
Advances in Immunotherapy for the Treatment of Adult Glioblastoma: Overcoming Chemical and Physical Barriers

 , , , ,
, , , , 

Simple Summary
Abstract
1. Introduction
2. Current Immunotherapy Options and Developments
2.1. Immune Checkpoint Inhibitors
2.2. T-Cell Transfer Therapies
2.3. Vaccination
2.4. Oncolytic Virus Therapy
3. Challenges to Immunotherapy
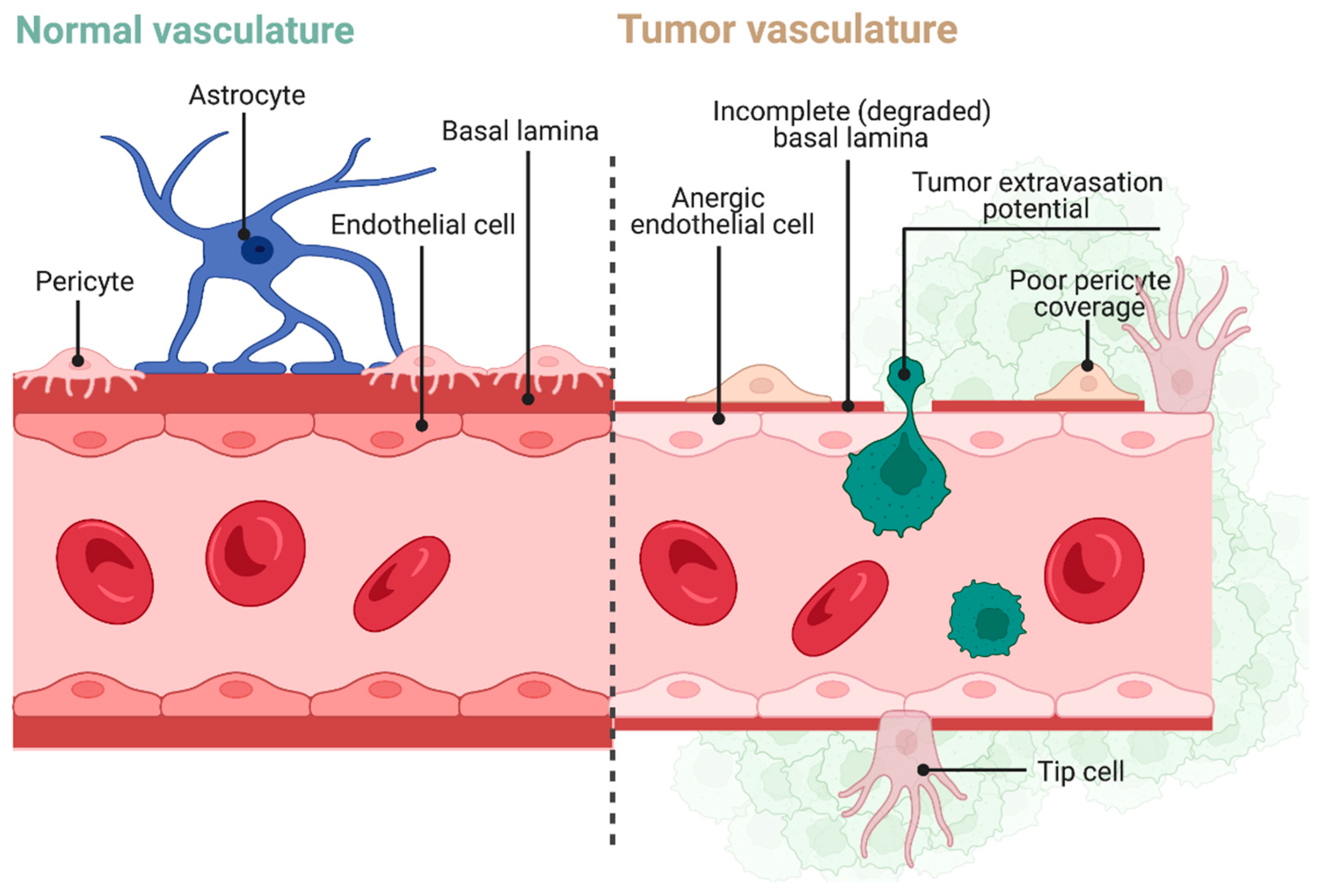
3.1. Blood–Brain/Brain–Tumor Barriers
3.2. The Immune-Suppressive Microenvironment
3.3. Low Tumor Mutational Burden
4. Strategies to Enhance Immunotherapy’s Effectiveness
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Melenhorst, J.J.; Chen, G.M.; Wang, M.; Porter, D.L.; Chen, C.; Collins, M.A.; Gao, P.; Bandyopadhyay, S.; Sun, H.; Zhao, Z.; et al. Decade-Long Leukaemia Remissions with Persistence of CD4+ CAR T Cells. Nature 2022, 602, 503–509. [Google Scholar] [CrossRef]
- Hynynen, K.; McDannold, N.; Vykhodtseva, N.; Jolesz, F.A. Noninvasive MR Imaging-Guided Focal Opening of the Blood-Brain Barrier in Rabbits. Radiology 2001, 220, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Mainprize, T.; Lipsman, N.; Huang, Y.; Meng, Y.; Bethune, A.; Ironside, S.; Heyn, C.; Alkins, R.; Trudeau, M.; Sahgal, A.; et al. Blood-Brain Barrier Opening in Primary Brain Tumors with Non-Invasive MR-Guided Focused Ultrasound: A Clinical Safety and Feasibility Study. Sci. Rep. 2019, 9, 321. [Google Scholar] [CrossRef] [PubMed]
- Abrahao, A.; Meng, Y.; Llinas, M.; Huang, Y.; Hamani, C.; Mainprize, T.; Aubert, I.; Heyn, C.; Black, S.E.; Hynynen, K.; et al. First-in-Human Trial of Blood–Brain Barrier Opening in Amyotrophic Lateral Sclerosis Using MR-Guided Focused Ultrasound. Nat. Commun. 2019, 10, 4373. [Google Scholar] [CrossRef]
- Chen, K.-T.; Chai, W.-Y.; Lin, Y.-J.; Lin, C.-J.; Chen, P.-Y.; Tsai, H.-C.; Huang, C.-Y.; Kuo, J.S.; Liu, H.-L.; Wei, K.-C. Neuronavigation-Guided Focused Ultrasound for Transcranial Blood-Brain Barrier Opening and Immunostimulation in Brain Tumors. Sci. Adv. 2021, 7, eabd0772. [Google Scholar] [CrossRef]
- Delves, P.J.; Roitt, I.M. The Immune System. N. Engl. J. Med. 2000, 343, 37–49. [Google Scholar] [CrossRef]
- Khattri, R.; Cox, T.; Yasayko, S.-A.; Ramsdell, F. An Essential Role for Scurfin in CD4+CD25+ T Regulatory Cells. Nat. Immunol. 2003, 4, 337–342. [Google Scholar] [CrossRef]
- He, X.; Xu, C. Immune Checkpoint Signaling and Cancer Immunotherapy. Cell Res. 2020, 30, 660–669. [Google Scholar] [CrossRef]
- Brahm, C.G.; van Linde, M.E.; Enting, R.H.; Schuur, M.; Otten, R.H.J.; Heymans, M.W.; Verheul, H.M.W.; Walenkamp, A.M.E. The Current Status of Immune Checkpoint Inhibitors in Neuro-Oncology: A Systematic Review. Cancers 2020, 12, 586. [Google Scholar] [CrossRef]
- Majd, N.; Dasgupta, P.; de Groot, J. Immunotherapy for Neuro-Oncology. Adv. Exp. Med. Biol. 2020, 1244, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Maggs, L.; Cattaneo, G.; Dal, A.E.; Moghaddam, A.S.; Ferrone, S. CAR T Cell-Based Immunotherapy for the Treatment of Glioblastoma. Front. Neurosci. 2021, 15, 662064. [Google Scholar] [CrossRef] [PubMed]
- Keskin, D.B.; Anandappa, A.J.; Sun, J.; Tirosh, I.; Mathewson, N.D.; Li, S.; Oliveira, G.; Giobbie-Hurder, A.; Felt, K.; Gjini, E.; et al. Neoantigen Vaccine Generates Intratumoral T Cell Responses in Phase Ib Glioblastoma Trial. Nature 2019, 565, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Chen, M.; Nie, H.; Yuan, Y. PD-1 and PD-L1 in Cancer Immunotherapy: Clinical Implications and Future Considerations. Hum. Vaccines Immunother. 2019, 15, 1111–1122. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 Pathway: Current Researches in Cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Khasraw, M.; Reardon, D.A.; Weller, M.; Sampson, J.H. PD-1 Inhibitors: Do They Have a Future in the Treatment of Glioblastoma? Clin. Cancer Res. 2020, 26, 5287–5296. [Google Scholar] [CrossRef]
- Liu, F.; Huang, J.; Liu, X.; Cheng, Q.; Luo, C.; Liu, Z. CTLA-4 Correlates with Immune and Clinical Characteristics of Glioma. Cancer Cell Int. 2020, 20, 7. [Google Scholar] [CrossRef]
- Reardon, D.A.; Omuro, A.; Brandes, A.A.; Rieger, J.; Wick, A.; Sepulveda, J.; Phuphanich, S.; de Souza, P.; Ahluwalia, M.S.; Lim, M.; et al. OS10.3 Randomized Phase 3 Study Evaluating the Efficacy and Safety of Nivolumab vs Bevacizumab in Patients with Recurrent Glioblastoma: CheckMate 143. Neuro-Oncology 2017, 19, iii21. [Google Scholar] [CrossRef]
- Sampson, J.H.; Omuro, A.M.P.; Preusser, M.; Lim, M.; Butowski, N.A.; Cloughesy, T.F.; Strauss, L.C.; Latek, R.R.; Paliwal, P.; Weller, M.; et al. A Randomized, Phase 3, Open-Label Study of Nivolumab versus Temozolomide (TMZ) in Combination with Radiotherapy (RT) in Adult Patients (Pts) with Newly Diagnosed, O-6-Methylguanine DNA Methyltransferase (MGMT)-Unmethylated Glioblastoma (GBM): CheckMate-498. J. Clin. Oncol. 2016, 34, TPS2079. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant Anti-PD-1 Immunotherapy Promotes a Survival Benefit with Intratumoral and Systemic Immune Responses in Recurrent Glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Neyns, B.; Ben Salama, L.; Awada, G.; De Cremer, J.; Schwarze, J.K.; Seynaeve, L.; Du Four, S.; Fischbuch, L.; Vanbinst, A.-M.; Everaert, H.; et al. GLIAVAX: A Stratified Phase II Clinical Trial of Avelumab and Axitinib in Patients with Recurrent Glioblastoma. J. Clin. Oncol. 2019, 37, 2034. [Google Scholar] [CrossRef]
- Reardon, D.A.; Kaley, T.J.; Dietrich, J.; Clarke, J.L.; Dunn, G.; Lim, M.; Cloughesy, T.F.; Gan, H.K.; Park, A.J.; Schwarzenberger, P.; et al. Phase II Study to Evaluate Safety and Efficacy of MEDI4736 (Durvalumab) + Radiotherapy in Patients with Newly Diagnosed Unmethylated MGMT Glioblastoma (New Unmeth GBM). J. Clin. Oncol. 2019, 37, 2032. [Google Scholar] [CrossRef]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.-C.; Lu, L.; Zheng, Z.; et al. Pilot Trial of Adoptive Transfer of Chimeric Antigen Receptor-Transduced T Cells Targeting EGFRvIII in Patients with Glioblastoma. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Platten, M.; Bunse, L.; Wick, A.; Bunse, T.; Le Cornet, L.; Harting, I.; Sahm, F.; Sanghvi, K.; Tan, C.L.; Poschke, I.; et al. A Vaccine Targeting Mutant IDH1 in Newly Diagnosed Glioma. Nature 2021, 592, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Fenstermaker, R.A.; Ciesielski, M.J.; Qiu, J.; Yang, N.; Frank, C.L.; Lee, K.P.; Mechtler, L.R.; Belal, A.; Ahluwalia, M.S.; Hutson, A.D. Clinical Study of a Survivin Long Peptide Vaccine (SurVaxM) in Patients with Recurrent Malignant Glioma. Cancer Immunol. Immunother. 2016, 65, 1339–1352. [Google Scholar] [CrossRef]
- MimiVax, LLC. Prospective Randomized Placebo-Controlled Trial of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma (SURVIVE). 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT05163080 (accessed on 28 February 2022).
