili, S.S. Autophagy and Intermittent Fasting: The Connection for Cancer Therapy? Clinics 2018, 73. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Mattson, M.P. Fasting: Molecular Mechanisms and Clinical Applications. Cell Metab. 2014, 19, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Mariño, G.; Levine, B. Autophagy and the Integrated Stress Response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alirezaei, M.; Kiosses, W.B.; Flynn, C.T.; Brady, N.R.; Fox, H.S. Disruption of Neuronal Autophagy by Infected Microglia Results in Neurodegeneration. PloS ONE 2008, 3, e2906. [Google Scholar] [CrossRef] [PubMed]
- Bagherniya, M.; Butler, A.E.; Barreto, G.E.; Sahebkar, A. The Effect of Fasting or Calorie Restriction on Autophagy Induction: A Review of the Literature. Ageing Res. Rev. 2018, 47, 183–197. [Google Scholar] [CrossRef]
- Loos, B.; Klionsky, D.J.; Wong, E. Augmenting Brain Metabolism to Increase Macro- and Chaperone-Mediated Autophagy for Decreasing Neuronal Proteotoxicity and Aging. Prog. Neurobiol. 2017, 156, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alirezaei, M.; Kemball, C.C.; Flynn, C.T.; Wood, M.R.; Whitton, J.L.; Kiosses, W.B. Short-Term Fasting Induces Profound Neuronal Autophagy. Autophagy 2010, 6, 702–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Waguri, S.; Ueno, T.; Iwata, J.; Murata, S.; Tanida, I.; Ezaki, J.; Mizushima, N.; Ohsumi, Y.; Uchiyama, Y.; et al. Impairment of Starvation-Induced and Constitutive Autophagy in Atg7-Deficient Mice. J. Cell Biol. 2005, 169, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Cuervo, A.M. Liver Autophagy: Much More than Just Taking out the Trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Lanza, I.R.; Zabielski, P.; Klaus, K.A.; Morse, D.M.; Heppelmann, C.J.; Bergen, H.R.; Dasari, S.; Walrand, S.; Short, K.R.; Johnson, M.L.; et al. Chronic Caloric Restriction Preserves Mitochondrial Function in Senescence without Increasing Mitochondrial Biogenesis. Cell Metab. 2012, 16, 777–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, X.; Turdi, S.; Hu, N.; Guo, R.; Zhang, Y.; Ren, J. Influence of Long-Term Caloric Restriction on Myocardial and Cardiomyocyte Contractile Function and Autophagy in Mice. J. Nutr. Biochem. 2012, 23, 1592–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donati, A.; Cavallini, G.; Bergamini, E. Effects of Aging, Antiaging Calorie Restriction and in Vivo Stimulation of Autophagy on the Urinary Excretion of 8OHdG in Male Sprague-Dawley Rats. Age 2013, 35, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Kovács, A.L.; László, L.; Fellinger, E.; Jakab, A.; Orosz, A.; Réz, G.; Kovács, J. Combined Effects of Fasting and Vinblastine Treatment on Serum Insulin Level, the Size of Autophagic-Lysosomal Compartment, Protein Content and Lysosomal Enzyme Activities of Liver and Exocrine Pancreatic Cells of the Mouse. Comp. Biochem. Physiol. B Comp. Biochem. 1989, 94, 505–510. [Google Scholar] [CrossRef]
- Krustew, L.P.; Tashev, T.A.; Popov, A.A.; Apostolov, J.Z.; Borov, B.J.; Stefanova, M.S. Histochemical, Biochemical and Ultrastructural Studies on the Liver of Fasted Rats. Acta Histochem. 1967, 57, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Luévano-Martínez, L.A.; Forni, M.F.; Peloggia, J.; Watanabe, I.S.; Kowaltowski, A.J. Calorie Restriction Promotes Cardiolipin Biosynthesis and Distribution between Mitochondrial Membranes. Mech. Ageing Dev. 2017, 162, 9–17. [Google Scholar] [CrossRef]
