
Ion Channel Drugs Suppress Cancer Phenotype in NG108-15 and U87 Cells: Toward Novel Electroceuticals for Glioblastoma

 ,
, 

Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Molecular Biology
2.4. Generation of Stable Lines
2.5. Growth and FUCCI Assays
2.6. Antibody Staining, BrdU and Senescence Assays
2.7. Dye Staining Protocols
2.8. Electrophysiology
3. Results
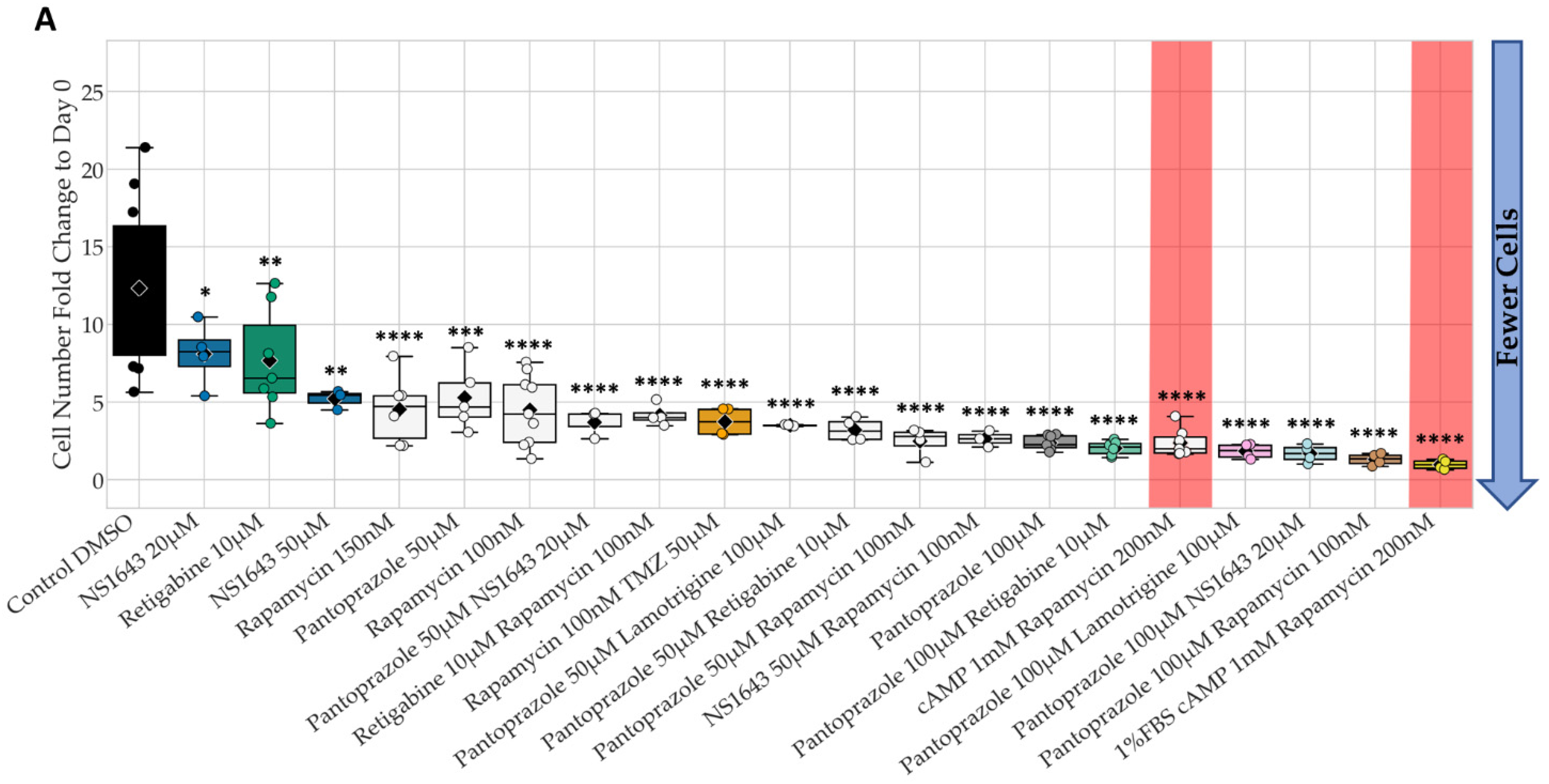
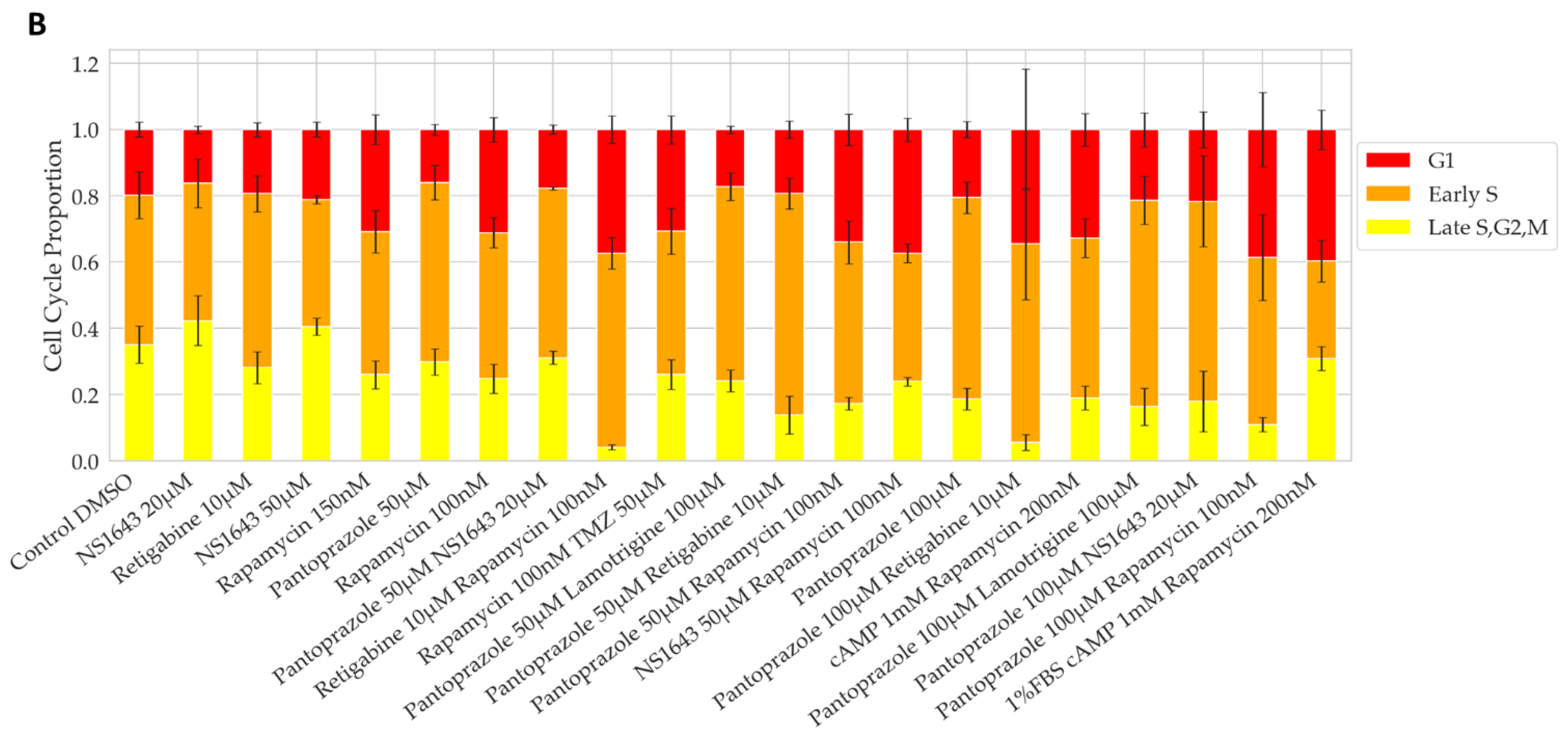
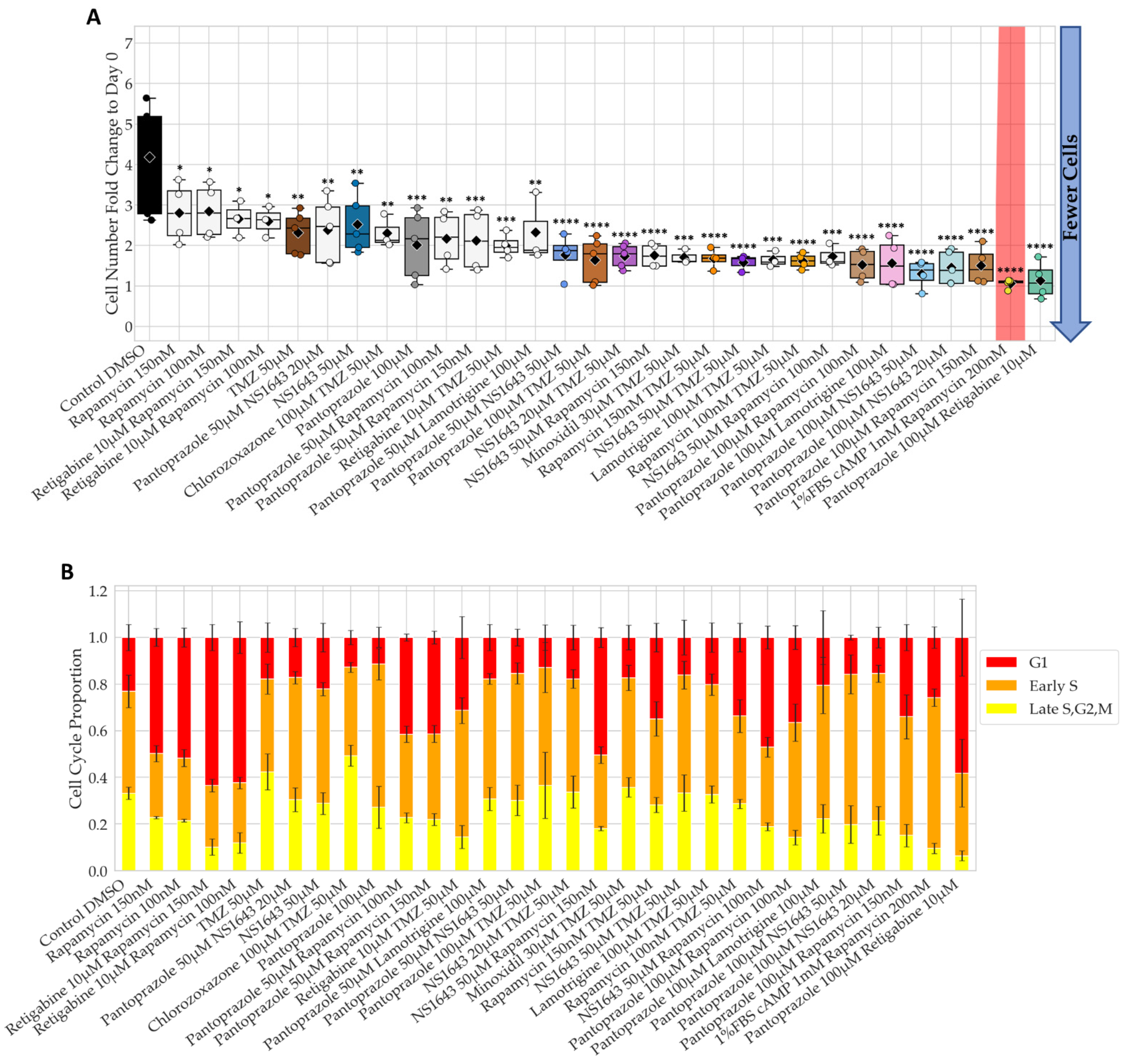
3.1. Bioelectric Compounds and Combinations with the Proton Pump Inhibitor, Pantoprazole Stop Proliferation of NG108-15 Cells in High Serum and Shift Proportion of Cells in Late S, G2, M
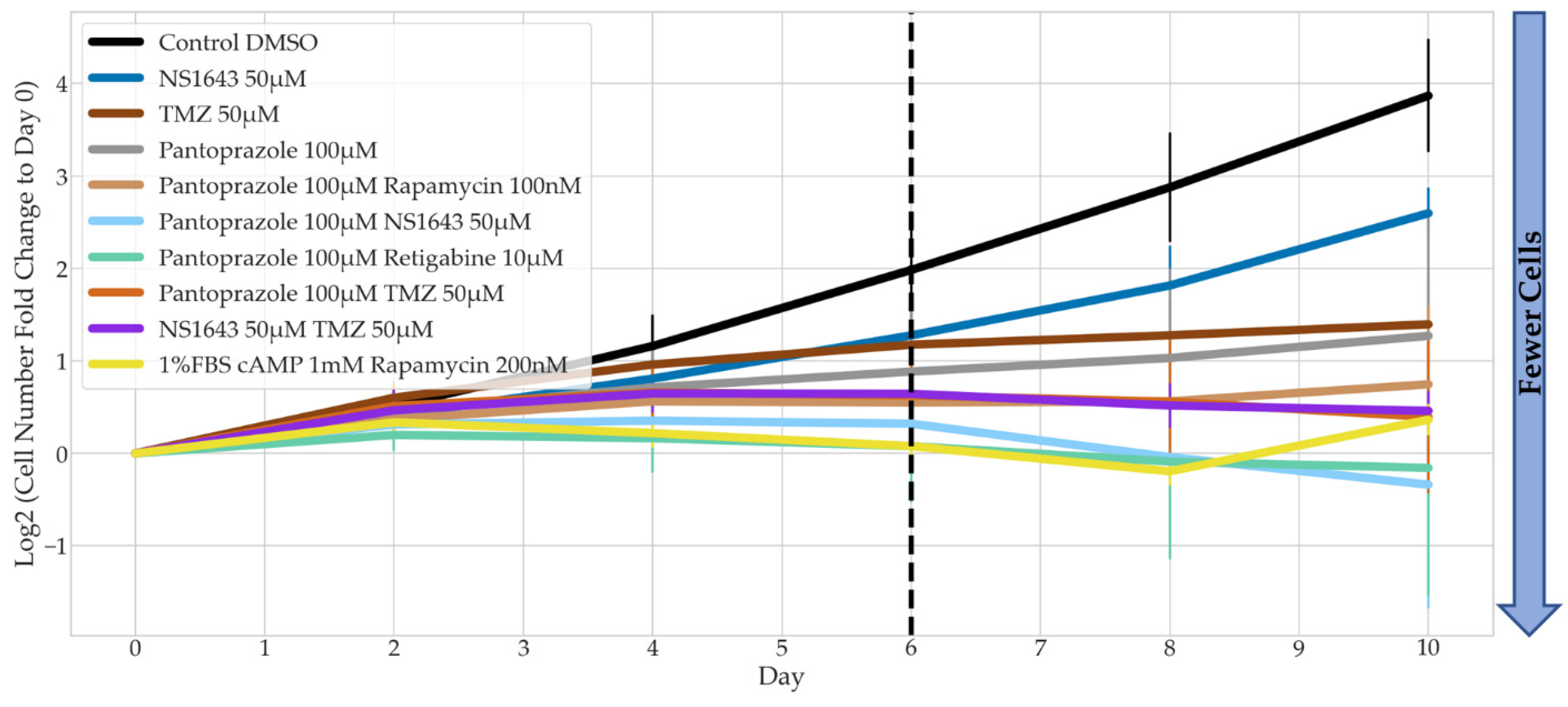
3.2. Bioelectric Drugs in Combination with Pantoprazole Have a Lasting Effect on NG108-15 Proliferation after Treatment Is Removed
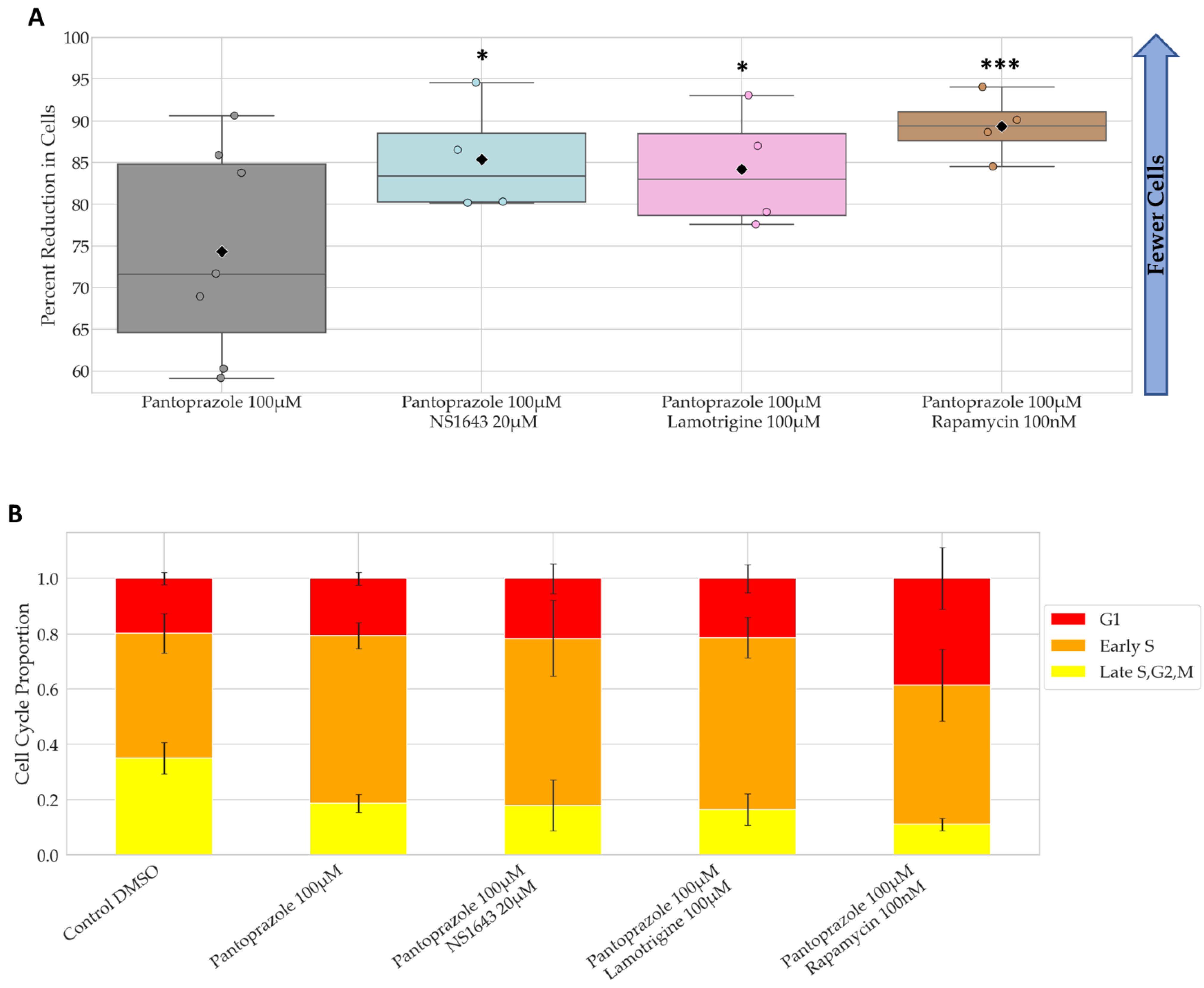
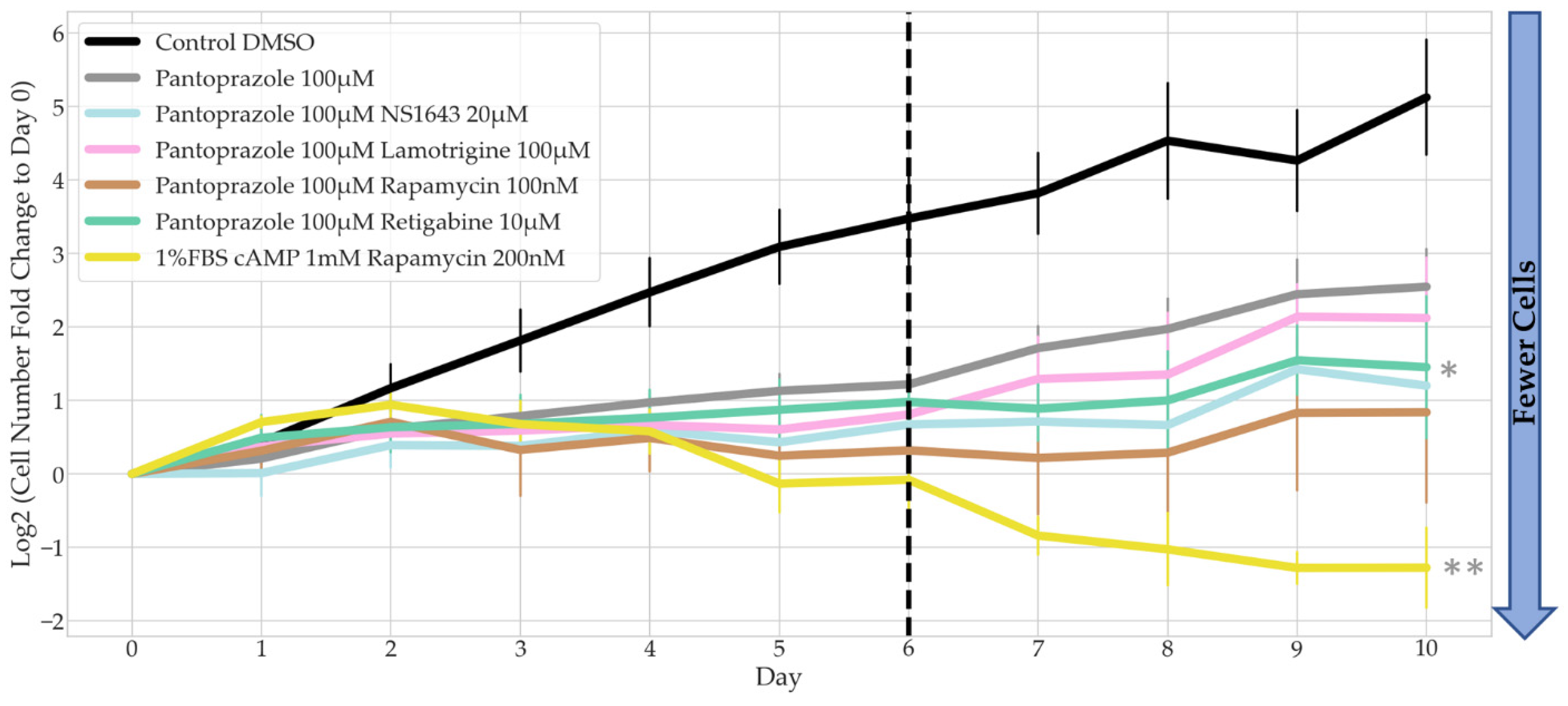
3.3. Treatments That Were Successful in Reducing the Proliferative Capacity of NG108-15 Cells Were Also Largely Successful in U87 Cells
3.4. Bioelectric Drug Combinations including Pantoprazole Reduce U87 Proliferation after Treatment Is Removed
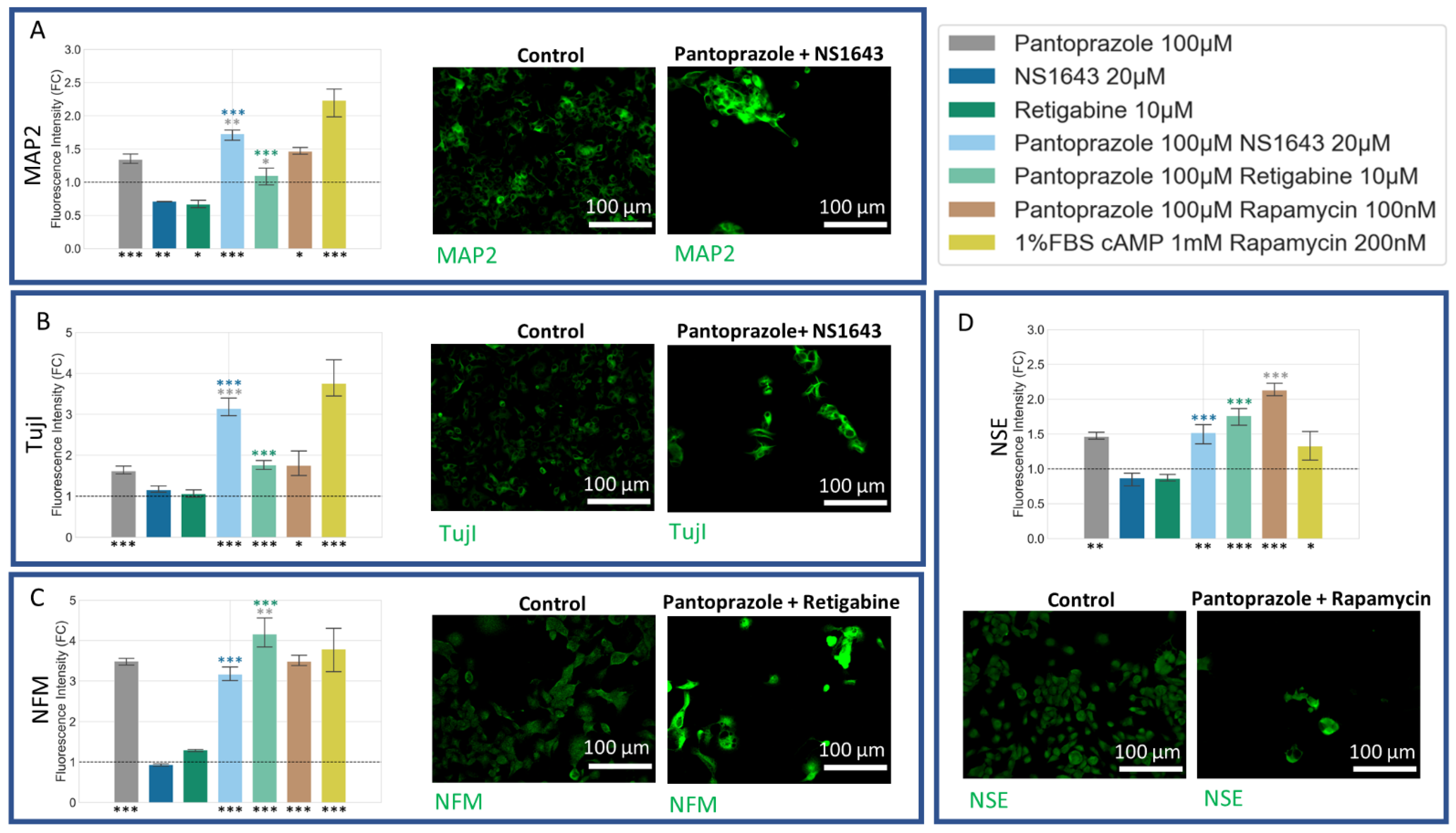
3.5. NG108-15 Cells Express Neuronal Markers with Drug Treatments after 6 Days
3.6. NG108-15 Cells Express Astrocyte and Other Differentiation Markers after Drug Treatments for 6 Days
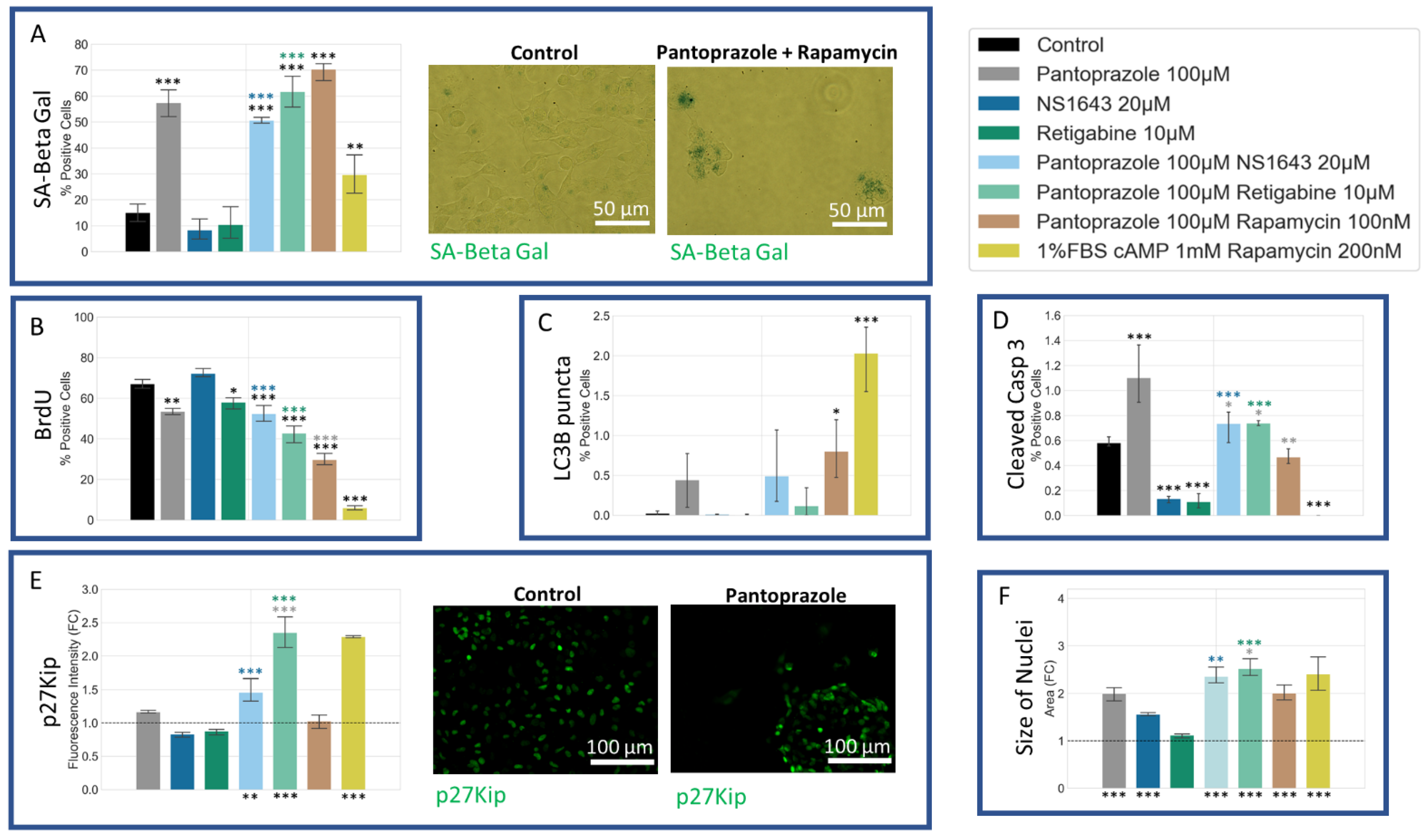
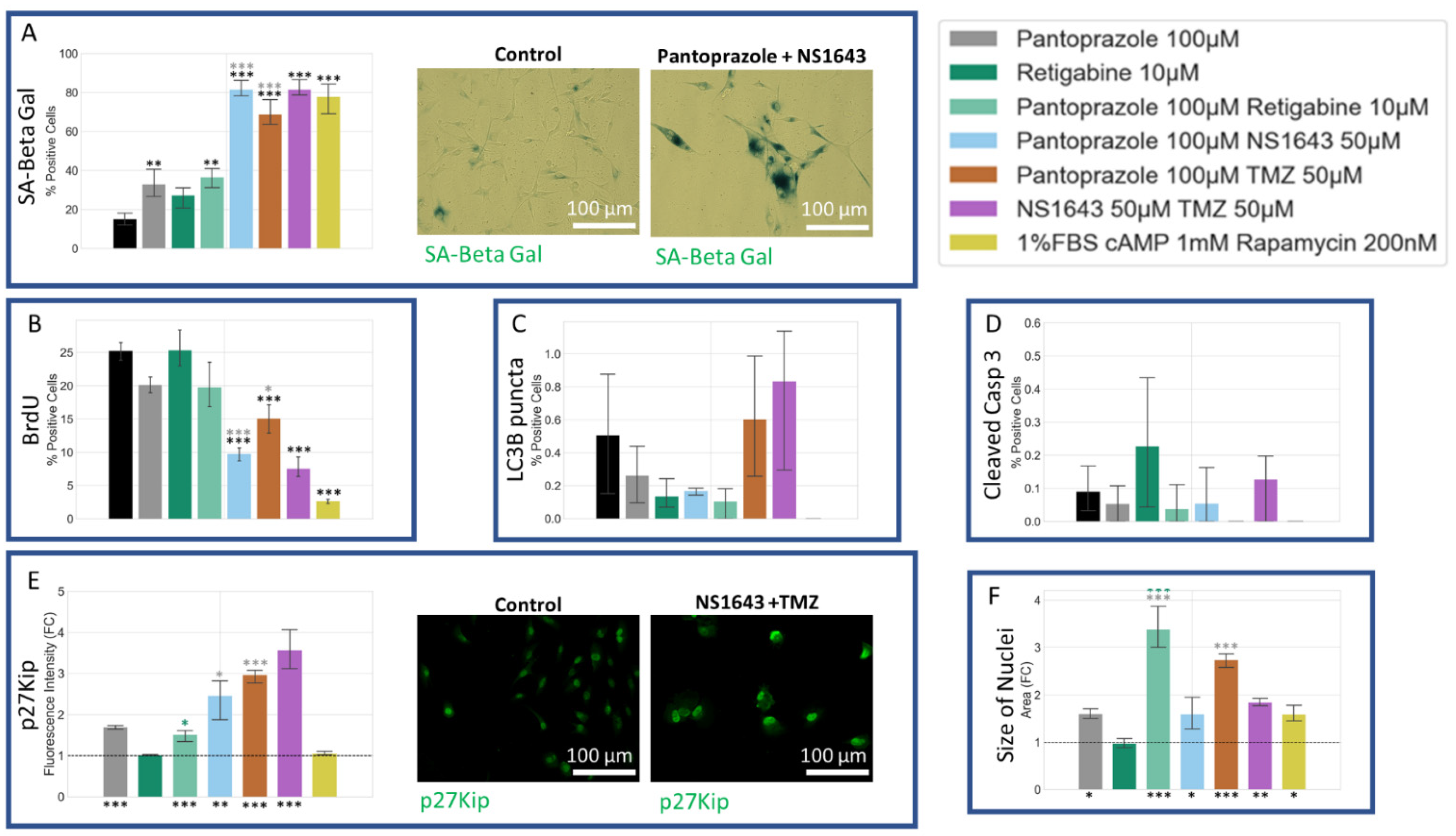
3.7. NG108-15 Cells Show an Increase in p27Kip1 and Senescence Markers When Treated with Pantoprazole Alone or in Combination with Hyperpolarizing Compounds