- Markert, J.M.; Razdan, S.N.; Kuo, H.-C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A Phase 1 Trial of Oncolytic HSV-1, G207, given in Combination with Radiation for Recurrent GBM Demonstrates Safety and Radiographic Responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef]
- Friedman, G.K.; Johnston, J.M.; Bag, A.K.; Bernstock, J.D.; Li, R.; Aban, I.; Kachurak, K.; Nan, L.; Kang, K.-D.; Totsch, S.; et al. Oncolytic HSV-1 G207 Immunovirotherapy for Pediatric High-Grade Gliomas. N. Engl. J. Med. 2021, 384, 1613–1622. [Google Scholar] [CrossRef]
- Wheeler, L.A.; Manzanera, A.G.; Bell, S.D.; Cavaliere, R.; McGregor, J.M.; Grecula, J.C.; Newton, H.B.; Lo, S.S.; Badie, B.; Portnow, J.; et al. Phase II Multicenter Study of Gene-Mediated Cytotoxic Immunotherapy as Adjuvant to Surgical Resection for Newly Diagnosed Malignant Glioma. Neuro-Oncology 2016, 18, 1137–1145. [Google Scholar] [CrossRef]
- Westphal, M.; Ylä-Herttuala, S.; Martin, J.; Warnke, P.; Menei, P.; Eckland, D.; Kinley, J.; Kay, R.; Ram, Z.; ASPECT Study Group. Adenovirus-Mediated Gene Therapy with Sitimagene Ceradenovec Followed by Intravenous Ganciclovir for Patients with Operable High-Grade Glioma (ASPECT): A Randomised, Open-Label, Phase 3 Trial. Lancet Oncol. 2013, 14, 823–833. [Google Scholar] [CrossRef]
- Desjardins, A.; Gromeier, M.; Herndon, J.E.; Beaubier, N.; Bolognesi, D.P.; Friedman, A.H.; Friedman, H.S.; McSherry, F.; Muscat, A.M.; Nair, S.; et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N. Engl. J. Med. 2018, 379, 150–161. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Landolfi, J.; Vogelbaum, M.A.; Ostertag, D.; Elder, J.B.; Bloomfield, S.; Carter, B.; Chen, C.C.; Kalkanis, S.N.; Kesari, S.; et al. Durable Complete Responses in Some Recurrent High-Grade Glioma Patients Treated with Toca 511 + Toca FC. Neuro-Oncology 2018, 20, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Petrecca, K.; Walbert, T.; Butowski, N.; Salacz, M.; Perry, J.; Damek, D.; Bota, D.; Bettegowda, C.; Zhu, J.-J.; et al. Effect of Vocimagene Amiretrorepvec in Combination with Flucytosine vs Standard of Care on Survival Following Tumor Resection in Patients with Recurrent High-Grade Glioma: A Randomized Clinical Trial. JAMA Oncol. 2020, 6, 1939–1946. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Cohen, C.J.; Gartner, J.J.; Horovitz-Fried, M.; Shamalov, K.; Trebska-McGowan, K.; Bliskovsky, V.V.; Parkhurst, M.R.; Ankri, C.; Prickett, T.D.; Crystal, J.S.; et al. Isolation of Neoantigen-Specific T Cells from Tumor and Peripheral Lymphocytes. J. Clin. Investig. 2015, 125, 3981–3991. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Packard, B.S.; Aebersold, P.M.; Solomon, D.; Topalian, S.L.; Toy, S.T.; Simon, P.; Lotze, M.T.; Yang, J.C.; Seipp, C.A.; et al. Use of Tumor-Infiltrating Lymphocytes and Interleukin-2 in the Immunotherapy of Patients with Metastatic Melanoma. N. Engl. J. Med. 1988, 319, 1676–1680. [Google Scholar] [CrossRef]
- Wang, S.; Sun, J.; Chen, K.; Ma, P.; Lei, Q.; Xing, S.; Cao, Z.; Sun, S.; Yu, Z.; Liu, Y.; et al. Perspectives of Tumor-Infiltrating Lymphocyte Treatment in Solid Tumors. BMC Med. 2021, 19, 140. [Google Scholar] [CrossRef]
- Mathewson, N.D.; Ashenberg, O.; Tirosh, I.; Gritsch, S.; Perez, E.M.; Marx, S.; Jerby-Arnon, L.; Chanoch-Myers, R.; Hara, T.; Richman, A.R.; et al. Inhibitory CD161 Receptor Identified in Glioma-Infiltrating T Cells by Single-Cell Analysis. Cell 2021, 184, 1281–1298. [Google Scholar] [CrossRef]
- Brown, C.E.; Mackall, C.L. CAR T Cell Therapy: Inroads to Response and Resistance. Nat. Rev. Immunol. 2019, 19, 73–74. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.; June, C.H. Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Clinical Lessons Learned from the First Leg of the CAR T Cell Journey. Nat. Med. 2019, 25, 1341–1355. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A Single Dose of Peripherally Infused EGFRvIII-Directed CAR T Cells Mediates Antigen Loss and Induces Adaptive Resistance in Patients with Recurrent Glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of Apoptosis and Mitotic Spindle Checkpoint by Survivin. Nature 1998, 396, 580–584. [Google Scholar] [CrossRef]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A Novel Anti-Apoptosis Gene, Survivin, Expressed in Cancer and Lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, Y.; Yamasaki, F.; Hama, S.; Yahara, K.; Yoshioka, H.; Sugiyama, K.; Arita, K.; Kurisu, K. Expression of Survivin in Astrocytic Tumors: Correlation with Malignant Grade and Prognosis. Cancer 2003, 97, 1077–1083. [Google Scholar] [CrossRef] [PubMed]
- Satoh, K.; Kaneko, K.; Hirota, M.; Masamune, A.; Satoh, A.; Shimosegawa, T. Expression of Survivin Is Correlated with Cancer Cell Apoptosis and Is Involved in the Development of Human Pancreatic Duct Cell Tumors. Cancer 2001, 92, 271–278. [Google Scholar] [CrossRef]
- Galbo, P.M.; Ciesielski, M.J.; Figel, S.; Maguire, O.; Qiu, J.; Wiltsie, L.; Minderman, H.; Fenstermaker, R.A. Circulating CD9+/GFAP+/Survivin+ Exosomes in Malignant Glioma Patients Following Survivin Vaccination. Oncotarget 2017, 8, 114722–114735. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Nassiri, F.; Wang, J.; Peruzzi, P.; Zadeh, G. Viral and Other Therapies for Recurrent Glioblastoma: Is a 24-Month Durable Response Unusual? Neuro-Oncology 2019, 21, 14–25. [Google Scholar] [CrossRef]
- Marelli, G.; Howells, A.; Lemoine, N.R.; Wang, Y. Oncolytic Viral Therapy and the Immune System: A Double-Edged Sword Against Cancer. Front. Immunol. 2018, 9, 866. [Google Scholar] [CrossRef]
- Rainov, N.G. A Phase III Clinical Evaluation of Herpes Simplex Virus Type 1 Thymidine Kinase and Ganciclovir Gene Therapy as an Adjuvant to Surgical Resection and Radiation in Adults with Previously Untreated Glioblastoma Multiforme. Hum. Gene Ther. 2000, 11, 2389–2401. [Google Scholar] [CrossRef]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated Multi-Mutated Herpes Simplex Virus-1 for the Treatment of Malignant Gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef]
- Markert, J.M.; Medlock, M.D.; Rabkin, S.D.; Gillespie, G.Y.; Todo, T.; Hunter, W.D.; Palmer, C.A.; Feigenbaum, F.; Tornatore, C.; Tufaro, F.; et al. Conditionally Replicating Herpes Simplex Virus Mutant, G207 for the Treatment of Malignant Glioma: Results of a Phase I Trial. Gene Ther. 2000, 7, 867–874. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Nakashima, H.; Kasai, K.; Fernandez, S.A.; Oglesbee, M. Preclinical Toxicology of RQNestin34.5v.2: An Oncolytic Herpes Virus with Transcriptional Regulation of the ICP34.5 Neurovirulence Gene. Mol. Ther.-Methods Clin. Dev. 2020, 17, 871–893. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A. A Phase I Study of the Treatment of Recurrent Malignant Glioma with RQNestin34.5v.2, a Genetically Engineered HSV-1 Virus, and Immunomodulation with Cyclophosphamide. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03152318 (accessed on 28 February 2022).