- Rickenbacher, A.; Jang, J.H.; Limani, P.; Ungethüm, U.; Lehmann, K.; Oberkofler, C.E.; Weber, A.; Graf, R.; Humar, B.; Clavien, P.A. Fasting Protects Liver from Ischemic Injury through Sirt1-Mediated Downregulation of Circulating HMGB1 in Mice. J. Hepatol. 2014, 61, 301–308. [Google Scholar] [CrossRef]
- Pietrocola, F.; Demont, Y.; Castoldi, F.; Enot, D.; Durand, S.; Semeraro, M.; Baracco, E.E.; Pol, J.; Bravo-San Pedro, J.M.; Bordenave, C.; et al. Metabolic Effects of Fasting on Human and Mouse Blood in Vivo. Autophagy 2017, 13, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.O.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of Antibacterial Autophagy by NADPH Oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How Mitochondria Produce Reactive Oxygen Species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherz-Shouval, R.; Elazar, Z. ROS, Mitochondria and the Regulation of Autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef]
- Oude Ophuis, R.J.A.; Wijers, M.; Bennink, M.B.; van de Loo, F.A.J.; Fransen, J.A.M.; Wieringa, B.; Wansink, D.G. A Tail-Anchored Myotonic Dystrophy Protein Kinase Isoform Induces Perinuclear Clustering of Mitochondria, Autophagy, and Apoptosis. PloS ONE 2009, 4, e8024. [Google Scholar] [CrossRef] [PubMed]
- Campello, S.; Strappazzon, F.; Cecconi, F. Mitochondrial Dismissal in Mammals, from Protein Degradation to Mitophagy. Biochim. Biophys. Acta 2014, 1837, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular Network Formation Protects Mitochondria from Autophagosomal Degradation during Nutrient Starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Gomes, L.C.; Benedetto, G.D.; Scorrano, L. During Autophagy Mitochondria Elongate, Are Spared from Degradation and Sustain Cell Viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zmijewski, J.W.; Banerjee, S.; Bae, H.; Friggeri, A.; Lazarowski, E.R.; Abraham, E. Exposure to Hydrogen Peroxide Induces Oxidation and Activation of AMP-Activated Protein Kinase. J. Biol. Chem. 2010, 285, 33154–33164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desideri, E.; Filomeni, G.; Ciriolo, M.R. Glutathione Participates in the Modulation of Starvation-Induced Autophagy in Carcinoma Cells. Autophagy 2012, 8, 1769–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Wang, X.; Sun, Y.; Berleth, N.; Deitersen, J.; Schlütermann, D.; Stuhldreier, F.; Wallot-Hieke, N.; José Mendiburo, M.; Cox, J.; et al. TNF-Induced Necroptosis Initiates Early Autophagy Events via RIPK3-Dependent AMPK Activation, but Inhibits Late Autophagy. Autophagy 2021, 17, 3992–4009. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular Mechanisms of Cell Death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Annibaldi, A.; Meier, P. Checkpoints in TNF-Induced Cell Death: Implications in Inflammation and Cancer. Trends Mol. Med. 2018, 24, 49–65. [Google Scholar] [CrossRef]
- Lin, Y.; Devin, A.; Rodriguez, Y.; Liu, Z.G. Cleavage of the Death Domain Kinase RIP by Caspase-8 Prompts TNF-Induced Apoptosis. Genes Dev. 1999, 13, 2514–2526. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Yang, Y.; Mei, Y.; Ma, L.; Zhu, D.E.; Hoti, N.; Castanares, M.; Wu, M. Cleavage of RIP3 Inactivates Its Caspase-Independent Apoptosis Pathway by Removal of Kinase Domain. Cell. Signal. 2007, 19, 2056–2067. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Yang, Z.; Xu, Y.; Chen, Y.; Yu, Q. Apoptosis, Autophagy, Necroptosis, and Cancer Metastasis. Mol. Cancer 2015, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an Energy Metabolism Regulator That Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Fulda, S. The Mechanism of Necroptosis in Normal and Cancer Cells. Cancer Biol. Ther. 2013, 14, 999–1004. [Google Scholar] [CrossRef] [Green Version]