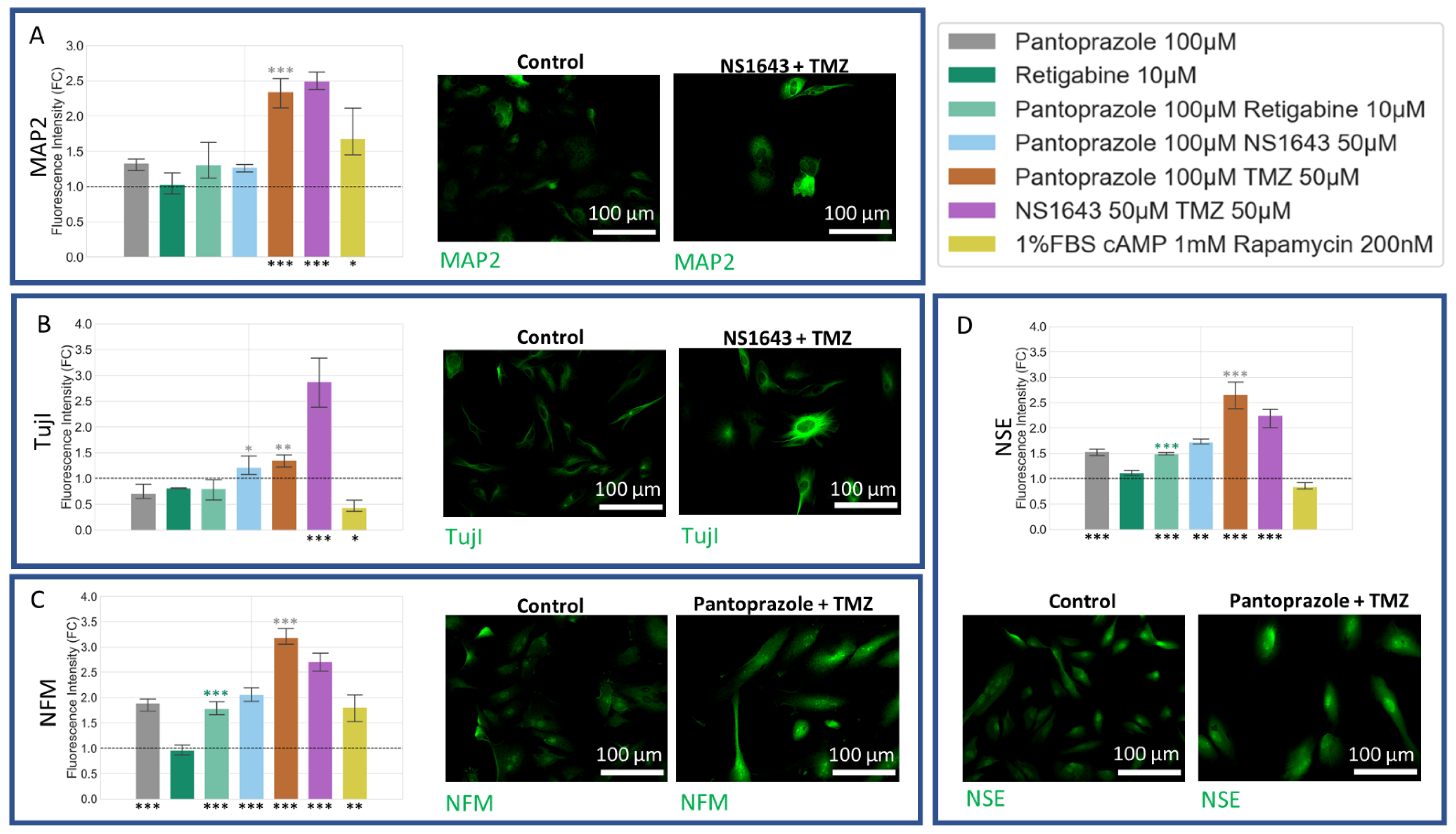
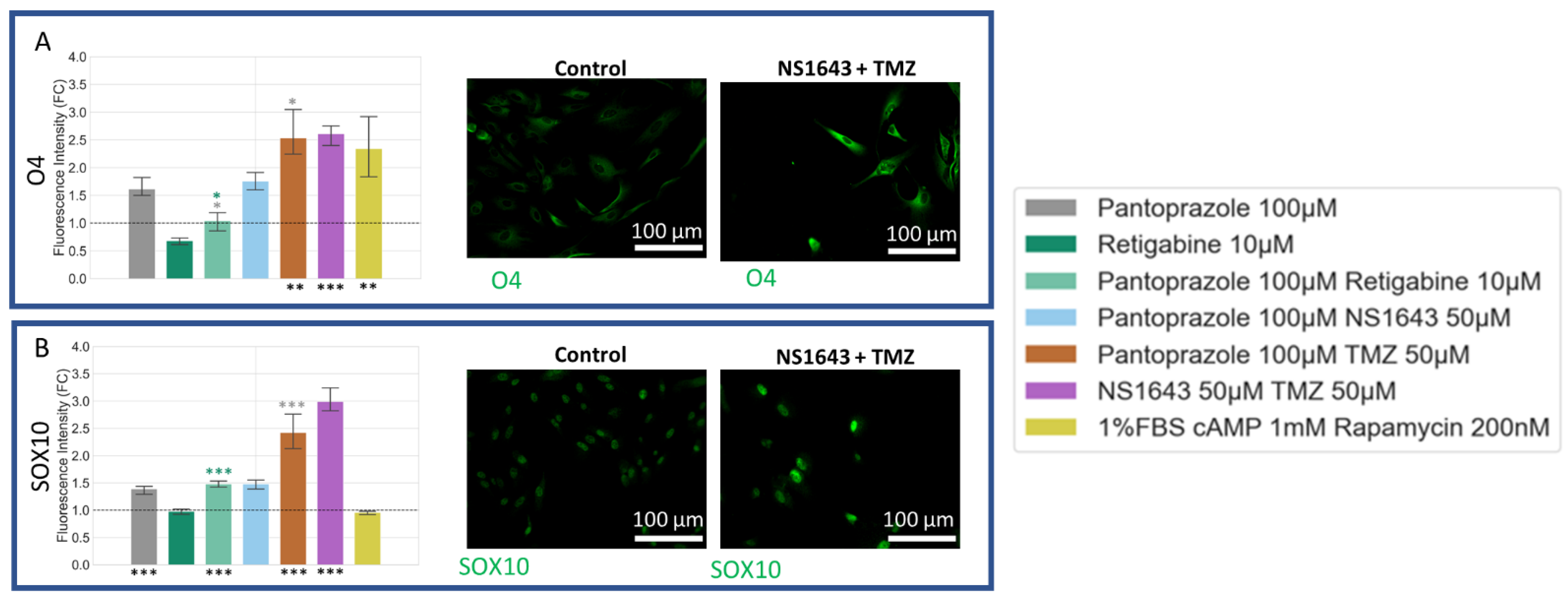
3.8. U87 Cells Show Increases in Neuronal Markers When Treated with Combination Treatments of Hyperpolarizing Drugs and Pantoprazole or NS1643 and TMZ
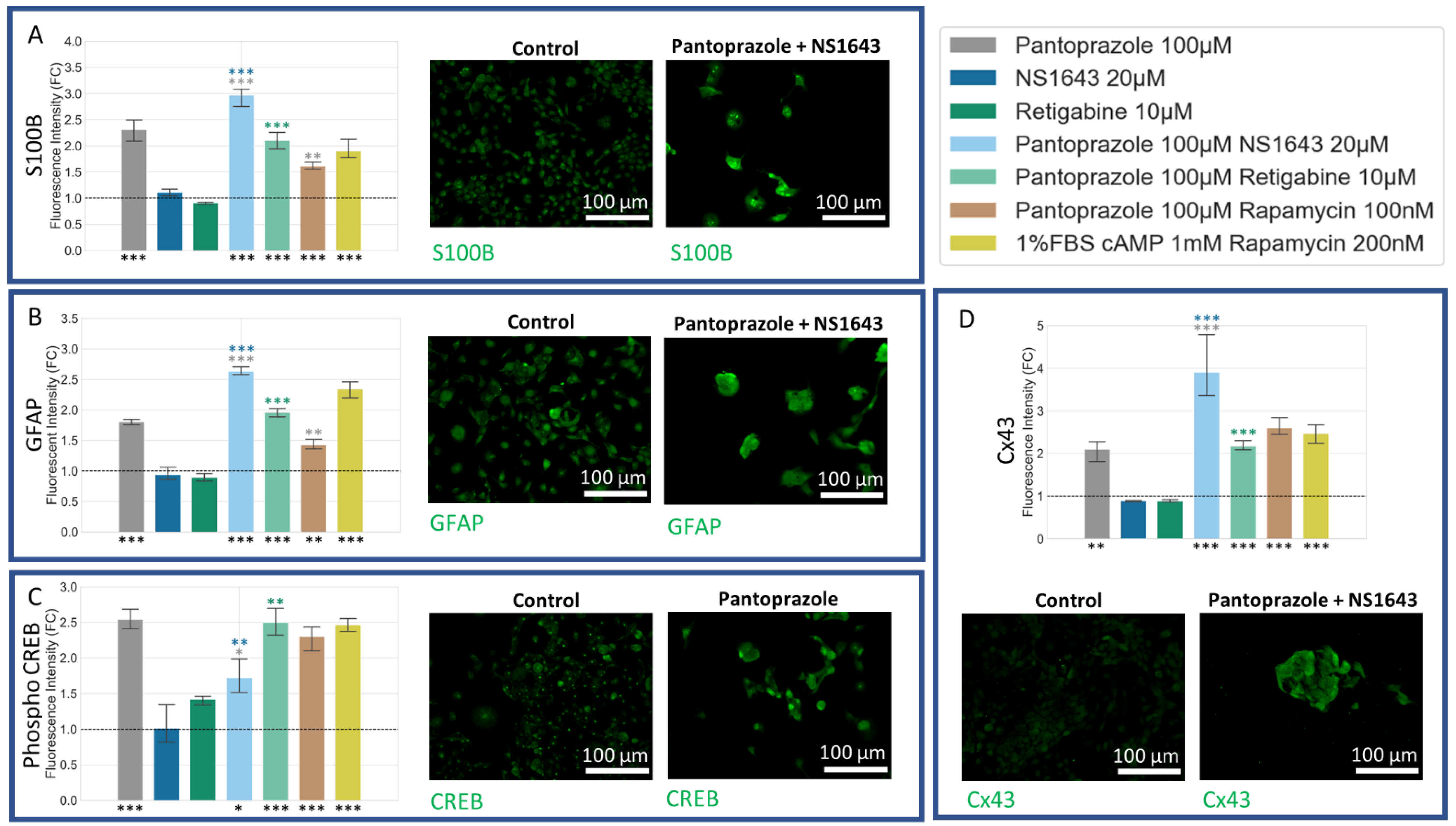
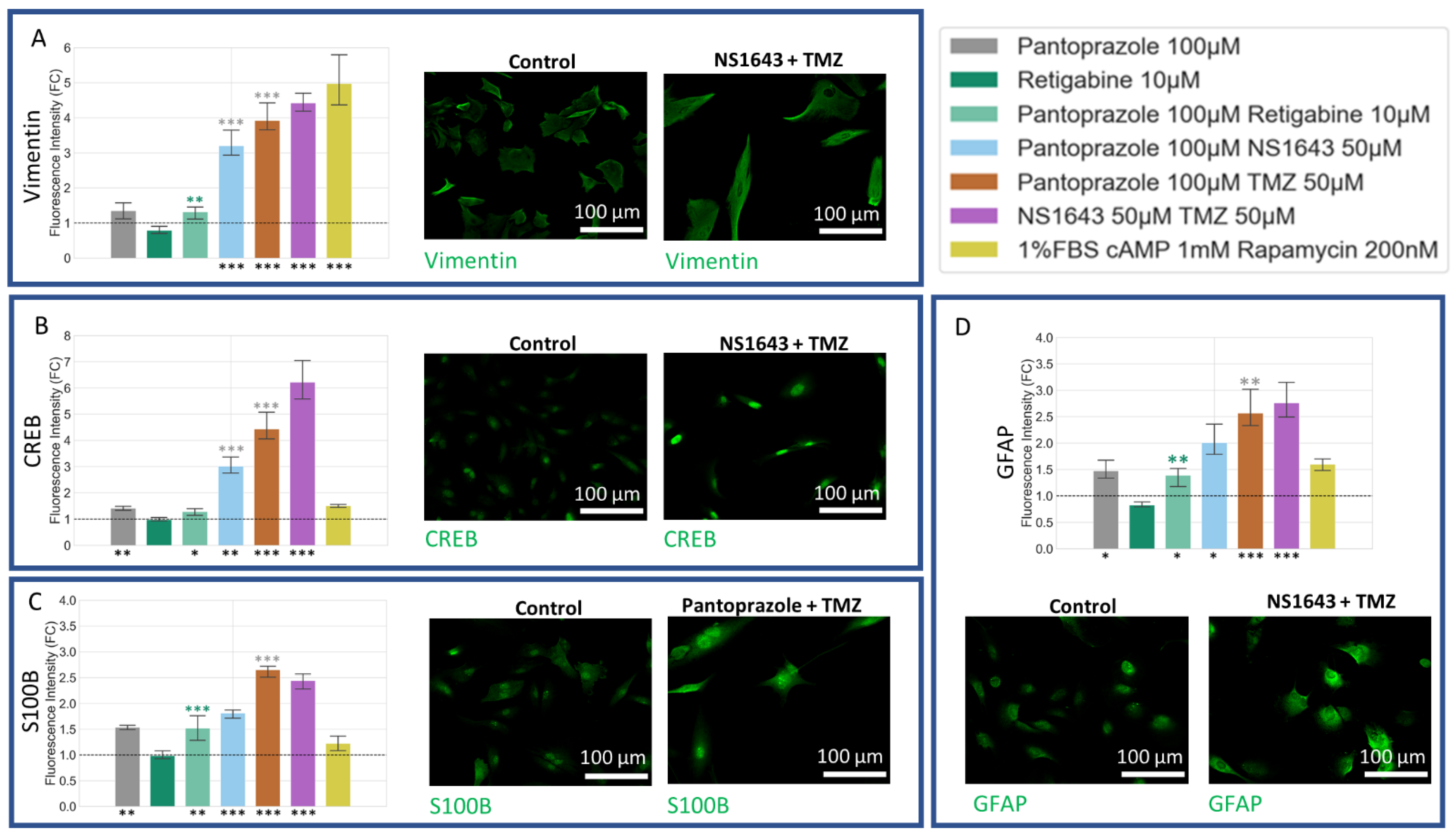
3.9. U87 Cells Also Show Increases in Astrocytic Markers When Treated with Combination Treatments of Hyperpolarizing Drugs and Pantoprazole or NS1643 and TMZ
3.10. U87 Cells Show Increase in Oligodendrocyte Markers When Treated with Pantoprazole in Combination with Hyperpolarizing Compounds or NS1643 and TMZ
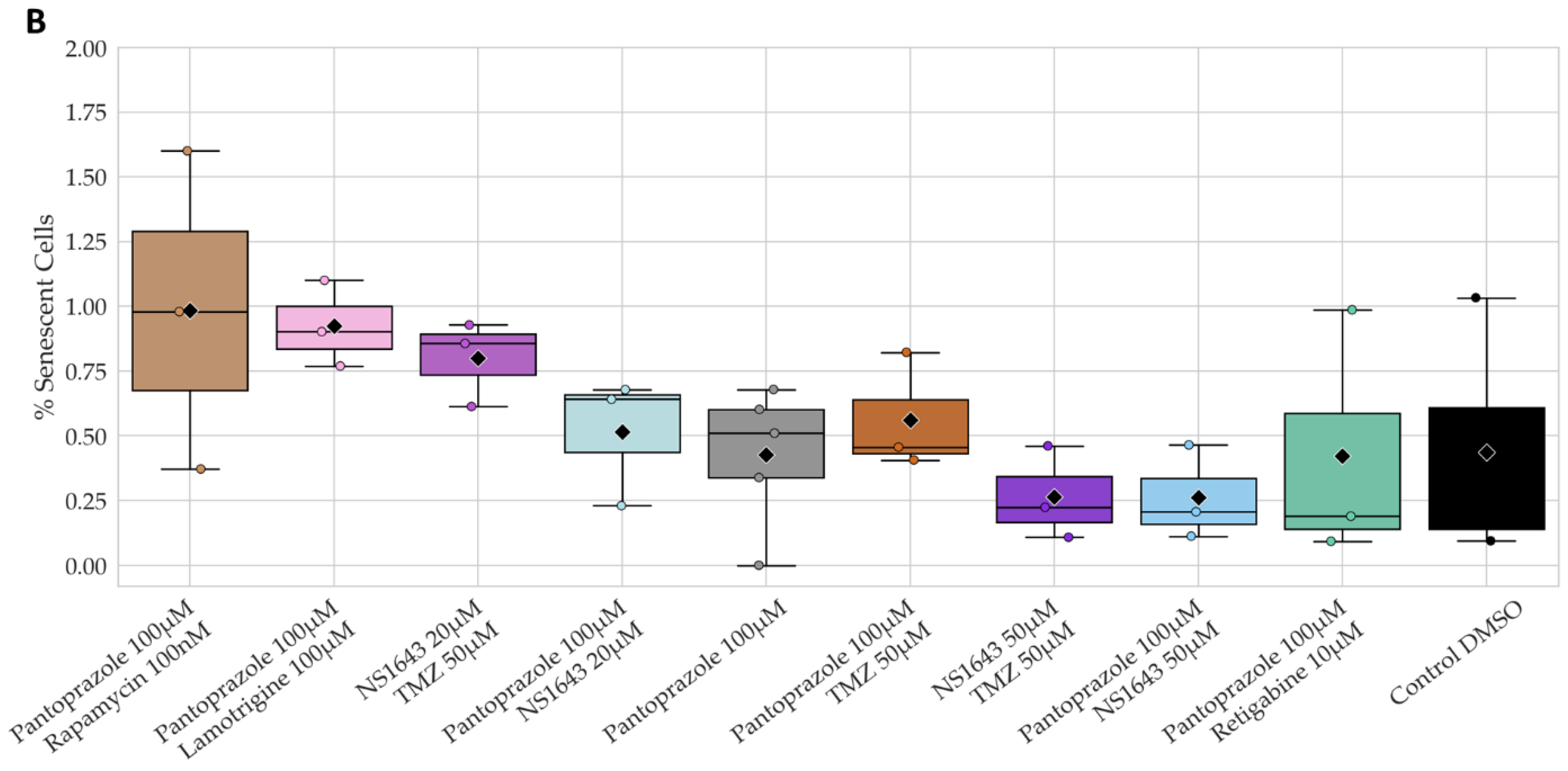
3.11. U87 Cells Also Show an Increase in p27Kip1 and Senescence Markers When Treated with Pantoprazole Alone or in Combination with Hyperpolarizing Compounds
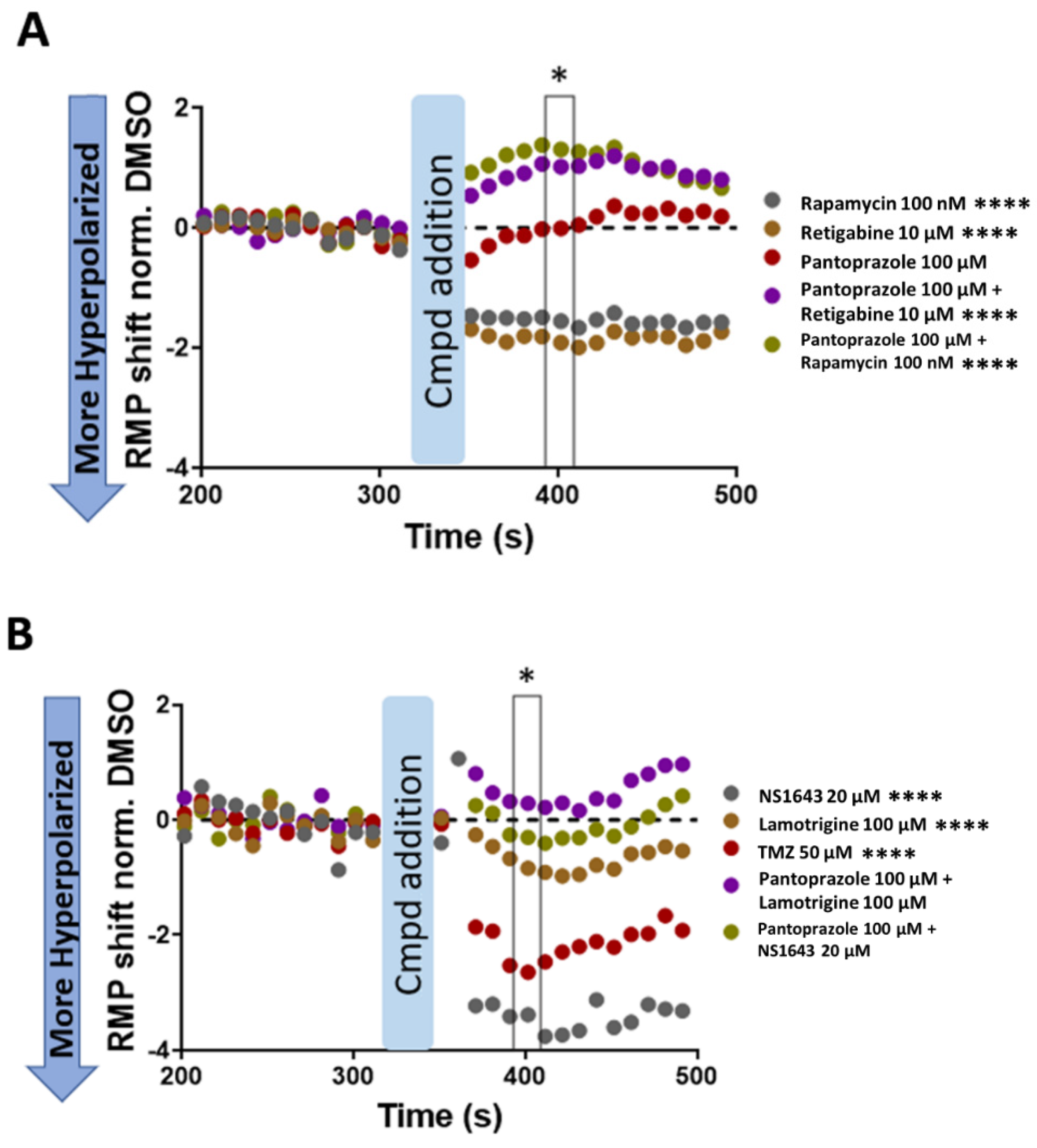
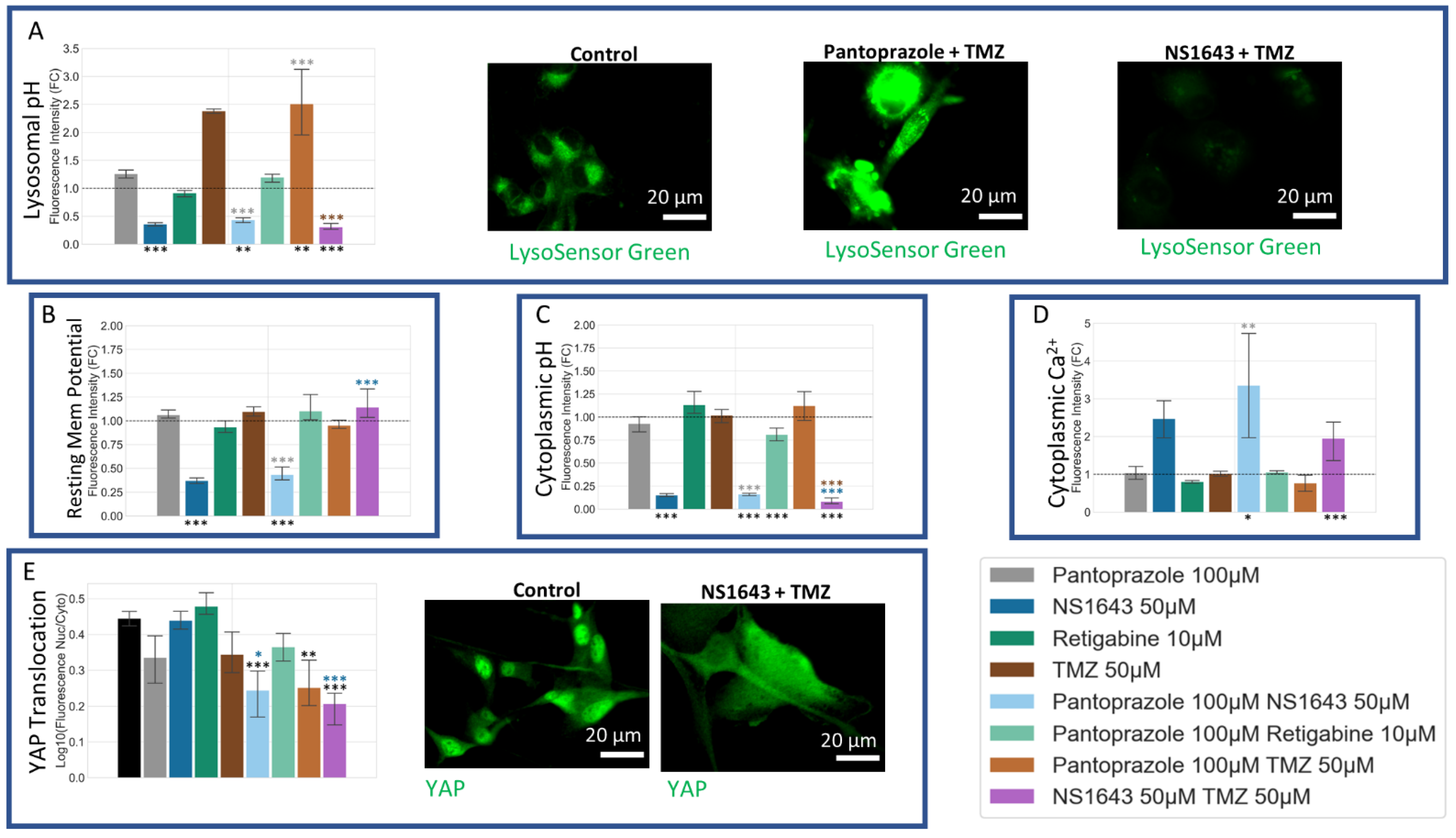
3.12. U87 Cells Are Hyperpolarized by Treatment with NS1643 and Its Combination with Pantoprazole and the Combination of NS1643 with Pantoprazole or TMZ Increases the Translocation of YAP to the Cytoplasm
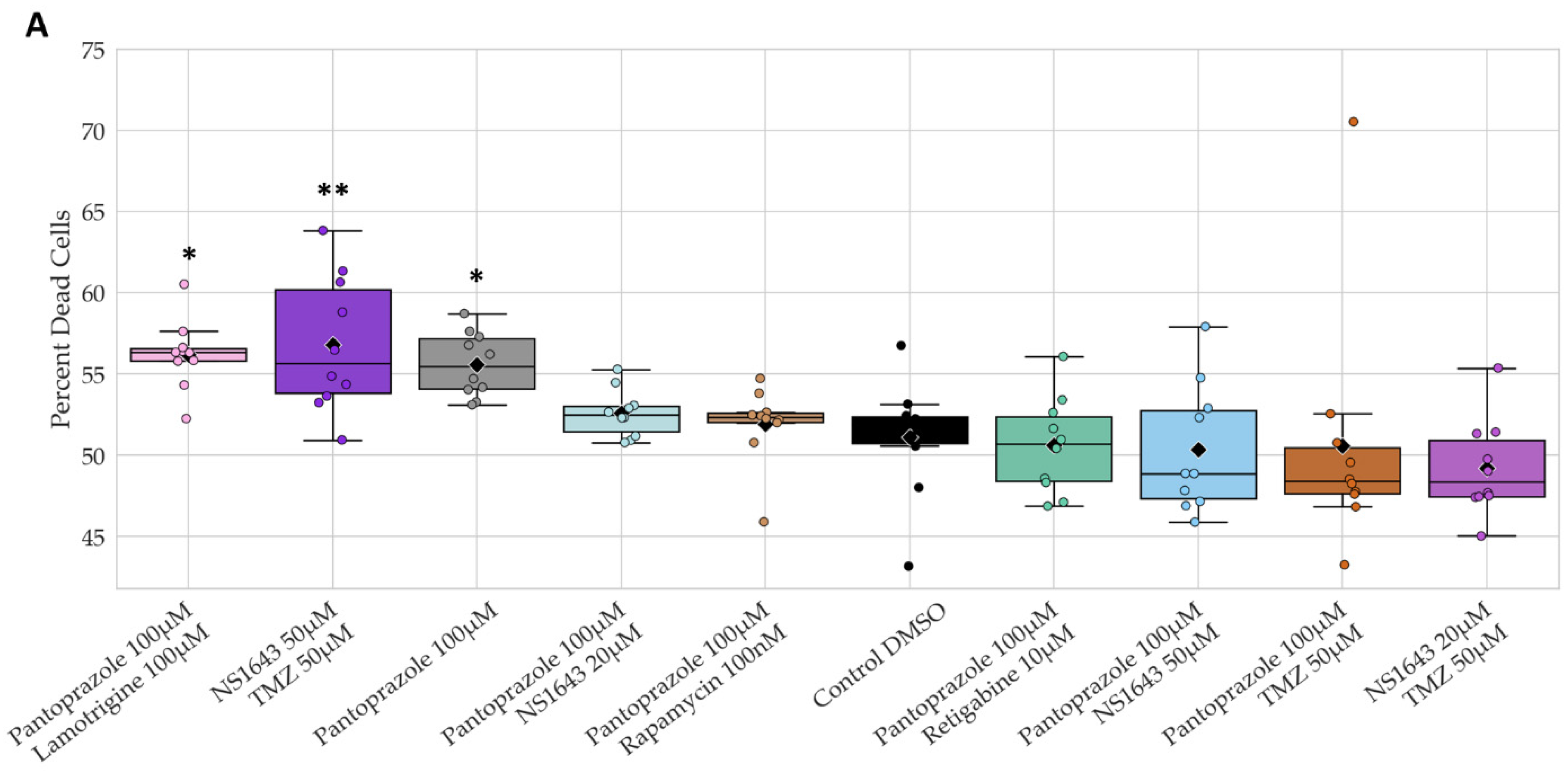
3.13. Neuronal Cell Toxicity Was Minimal after a Three-Day Treatment with the Top Performing Drugs and Drug Combinations
4. Discussion
4.1. Putative Electroceuticals for Cancer
4.2. Effects on Proliferation
4.3. Effects on Differentiation
4.4. Electrophysiology, pH, and Cell Cycle Regulation
4.5. Drug Concentrations and FDA Status: Paths towards Clinical Use
4.6. Limitations of the Study