- Chiocca, E.A.; Aguilar, L.K.; Bell, S.D.; Kaur, B.; Hardcastle, J.; Cavaliere, R.; McGregor, J.; Lo, S.; Ray-Chaudhuri, A.; Chakravarti, A.; et al. Phase IB Study of Gene-Mediated Cytotoxic Immunotherapy Adjuvant to up-Front Surgery and Intensive Timing Radiation for Malignant Glioma. J. Clin. Oncol. 2011, 29, 3611–3619. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Smith, K.M.; McKinney, B.; Palmer, C.A.; Rosenfeld, S.; Lillehei, K.; Hamilton, A.; DeMasters, B.K.; Judy, K.; Kirn, D. A Phase I Trial of Ad.HIFN-β Gene Therapy for Glioma. Mol. Ther. 2008, 16, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Gromeier, M.; Alexander, L.; Wimmer, E. Internal Ribosomal Entry Site Substitution Eliminates Neurovirulence in Intergeneric Poliovirus Recombinants. Proc. Natl. Acad. Sci. USA 1996, 93, 2370–2375. [Google Scholar] [CrossRef]
- Sloan, K.E.; Eustace, B.K.; Stewart, J.K.; Zehetmeier, C.; Torella, C.; Simeone, M.; Roy, J.E.; Unger, C.; Louis, D.N.; Ilag, L.L.; et al. CD155/PVR Plays a Key Role in Cell Motility during Tumor Cell Invasion and Migration. BMC Cancer 2004, 4, 73. [Google Scholar] [CrossRef]
- Lupo, K.B.; Matosevic, S. CD155 Immunoregulation as a Target for Natural Killer Cell Immunotherapy in Glioblastoma. J. Hematol. Oncol. 2020, 13, 76. [Google Scholar] [CrossRef]
- Brown, M.C.; Gromeier, M. Cytotoxic and Immunogenic Mechanisms of Recombinant Oncolytic Poliovirus. Curr. Opin. Virol. 2015, 13, 81–85. [Google Scholar] [CrossRef]
- Carlsten, M.; Norell, H.; Bryceson, Y.T.; Poschke, I.; Schedvins, K.; Ljunggren, H.-G.; Kiessling, R.; Malmberg, K.-J. Primary Human Tumor Cells Expressing CD155 Impair Tumor Targeting by Down-Regulating DNAM-1 on NK Cells. J. Immunol. 2009, 183, 4921–4930. [Google Scholar] [CrossRef]
- Strauss, M.; Filman, D.J.; Belnap, D.M.; Cheng, N.; Noel, R.T.; Hogle, J.M. Nectin-like Interactions between Poliovirus and Its Receptor Trigger Conformational Changes Associated with Cell Entry. J. Virol. 2015, 89, 4143–4157. [Google Scholar] [CrossRef]
- Wong, H.H.; Lemoine, N.R.; Wang, Y. Oncolytic Viruses for Cancer Therapy: Overcoming the Obstacles. Viruses 2010, 2, 78–106. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.S.; Foster, C.G.; Courtney, J.-M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef]
- Cabezas, R.; Ávila, M.; Gonzalez, J.; El-Bachá, R.S.; Báez, E.; García-Segura, L.M.; Jurado Coronel, J.C.; Capani, F.; Cardona-Gomez, G.P.; Barreto, G.E. Astrocytic Modulation of Blood Brain Barrier: Perspectives on Parkinson’s Disease. Front. Cell Neurosci. 2014, 8, 211. [Google Scholar] [CrossRef] [PubMed]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, Maintenance and Disruption of the Blood-Brain Barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; de Sampaio e Spohr, T.C.L.; Porto-Carreiro, I.; Pereira, C.M.; Balça-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the Vascular Fragility of the Blood Brain Barrier. Front. Cell Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef]
- Kamran, N.; Kadiyala, P.; Saxena, M.; Candolfi, M.; Li, Y.; Moreno-Ayala, M.A.; Raja, N.; Shah, D.; Lowenstein, P.R.; Castro, M.G. Immunosuppressive Myeloid Cells’ Blockade in the Glioma Microenvironment Enhances the Efficacy of Immune-Stimulatory Gene Therapy. Mol. Ther. 2017, 25, 232–248. [Google Scholar] [CrossRef]
- Bouffet, E.; Larouche, V.; Campbell, B.B.; Merico, D.; de Borja, R.; Aronson, M.; Durno, C.; Krueger, J.; Cabric, V.; Ramaswamy, V.; et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. J. Clin. Oncol. 2016, 34, 2206–2211. [Google Scholar] [CrossRef]
- Ott, M.; Tomaszowski, K.-H.; Marisetty, A.; Kong, L.-Y.; Wei, J.; Duna, M.; Blumberg, K.; Ji, X.; Jacobs, C.; Fuller, G.N.; et al. Profiling of Patients with Glioma Reveals the Dominant Immunosuppressive Axis Is Refractory to Immune Function Restoration. JCI Insight 2020, 5, 134386. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.-C.; Gutiérrez-Vázquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of Tumor-Associated Macrophages and T Cells in Glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef]
- Yang, I.; Tihan, T.; Han, S.J.; Wrensch, M.R.; Wiencke, J.; Sughrue, M.E.; Parsa, A.T. CD8+ T-Cell Infiltrate in Newly Diagnosed Glioblastoma Is Associated with Long-Term Survival. J. Clin. Neurosci. 2010, 17, 1381–1385. [Google Scholar] [CrossRef]
- Kmiecik, J.; Poli, A.; Brons, N.H.C.; Waha, A.; Eide, G.E.; Enger, P.Ø.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ Tumor-Infiltrating Immune Cells Correlate with Prolonged Survival in Glioblastoma Patients despite Integrated Immunosuppressive Mechanisms in the Tumor Microenvironment and at the Systemic Level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, H.; Pala, A.; Högel, J.; Hlavac, M.; Dietrich, E.; Westhoff, M.A.; Nonnenmacher, L.; Burster, T.; Georgieff, M.; Wirtz, C.R.; et al. Immune Phenotypes Predict Survival in Patients with Glioblastoma Multiforme. J. Hematol. Oncol. 2016, 9, 77. [Google Scholar] [CrossRef] [PubMed]
- Varn, F.S.; Wang, Y.; Mullins, D.W.; Fiering, S.; Cheng, C. Systematic Pan-Cancer Analysis Reveals Immune Cell Interactions in the Tumor Microenvironment. Cancer Res. 2017, 77, 1271–1282. [Google Scholar] [CrossRef] [PubMed]
- Orrego, E.; Castaneda, C.A.; Castillo, M.; Bernabe, L.A.; Casavilca, S.; Chakravarti, A.; Meng, W.; Garcia-Corrochano, P.; Villa-Robles, M.R.; Zevallos, R.; et al. Distribution of Tumor-Infiltrating Immune Cells in Glioblastoma. CNS Oncol. 2018, 7, CNS21. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer Stem Cells in Glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Wang, Z.-F.; Liao, F.; Wu, H.; Dai, J. Glioma Stem Cells-Derived Exosomal MiR-26a Promotes Angiogenesis of Microvessel Endothelial Cells in Glioma. J. Exp. Clin. Cancer Res. 2019, 38, 201. [Google Scholar] [CrossRef]
- Taniguchi, H.; Suzuki, Y.; Natori, Y. The Evolving Landscape of Cancer Stem Cells and Ways to Overcome Cancer Heterogeneity. Cancers 2019, 11, 532. [Google Scholar] [CrossRef]
- Lei, M.M.L.; Lee, T.K.W. Cancer Stem Cells: Emerging Key Players in Immune Evasion of Cancers. Front. Cell Dev. Biol. 2021, 9, 692940. [Google Scholar] [CrossRef]
- Sainz, B.; Carron, E.; Vallespinós, M.; Machado, H.L. Cancer Stem Cells and Macrophages: Implications in Tumor Biology and Therapeutic Strategies. Mediat. Inflamm. 2016, 2016, 9012369. [Google Scholar] [CrossRef]