- Liu, P.; Xu, B.; Shen, W.; Zhu, H.; Wu, W.; Fu, Y.; Chen, H.; Dong, H.; Zhu, Y.; Miao, K.; et al. Dysregulation of TNFα-Induced Necroptotic Signaling in Chronic Lymphocytic Leukemia: Suppression of CYLD Gene by LEF1. Leukemia 2012, 26, 1293–1300. [Google Scholar] [CrossRef] [Green Version]
- Cerhan, J.R.; Ansell, S.M.; Fredericksen, Z.S.; Kay, N.E.; Liebow, M.; Call, T.G.; Dogan, A.; Cunningham, J.M.; Wang, A.H.; Liu-Mares, W.; et al. Genetic Variation in 1253 Immune and Inflammation Genes and Risk of Non-Hodgkin Lymphoma. Blood 2007, 110, 4455–4463. [Google Scholar] [CrossRef] [Green Version]
- Fu, Z.; Deng, B.; Liao, Y.; Shan, L.; Yin, F.; Wang, Z.; Zeng, H.; Zuo, D.; Hua, Y.; Cai, Z. The Anti-Tumor Effect of Shikonin on Osteosarcoma by Inducing RIP1 and RIP3 Dependent Necroptosis. BMC Cancer 2013, 13. [Google Scholar] [CrossRef] [Green Version]
- Buchheit, C.L.; Rayavarapu, R.R.; Schafer, Z.T. The Regulation of Cancer Cell Death and Metabolism by Extracellular Matrix Attachment. Semin. Cell Dev. Biol. 2012, 23, 402–411. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K.M. Phosphorylation-Driven Assembly of the RIP1-RIP3 Complex Regulates Programmed Necrosis and Virus-Induced Inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [Green Version]
- Jouan-Lanhouet, S.; Arshad, M.I.; Piquet-Pellorce, C.; Martin-Chouly, C.; le Moigne-Muller, G.; van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Vandenabeele, P.; et al. TRAIL Induces Necroptosis Involving RIPK1/RIPK3-Dependent PARP-1 Activation. Cell Death Differ. 2012, 19, 2003–2014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodall, M.L.; Fitzwalter, B.E.; Zahedi, S.; Wu, M.; Rodriguez, D.; Mulcahy-Levy, J.M.; Green, D.R.; Morgan, M.; Cramer, S.D.; Thorburn, A. The Autophagy Machinery Controls Cell Death Switching between Apoptosis and Necroptosis. Dev. Cell 2016, 37, 337–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.; Koh, J.Y.; Price, S.; White, E.; Mehnert, J.M. Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov. 2015, 5, 410–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of Autophagy and Inhibition of Tumorigenesis by Beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef] [PubMed]
- Aita, V.M.; Liang, X.H.; Murty, V.V.V.S.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.C.; Levine, B. Cloning and Genomic Organization of Beclin 1, a Candidate Tumor Suppressor Gene on Chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef] [PubMed]
- White, E. Deconvoluting the Context-Dependent Role for Autophagy in Cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamura, A.; Komatsu, M.; Hara, T.; Sakamoto, A.; Kishi, C.; Waguri, S.; Eishi, Y.; Hino, O.; Tanaka, K.; Mizushima, N. Autophagy-Deficient Mice Develop Multiple Liver Tumors. Genes Dev. 2011, 25, 795–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; Mackay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. P53 Status Determines the Role of Autophagy in Pancreatic Tumour Development. Nature 2013, 504, 296–300. [Google Scholar] [CrossRef]