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee Sh, U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20 (Suppl. 5), S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Lemee, J.M.; Clavreul, A.; Aubry, M.; Com, E.; de Tayrac, M.; Eliat, P.A.; Henry, C.; Rousseau, A.; Mosser, J.; Menei, P. Characterizing the peritumoral brain zone in glioblastoma: A multidisciplinary analysis. J. Neurooncol. 2015, 122, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Xiong, Y.; Wang, Y. A reignited debate over the cell(s) of origin for glioblastoma and its clinical implications. Front. Med. 2019, 13, 531–539. [Google Scholar] [CrossRef]
- Liu, C.; Sage, J.C.; Miller, M.R.; Verhaak, R.G.; Hippenmeyer, S.; Vogel, H.; Foreman, O.; Bronson, R.T.; Nishiyama, A.; Luo, L.; et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell 2011, 146, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, F.; Ceresa, D.; Appolloni, I.; Pagani, F.; Poliani, P.L.; Marubbi, D.; Malatesta, P. Glioblastoma models driven by different mutations converge to the proneural subtype. Cancer Lett. 2020, 469, 447–455. [Google Scholar] [CrossRef]
- Alcantara Llaguno, S.R.; Wang, Z.; Sun, D.; Chen, J.; Xu, J.; Kim, E.; Hatanpaa, K.J.; Raisanen, J.M.; Burns, D.K.; Johnson, J.E.; et al. Adult Lineage-Restricted CNS Progenitors Specify Distinct Glioblastoma Subtypes. Cancer Cell 2015, 28, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, N.; Jiang, Y.; Xie, Y.; Bolouri, H.; Kastemar, M.; Olofsson, T.; Holland, E.C.; Uhrbom, L. Oncogenic signaling is dominant to cell of origin and dictates astrocytic or oligodendroglial tumor development from oligodendrocyte precursor cells. J. Neurosci. 2014, 34, 14644–14651. [Google Scholar] [CrossRef]