- Xu, Q.; Liu, G.; Yuan, X.; Xu, M.; Wang, H.; Ji, J.; Konda, B.; Black, K.L.; Yu, J.S. Antigen-Specific T-Cell Response from Dendritic Cell Vaccination Using Cancer Stem-like Cell-Associated Antigens. Stem Cells 2009, 27, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- de Frel, D.L.; Atsma, D.E.; Pijl, H.; Seidell, J.C.; Leenen, P.J.M.; Dik, W.A.; van Rossum, E.F.C. The Impact of Obesity and Lifestyle on the Immune System and Susceptibility to Infections Such as COVID-19. Front. Nutr. 2020, 7, 597600. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, R.P.; Sharma, S.; Watabe, K. The Confounders of Cancer Immunotherapy: Roles of Lifestyle, Metabolic Disorders and Sociological Factors. Cancers 2020, 12, 2983. [Google Scholar] [CrossRef] [PubMed]
- Lewis, D.E.; Lysaght, J.; Wu, H. Editorial: T Cell Alterations in Adipose Tissue During Obesity, HIV, and Cancer. Front. Immunol. 2019, 10, 1190. [Google Scholar] [CrossRef]
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M.; et al. Association of Body-Mass Index and Outcomes in Patients with Metastatic Melanoma Treated with Targeted Therapy, Immunotherapy, or Chemotherapy: A Retrospective, Multicohort Analysis. Lancet Oncol. 2018, 19, 310–322. [Google Scholar] [CrossRef]
- Desrichard, A.; Kuo, F.; Chowell, D.; Lee, K.-W.; Riaz, N.; Wong, R.J.; Chan, T.A.; Morris, L.G.T. Tobacco Smoking-Associated Alterations in the Immune Microenvironment of Squamous Cell Carcinomas. J. Natl. Cancer Inst. 2018, 110, 1386–1392. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Mutational Landscape Determines Sensitivity to PD-1 Blockade in Non–Small Cell Lung Cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef]
- Norum, J.; Nieder, C. Tobacco Smoking and Cessation and PD-L1 Inhibitors in Non-Small Cell Lung Cancer (NSCLC): A Review of the Literature. ESMO Open 2018, 3, e000406. [Google Scholar] [CrossRef]
- Devarakonda, S.; Rotolo, F.; Tsao, M.-S.; Lanc, I.; Brambilla, E.; Masood, A.; Olaussen, K.A.; Fulton, R.; Sakashita, S.; McLeer-Florin, A.; et al. Tumor Mutation Burden as a Biomarker in Resected Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2018, 36, 2995–3006. [Google Scholar] [CrossRef]
- Hwang, W.L.; Wolfson, R.L.; Niemierko, A.; Marcus, K.J.; DuBois, S.G.; Haas-Kogan, D. Clinical Impact of Tumor Mutational Burden in Neuroblastoma. J. Natl. Cancer Inst. 2018, 111, 695–699. [Google Scholar] [CrossRef]
- Meléndez, B.; Van Campenhout, C.; Rorive, S.; Remmelink, M.; Salmon, I.; D’Haene, N. Methods of Measurement for Tumor Mutational Burden in Tumor Tissue. Transl. Lung Cancer Res. 2018, 7, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Serratì, S.; De Summa, S.; Pilato, B.; Petriella, D.; Lacalamita, R.; Tommasi, S.; Pinto, R. Next-Generation Sequencing: Advances and Applications in Cancer Diagnosis. OncoTargets Ther. 2016, 9, 7355–7365. [Google Scholar] [CrossRef] [PubMed]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-Cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature 2019, 565, 240–245. [Google Scholar] [CrossRef] [PubMed]
- Weenink, B.; Draaisma, K.; Ooi, H.Z.; Kros, J.M.; Sillevis Smitt, P.A.E.; Debets, R.; French, P.J. Low-Grade Glioma Harbors Few CD8 T Cells, Which Is Accompanied by Decreased Expression of Chemo-Attractants, Not Immunogenic Antigens. Sci. Rep. 2019, 9, 14643. [Google Scholar] [CrossRef]
- Weenink, B.; French, P.J.; Sillevis Smitt, P.A.E.; Debets, R.; Geurts, M. Immunotherapy in Glioblastoma: Current Shortcomings and Future Perspectives. Cancers 2020, 12, 751. [Google Scholar] [CrossRef]
- Wang, L.; Ge, J.; Lan, Y.; Shi, Y.; Luo, Y.; Tan, Y.; Liang, M.; Deng, S.; Zhang, X.; Wang, W.; et al. Tumor Mutational Burden Is Associated with Poor Outcomes in Diffuse Glioma. BMC Cancer 2020, 20, 213. [Google Scholar] [CrossRef]
- Valero, C.; Lee, M.; Hoen, D.; Wang, J.; Nadeem, Z.; Patel, N.; Postow, M.A.; Shoushtari, A.N.; Plitas, G.; Balachandran, V.P.; et al. The Association between Tumor Mutational Burden and Prognosis Is Dependent on Treatment Context. Nat. Genet. 2021, 53, 11–15. [Google Scholar] [CrossRef]
- Gromeier, M.; Brown, M.C.; Zhang, G.; Lin, X.; Chen, Y.; Wei, Z.; Beaubier, N.; Yan, H.; He, Y.; Desjardins, A.; et al. Very Low Mutation Burden Is a Feature of Inflamed Recurrent Glioblastomas Responsive to Cancer Immunotherapy. Nat. Commun. 2021, 12, 352. [Google Scholar] [CrossRef]
- Schiff, D.; Lee, E.Q.; Nayak, L.; Norden, A.D.; Reardon, D.A.; Wen, P.Y. Medical Management of Brain Tumors and the Sequelae of Treatment. Neuro-Oncology 2015, 17, 488–504. [Google Scholar] [CrossRef]
- Murayi, R.; Chittiboina, P. Glucocorticoids in the Management of Peritumoral Brain Edema: A Review of Molecular Mechanisms. Child’s Nerv. Syst. 2016, 32, 2293–2302. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the Blood-Brain Barrier Really Disrupted in All Glioblastomas? A Critical Assessment of Existing Clinical Data. Neuro-Oncology 2018, 20, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. Characteristics of Compounds That Cross the Blood-Brain Barrier. BMC Neurol. 2009, 9, S3. [Google Scholar] [CrossRef] [PubMed]
- Arvanitis, C.D.; Ferraro, G.B.; Jain, R.K. The Blood–Brain Barrier and Blood–Tumour Barrier in Brain Tumours and Metastases. Nat. Rev. Cancer 2020, 20, 26–41. [Google Scholar] [CrossRef]
- Poon, C.; McMahon, D.; Hynynen, K. Noninvasive and Targeted Delivery of Therapeutics to the Brain Using Focused Ultrasound. Neuropharmacology 2017, 120, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Aryal, M.; Vykhodtseva, N.; Zhang, Y.-Z.; Park, J.; McDannold, N. Multiple Treatments with Liposomal Doxorubicin and Ultrasound-Induced Disruption of Blood-Tumor and Blood-Brain Barriers Improve Outcomes in a Rat Glioma Model. J. Control. Release 2013, 169, 103–111. [Google Scholar] [CrossRef]
- McDannold, N.; Arvanitis, C.D.; Vykhodtseva, N.; Livingstone, M.S. Temporary Disruption of the Blood-Brain Barrier by Use of Ultrasound and Microbubbles: Safety and Efficacy Evaluation in Rhesus Macaques. Cancer Res. 2012, 72, 3652–3663. [Google Scholar] [CrossRef]
- Lynch, M.; Heinen, S.; Markham-Coultes, K.; O’Reilly, M.; Van Slyke, P.; Dumont, D.J.; Hynynen, K.; Aubert, I. Vasculotide Restores the Blood-Brain Barrier after Focused Ultrasound-Induced Permeability in a Mouse Model of Alzheimer’s Disease. Int. J. Med. Sci. 2021, 18, 482–493. [Google Scholar] [CrossRef]
- Kovacs, Z.I.; Kim, S.; Jikaria, N.; Qureshi, F.; Milo, B.; Lewis, B.K.; Bresler, M.; Burks, S.R.; Frank, J.A. Disrupting the Blood-Brain Barrier by Focused Ultrasound Induces Sterile Inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, E75–E84. [Google Scholar] [CrossRef]
- Shi, L.; Palacio-Mancheno, P.; Badami, J.; Shin, D.W.; Zeng, M.; Cardoso, L.; Tu, R.; Fu, B.M. Quantification of Transient Increase of the Blood-Brain Barrier Permeability to Macromolecules by Optimized Focused Ultrasound Combined with Microbubbles. Int. J. Nanomed. 2014, 9, 4437–4448. [Google Scholar] [CrossRef]
