
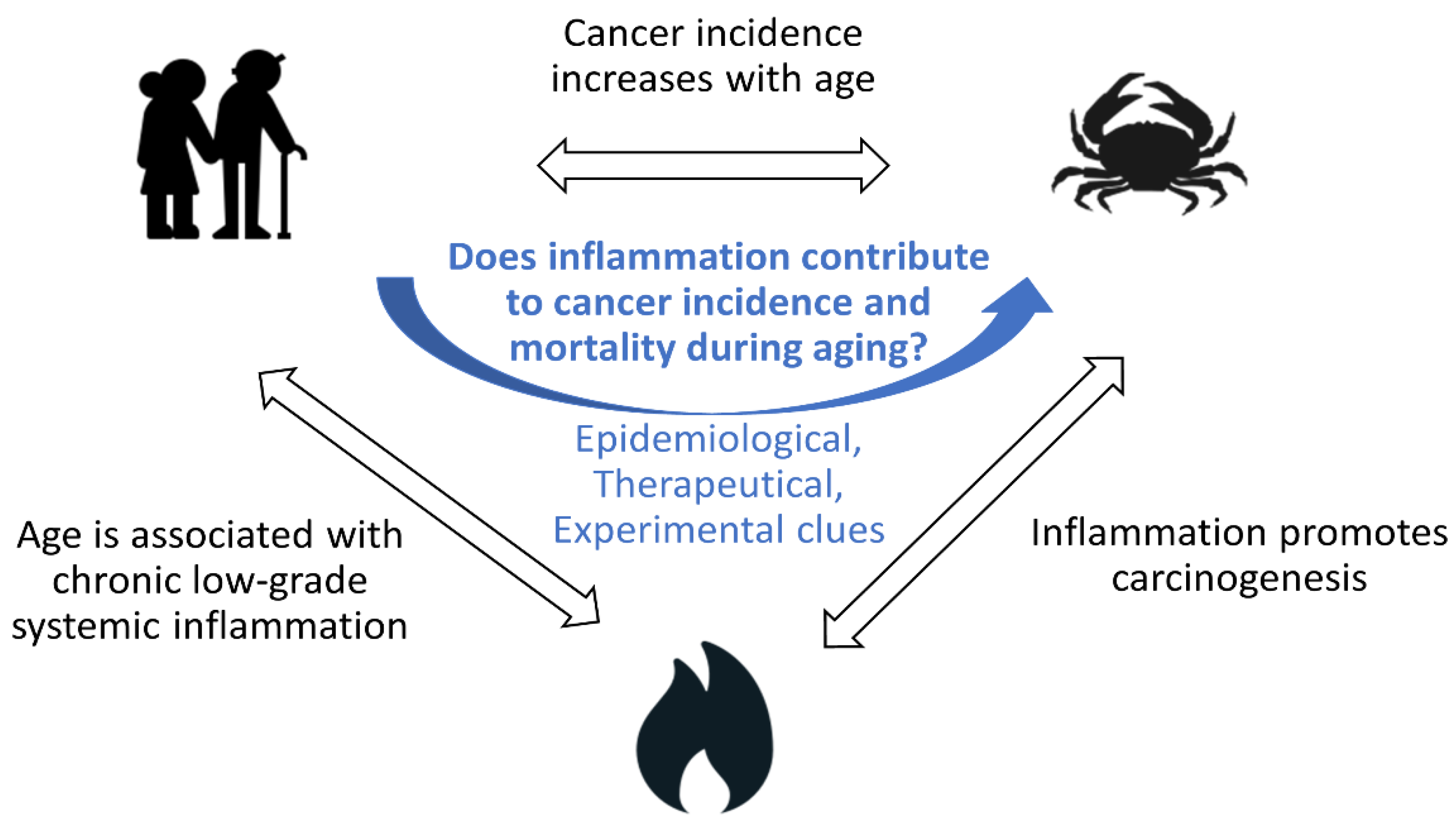
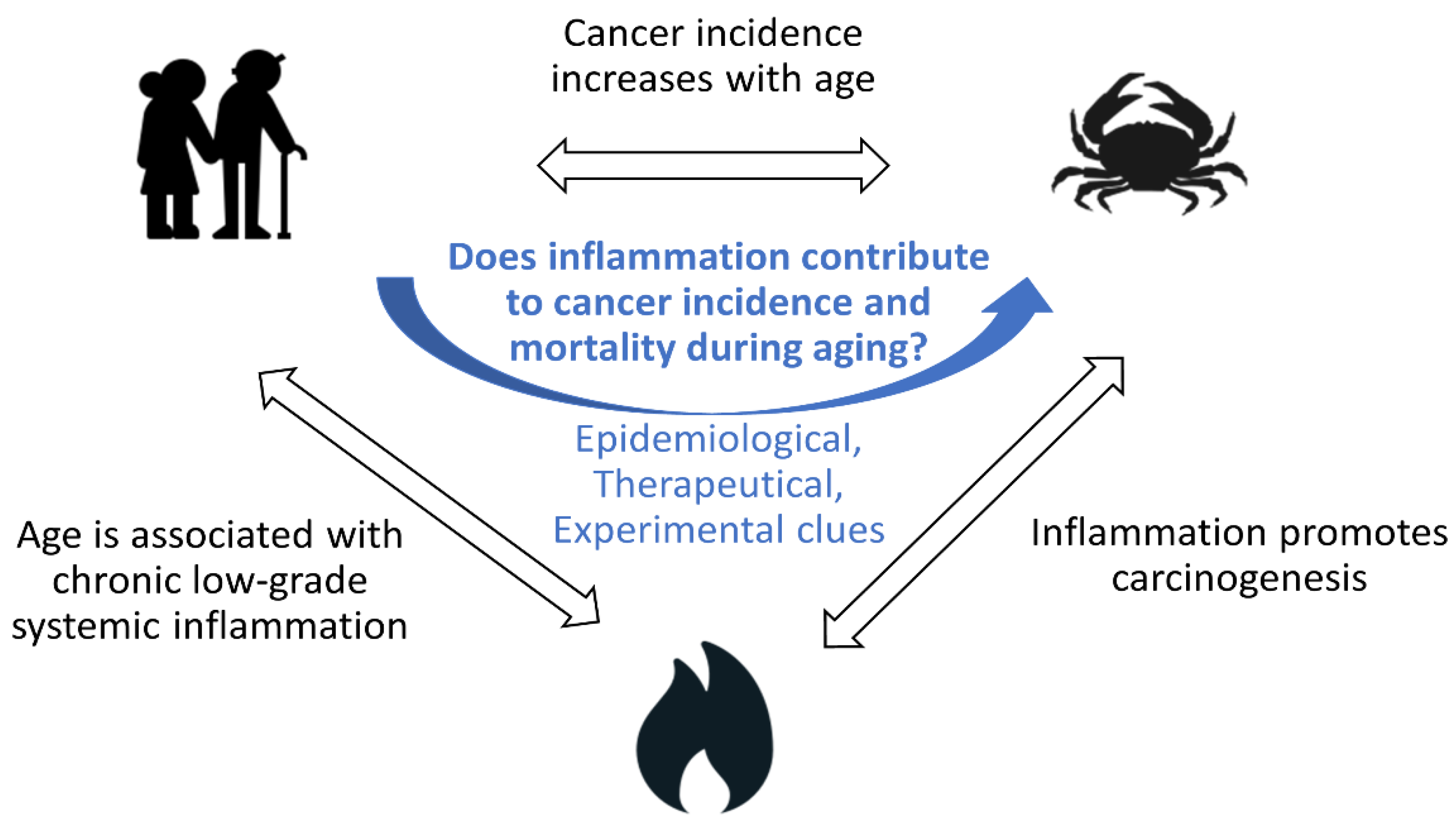
Does Inflammation Contribute to Cancer Incidence and Mortality during Aging? A Conceptual Review

 , ,
, ,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Cancer Incidence and Mortality Increase with Age
1.2. Aging Is Associated with Chronic Low-Grade Inflammation
1.3. Inflammation Contributes to Cancer Incidence
1.4. Does Inflammation Contribute to Cancer Incidence and Mortality during Aging?