- Yang, A.; Kimmelman, A.C. Inhibition of Autophagy Attenuates Pancreatic Cancer Growth Independent of TP53/TRP53 Status. Autophagy 2014, 10, 1683–1684. [Google Scholar] [CrossRef] [Green Version]
- Folkerts, H.; Hilgendorf, S.; Vellenga, E.; Bremer, E.; Wiersma, V.R. The Multifaceted Role of Autophagy in Cancer and the Microenvironment. Med. Res. Rev. 2019, 39, 517–560. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an Autophagy Gene Essential for Early Embryonic Development, Is a Haploinsufficient Tumor Suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eccles, D.M.; Russell, S.E.; Haites, N.E.; Atkinson, R.; Bell, D.W.; Gruber, L.; Hickey, I.; Kelly, K.; Kitchener, H.; Leonard, R.; et al. Early Loss of Heterozygosity on 17q in Ovarian Cancer. The Abe Ovarian Cancer Genetics Group. Oncogene 1992, 7, 2069–2072. [Google Scholar] [PubMed]
- Egan, D.F.; Chun, M.G.H.; Vamos, M.; Zou, H.; Rong, J.; Miller, C.J.; Lou, H.J.; Raveendra-Panickar, D.; Yang, C.C.; Sheffler, D.J.; et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol. Cell 2015, 59, 285–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maria Fimia, G.; Stoykova, A.; Romagnoli, A.; Giunta, L.; di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 Regulates Autophagy and Development of the Nervous System. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukhopadhyay, S.; vander Heiden, M.G.; McCormick, F. The Metabolic Landscape of RAS-Driven Cancers from Biology to Therapy. Nat. Cancer 2021, 2, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting Autophagy in Cancer: Recent Advances and Future Directions. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.S.; Karantza, V.; et al. Activated Ras Requires Autophagy to Maintain Oxidative Metabolism and Tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [Green Version]
- Bryant, K.L.; Stalnecker, C.A.; Zeitouni, D.; Klomp, J.E.; Peng, S.; Tikunov, A.P.; Gunda, V.; Pierobon, M.; Waters, A.M.; George, S.D.; et al. Combination of ERK and Autophagy Inhibition as a Treatment Approach for Pancreatic Cancer. Nat. Med. 2019, 25, 628–640. [Google Scholar] [CrossRef]
- Guo, J.Y.; White, E. Autophagy Is Required for Mitochondrial Function, Lipid Metabolism, Growth, and Fate of KRAS(G12D)-Driven Lung Tumors. Autophagy 2013, 9, 1636–1638. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, C.G.; Camolotto, S.A.; Boespflug, A.M.; Guillen, K.P.; Foth, M.; Truong, A.; Schuman, S.S.; Shea, J.E.; Seipp, M.T.; Yap, J.T.; et al. Protective Autophagy Elicited by RAF→MEK→ERK Inhibition Suggests a Treatment Strategy for RAS-Driven Cancers. Nat. Med. 2019, 25, 620–627. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Thompson, J.C.; Griesinger, A.M.; Amani, V.; Donson, A.M.; Birks, D.K.; Morgan, M.J.; Mirsky, D.M.; Handler, M.H.; Foreman, N.K.; et al. Autophagy Inhibition Improves Chemosensitivity in BRAF(V600E) Brain Tumors. Cancer Discov. 2014, 4, 773–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, J.M.M.; Zahedi, S.; Griesinger, A.M.; Morin, A.; Davies, K.D.; Aisner, D.L.; Kleinschmidt-DeMasters, B.K.; Fitzwalter, B.E.; Goodall, M.L.; Thorburn, J.; et al. Autophagy Inhibition Overcomes Multiple Mechanisms of Resistance to BRAF Inhibition in Brain Tumors. eLife 2017, 6. [Google Scholar] [CrossRef]
- Levy, J.M.M.; Towers, C.G.; Thorburn, A. Targeting Autophagy in Cancer. Nat. Rev. Cancer 2017, 17, 528–542. [Google Scholar] [CrossRef] [PubMed]