- Marquet, F.; Tung, Y.-S.; Teichert, T.; Ferrera, V.P.; Konofagou, E.E. Noninvasive, Transient and Selective Blood-Brain Barrier Opening in Non-Human Primates In Vivo. PLoS ONE 2011, 6, e22598. [Google Scholar] [CrossRef] [PubMed]
- McDannold, N.; Zhang, Y.; Supko, J.G.; Power, C.; Sun, T.; Vykhodtseva, N.; Golby, A.J.; Reardon, D.A. Blood-Brain Barrier Disruption and Delivery of Irinotecan in a Rat Model Using a Clinical Transcranial MRI-Guided Focused Ultrasound System. Sci. Rep. 2020, 10, 8766. [Google Scholar] [CrossRef] [PubMed]
- Todd, N.; Angolano, C.; Ferran, C.; Devor, A.; Borsook, D.; McDannold, N. Secondary Effects on Brain Physiology Caused by Focused Ultrasound-Mediated Disruption of the Blood-Brain Barrier. J. Control. Release 2020, 324, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Canney, M.; Vignot, A.; Reina, V.; Beccaria, K.; Horodyckid, C.; Karachi, C.; Leclercq, D.; Lafon, C.; Chapelon, J.-Y.; et al. Clinical Trial of Blood-Brain Barrier Disruption by Pulsed Ultrasound. Sci. Transl. Med. 2016, 8, 343re2. [Google Scholar] [CrossRef] [PubMed]
- Idbaih, A.; Canney, M.; Belin, L.; Desseaux, C.; Vignot, A.; Bouchoux, G.; Asquier, N.; Law-Ye, B.; Leclercq, D.; Bissery, A.; et al. Safety and Feasibility of Repeated and Transient Blood-Brain Barrier Disruption by Pulsed Ultrasound in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2019, 25, 3793–3801. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; MacIntosh, B.J.; Shirzadi, Z.; Kiss, A.; Bethune, A.; Heyn, C.; Mithani, K.; Hamani, C.; Black, S.E.; Hynynen, K.; et al. Resting State Functional Connectivity Changes after MR-Guided Focused Ultrasound Mediated Blood-Brain Barrier Opening in Patients with Alzheimer’s Disease. Neuroimage 2019, 200, 275–280. [Google Scholar] [CrossRef]
- Rezai, A.R.; Ranjan, M.; D’Haese, P.-F.; Haut, M.W.; Carpenter, J.; Najib, U.; Mehta, R.I.; Chazen, J.L.; Zibly, Z.; Yates, J.R.; et al. Noninvasive Hippocampal Blood−brain Barrier Opening in Alzheimer’s Disease with Focused Ultrasound. Proc. Natl. Acad. Sci. USA 2020, 117, 9180–9182. [Google Scholar] [CrossRef]
- Deng, Z.; Sheng, Z.; Yan, F. Ultrasound-Induced Blood-Brain-Barrier Opening Enhances Anticancer Efficacy in the Treatment of Glioblastoma: Current Status and Future Prospects. J. Oncol. 2019, 2019, e2345203. [Google Scholar] [CrossRef]
- Alli, S.; Figueiredo, C.A.; Golbourn, B.; Sabha, N.; Wu, M.Y.; Bondoc, A.; Luck, A.; Coluccia, D.; Maslink, C.; Smith, C.; et al. Brainstem Blood Brain Barrier Disruption Using Focused Ultrasound: A Demonstration of Feasibility and Enhanced Doxorubicin Delivery. J. Control. Release 2018, 281, 29–41. [Google Scholar] [CrossRef]
- Gasca-Salas, C.; Fernández-Rodríguez, B.; Pineda-Pardo, J.A.; Rodríguez-Rojas, R.; Obeso, I.; Hernández-Fernández, F.; del Álamo, M.; Mata, D.; Guida, P.; Ordás-Bandera, C.; et al. Blood-Brain Barrier Opening with Focused Ultrasound in Parkinson’s Disease Dementia. Nat. Commun. 2021, 12, 779. [Google Scholar] [CrossRef]
- Lipsman, N.; Meng, Y.; Bethune, A.J.; Huang, Y.; Lam, B.; Masellis, M.; Herrmann, N.; Heyn, C.; Aubert, I.; Boutet, A.; et al. Blood–Brain Barrier Opening in Alzheimer’s Disease Using MR-Guided Focused Ultrasound. Nat. Commun. 2018, 9, 2336. [Google Scholar] [CrossRef]
- Pouliopoulos, A.N.; Kwon, N.; Jensen, G.; Meaney, A.; Niimi, Y.; Burgess, M.T.; Ji, R.; McLuckie, A.J.; Munoz, F.A.; Kamimura, H.A.S.; et al. Safety Evaluation of a Clinical Focused Ultrasound System for Neuronavigation Guided Blood-Brain Barrier Opening in Non-Human Primates. Sci. Rep. 2021, 11, 15043. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Mériaux, S.; Larrat, B. About the Marty Model of Blood-Brain Barrier Closure after Its Disruption Using Focused Ultrasound. Phys. Med. Biol. 2019, 64, 14NT02. [Google Scholar] [CrossRef] [PubMed]
- McDannold, N.; Vykhodtseva, N.; Hynynen, K. Effects of Acoustic Parameters and Ultrasound Contrast Agent Dose on Focused-Ultrasound Induced Blood-Brain Barrier Disruption. Ultrasound Med. Biol. 2008, 34, 930–937. [Google Scholar] [CrossRef] [PubMed]
- Marty, B.; Larrat, B.; Van Landeghem, M.; Robic, C.; Robert, P.; Port, M.; Le Bihan, D.; Pernot, M.; Tanter, M.; Lethimonnier, F.; et al. Dynamic Study of Blood-Brain Barrier Closure after Its Disruption Using Ultrasound: A Quantitative Analysis. J. Cereb. Blood Flow Metab. 2012, 32, 1948–1958. [Google Scholar] [CrossRef]
- Park, J.; Zhang, Y.; Vykhodtseva, N.; Jolesz, F.A.; McDannold, N.J. The Kinetics of Blood Brain Barrier Permeability and Targeted Doxorubicin Delivery into Brain Induced by Focused Ultrasound. J. Control. Release 2012, 162, 134–142. [Google Scholar] [CrossRef]
- O’Reilly, M.A.; Waspe, A.C.; Ganguly, M.; Hynynen, K. Focused-Ultrasound Disruption of the Blood-Brain Barrier Using Closely-Timed Short Pulses: Influence of Sonication Parameters and Injection Rate. Ultrasound Med. Biol. 2011, 37, 587–594. [Google Scholar] [CrossRef]
- Samiotaki, G.; Vlachos, F.; Tung, Y.-S.; Konofagou, E.E. A Quantitative Pressure and Microbubble-Size Dependence Study of Focused Ultrasound-Induced Blood-Brain Barrier Opening Reversibility in Vivo Using MRI. Magn. Reson. Med. 2012, 67, 769–777. [Google Scholar] [CrossRef]
- Chen, K.-T.; Wei, K.-C.; Liu, H.-L. Theranostic Strategy of Focused Ultrasound Induced Blood-Brain Barrier Opening for CNS Disease Treatment. Front. Pharmacol. 2019, 10, 86. [Google Scholar] [CrossRef]
- Aryal, M.; Arvanitis, C.D.; Alexander, P.M.; McDannold, N. Ultrasound-Mediated Blood-Brain Barrier Disruption for Targeted Drug Delivery in the Central Nervous System. Adv. Drug Deliv. Rev. 2014, 72, 94–109. [Google Scholar] [CrossRef]
- McDannold, N.; Vykhodtseva, N.; Hynynen, K. Use of Ultrasound Pulses Combined with Definity for Targeted Blood-Brain Barrier Disruption: A Feasibility Study. Ultrasound Med. Biol. 2007, 33, 584–590. [Google Scholar] [CrossRef]
- Chu, P.-C.; Liu, H.-L.; Lai, H.-Y.; Lin, C.-Y.; Tsai, H.-C.; Pei, Y.-C. Neuromodulation Accompanying Focused Ultrasound-Induced Blood-Brain Barrier Opening. Sci. Rep. 2015, 5, 15477. [Google Scholar] [CrossRef] [PubMed]
- McDannold, N.; Vykhodtseva, N.; Hynynen, K. Targeted Disruption of the Blood-Brain Barrier with Focused Ultrasound: Association with Cavitation Activity. Phys. Med. Biol. 2006, 51, 793–807. [Google Scholar] [CrossRef]