2. Epidemiological Associations between Systemic Inflammation and Cancer Incidence in Older People
2.1. Inflammatory Biomarkers and Cancer Incidence: Summary of Meta-Analyses
2.2. Inflammatory Biomarkers and Cancer Mortality: Summary of Single Studies
2.3. Limitations of These Studies
2.4. Inflammatory Markers Are Associated with Clinical Outcomes among Cancer Patients
3. Therapeutic Approaches Targeting Inflammation to Reduce Cancer Incidence
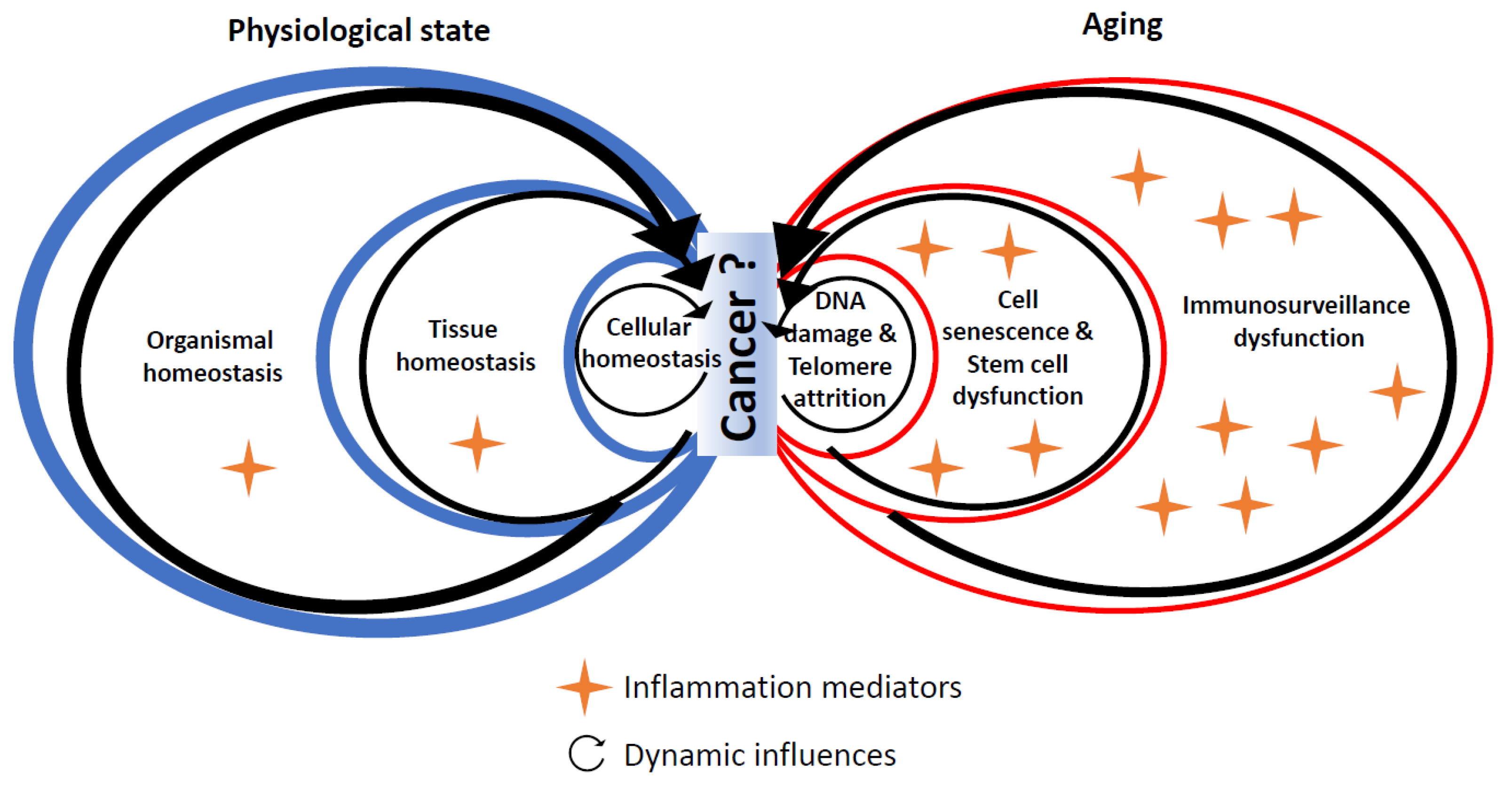
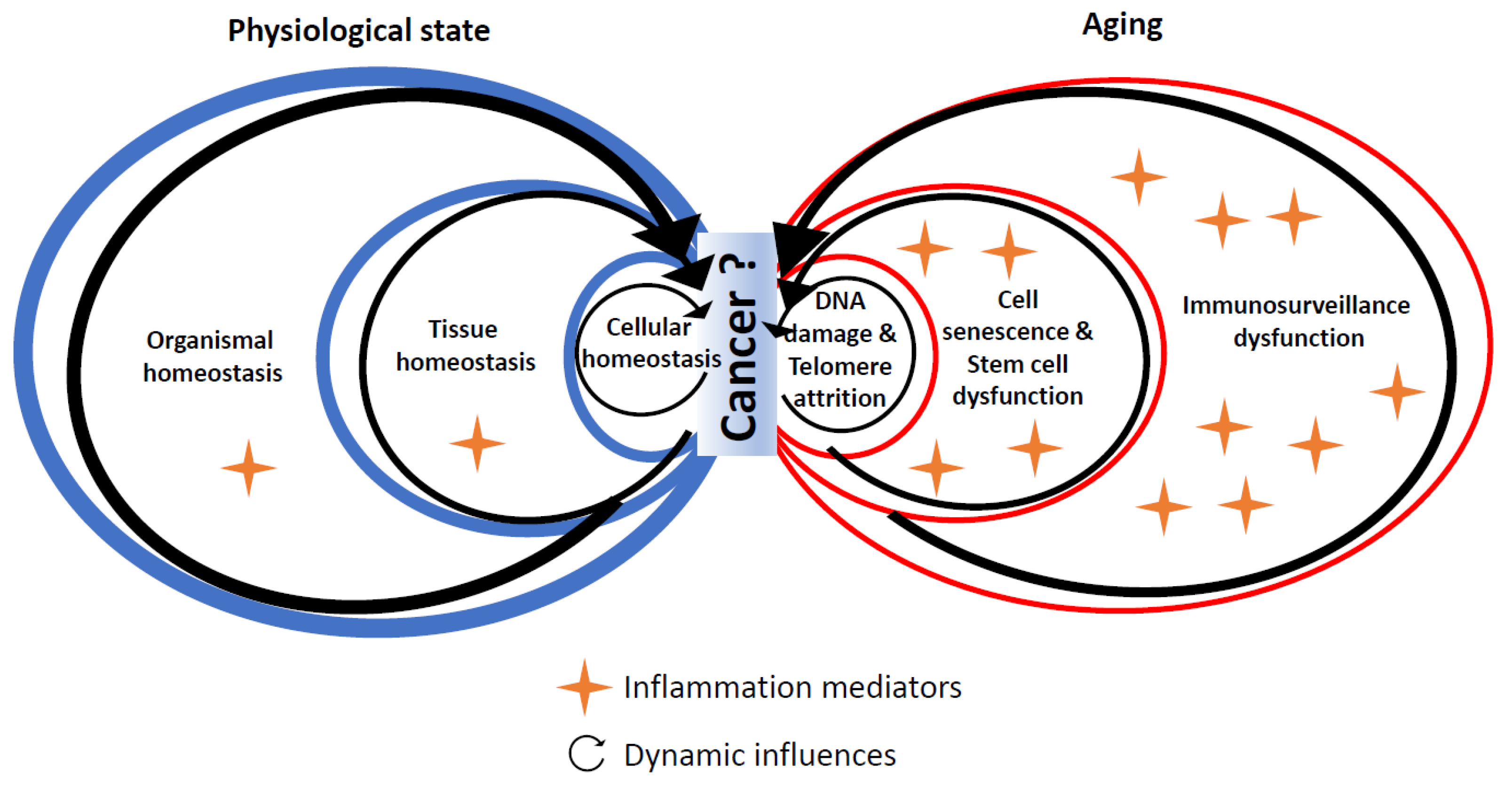
4. Experimental Evidence Linking Hallmarks of Biological Aging and Cancer through Inflammation
4.1. DNA Damage
4.2. Telomere Shortening
4.3. Cell Senescence
4.4. Stem Cell Dysfunction
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- SEER Cancer Stat Facts. Available online: https://seer.cancer.gov/statfacts/index.html (accessed on 6 August 2021).
- Global Health Estimates. Available online: https://www.who.int/data/global-health-estimates (accessed on 16 December 2021).