- Levy, J.M.M.; Thorburn, A. Targeting Autophagy during Cancer Therapy to Improve Clinical Outcomes. Pharmacol. Ther. 2011, 131, 130–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorburn, A.; Thamm, D.H.; Gustafson, D.L. Autophagy and Cancer Therapy. Mol. Pharmacol. 2014, 85, 830–838. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.; Kimmelman, A.C.; White, E. Recent Insights into the Function of Autophagy in Cancer. Genes Dev. 2016, 30, 1913–1930. [Google Scholar] [CrossRef]
- Yang, Y.P.; Hu, L.F.; Zheng, H.F.; Mao, C.J.; Hu, W.D.; Xiong, K.P.; Wang, F.; Liu, C.F. Application and Interpretation of Current Autophagy Inhibitors and Activators. Acta Pharmacol. Sin. 2013, 34, 625–635. [Google Scholar] [CrossRef] [Green Version]
- Maycotte, P.; Aryal, S.; Cummings, C.T.; Thorburn, J.; Morgan, M.J.; Thorburn, A. Chloroquine Sensitizes Breast Cancer Cells to Chemotherapy Independent of Autophagy. Autophagy 2012, 8, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.H.; Wang, Z.; Tkach, D.; Toral-Barza, L.; Ugwonali, S.; Liu, S.; Fitzgerald, S.L.; George, E.; Frias, E.; Cochran, N.; et al. Macroautophagy Is Dispensable for Growth of KRAS Mutant Tumors and Chloroquine Efficacy. Proc. Natl. Acad. Sci. USA 2016, 113, 182–187. [Google Scholar] [CrossRef] [Green Version]
- Maes, H.; Kuchnio, A.; Peric, A.; Moens, S.; Nys, K.; DeBock, K.; Quaegebeur, A.; Schoors, S.; Georgiadou, M.; Wouters, J.; et al. Tumor Vessel Normalization by Chloroquine Independent of Autophagy. Cancer Cell 2014, 26, 190–206. [Google Scholar] [CrossRef] [Green Version]
- Briceño, E.; Reyes, S.; Sotelo, J. Therapy of Glioblastoma Multiforme Improved by the Antimutagenic Chloroquine. Neurosurg. Focus 2003, 14. [Google Scholar] [CrossRef] [PubMed]
- Eldredge, H.B.; DeNittis, A.; DuHadaway, J.B.; Chernick, M.; Metz, R.; Prendergast, G.C. Concurrent whole brain radiotherapy and short-course chloroquine in patients with brain metastases: A pilot triaL. J. Radiat. Oncol. 2013, 2, 315–321. [Google Scholar] [CrossRef]
- Rojas-Puentes, L.L.; Gonzalez-Pinedo, M.; Crismatt, A.; Ortega-Gomez, A.; Gamboa-Vignolle, C.; Nuñez-Gomez, R.; Dorantes-Gallareta, Y.; Arce-Salinas, C.; Arrieta, O. Phase II Randomized, Double-Blind, Placebo-Controlled Study of Whole-Brain Irradiation with Concomitant Chloroquine for Brain Metastases. Radiat. Oncol. 2013, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masui, A.; Hamada, M.; Kameyama, H.; Wakabayashi, K.; Takasu, A.; Imai, T.; Iwai, S.; Yura, Y. Autophagy as a Survival Mechanism for Squamous Cell Carcinoma Cells in Endonuclease G-Mediated Apoptosis. PloS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, J.K.; Szilard, A.; Goussetis, D.J.; Sassano, A.; Colamonici, M.; Gounaris, E.; Frankfurt, O.; Giles, F.J.; Eklund, E.A.; Beauchamp, E.M.; et al. Autophagy Is a Survival Mechanism of Acute Myelogenous Leukemia Precursors during Dual MTORC2/MTORC1 Targeting. Clin. Cancer Res. 2014, 20, 2400–2409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzwalter, B.E.; Thorburn, A. Recent Insights into Cell Death and Autophagy. FEBS J. 2015, 282, 4279–4288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, K.; Guo, H.; Tang, H.; Yan, Y.; Zhang, J.; Zhang, L.; Han, K.; Zhang, X. Gastrin Enhances Autophagy and Promotes Gastric Carcinoma Proliferation via Inducing AMPKα. Oncol. Res. 2017, 25, 1399–1407. [Google Scholar] [CrossRef]