- Hide, T.; Shibahara, I.; Kumabe, T. Novel concept of the border niche: Glioblastoma cells use oligodendrocytes progenitor cells (GAOs) and microglia to acquire stem cell-like features. Brain Tumor Pathol. 2019, 36, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Liu, Q. Differentiation therapy: A promising strategy for cancer treatment. Chin. J. Cancer 2016, 35, 3. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.; Pezzulo, G.; Finkelstein, J.M. Endogenous Bioelectric Signaling Networks: Exploiting Voltage Gradients for Control of Growth and Form. Annu. Rev. Biomed. Eng. 2017, 19, 353–387. [Google Scholar] [CrossRef]
- Panicker, S.P.; Raychaudhuri, B.; Sharma, P.; Tipps, R.; Mazumdar, T.; Mal, A.K.; Palomo, J.M.; Vogelbaum, M.A.; Haque, S.J. p300- and Myc-mediated regulation of glioblastoma multiforme cell differentiation. Oncotarget 2010, 1, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.C.; Jung, T.H.; Lee, S.; Kim, H.Y.; Choi, G.; Jung, M.; Jung, D.; Lee, H.K.; Lee, J.O.; Park, J.H.; et al. Effect of bexarotene on differentiation of glioblastoma multiforme compared with ATRA. Clin. Exp. Metastasis 2016, 33, 417–429. [Google Scholar] [CrossRef]
- Kang, T.W.; Choi, S.W.; Yang, S.R.; Shin, T.H.; Kim, H.S.; Yu, K.R.; Hong, I.S.; Ro, S.; Cho, J.M.; Kang, K.S. Growth arrest and forced differentiation of human primary glioblastoma multiforme by a novel small molecule. Sci. Rep. 2014, 4, 5546. [Google Scholar] [CrossRef][Green Version]