- Niccoli, T.; Partridge, L. Ageing as a Risk Factor for Disease. Curr. Biol. 2012, 22, R741–R752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partridge, L.; Deelen, J.; Slagboom, P.E. Facing up to the Global Challenges of Ageing. Nature 2018, 561, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.K.; Berger, S.L.; Brunet, A.; Campisi, J.; Cuervo, A.M.; Epel, E.S.; Franceschi, C.; Lithgow, G.J.; Morimoto, R.I.; Pessin, J.E.; et al. Geroscience: Linking Aging to Chronic Disease. Cell 2014, 159, 709–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, G.C.; Accardi, G.; Monastero, R.; Nicoletti, F.; Libra, M. Ageing: From Inflammation to Cancer. Immun. Ageing 2018, 15, 1. [Google Scholar] [CrossRef]
- Bottazzi, B.; Riboli, E.; Mantovani, A. Aging, Inflammation and Cancer. Semin. Immunol. 2018, 40, 74–82. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and Physiological Roles of Inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic Inflammation in the Etiology of Disease across the Life Span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [Green Version]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging. An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Roubenoff, R.; Harris, T.B.; Abad, L.W.; Wilson, P.W.F.; Dallal, G.E.; Dinarello, C.A. Monocyte Cytokine Production in an Elderly Population: Effect of Age and Inflammation. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 1998, 53A, M20–M26. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The Origins of Age-Related Proinflammatory State. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahluwalia, N.; Mastro, A.M.; Ball, R.; Miles, M.P.; Rajendra, R.; Handte, G. Cytokine Production by Stimulated Mononuclear Cells Did Not Change with Aging in Apparently Healthy, Well-Nourished Women. Mech. Ageing Dev. 2001, 122, 1269–1279. [Google Scholar] [CrossRef] [Green Version]
- Beharka, A.A.; Meydani, M.; Wu, D.; Leka, L.S.; Meydani, A.; Meydani, S.N. Interleukin-6 Production Does Not Increase with Age. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B81–B88. [Google Scholar] [CrossRef] [PubMed]
- Puzianowska-Kuźnicka, M.; Owczarz, M.; Wieczorowska-Tobis, K.; Nadrowski, P.; Chudek, J.; Slusarczyk, P.; Skalska, A.; Jonas, M.; Franek, E.; Mossakowska, M. Interleukin-6 and C-Reactive Protein, Successful Aging, and Mortality: The PolSenior Study. Immun. Ageing 2016, 13, 21. [Google Scholar] [CrossRef] [Green Version]
- Morrisette-Thomas, V.; Cohen, A.A.; Fülöp, T.; Riesco, É.; Legault, V.; Li, Q.; Milot, E.; Dusseault-Bélanger, F.; Ferrucci, L. Inflamm-Aging Does Not Simply Reflect Increases in pro-Inflammatory Markers. Mech. Ageing Dev. 2014, 139, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Varadhan, R.; Yao, W.; Matteini, A.; Beamer, B.A.; Xue, Q.-L.; Yang, H.; Manwani, B.; Reiner, A.; Jenny, N.; Parekh, N.; et al. Simple Biologically Informed Inflammatory Index of Two Serum Cytokines Predicts 10 Year All-Cause Mortality in Older Adults. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 165–173. [Google Scholar] [CrossRef]
- Ferrucci, L.; Fabbri, E. Inflammageing: Chronic Inflammation in Ageing, Cardiovascular Disease, and Frailty. Nat. Rev. Cardiol. 2018, 15, 505–522. [Google Scholar] [CrossRef]
- Soysal, P.; Stubbs, B.; Lucato, P.; Luchini, C.; Solmi, M.; Peluso, R.; Sergi, G.; Isik, A.T.; Manzato, E.; Maggi, S.; et al. Inflammation and Frailty in the Elderly: A Systematic Review and Meta-Analysis. Ageing Res. Rev. 2016, 31, 1–8. [Google Scholar] [CrossRef]
- Cohen, H.J.; Pieper, C.F.; Harris, T.; Rao, K.M.; Currie, M.S. The Association of Plasma IL-6 Levels with Functional Disability in Community-Dwelling Elderly. J. Gerontol. A Biol. Sci. Med. Sci. 1997, 52, M201–M208. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, L.; Harris, T.B.; Guralnik, J.M.; Tracy, R.P.; Corti, M.C.; Cohen, H.J.; Penninx, B.; Pahor, M.; Wallace, R.; Havlik, R.J. Serum IL-6 Level and the Development of Disability in Older Persons. J. Am. Geriatr. Soc. 1999, 47, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Kuo, H.-K.; Bean, J.F.; Yen, C.-J.; Leveille, S.G. Linking C-Reactive Protein to Late-Life Disability in the National Health and Nutrition Examination Survey (NHANES) 1999–2002. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 380–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youm, Y.-H.; Kanneganti, T.-D.; Vandanmagsar, B.; Zhu, X.; Ravussin, A.; Adijiang, A.; Owen, J.S.; Thomas, M.J.; Francis, J.; Parks, J.S.; et al. The Nlrp3 Inflammasome Promotes Age-Related Thymic Demise and Immunosenescence. Cell Rep. 2012, 1, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Colotta, F.; Allavena, P.; Sica, A.; Garlanda, C.; Mantovani, A. Cancer-Related Inflammation, the Seventh Hallmark of Cancer: Links to Genetic Instability. Carcinogenesis 2009, 30, 1073–1081. [Google Scholar] [CrossRef] [Green Version]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, Inflammation, and Cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [Green Version]