- O’Reilly, M.A.; Hynynen, K. Blood-Brain Barrier: Real-Time Feedback-Controlled Focused Ultrasound Disruption by Using an Acoustic Emissions-Based Controller. Radiology 2012, 263, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Samiotaki, G.; Wang, S.; Acosta, C.; Chen, C.C.; Konofagou, E.E. Acoustic Cavitation-Based Monitoring of the Reversibility and Permeability of Ultrasound-Induced Blood-Brain Barrier Opening. Phys. Med. Biol. 2015, 60, 9079–9094. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhang, Y.; Power, C.; Alexander, P.M.; Sutton, J.T.; Aryal, M.; Vykhodtseva, N.; Miller, E.L.; McDannold, N.J. Closed-Loop Control of Targeted Ultrasound Drug Delivery across the Blood–Brain/Tumor Barriers in a Rat Glioma Model. Proc. Natl. Acad. Sci. USA 2017, 114, E10281–E10290. [Google Scholar] [CrossRef]
- Patel, A.; Schoen, S.J.; Arvanitis, C.D. Closed-Loop Spatial and Temporal Control of Cavitation Activity with Passive Acoustic Mapping. IEEE Trans. Biomed. Eng. 2019, 66, 2022–2031. [Google Scholar] [CrossRef]
- Collis, J.; Manasseh, R.; Liovic, P.; Tho, P.; Ooi, A.; Petkovic-Duran, K.; Zhu, Y. Cavitation Microstreaming and Stress Fields Created by Microbubbles. Ultrasonics 2010, 50, 273–279. [Google Scholar] [CrossRef]
- Sutton, J.T.; Haworth, K.J.; Pyne-Geithman, G.; Holland, C.K. Ultrasound-Mediated Drug Delivery for Cardiovascular Disease. Expert Opin. Drug Deliv. 2013, 10, 573–592. [Google Scholar] [CrossRef]
- Guo, Y.; Lee, H.; Fang, Z.; Velalopoulou, A.; Kim, J.; Ben Thomas, M.; Liu, J.; Abramowitz, R.G.; Kim, Y.; Coskun, A.F.; et al. Single-Cell Analysis Reveals Effective SiRNA Delivery in Brain Tumors with Microbubble-Enhanced Ultrasound and Cationic Nanoparticles. Sci. Adv. 2021, 7, eabf7390. [Google Scholar] [CrossRef]
- Anastasiadis, P.; Gandhi, D.; Guo, Y.; Ahmed, A.-K.; Bentzen, S.M.; Arvanitis, C.; Woodworth, G.F. Localized Blood–Brain Barrier Opening in Infiltrating Gliomas with MRI-Guided Acoustic Emissions–Controlled Focused Ultrasound. Proc. Natl. Acad. Sci. USA 2021, 118, e2103280118. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.J.; Pernot, M.; Small, S.A.; Konofagou, E.E. Noninvasive, Transcranial and Localized Opening of the Blood-Brain Barrier Using Focused Ultrasound in Mice. Ultrasound Med. Biol. 2007, 33, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Morse, S.V.; Pouliopoulos, A.N.; Chan, T.G.; Copping, M.J.; Lin, J.; Long, N.J.; Choi, J.J. Rapid Short-Pulse Ultrasound Delivers Drugs Uniformly across the Murine Blood-Brain Barrier with Negligible Disruption. Radiology 2019, 291, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Beccaria, K.; Sabbagh, A.; de Groot, J.; Canney, M.; Carpentier, A.; Heimberger, A.B. Blood-Brain Barrier Opening with Low Intensity Pulsed Ultrasound for Immune Modulation and Immune Therapeutic Delivery to CNS Tumors. J. Neuro-Oncol. 2021, 151, 65–73. [Google Scholar] [CrossRef]
- Hynynen, K.; McDannold, N.; Clement, G.; Jolesz, F.A.; Zadicario, E.; Killiany, R.; Moore, T.; Rosen, D. Pre-Clinical Testing of a Phased Array Ultrasound System for MRI-Guided Noninvasive Surgery of the Brain—A Primate Study. Eur. J. Radiol. 2006, 59, 149–156. [Google Scholar] [CrossRef]
- Song, J.; Hynynen, K. Feasibility of Using Lateral Mode Coupling Method for a Large Scale Ultrasound Phased Array for Noninvasive Transcranial Therapy. IEEE Trans. Biomed. Eng. 2010, 57, 124–133. [Google Scholar] [CrossRef]
- O’Reilly, M.A.; Jones, R.M.; Hynynen, K. Three-Dimensional Transcranial Ultrasound Imaging of Microbubble Clouds Using a Sparse Hemispherical Array. IEEE Trans. Biomed. Eng. 2014, 61, 1285–1294. [Google Scholar] [CrossRef]
- Park, S.H.; Kim, M.J.; Jung, H.H.; Chang, W.S.; Choi, H.S.; Rachmilevitch, I.; Zadicario, E.; Chang, J.W. One-Year Outcome of Multiple Blood–Brain Barrier Disruptions with Temozolomide for the Treatment of Glioblastoma. Front. Oncol. 2020, 10, 1663. [Google Scholar] [CrossRef]
- Wei, K.-C.; Tsai, H.-C.; Lu, Y.-J.; Yang, H.-W.; Hua, M.-Y.; Wu, M.-F.; Chen, P.-Y.; Huang, C.-Y.; Yen, T.-C.; Liu, H.-L. Neuronavigation-Guided Focused Ultrasound-Induced Blood-Brain Barrier Opening: A Preliminary Study in Swine. AJNR Am. J. Neuroradiol. 2013, 34, 115–120. [Google Scholar] [CrossRef]
- Wu, S.-Y.; Aurup, C.; Sanchez, C.S.; Grondin, J.; Zheng, W.; Kamimura, H.; Ferrera, V.P.; Konofagou, E.E. Efficient Blood-Brain Barrier Opening in Primates with Neuronavigation-Guided Ultrasound and Real-Time Acoustic Mapping. Sci. Rep. 2018, 8, 7978. [Google Scholar] [CrossRef]
- Pouliopoulos, A.N.; Wu, S.-Y.; Burgess, M.T.; Karakatsani, M.E.; Kamimura, H.A.S.; Konofagou, E.E. A Clinical System for Non-Invasive Blood-Brain Barrier Opening Using a Neuronavigation-Guided Single-Element Focused Ultrasound Transducer. Ultrasound Med. Biol. 2020, 46, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.-T.; Lin, Y.-J.; Chai, W.-Y.; Lin, C.-J.; Chen, P.-Y.; Huang, C.-Y.; Kuo, J.S.; Liu, H.-L.; Wei, K.-C. Neuronavigation-Guided Focused Ultrasound (NaviFUS) for Transcranial Blood-Brain Barrier Opening in Recurrent Glioblastoma Patients: Clinical Trial Protocol. Ann. Transl. Med. 2020, 8, 673. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C. A Feasibility Study Examining the Use of Non-Invasive Focused Ultrasound (FUS) with Oral Panobinostat Administration in Children with Progressive Diffuse Midline Glioma (DMG). 2022. Available online: https://clinicaltrials.gov/ct2/show/NCT04804709 (accessed on 28 February 2022).
- Ji, R.; Karakatsani, M.E.; Burgess, M.; Smith, M.; Murillo, M.F.; Konofagou, E.E. Cavitation-Modulated Inflammatory Response Following Focused Ultrasound Blood-Brain Barrier Opening. J. Control. Release 2021, 337, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Assistance Publique—Hôpitaux de Paris. A Study to Evaluate the Safety of Transient Opening of the Blood-Brain Barrier by Low Intensity Pulsed Ultrasound with the SonoCloud Implantable Device in Patients with Recurrent Glioblastoma before Chemotherapy Administration. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT02253212 (accessed on 28 February 2022).
- CarThera. A Study to Evaluate the Safety and the Efficacy of Transient Opening of the Blood-Brain Barrier (BBB) by Low Intensity Pulsed Ultrasound with the SonoCloud-9 Implantable Device in Recurrent Glioblastoma Patients Eligible for Surgery and for Carboplatin Chemotherapy. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT03744026 (accessed on 28 February 2022).
- Assistance Publique–Hôpitaux de Paris. Safety and Efficacy of Blood Brain Barrier Opening with Implantable Device Sonocloud® Combined with Nivolumab Used Alone or an Association with Ipilimumab in Brain Metastases from Patients with Malignant Melanoma. 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04021420 (accessed on 28 February 2022).