- Xing, F.; Luan, Y.; Cai, J.; Wu, S.; Mai, J.; Gu, J.; Zhang, H.; Li, K.; Lin, Y.; Xiao, X.; et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1alpha Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. 2017, 18, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Piccirillo, S.G.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar] [CrossRef] [PubMed]
- Park, D.M.; Li, J.; Okamoto, H.; Akeju, O.; Kim, S.H.; Lubensky, I.; Vortmeyer, A.; Dambrosia, J.; Weil, R.J.; Oldfield, E.H.; et al. N-CoR pathway targeting induces glioblastoma derived cancer stem cell differentiation. Cell Cycle 2007, 6, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Han, Q.; Zou, Y.; Deng, Z.; Lu, X.; Wang, X.; Zhang, W.; Jin, H.; Su, J.; Jiang, T.; et al. Long-term exposure to imatinib reduced cancer stem cell ability through induction of cell differentiation via activation of MAPK signaling in glioblastoma cells. Mol. Cell. Biochem. 2012, 370, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Chlapek, P.; Slavikova, V.; Mazanek, P.; Sterba, J.; Veselska, R. Why Differentiation Therapy Sometimes Fails: Molecular Mechanisms of Resistance to Retinoids. Int. J. Mol. Sci. 2018, 19, 132. [Google Scholar] [CrossRef]
- Mathews, J.; Levin, M. The body electric 2.0: Recent advances in developmental bioelectricity for regenerative and synthetic bioengineering. Curr. Opin. Biotechnol. 2018, 52, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Bates, E. Ion channels in development and cancer. Annu. Rev. Cell Dev. Biol. 2015, 31, 231–247. [Google Scholar] [CrossRef] [PubMed]
- Levin, M. Bioelectric signaling: Reprogrammable circuits underlying embryogenesis, regeneration, and cancer. Cell 2021, 184, 1971–1989. [Google Scholar] [CrossRef]