- Meira, L.B.; Bugni, J.M.; Green, S.L.; Lee, C.-W.; Pang, B.; Borenshtein, D.; Rickman, B.H.; Rogers, A.B.; Moroski-Erkul, C.A.; McFaline, J.L.; et al. DNA Damage Induced by Chronic Inflammation Contributes to Colon Carcinogenesis in Mice. J. Clin. Investig. 2008, 118, 2516–2525. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-Related Inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Karin, M. Dangerous Liaisons: STAT3 and NF-KappaB Collaboration and Crosstalk in Cancer. Cytokine Growth Factor Rev. 2010, 21, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, D.X.; Bos, P.D.; Massagué, J. Metastasis: From Dissemination to Organ-Specific Colonization. Nat. Rev. Cancer. 2009, 9, 274–284. [Google Scholar] [CrossRef]
- Caruso, C.; Lio, D.; Cavallone, L.; Franceschi, C. Aging, Longevity, Inflammation, and Cancer. Ann. N. Y. Acad. Sci. 2004, 1028, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, Cellular Senescence, and Cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, D.S.M.; Bandera, E.V.; Greenwood, D.C.; Norat, T. Circulating C-Reactive Protein and Breast Cancer Risk—Systematic Literature Review and Meta-Analysis of Prospective Cohort Studies. Cancer Epidemiol. Biomark. Prev. 2015, 24, 1439–1449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, L.; Liu, S.; Zhang, S.; Chen, Q.; Zhang, M.; Quan, P.; Lu, J.; Sun, X. C-Reactive Protein and Risk of Breast Cancer: A Systematic Review and Meta-Analysis. Sci. Rep. 2015, 5, 10508. [Google Scholar] [CrossRef] [PubMed]
- Kakourou, A.; Koutsioumpa, C.; Lopez, D.S.; Hoffman-Bolton, J.; Bradwin, G.; Rifai, N.; Helzlsouer, K.J.; Platz, E.A.; Tsilidis, K.K. Interleukin-6 and Risk of Colorectal Cancer: Results from the CLUE II Cohort and a Meta-Analysis of Prospective Studies. Cancer Causes Control 2015, 26, 1449–1460. [Google Scholar] [CrossRef] [Green Version]
- Tsilidis, K.K.; Branchini, C.; Guallar, E.; Helzlsouer, K.J.; Erlinger, T.P.; Platz, E.A. C-Reactive Protein and Colorectal Cancer Risk: A Systematic Review of Prospective Studies. Int. J. Cancer 2008, 123, 1133–1140. [Google Scholar] [CrossRef]
- Guo, Y.-Z.; Pan, L.; Du, C.-J.; Ren, D.-Q.; Xie, X.-M. Association Between C-Reactive Protein and Risk of Cancer: A Meta-Analysis of Prospective Cohort Studies. Asian Pac. J. Cancer Prev. 2013, 14, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Ghuman, S.; Van Hemelrijck, M.; Garmo, H.; Holmberg, L.; Malmström, H.; Lambe, M.; Hammar, N.; Walldius, G.; Jungner, I.; Wulaningsih, W. Serum Inflammatory Markers and Colorectal Cancer Risk and Survival. Br. J. Cancer 2017, 116, 1358–1365. [Google Scholar] [CrossRef] [Green Version]
- Wulaningsih, W.; Holmberg, L.; Garmo, H.; Malmstrom, H.; Lambe, M.; Hammar, N.; Walldius, G.; Jungner, I.; Van Hemelrijck, M. Prediagnostic Serum Inflammatory Markers in Relation to Breast Cancer Risk, Severity at Diagnosis and Survival in Breast Cancer Patients. Carcinogenesis 2015, 36, 1121–1128. [Google Scholar] [CrossRef]
- Margolis, K.L.; Rodabough, R.J.; Thomson, C.A.; Lopez, A.M.; McTiernan, A. Women’s Health Initiative Research Group Prospective Study of Leukocyte Count as a Predictor of Incident Breast, Colorectal, Endometrial, and Lung Cancer and Mortality in Postmenopausal Women. Arch. Intern. Med. 2007, 167, 1837–1844. [Google Scholar] [CrossRef]
- Il’yasova, D.; Colbert, L.H.; Harris, T.B.; Newman, A.B.; Bauer, D.C.; Satterfield, S.; Kritchevsky, S.B. Circulating Levels of Inflammatory Markers and Cancer Risk in the Health Aging and Body Composition Cohort. Cancer Epidemiol. Biomark. Prev. 2005, 14, 2413–2418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proctor, M.J.; McMillan, D.C.; Horgan, P.G.; Fletcher, C.D.; Talwar, D.; Morrison, D.S. Systemic Inflammation Predicts All-Cause Mortality: A Glasgow Inflammation Outcome Study. PLoS ONE 2015, 10, e0116206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watts, E.L.; Perez-Cornago, A.; Kothari, J.; Allen, N.E.; Travis, R.C.; Key, T.J. Hematologic Markers and Prostate Cancer Risk: A Prospective Analysis in UK Biobank. Cancer Epidemiol. Biomark. Prev. 2020, 29, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Goyal, A.; Terry, M.B.; Jin, Z.; Siegel, A.B. C-Reactive Protein and Colorectal Cancer Mortality in U.S. Adults. Cancer Epidemiol. Biomark. Prev. 2014, 23, 1609–1618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agnoli, C.; Grioni, S.; Pala, V.; Allione, A.; Matullo, G.; Gaetano, C.D.; Tagliabue, G.; Sieri, S.; Krogh, V. Biomarkers of Inflammation and Breast Cancer Risk: A Case-Control Study Nested in the EPIC-Varese Cohort. Sci. Rep. 2017, 7, 12708. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Ridker, P.; Lorenzatti, A.; Krum, H.; Varigos, J.; et al. Effect of Interleukin-1β Inhibition with Canakinumab on Incident Lung Cancer in Patients with Atherosclerosis: Exploratory Results from a Randomised, Double-Blind, Placebo-Controlled Trial. Lancet 2017, 390, 1833–1842. [Google Scholar] [CrossRef]
- Wong, C.C.; Baum, J.; Silvestro, A.; Beste, M.T.; Bharani-Dharan, B.; Xu, S.; Wang, Y.A.; Wang, X.; Prescott, M.F.; Krajkovich, L.; et al. Inhibition of IL1β by Canakinumab May Be Effective against Diverse Molecular Subtypes of Lung Cancer: An Exploratory Analysis of the CANTOS Trial. Cancer Res. 2020, 80, 5597–5605. [Google Scholar] [CrossRef]
- McMillan, D.C. The Systemic Inflammation-Based Glasgow Prognostic Score: A Decade of Experience in Patients with Cancer. Cancer Treat. Rev. 2013, 39, 534–540. [Google Scholar] [CrossRef]