- Asquier, N.; Bouchoux, G.; Canney, M.; Martin, C.; Law-Ye, B.; Leclercq, D.; Chapelon, J.-Y.; Lafon, C.; Idbaih, A.; Carpentier, A. Blood-Brain Barrier Disruption in Humans Using an Implantable Ultrasound Device: Quantification with MR Images and Correlation with Local Acoustic Pressure. J. Neurosurg. 2019, 132, 875–883. [Google Scholar] [CrossRef]
- Liu, H.-L.; Hsu, P.-H.; Lin, C.-Y.; Huang, C.-W.; Chai, W.-Y.; Chu, P.-C.; Huang, C.-Y.; Chen, P.-Y.; Yang, L.-Y.; Kuo, J.S.; et al. Focused Ultrasound Enhances Central Nervous System Delivery of Bevacizumab for Malignant Glioma Treatment. Radiology 2016, 281, 99–108. [Google Scholar] [CrossRef]
- Coluccia, D.; Figueiredo, C.A.; Wu, M.Y.; Riemenschneider, A.N.; Diaz, R.; Luck, A.; Smith, C.; Das, S.; Ackerley, C.; O’Reilly, M.; et al. Enhancing Glioblastoma Treatment Using Cisplatin-Gold-Nanoparticle Conjugates and Targeted Delivery with Magnetic Resonance-Guided Focused Ultrasound. Nanomedicine 2018, 14, 1137–1148. [Google Scholar] [CrossRef]
- Thévenot, E.; Jordão, J.F.; O’Reilly, M.A.; Markham, K.; Weng, Y.-Q.; Foust, K.D.; Kaspar, B.K.; Hynynen, K.; Aubert, I. Targeted Delivery of Self-Complementary Adeno-Associated Virus Serotype 9 to the Brain, Using Magnetic Resonance Imaging-Guided Focused Ultrasound. Hum. Gene Ther. 2012, 23, 1144–1155. [Google Scholar] [CrossRef]
- Noroozian, Z.; Xhima, K.; Huang, Y.; Kaspar, B.K.; Kügler, S.; Hynynen, K.; Aubert, I. MRI-Guided Focused Ultrasound for Targeted Delivery of RAAV to the Brain. Methods Mol. Biol. 2019, 1950, 177–197. [Google Scholar] [CrossRef]
- Jordão, J.F.; Ayala-Grosso, C.A.; Markham, K.; Huang, Y.; Chopra, R.; McLaurin, J.; Hynynen, K.; Aubert, I. Antibodies Targeted to the Brain with Image-Guided Focused Ultrasound Reduces Amyloid-Beta Plaque Load in the TgCRND8 Mouse Model of Alzheimer’s Disease. PLoS ONE 2010, 5, e10549. [Google Scholar] [CrossRef]
- Kobus, T.; Zervantonakis, I.K.; Zhang, Y.; McDannold, N.J. Growth Inhibition in a Brain Metastasis Model by Antibody Delivery Using Focused Ultrasound-Mediated Blood-Brain Barrier Disruption. J. Control. Release 2016, 238, 281–288. [Google Scholar] [CrossRef]
- Morse, S.V.; Mishra, A.; Chan, T.G.; de Rosales, R.M.; Choi, J.J.; Choi, J.J. Liposome Delivery to the Brain with Rapid Short-Pulses of Focused Ultrasound and Microbubbles. J. Control. Release 2022, 341, 605–615. [Google Scholar] [CrossRef]
- Chan, T.G.; Morse, S.V.; Copping, M.J.; Choi, J.J.; Vilar, R. Targeted Delivery of DNA-Au Nanoparticles across the Blood-Brain Barrier Using Focused Ultrasound. ChemMedChem 2018, 13, 1311–1314. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Huang, Q.; Wang, F.; Zhang, X.; Hu, J.; Tan, Y.; Huang, N.; Wang, Z.; Wang, Z.; Cheng, Y. Targeted ShRNA-Loaded Liposome Complex Combined with Focused Ultrasound for Blood Brain Barrier Disruption and Suppressing Glioma Growth. Cancer Lett. 2018, 418, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Ayala-Grosso, C.A.; Ganguly, M.; Jordão, J.F.; Aubert, I.; Hynynen, K. Targeted Delivery of Neural Stem Cells to the Brain Using MRI-Guided Focused Ultrasound to Disrupt the Blood-Brain Barrier. PLoS ONE 2011, 6, e27877. [Google Scholar] [CrossRef] [PubMed]
- Alkins, R.; Burgess, A.; Kerbel, R.; Wels, W.S.; Hynynen, K. Early Treatment of HER2-Amplified Brain Tumors with Targeted NK-92 Cells and Focused Ultrasound Improves Survival. Neuro-Oncology 2016, 18, 974–981. [Google Scholar] [CrossRef] [PubMed]
- McDannold, N.; Zhang, Y.; Supko, J.G.; Power, C.; Sun, T.; Peng, C.; Vykhodtseva, N.; Golby, A.J.; Reardon, D.A. Acoustic Feedback Enables Safe and Reliable Carboplatin Delivery across the Blood-Brain Barrier with a Clinical Focused Ultrasound System and Improves Survival in a Rat Glioma Model. Theranostics 2019, 9, 6284–6299. [Google Scholar] [CrossRef]
- Zhang, P.; Miska, J.; Lee-Chang, C.; Rashidi, A.; Panek, W.K.; An, S.; Zannikou, M.; Lopez-Rosas, A.; Han, Y.; Xiao, T.; et al. Therapeutic Targeting of Tumor-Associated Myeloid Cells Synergizes with Radiation Therapy for Glioblastoma. Proc. Natl. Acad. Sci. USA 2019, 116, 23714–23723. [Google Scholar] [CrossRef]
- Taiarol, L.; Formicola, B.; Magro, R.D.; Sesana, S.; Re, F. An Update of Nanoparticle-Based Approaches for Glioblastoma Multiforme Immunotherapy. Nanomedicine 2020, 15, 1861–1871. [Google Scholar] [CrossRef]
- Mathew, E.N.; Berry, B.C.; Yang, H.W.; Carroll, R.S.; Johnson, M.D. Delivering Therapeutics to Glioblastoma: Overcoming Biological Constraints. Int. J. Mol. Sci. 2022, 23, 1711. [Google Scholar] [CrossRef]
- Zhang, F.; Stephan, S.B.; Ene, C.I.; Smith, T.T.; Holland, E.C.; Stephan, M.T. Nanoparticles That Reshape the Tumor Milieu Create a Therapeutic Window for Effective T-Cell Therapy in Solid Malignancies. Cancer Res. 2018, 78, 3718–3730. [Google Scholar] [CrossRef]
- Chen, P.-Y.; Hsieh, H.-Y.; Huang, C.-Y.; Lin, C.-Y.; Wei, K.-C.; Liu, H.-L. Focused Ultrasound-Induced Blood–Brain Barrier Opening to Enhance Interleukin-12 Delivery for Brain Tumor Immunotherapy: A Preclinical Feasibility Study. J. Transl. Med. 2015, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Curley, C.T.; Sheybani, N.D.; Bullock, T.N.; Price, R.J. Focused Ultrasound Immunotherapy for Central Nervous System Pathologies: Challenges and Opportunities. Theranostics 2017, 7, 3608–3623. [Google Scholar] [CrossRef] [PubMed]
- Sevenich, L. Turning “Cold” Into “Hot” Tumors-Opportunities and Challenges for Radio-Immunotherapy Against Primary and Metastatic Brain Cancers. Front. Oncol. 2019, 9, 163. [Google Scholar] [CrossRef] [PubMed]
- Steeg, P.S. The Blood-Tumour Barrier in Cancer Biology and Therapy. Nat. Rev. Clin. Oncol. 2021, 18, 696–714. [Google Scholar] [CrossRef]
- Sabbagh, A.; Beccaria, K.; Ling, X.; Marisetty, A.; Ott, M.; Caruso, H.; Barton, E.; Kong, L.-Y.; Fang, D.; Latha, K.; et al. Opening of the Blood-Brain Barrier Using Low-Intensity Pulsed Ultrasound Enhances Responses to Immunotherapy in Preclinical Glioma Models. Clin. Cancer Res. 2021, 27, 4325–4337. [Google Scholar] [CrossRef]
- Alkins, R.; Burgess, A.; Ganguly, M.; Francia, G.; Kerbel, R.; Wels, W.S.; Hynynen, K. Focused Ultrasound Delivers Targeted Immune Cells to Metastatic Brain Tumors. Cancer Res. 2013, 73, 1892–1899. [Google Scholar] [CrossRef]
- Pacia, C.P.; Yuan, J.; Yue, Y.; Xu, L.; Nazeri, A.; Desai, R.; Gach, H.M.; Wang, X.; Talcott, M.R.; Chaudhuri, A.A.; et al. Sonobiopsy for Minimally Invasive, Spatiotemporally-Controlled, and Sensitive Detection of Glioblastoma-Derived Circulating Tumor DNA. Theranostics 2022, 12, 362–378. [Google Scholar] [CrossRef]
- Liu, H.-L.; Hsu, P.-H.; Chu, P.-C.; Wai, Y.-Y.; Chen, J.-C.; Shen, C.-R.; Yen, T.-C.; Wang, J.-J. Magnetic Resonance Imaging Enhanced by Superparamagnetic Iron Oxide Particles: Usefulness for Distinguishing between Focused Ultrasound-Induced Blood-Brain Barrier Disruption and Brain Hemorrhage. J. Magn. Reson. Imaging 2009, 29, 31–38. [Google Scholar] [CrossRef]
- Wu, S.-K.; Tsai, C.-L.; Huang, Y.; Hynynen, K. Focused Ultrasound and Microbubbles-Mediated Drug Delivery to Brain Tumor. Pharmaceutics 2020, 13, 15. [Google Scholar] [CrossRef]
- McMahon, D.; Hynynen, K. Acute Inflammatory Response Following Increased Blood-Brain Barrier Permeability Induced by Focused Ultrasound Is Dependent on Microbubble Dose. Theranostics 2017, 7, 3989–4000. [Google Scholar] [CrossRef]
- McMahon, D.; Mah, E.; Hynynen, K. Angiogenic Response of Rat Hippocampal Vasculature to Focused Ultrasound-Mediated Increases in Blood-Brain Barrier Permeability. Sci. Rep. 2018, 8, 12178. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Ye, D.; Chen, S.; Chen, H. Therapeutic Ultrasound-Enhanced Immune Checkpoint Inhibitor Therapy. Front. Phys. 2021, 9, 102. [Google Scholar] [CrossRef]
- Elias, W.J.; Huss, D.; Voss, T.; Loomba, J.; Khaled, M.; Zadicario, E.; Frysinger, R.C.; Sperling, S.A.; Wylie, S.; Monteith, S.J.; et al. A Pilot Study of Focused Ultrasound Thalamotomy for Essential Tremor. N. Engl. J. Med. 2013, 369, 640–648. [Google Scholar] [CrossRef] [PubMed]
- Elias, W.J.; Lipsman, N.; Ondo, W.G.; Ghanouni, P.; Kim, Y.G.; Lee, W.; Schwartz, M.; Hynynen, K.; Lozano, A.M.; Shah, B.B.; et al. A Randomized Trial of Focused Ultrasound Thalamotomy for Essential Tremor. N. Engl. J. Med. 2016, 375, 730–739. [Google Scholar] [CrossRef]