- Srivastava, P.; Kane, A.; Harrison, C.; Levin, M. A Meta-Analysis of Bioelectric Data in Cancer, Embryogenesis, and Regeneration. Bioelectricity 2021, 3, 42–67. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.R.; Perez-Neut, M.; Kaja, S.; Gentile, S. Voltage-gated ion channels in cancer cell proliferation. Cancers 2015, 7, 849–875. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, A.; Becchetti, A. New Trends in Cancer Therapy: Targeting Ion Channels and Transporters. Pharmaceuticals 2010, 3, 1202–1224. [Google Scholar] [CrossRef] [PubMed]
- Litan, A.; Langhans, S.A. Cancer as a channelopathy: Ion channels and pumps in tumor development and progression. Front. Cell. Neurosci. 2015, 9, 86. [Google Scholar] [CrossRef] [PubMed]
- Rapetti-Mauss, R.; Berenguier, C.; Allegrini, B.; Soriani, O. Interplay between Ion Channels and the Wnt/beta-Catenin Signaling Pathway in Cancers. Front. Pharmacol. 2020, 11, 525020. [Google Scholar] [CrossRef]
- Dowd, J.; Hendin, J.; Fukushiro-Lopes, D.F.; Laczynski, D.; Gentile, S. Ion Channels in Breast Cancer: From Signaling to Therapy; InTech: London, UK, 2017. [Google Scholar]
- Lang, F.; Stournaras, C. Ion channels in cancer: Future perspectives and clinical potential. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130108. [Google Scholar] [CrossRef]
- Yang, M.; Brackenbury, W.J. Membrane potential and cancer progression. Front. Physiol. 2013, 4, 185. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Chen, A.; Du, Q.; Liao, Q.; Shuai, Z.; Chen, C.; Yang, X.; Hu, Y.; Zhao, J.; Liu, S.; et al. Novel insights into ion channels in cancer stem cells (Review). Int. J. Oncol. 