- Martin, L.; Muscaritoli, M.; Bourdel-Marchasson, I.; Kubrak, C.; Laird, B.; Gagnon, B.; Chasen, M.; Gioulbasanis, I.; Wallengren, O.; Voss, A.C.; et al. Diagnostic Criteria for Cancer Cachexia: Reduced Food Intake and Inflammation Predict Weight Loss and Survival in an International, Multi-Cohort Analysis. J. Cachexia Sarcopenia Muscle 2021, 12, 1189–1202. [Google Scholar] [CrossRef]
- Brattinga, B.; Rutgers, A.; De Haan, J.J.; Absalom, A.R.; van der Wal-Huisman, H.; de Bock, G.H.; van Leeuwen, B.L. Preoperative Inflammatory Markers as a Predictor of Three-Year Overall Survival in Older Cancer Patients Undergoing Oncologic Surgery. Cancers 2021, 13, 1824. [Google Scholar] [CrossRef]
- Flossmann, E.; Rothwell, P.M. British Doctors Aspirin Trial and the UK-TIA Aspirin Trial Effect of Aspirin on Long-Term Risk of Colorectal Cancer: Consistent Evidence from Randomised and Observational Studies. Lancet 2007, 369, 1603–1613. [Google Scholar] [CrossRef]
- Taketo, M.M. Cyclooxygenase-2 Inhibitors in Tumorigenesis (Part II). J. Natl. Cancer Inst. 1998, 90, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; DuBois, R.N. Up-Regulation of Cyclooxygenase 2 Gene Expression in Human Colorectal Adenomas and Adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar] [CrossRef]
- Brasky, T.M.; Liu, J.; White, E.; Peters, U.; Potter, J.D.; Walter, R.B.; Baik, C.S.; Lane, D.S.; Manson, J.E.; Vitolins, M.Z.; et al. Non-Steroidal Anti-Inflammatory Drugs and Cancer Risk in Women: Results from the Women’s Health Initiative. Int. J. Cancer 2014, 135, 1869–1883. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Wilson, M.; Elwin, C.-E.; Norrving, B.; Algra, A.; Warlow, C.P.; Meade, T.W. Long-Term Effect of Aspirin on Colorectal Cancer Incidence and Mortality: 20-Year Follow-up of Five Randomised Trials. Lancet 2010, 376, 1741–1750. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Price, J.F.; Fowkes, F.G.R.; Zanchetti, A.; Roncaglioni, M.C.; Tognoni, G.; Lee, R.; Belch, J.F.F.; Wilson, M.; Mehta, Z.; et al. Short-Term Effects of Daily Aspirin on Cancer Incidence, Mortality, and Non-Vascular Death: Analysis of the Time Course of Risks and Benefits in 51 Randomised Controlled Trials. Lancet 2012, 379, 1602–1612. [Google Scholar] [CrossRef]
- Rothwell, P.M.; Fowkes, F.G.R.; Belch, J.F.F.; Ogawa, H.; Warlow, C.P.; Meade, T.W. Effect of Daily Aspirin on Long-Term Risk of Death Due to Cancer: Analysis of Individual Patient Data from Randomised Trials. Lancet 2011, 377, 31–41. [Google Scholar] [CrossRef]
- Algra, A.M.; Rothwell, P.M. Effects of Regular Aspirin on Long-Term Cancer Incidence and Metastasis: A Systematic Comparison of Evidence from Observational Studies versus Randomised Trials. Lancet Oncol. 2012, 13, 518–527. [Google Scholar] [CrossRef]
- Langley, R.E.; Burdett, S.; Tierney, J.F.; Cafferty, F.; Parmar, M.K.B.; Venning, G. Aspirin and Cancer: Has Aspirin Been Overlooked as an Adjuvant Therapy? Br. J. Cancer 2011, 105, 1107–1113. [Google Scholar] [CrossRef] [Green Version]
- Sankaranarayanan, R.; Kumar, D.R.; Altinoz, M.A.; Bhat, G.J. Mechanisms of Colorectal Cancer Prevention by Aspirin-A Literature Review and Perspective on the Role of COX-Dependent and -Independent Pathways. Int. J. Mol.Sci. 2020, 21, 9018. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem Cell Divisions, Somatic Mutations, Cancer Etiology, and Cancer Prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soria-Valles, C.; López-Soto, A.; Osorio, F.G.; López-Otín, C. Immune and Inflammatory Responses to DNA Damage in Cancer and Aging. Mech. Ageing Dev. 2017, 165, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Blasco, M.A. Telomere Length, Stem Cells and Aging. Nat. Chem. Biol. 2007, 3, 640–649. [Google Scholar] [CrossRef]
- Armanios, M.; Alder, J.K.; Parry, E.M.; Karim, B.; Strong, M.A.; Greider, C.W. Short Telomeres Are Sufficient to Cause the Degenerative Defects Associated with Aging. Am. J. Hum. Genet 2009, 85, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Tomás-Loba, A.; Flores, I.; Fernández-Marcos, P.J.; Cayuela, M.L.; Maraver, A.; Tejera, A.; Borrás, C.; Matheu, A.; Klatt, P.; Flores, J.M.; et al. Telomerase Reverse Transcriptase Delays Aging in Cancer-Resistant Mice. Cell 2008, 135, 609–622. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, K.L.; Chang, S.; Lee, H.W.; Blasco, M.; Gottlieb, G.J.; Greider, C.; DePinho, R.A. Longevity, Stress Response, and Cancer in Aging Telomerase-Deficient Mice. Cell 1999, 96, 701–712. [Google Scholar] [CrossRef] [Green Version]
- Artandi, S.E.; Chang, S.; Lee, S.L.; Alson, S.; Gottlieb, G.J.; Chin, L.; DePinho, R.A. Telomere Dysfunction Promotes Non-Reciprocal Translocations and Epithelial Cancers in Mice. Nature 2000, 406, 641–645. [Google Scholar] [CrossRef]
- Lex, K.; Maia Gil, M.; Lopes-Bastos, B.; Figueira, M.; Marzullo, M.; Giannetti, K.; Carvalho, T.; Ferreira, M.G. Telomere Shortening Produces an Inflammatory Environment That Increases Tumor Incidence in Zebrafish. Proc. Natl. Acad. Sci. USA 2020, 117, 15066–15074. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally Occurring P16(Ink4a)-Positive Cells Shorten Healthy Lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Deursen, J.M. Senolytic Therapies for Healthy Longevity. Science 2019, 364, 636–637. [Google Scholar] [CrossRef] [PubMed]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.-H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal Senescence Establishes an Immunosuppressive Microenvironment That Drives Tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef] [PubMed]