- Gump, J.M.; Staskiewicz, L.; Morgan, M.J.; Bamberg, A.; Riches, D.W.H.; Thorburn, A. Autophagy Variation within a Cell Population Determines Cell Fate through Selective Degradation of Fap-1. Nat. Cell Biol. 2014, 16, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Su, H.; Yang, F.; Fu, R.; Li, X.; French, R.; Mose, E.; Pu, X.; Trinh, B.; Kumar, A.; Liu, J.; et al. Cancer Cells Escape Autophagy Inhibition via NRF2-Induced Macropinocytosis. Cancer Cell 2021, 39, 678–693.e11. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Goswami, D.; Adiseshaiah, P.P.; Burgan, W.; Yi, M.; Guerin, T.M.; Kozlov, S.V.; Nissley, D.V.; McCormick, F. Undermining Glutaminolysis Bolsters Chemotherapy While NRF2 Promotes Chemoresistance in KRAS-Driven Pancreatic Cancers. Cancer Res. 2020, 80, 1630–1643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-Induced Nrf2 Transcription Promotes ROS Detoxification and Tumorigenesis. Nature 2011, 475, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Redding, A.; Aplin, A.E.; Grabocka, E. RAS-Mediated Tumor Stress Adaptation and the Targeting Opportunities It Presents. Dis. Models Mech. 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Young, T.W.; Mei, F.C.; Yang, G.; Thompson-Lanza, J.A.; Liu, J.; Cheng, X. Activation of Antioxidant Pathways in Ras-Mediated Oncogenic Transformation of Human Surface Ovarian Epithelial Cells Revealed by Functional Proteomics and Mass Spectrometry. Cancer Res. 2004, 64, 4577–4584. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Wu, Y.; Zhao, G.L.; Ye, Z.Y.; Xing, C.G.; Yang, X.D. Inhibition of Autophagy by 3-MA Promotes Hypoxia-Induced Apoptosis in Human Colorectal Cancer Cells. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 1047–1054. [Google Scholar] [CrossRef]
- Liu, L.; Li, L.; Li, M.; Luo, Z. Autophagy-Dependent Ferroptosis as a Therapeutic Target in Cancer. ChemMedChem 2021, 16, 2942–2950. [Google Scholar] [CrossRef] [PubMed]
- Piffoux, M.; Eriau, E.; Cassier, P.A. Autophagy as a Therapeutic Target in Pancreatic Cancer. Br. J. Cancer 2021, 124, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Wolpin, B.M.; Hezel, A.F.; Abrams, T.; Blaszkowsky, L.S.; Meyerhardt, J.A.; Chan, J.A.; Enzinger, P.C.; Allen, B.; Clark, J.W.; Ryan, D.P.; et al. Oral MTOR Inhibitor Everolimus in Patients with Gemcitabine-Refractory Metastatic Pancreatic Cancer. J. Clin. Oncol. 2009, 27, 193–198. [Google Scholar] [CrossRef]
- Samaras, P.; Tusup, M.; Nguyen-Kim, T.D.L.; Seifert, B.; Bachmann, H.; von Moos, R.; Knuth, A.; Pascolo, S. Phase I Study of a Chloroquine-Gemcitabine Combination in Patients with Metastatic or Unresectable Pancreatic Cancer. Cancer Chemother. Pharmacol. 2017, 80, 1005–1012. [Google Scholar] [CrossRef] [PubMed]
- Boone, B.A.; Bahary, N.; Zureikat, A.H.; Moser, A.J.; Normolle, D.P.; Wu, W.C.; Singhi, A.D.; Bao, P.; Bartlett, D.L.; Liotta, L.A.; et al. Safety and Biologic Response of Pre-Operative Autophagy Inhibition in Combination with Gemcitabine in Patients with Pancreatic Adenocarcinoma. Ann. Surg. Oncol. 2015, 22, 4402–4410. [Google Scholar] [CrossRef] [PubMed]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; de Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and Nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef]