- Iorio-Morin, C.; Yamamoto, K.; Sarica, C.; Zemmar, A.; Levesque, M.; Brisebois, S.; Germann, J.; Loh, A.; Boutet, A.; Elias, G.J.B.; et al. Bilateral Focused Ultrasound Thalamotomy for Essential Tremor (BEST-FUS Phase 2 Trial). Mov. Disord. 2021, 36, 2653–2662. [Google Scholar] [CrossRef]
- Martin, E.; Jeanmonod, D.; Morel, A.; Zadicario, E.; Werner, B. High-Intensity Focused Ultrasound for Noninvasive Functional Neurosurgery. Ann. Neurol. 2009, 66, 858–861. [Google Scholar] [CrossRef]
- Silvestrini, M.T.; Ingham, E.S.; Mahakian, L.M.; Kheirolomoom, A.; Liu, Y.; Fite, B.Z.; Tam, S.M.; Tucci, S.T.; Watson, K.D.; Wong, A.W.; et al. Priming Is Key to Effective Incorporation of Image-Guided Thermal Ablation into Immunotherapy Protocols. JCI Insight 2017, 2, e90521. [Google Scholar] [CrossRef]
- Chavez, M.; Silvestrini, M.T.; Ingham, E.S.; Fite, B.Z.; Mahakian, L.M.; Tam, S.M.; Ilovitsh, A.; Monjazeb, A.M.; Murphy, W.J.; Hubbard, N.E.; et al. Distinct Immune Signatures in Directly Treated and Distant Tumors Result from TLR Adjuvants and Focal Ablation. Theranostics 2018, 8, 3611–3628. [Google Scholar] [CrossRef]
- Eranki, A.; Srinivasan, P.; Ries, M.; Kim, A.; Lazarski, C.A.; Rossi, C.T.; Khokhlova, T.D.; Wilson, E.; Knoblach, S.M.; Sharma, K.V.; et al. High-Intensity Focused Ultrasound (HIFU) Triggers Immune Sensitization of Refractory Murine Neuroblastoma to Checkpoint Inhibitor Therapy. Clin. Cancer Res. 2020, 26, 1152–1161. [Google Scholar] [CrossRef]
- Qu, S.; Worlikar, T.; Felsted, A.E.; Ganguly, A.; Beems, M.V.; Hubbard, R.; Pepple, A.L.; Kevelin, A.A.; Garavaglia, H.; Dib, J.; et al. Non-Thermal Histotripsy Tumor Ablation Promotes Abscopal Immune Responses That Enhance Cancer Immunotherapy. J. Immunother. Cancer 2020, 8, e000200. [Google Scholar] [CrossRef]
- Elhelf, I.A.S.; Albahar, H.; Shah, U.; Oto, A.; Cressman, E.; Almekkawy, M. High Intensity Focused Ultrasound: The Fundamentals, Clinical Applications and Research Trends. Diagn. Interv. Imaging 2018, 99, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Li, X.; Li, Y.; Zhang, J.; Zong, Z.; Zhang, H. Current Immunotherapies for Glioblastoma Multiforme. Front. Immunol. 2021, 11, 3890. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem Cell-Associated Heterogeneity in Glioblastoma Results from Intrinsic Tumor Plasticity Shaped by the Microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Wei, T.; Farbiak, L.; Johnson, L.T.; Dilliard, S.A.; Siegwart, D.J. Selective Organ Targeting (SORT) Nanoparticles for Tissue-Specific MRNA Delivery and CRISPR–Cas Gene Editing. Nat. Nanotechnol. 2020, 15, 313–320. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Trial Number | Study Design | Trial Details | Patient Number | Reference |
|---|---|---|---|---|
| NCT02017717 | Phase III R, PA | Comparison of the PD-1 inhibitor nivolumab, with and without the CTLA-4 inhibitor ipilimumab, versus the VEGF inhibitor bevacizumab. The primary outcome is overall survival. Study is in progress. | 530 | [18] |
| NCT03291314 | Phase II PA | A study of the combination of the anti-PD-L1 molecule avelumab and the VEGF inhibitor axitinib on the progression of GBM. The 6-month progression-free survival (PFS) was 18% (95% CI 4–33, n = 27), which did not meet the threshold for justifying further investigation. | 52 | [21] |
| NCT02336165 | Phase II PA | A study of the anti-PD-L1 molecule durvalumab in subjects with glioblastoma. Patients were enrolled into 5 non-comparative cohorts receiving either durvalumab monotherapy or durvalumab and bevacizumab combotherapy. MOS was 15.1 months (95% CI 12.0–18.4). | 159 | [22] |
| NCT01454596 | Phase I/II SA | A study to determine the safety and effects of CART-EGFRvIII therapy in patients with recurrent GBM. CAR T-cell therapy was given in combination with a synthetic IL-2 molecule, aldesleukin, and a lymphodepleting preparative regimen of cyclophosphamide and fludarabine. MOS was 6.9 months (IQR 2.8–10.0), with 3 instances of adverse effects. | 18 | [23] |
| NCT02454634 | Phase I SGA | A study to identify the safety and tolerability of the first in-human mutant IDH1 peptide vaccine in patients with WHO Grade III–IV gliomas. Vaccine-induced immune responses were observed in 93.3% of patients. No regime-limiting toxicity was observed. | 32 | [24] |
| NCT01250470 | Phase I SGA | A study of the side effects of a vaccine therapy directed against the tumorigenic molecule survivin, in combination with the synthetic granulocyte-macrophage colony-stimulating factor sargramostim. The therapy was well tolerated with no serious adverse events attributable to the therapy; 6 out of 8 immunologically evaluable patients developed immune responses to the vaccine. | 9 | [25] |
| NCT05163080 | Phase II R, PA, DB | A Phase II clinical trial analyzing whether an antisurvivin vaccine treatment combined with SOC TMZ treatment is better than TMZ treatment alone for GBM patients. The primary outcome is OS. The trial is in progress. | 265 | [26] |
| NCT00157703 | Phase I SGA | A study to determine the safety of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM. Three serious AEs were reported (seizures after administration), possibly related to G207 administration. The estimated median survival time from G207 inoculation was 7.5 months (95% CI 3.0–12.7). | 9 | [27] |
| NCT02457845 | Phase I SGA | A study to determine the safety of G207 treatment in combination with radiotherapy for pediatric patients with recurrent supratentorial brain tumors. Twenty Grade 1 AEs were reported, possibly related to G207. MOS was 12.2 months (95% CI 8.0–16.4) as of 6/2020. The trial is in progress. | 12 | [28] |
| NCT00589875 | Phase II SGA | A study to determine the safety and potential efficacy of adenoviral vector expressing HSV1-tk (aglatimagene besadonevac, AdV-tk) followed by valacyclovir in combination with the SOC treatment. MOS was 17.1 month for treatment + SOC vs. 13.5 months for SOC alone (p = 0.0417) | 52 | [29] |
| EudraCT 2004-000464-28 | Phase III R, PA | A study comparing the adenovirus vector-mediated delivery of HSV1-tk (AdV-tk) followed by IV ganciclovir with the SOC treatment versus SOC treatment alone in newly diagnosed GBM. No difference in MOS was found in the experimental (497 days, 95% CI 369–574) versus the control group (452 days, 95% CI 437–558) (HR 1.18, 95% CI 0.86–1.61, p = 0.31). | 250 | [30] |
| NCT01491893 | Phase I SA | A study to determine the maximum tolerated dose of a live attenuated polio–rhinovirus chimera (PVSRIPO) on GBM; 19% of patients treated with PVSRIPO had a Grade 3 or higher adverse event. | 61 | [31] |
| NCT02414165 | Phase II/III R, PA | A study of a gamma retroviral replicating vector encoding a yeast cytosine deaminase, vocimagene amiretrorepvec, combined with 5-fluorocytosine treatment versus SOC in recurrent GBM. MOS was 11.10 months for the experimental group compared to 12.22 months for the control group (HR = 1.06; 95% CI 0.83, 1.35; p = 0.62). | 58 | [32,33] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lechpammer, M.; Rao, R.; Shah, S.; Mirheydari, M.; Bhattacharya, D.; Koehler, A.; Toukam, D.K.; Haworth, K.J.; Pomeranz Krummel, D.; Sengupta, S. Advances in Immunotherapy for the Treatment of Adult Glioblastoma: Overcoming Chemical and Physical Barriers. Cancers 2022, 14, 1627. https://doi.org/10.3390/cancers14071627
Lechpammer M, Rao R, Shah S, Mirheydari M, Bhattacharya D, Koehler A, Toukam DK, Haworth KJ, Pomeranz Krummel D, Sengupta S. Advances in Immunotherapy for the Treatment of Adult Glioblastoma: Overcoming Chemical and Physical Barriers. Cancers. 2022; 14(7):1627. https://doi.org/10.3390/cancers14071627
Chicago/Turabian StyleLechpammer, Mirna, Rohan Rao, Sanjit Shah, Mona Mirheydari, Debanjan Bhattacharya, Abigail Koehler, Donatien Kamdem Toukam, Kevin J. Haworth, Daniel Pomeranz Krummel, and Soma Sengupta. 2022. "Advances in Immunotherapy for the Treatment of Adult Glioblastoma: Overcoming Chemical and Physical Barriers" Cancers 14, no. 7: 1627. https://doi.org/10.3390/cancers14071627
APA StyleLechpammer, M., Rao, R., Shah, S., Mirheydari, M., Bhattacharya, D., Koehler, A., Toukam, D. K., Haworth, K. J., Pomeranz Krummel, D., & Sengupta, S. (2022). Advances in Immunotherapy for the Treatment of Adult Glioblastoma: Overcoming Chemical and Physical Barriers. Cancers, 14(7), 1627. https://doi.org/10.3390/cancers14071627