- Pribluda, A.; Elyada, E.; Wiener, Z.; Hamza, H.; Goldstein, R.E.; Biton, M.; Burstain, I.; Morgenstern, Y.; Brachya, G.; Billauer, H.; et al. A Senescence-Inflammatory Switch from Cancer-Inhibitory to Cancer-Promoting Mechanism. Cancer Cell 2013, 24, 242–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondar, T.; Medzhitov, R. The Origins of Tumor-Promoting Inflammation. Cancer Cell 2013, 24, 143–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grants, J.M.; Wegrzyn, J.; Hui, T.; O’Neill, K.; Shadbolt, M.; Knapp, D.J.H.F.; Parker, J.; Deng, Y.; Gopal, A.; Docking, T.R.; et al. Altered MicroRNA Expression Links IL6 and TNF-Induced Inflammaging with Myeloid Malignancy in Humans and Mice. Blood 2020, 135, 2235–2251. [Google Scholar] [CrossRef]
- Henry, C.J.; Casás-Selves, M.; Kim, J.; Zaberezhnyy, V.; Aghili, L.; Daniel, A.E.; Jimenez, L.; Azam, T.; McNamee, E.N.; Clambey, E.T.; et al. Aging-Associated Inflammation Promotes Selection for Adaptive Oncogenic Events in B Cell Progenitors. J. Clin. Investig. 2015, 125, 4666–4680. [Google Scholar] [CrossRef]
- Farber, D.L. Tissues, Not Blood, Are Where Immune Cells Function. Nature 2021, 593, 506–509. [Google Scholar] [CrossRef]
- Reuter, S.; Gupta, S.C.; Chaturvedi, M.M.; Aggarwal, B.B. Oxidative Stress, Inflammation, and Cancer: How Are They Linked? Free Radic. Biol. Med. 2010, 49, 1603–1616. [Google Scholar] [CrossRef] [Green Version]
- Kepp, O.; Galluzzi, L.; Kroemer, G. Mitochondrial Control of the NLRP3 Inflammasome. Nat. Immunol. 2011, 12, 199–200. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The Immunobiology of Cancer Immunosurveillance and Immunoediting. Immunity 2004, 21, 137–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fulop, T.; Dupuis, G.; Baehl, S.; Le Page, A.; Bourgade, K.; Frost, E.; Witkowski, J.M.; Pawelec, G.; Larbi, A.; Cunnane, S. From Inflamm-Aging to Immune-Paralysis: A Slippery Slope during Aging for Immune-Adaptation. Biogerontology 2016, 17, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Nikolich-Žugich, J. The Twilight of Immunity: Emerging Concepts in Aging of the Immune System. Nat. Immunol. 2018, 19, 10–19. [Google Scholar] [CrossRef]
- Briceño, O.; Lissina, A.; Wanke, K.; Afonso, G.; von Braun, A.; Ragon, K.; Miquel, T.; Gostick, E.; Papagno, L.; Stiasny, K.; et al. Reduced Naïve CD8(+) T-Cell Priming Efficacy in Elderly Adults. Aging Cell 2016, 15, 14–21. [Google Scholar] [CrossRef]
- Fulop, T.; Kotb, R.; Fortin, C.F.; Pawelec, G.; de Angelis, F.; Larbi, A. Potential Role of Immunosenescence in Cancer Development. Ann. N. Y. Acad. Sci. 2010, 1197, 158–165. [Google Scholar] [CrossRef]
- Iannello, A.; Thompson, T.W.; Ardolino, M.; Lowe, S.W.; Raulet, D.H. P53-Dependent Chemokine Production by Senescent Tumor Cells Supports NKG2D-Dependent Tumor Elimination by Natural Killer Cells. J. Exp. Med. 2013, 210, 2057–2069. [Google Scholar] [CrossRef]
- Robert, C. A Decade of Immune-Checkpoint Inhibitors in Cancer Therapy. Nat. Commun. 2020, 11, 3801. [Google Scholar] [CrossRef]
- Daste, A.; Domblides, C.; Gross-Goupil, M.; Chakiba, C.; Quivy, A.; Cochin, V.; de Mones, E.; Larmonier, N.; Soubeyran, P.; Ravaud, A. Immune Checkpoint Inhibitors and Elderly People: A Review. Eur. J. Cancer 2017, 82, 155–166. [Google Scholar] [CrossRef]
- Van Herck, Y.; Feyaerts, A.; Alibhai, S.; Papamichael, D.; Decoster, L.; Lambrechts, Y.; Pinchuk, M.; Bechter, O.; Herrera-Caceres, J.; Bibeau, F.; et al. Is Cancer Biology Different in Older Patients? Lancet Healthy Longev. 2021, 2, e663–e677. [Google Scholar] [CrossRef]
- Golomb, L.; Sagiv, A.; Pateras, I.S.; Maly, A.; Krizhanovsky, V.; Gorgoulis, V.G.; Oren, M.; Ben-Yehuda, A. Age-Associated Inflammation Connects RAS-Induced Senescence to Stem Cell Dysfunction and Epidermal Malignancy. Cell Death Differ. 2015, 22, 1764–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Cancer | Biomarker | N Participants/n Cases | Countries | Design | Mean of Median Age at Inclusion | Effect Size | Heterogeneity and Bias (for Meta-Analysis) | Follow-Up | Reference |
|---|---|---|---|---|---|---|---|---|---|
| Meta-analyses focused on incidence | |||||||||
| Breast | CRP | 69,610/3522 | America (n = 6)/Europe (n = 5)/Asia (n = 1) | 12 studies | 49–73 y 9 studies restricted to post-menopausal women | RR per doubling CRP concentration: 1.07 (95% CI: 1.02–1.12) 1.06 (1.01–1.11) for post-menopausal women | I² = 47%, Ph = 0.04 Egger’s tests showed some evidence of publication or small study bias (p = 0.08) | 5–13 y | [35] |
| Breast | CRP | /5286 | America (n = 6) Europe (n = 6)/Asia (n = 3) | 15 cohorts + case-control studies | 45–73 y | Combined OR per natural log unit change in CRP: 1.16 (95% CI: 1.06–1.27) Only significant in persons >60 y (6 studies) | I2 = 45.9% No evidence of publication bias, Begg’s (0.805) and Egger’s tests (0.172) | [36] | |
| Colorectal | IL6 | 8420/1308 | USA (n = 5), UK (n = 2) | 3 cohorts and 3 nested case-control studies | summary RR per natural log unit change in IL6: 1.22 (95 % CI 1.00–1.49) Inverse association was noted for rectal cancer (RR 0.69; 95 % CI 0.54–0.88) | Some evidence of heterogeneity (I2 = 46 %). No evidence of small study effect, Egger’s p 0.77 I2 = 0 %. Evidence for small-study effects (p 0.02). | [37] | ||