- Javle, M.M.; Shroff, R.T.; Xiong, H.; Varadhachary, G.A.; Fogelman, D.; Reddy, S.A.; Davis, D.; Zhang, Y.; Wolff, R.A.; Abbruzzese, J.L. Inhibition of the Mammalian Target of Rapamycin (MTOR) in Advanced Pancreatic Cancer: Results of Two Phase II Studies. BMC Cancer 2010, 10, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, A.J.; Malinowska-Kolodziej, I.; Morgan, J.A.; Qin, W.; Fletcher, C.D.M.; Vena, N.; Ligon, A.H.; Antonescu, C.R.; Ramaiya, N.H.; Demetri, G.D.; et al. Clinical Activity of MTOR Inhibition with Sirolimus in Malignant Perivascular Epithelioid Cell Tumors: Targeting the Pathogenic Activation of MTORC1 in Tumors. J. Clin. Oncol. 2010, 28, 835–840. [Google Scholar] [CrossRef]
- McCormack, F.X.; Inoue, Y.; Moss, J.; Singer, L.G.; Strange, C.; Nakata, K.; Barker, A.F.; Chapman, J.T.; Brantly, M.L.; Stocks, J.M.; et al. Efficacy and Safety of Sirolimus in Lymphangioleiomyomatosis. N. Engl. J. Med. 2011, 364, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Iyer, G.; Hanrahan, A.J.; Milowsky, M.I.; Al-Ahmadie, H.; Scott, S.N.; Janakiraman, M.; Pirun, M.; Sander, C.; Socci, N.D.; Ostrovnaya, I.; et al. Genome Sequencing Identifies a Basis for Everolimus Sensitivity. Science 2012, 338, 221. [Google Scholar] [CrossRef] [Green Version]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A Highly Potent and Selective Vps34 Inhibitor Alters Vesicle Trafficking and Autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.H.; Choi, K.Y.; Min, D.S. Phospholipase D-Mediated Autophagic Regulation Is a Potential Target for Cancer Therapy. Cell Death Differ. 2014, 21, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Karsli-Uzunbas, G.; Guo, J.Y.; Price, S.; Teng, X.; Laddha, S.V.; Khor, S.; Kalaany, N.Y.; Jacks, T.; Chan, C.S.; Rabinowitz, J.D.; et al. Autophagy Is Required for Glucose Homeostasis and Lung Tumor Maintenance. Cancer Discov. 2014, 4, 915–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakai, A.; Yamaguchi, O.; Takeda, T.; Higuchi, Y.; Hikoso, S.; Taniike, M.; Omiya, S.; Mizote, I.; Matsumura, Y.; Asahi, M.; et al. The Role of Autophagy in Cardiomyocytes in the Basal State and in Response to Hemodynamic Stress. Nat. Med. 2007, 13, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Cell Biology. Metabolic Control of Cell Death. Science 2014, 345, 1250256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Sumpter, R.; Levine, B. Exercise Induces Autophagy in Peripheral Tissues and in the Brain. Autophagy 2012, 8, 1548–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnard, R.A.; Wittenburg, L.A.; Amaravadi, R.K.; Gustafson, D.L.; Thorburn, A.; Thamm, D.H. Phase I Clinical Trial and Pharmacodynamic Evaluation of Combination Hydroxychloroquine and Doxorubicin Treatment in Pet Dogs Treated for Spontaneously Occurring Lymphoma. Autophagy 2014, 10, 1415–1425. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, D.; Mita, M.; Sarantopoulos, J.; Wood, L.; Amaravadi, R.K.; Davis, L.E.; Mita, A.C.; Curiel, T.J.; Espitia, C.M.; Nawrocki, S.T.; et al. Combined Autophagy and HDAC Inhibition: A Phase I Safety, Tolerability, Pharmacokinetic, and Pharmacodynamic Analysis of Hydroxychloroquine in Combination with the HDAC Inhibitor Vorinostat in Patients with Advanced Solid Tumors. Autophagy 2014, 10, 1403–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAfee, Q.; Zhang, Z.; Samanta, A.; Levi, S.M.; Ma, X.H.; Piao, S.; Lynch, J.P.; Uehara, T.; Sepulveda, A.R.; Davis, L.E.; et al. Autophagy Inhibitor Lys05 Has Single-Agent Antitumor Activity and Reproduces the Phenotype of a Genetic Autophagy Deficiency. Proc. Natl. Acad. Sci. USA 2012, 109, 8253–8258. [Google Scholar] [CrossRef] [Green Version]