2018, 53, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Leanza, L.; Manago, A.; Zoratti, M.; Gulbins, E.; Szabo, I. Pharmacological targeting of ion channels for cancer therapy: In Vivo evidences. Biochim. Biophys. Acta 2016, 1863 6 Pt B, 1385–1397. [Google Scholar] [CrossRef]
- Lobikin, M.; Chernet, B.; Lobo, D.; Levin, M. Resting potential, oncogene-induced tumorigenesis, and metastasis: The bioelectric basis of cancer in vivo. Phys. Biol. 2012, 9, 065002. [Google Scholar] [CrossRef]
- Becchetti, A.; Munaron, L.; Arcangeli, A. The role of ion channels and transporters in cell proliferation and cancer. Front. Physiol. 2013, 4, 312. [Google Scholar] [CrossRef] [PubMed]
- Sontheimer, H. An unexpected role for ion channels in brain tumor metastasis. Exp. Biol. Med. 2008, 233, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Binggeli, R.; Weinstein, R.C. Membrane potentials and sodium channels: Hypotheses for growth regulation and cancer formation based on changes in sodium channels and gap junctions. J. Theor. Biol. 1986, 123, 377–401. [Google Scholar] [CrossRef]
- Chernet, B.T.; Levin, M. Transmembrane voltage potential is an essential cellular parameter for the detection and control of tumor development in a Xenopus model. Dis. Model. Mech. 2013, 6, 595–607. [Google Scholar] [CrossRef] [PubMed]
- Cone, C.D., Jr.; Tongier, M., Jr. Control of somatic cell mitosis by simulated changes in the transmembrane potential level. Oncology 1971, 25, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Stillwell, E.F.; Cone, C.M.; Cone, C.D., Jr. Stimulation of DNA synthesis in CNS neurones by sustained depolarisation. Nat. New Biol. 1973, 246, 110–111. [Google Scholar] [CrossRef] [PubMed]