| Colorectal | CRP | 1159 cases/37,986 controls | USA (n = 3), Europe (n = 3), Japan (n = 2) | 3 cohort and 5 nested case-control studies | 53–73 y | Summary RR per one unit change in natural log-transformed high-sensitivity CRP: 1.13 (95% CI, 1.00–1.27) for colon cancer, and 1.06 (95% CI, 0.86–1.30) for rectal cancer Only significant in persons >60 y (3 studies) | Ph 0.16 (colon) and 0.02 (rectal) | 5.5–14 y | [38] |
| All | CRP | 194,796/11,459 | USA (n = 5), Europe (n = 6) | 11 cohort studies | 45–73 y | Pooled HR per natural log unit change in CRP: 1.105 (95% (CI): 1.033–1.178) for all cancers 1.308 (95% CI: 1.097–1.519) for lung cancer Not significant for breast, prostate and colorectal separately Only significant in persons >60 y (n = 24,000) | Substantial heterogeneity across studies (Ph = 0.000, I2 = 70.10%) | [39] | |
| Single studies focused on cancer mortality | |||||||||
| Colorectal | haptoglobin | 325,599/1467 | Sweden | Cohort | Mean (SD) 46 (14) | Adjusted HR (>1.2 vs. <0.9 g/L): 1.19; 95% CI: 1.01–1.41 | No significant association with CRP and albumin | 18 y (mean) | [40] |
| Breast | haptoglobin | 155,179/736 | Sweden | Cohort | Mean (SD) 50 (11) | Adjusted HR (>1.4 vs. <1.4 g/L): 1.27, 95% CI: 1.02–1.59 | No significant association with CRP and albumin | 18 y (mean) | [41] |
| Breast, lung, all | leukocytes | 143,748/3062 | USA | Cohort | 63 (50–79) | Adjusted HR: (higher vs. lower quartile) 2.16 (1.33–3.50) for breast 1.65 (1.29–2.12) for lung 1.33 (1.17–1.51) for all | Not significant for endometrial and colorectal | 8 y (mean) | [42] |
| All | CRP, IL6, TNF | 2438 | USA | Cohort | 73 (70–79) | Adjusted HR (1-unit increase on natural log scale): 1.63 (1.19–2.23) for IL-6 1.64 (1.20–2.24) for CRP 1.82 (1.14–2.92) for TNF | 5.5 y (mean) | [43] | |
| All | CRP, albumin, neutrophils | 160,481/13,173 | UK | Cohort | 35% > 65 y | Adjusted HR 1.85 for CRP > vs. <10 mg/L (p < 0.001) 2.08 for albumin < vs. >35 g/L (p < 0.001) Neutrophils > vs. < 7.5 × 109/L (p < 0.001) | 69 months (mean) | [44] | |
| Prostate | Leukocytes, neutrophils | 210,000/323 | UK | Cohort | Mean (SD) 57 (8) | Per one SD increase: HR = 1.14, 1.05–1.24 for leukocytes HR = 1.27, 1.09–1.48 for neutrophils | 6.8 y | [45] | |
| Colorectal | CRP | 16,000/92 | USA | Cohort | 50 | Adjusted HR = 3.96 (95% CI, 1.64–9.52) for levels >1.00 vs. <0.22 mg/dL | 14.2 y | [46] | |
| Cancer | Drug | Outcomes | N | Country | Design | Age at Inclusion | Reference |
|---|---|---|---|---|---|---|---|
| Randomized controlled trials | |||||||
| Lung/ All | Canakinumab | Reduction in
| 10,061 | Secondary analyses of a RCT | Mean (sd) 61 (10) | [48] | |
| Colorectal | Aspirin | Cancer incidence reduction (only after 10 years) No effect on incidence of other cancers | 5000 + 2500 | UK | 2 RCT | Mean (sd) 62 (7)/ 60 (9) | [53] |
| Colorectal | Aspirin | 20 y incidence and mortality reduction (colon but not rectum) Effect when treatment duration >5 years. No increase in benefit for dose >75 mg/d 2% absolute reduction of 20 y fatal cancer risk | 14,000 | UK, Sweden | Meta-analysis of 4 RCT | [57] | |
| All | Aspirin | Reduction in
| 51 RCT | [58] | |||
| All | Aspirin | Reduction in 20-year cancer death Benefit increased with age and duration of treatment (but unrelated to aspirin dose, starting at 75 mg) | 25,000 | 8 RCT | [59] | ||
| Observational studies | |||||||
| Colorectal | Aspirin or NSAID | Associated with lower colorectal cancer incidence Especially after 10 y of use, Aspirin dose: consistent effect only when dose > 300 mg/d No systematic effect of age on associations | 21,000 cases + 1,136,110 (6000 cases) | Meta- analysis 19 case-control studies + 11 cohorts | [53] | ||
| All (women) | NSAID (0 and 3 years) | Associated with lower colorectal, ovarian cancer and melanoma incidence (but not with total cancer incidence) | 1,290,000 | USA | Cohort | 50–79 yo | [56] |
| Observational studies and randomized controlled trials | |||||||
| All | Aspirin | Associated with reduced proportion of cancers with distant metastasis but not with any reduction in regional spread | 329 + 5984 | Meta-analyses of 3 RCT and 5 observational studies | [60] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerville, F.; Bourdel-Marchasson, I.; Déchanet-Merville, J.; Pellegrin, I.; Soubeyran, P.; Appay, V.; Lemoine, M. Does Inflammation Contribute to Cancer Incidence and Mortality during Aging? A Conceptual Review. Cancers 2022, 14, 1622. https://doi.org/10.3390/cancers14071622
Guerville F, Bourdel-Marchasson I, Déchanet-Merville J, Pellegrin I, Soubeyran P, Appay V, Lemoine M. Does Inflammation Contribute to Cancer Incidence and Mortality during Aging? A Conceptual Review. Cancers. 2022; 14(7):1622. https://doi.org/10.3390/cancers14071622
Chicago/Turabian StyleGuerville, Florent, Isabelle Bourdel-Marchasson, Julie Déchanet-Merville, Isabelle Pellegrin, Pierre Soubeyran, Victor Appay, and Maël Lemoine. 2022. "Does Inflammation Contribute to Cancer Incidence and Mortality during Aging? A Conceptual Review" Cancers 14, no. 7: 1622. https://doi.org/10.3390/cancers14071622
APA StyleGuerville, F., Bourdel-Marchasson, I., Déchanet-Merville, J., Pellegrin, I., Soubeyran, P., Appay, V., & Lemoine, M. (2022). Does Inflammation Contribute to Cancer Incidence and Mortality during Aging? A Conceptual Review. Cancers, 14(7), 1622. https://doi.org/10.3390/cancers14071622