- Füllgrabe, J.; Heldring, N.; Hermanson, O.; Joseph, B. Cracking the Survival Code: Autophagy-Related Histone Modifications. Autophagy 2014, 10, 556–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yu, Y.; Jiang, Z.; Cao, W.M.; Wang, Z.; Dou, J.; Zhao, Y.; Cui, Y.; Zhang, H. Next-Generation Proteasome Inhibitor MLN9708 Sensitizes Breast Cancer Cells to Doxorubicin-Induced Apoptosis. Sci. Rep. 2016, 6, 1–13. [Google Scholar] [CrossRef] [PubMed]
- US National Library of Medicine. Available online: Https://Clinicaltrials.Gov/Ct2/Show/NCT01881451?Term=NCT01881451&rank=1 (accessed on 14 February 2022).
- US National Library of Medicine. Available online: Https://Clinicaltrials.Gov/Ct2/Show/NCT02233387?Term=NCT02233387&rank=1 (accessed on 14 February 2022).
- US National Library of Medicine. Available online: Https://Clinicaltrials.Gov/Ct2/Show/NCT01594242?Term=NCT01594242&rank=1 (accessed on 14 February 2022).
- US National Library of Medicine. Available online: Https://Clinicaltrials.Gov/Ct2/Show/NCT01206530?Term=NCT01206530&rank=1 (accessed on 14 February 2022).
- US National Library of Medicine. Available online: Https://Clinicaltrials.Gov/Ct2/Show/NCT02042989?Term=NCT02042989&rank=1 (accessed on 14 February 2022).
- US National Library of Medicine. Available online: Https://Clinicaltrials.Gov/Ct2/Show/NCT01874548?Term=NCT01874548&rank=1 (accessed on 14 February 2022).
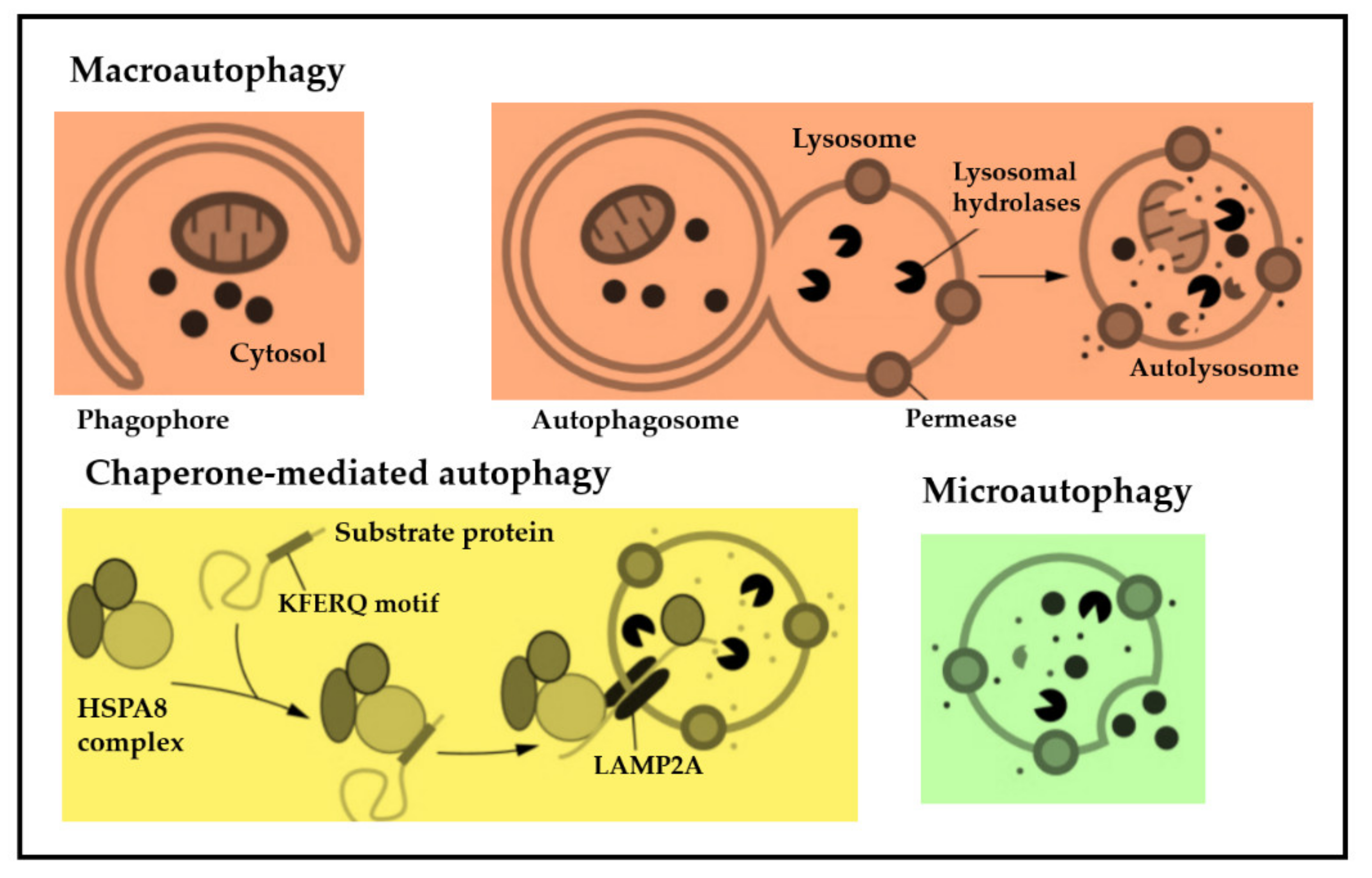

{kind=link}
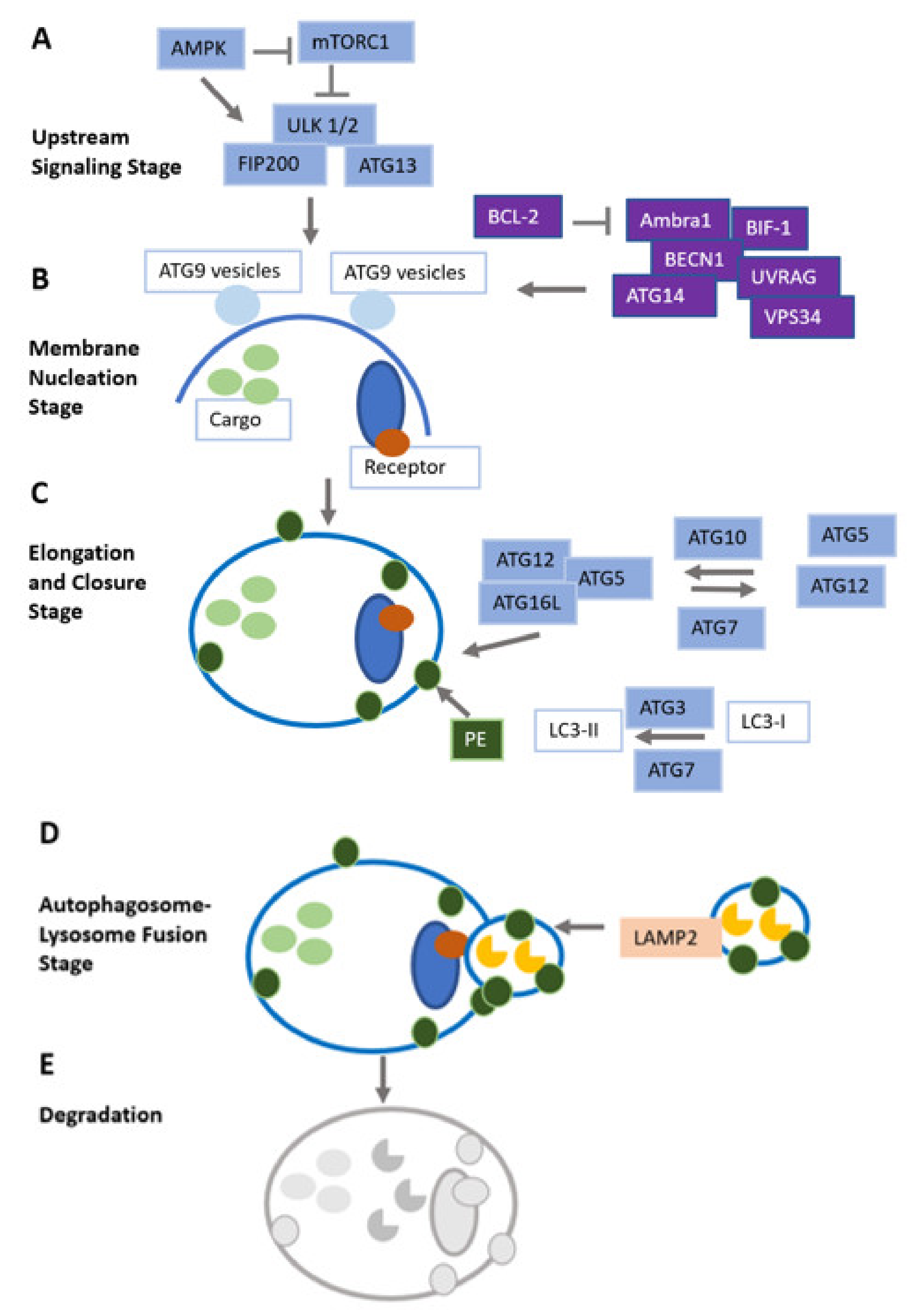
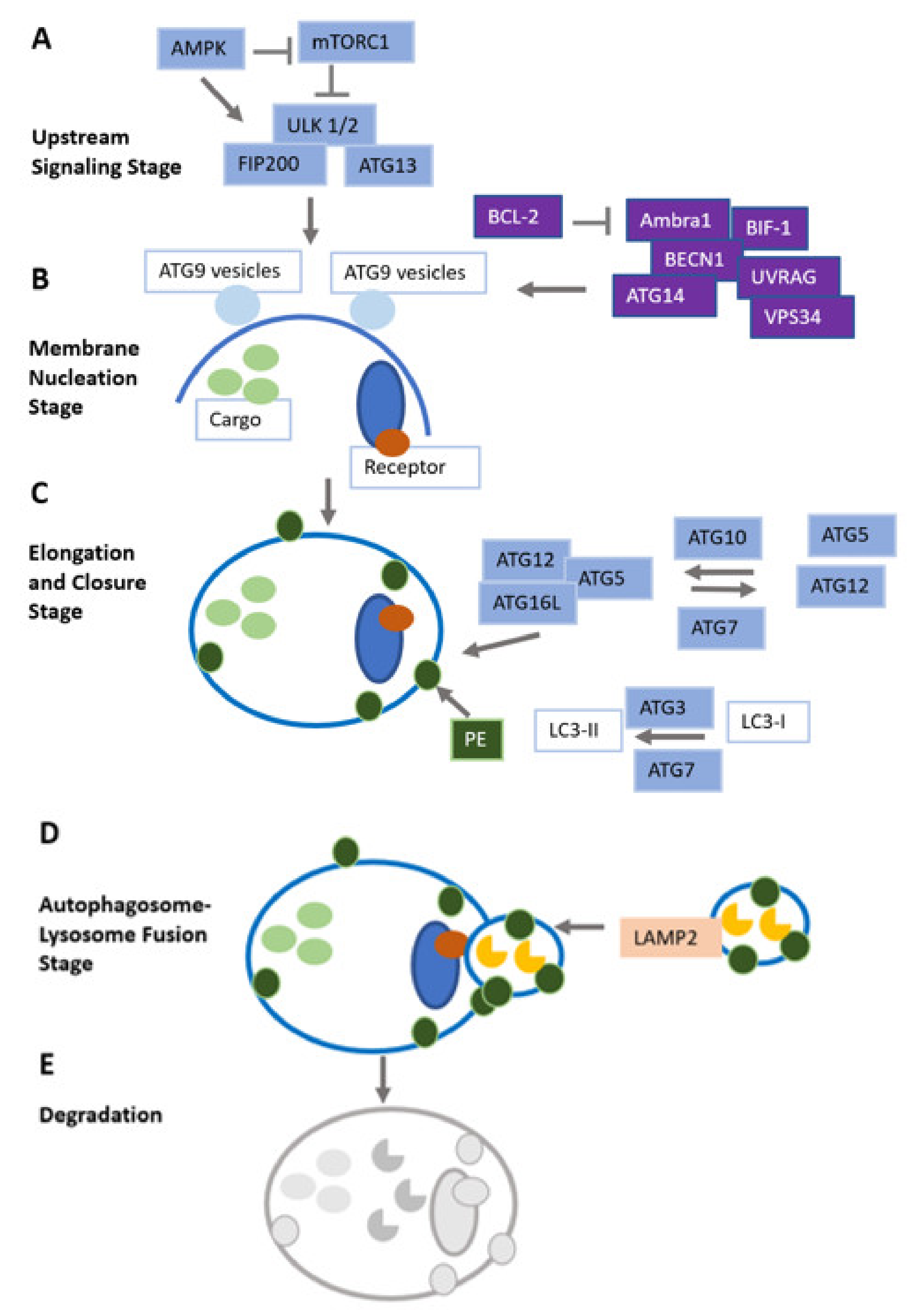{kind=link}
| Biomarkers of Autophagy | Assessment Mode | Tumor Entity | ID of Clinical Trial |
|---|---|---|---|
| Hypoxia | 18F-EF5 PET LC3-II staining of tumor tissue Expression analysis of autophagy genes | Clear cell ovarian cancer | NCT01881451 [176] |
| Hypoxia | 18F-HX4 PET Expression analysis of autophagy genes | Cervical cancer | NCT02233387 [177] |
| Autophagosome count | Number of autophagy vesicles in peripheral blood mononuclear cells | Multiple myeloma | NCT01594242 [178] |
| Aberrations in cell metabolism | 18FDG PET Number of autophagy vesicles in peripheral blood mononuclear cells | Colorectal cancer | NCT01206530 [179] |
| Aberrations in cell metabolism | Analysis of metabolism-related serum parameters | Advanced-stage p53 mutated cancers | NCT02042989 [180] |
| Aberrations in cell metabolism | MRI study including MR spectroscopy and diffusion weighted imaging | Cervical cancer | NCT01874548 [181] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taucher, E.; Mykoliuk, I.; Fediuk, M.; Smolle-Juettner, F.-M. Autophagy, Oxidative Stress and Cancer Development. Cancers 2022, 14, 1637. https://doi.org/10.3390/cancers14071637
Taucher E, Mykoliuk I, Fediuk M, Smolle-Juettner F-M. Autophagy, Oxidative Stress and Cancer Development. Cancers. 2022; 14(7):1637. https://doi.org/10.3390/cancers14071637
Chicago/Turabian StyleTaucher, Elisabeth, Iurii Mykoliuk, Melanie Fediuk, and Freyja-Maria Smolle-Juettner. 2022. "Autophagy, Oxidative Stress and Cancer Development" Cancers 14, no. 7: 1637. https://doi.org/10.3390/cancers14071637
APA StyleTaucher, E., Mykoliuk, I., Fediuk, M., & Smolle-Juettner, F.-M. (2022). Autophagy, Oxidative Stress and Cancer Development. Cancers, 14(7), 1637. https://doi.org/10.3390/cancers14071637