- Cone, C.D. Unified theory on the basic mechanism of normal mitotic control and oncogenesis. J. Theor. Biol. 1971, 30, 151–181. [Google Scholar] [CrossRef]
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Depolarization alters phenotype, maintains plasticity of predifferentiated mesenchymal stem cells. Tissue Eng. Part A 2013, 19, 1889–1908. [Google Scholar] [CrossRef]
- Sundelacruz, S.; Levin, M.; Kaplan, D.L. Membrane potential controls adipogenic and osteogenic differentiation of mesenchymal stem cells. PLoS ONE 2008, 3, e3737. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Yue, J.; Che, H.; Sun, H.Y.; Tse, H.F.; Li, G.R. BKCa and hEag1 channels regulate cell proliferation and differentiation in human bone marrow-derived mesenchymal stem cells. J. Cell. Physiol. 2014, 229, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Papp, F.; Mocsar, G.; Kokai, E.; Kovacs, K.; Tajti, G.; Panyi, G. The voltage-gated proton channel hHv1 is functionally expressed in human chorion-derived mesenchymal stem cells. Sci. Rep. 2020, 10, 7100. [Google Scholar] [CrossRef] [PubMed]
- Pillozzi, S.; Becchetti, A. Ion Channels in Hematopoietic and Mesenchymal Stem Cells. Stem Cells Int. 2012, 2012, 217910. [Google Scholar] [CrossRef] [PubMed]
- Chernet, B.; Levin, M. Endogenous Voltage Potentials and the Microenvironment: Bioelectric Signals that Reveal, Induce and Normalize Cancer. J. Clin. Exp. Oncol. 2013, (Suppl. 1), S1-002. [Google Scholar] [CrossRef]
- Chernet, B.T.; Adams, D.S.; Lobikin, M.; Levin, M. Use of genetically encoded, light-gated ion translocators to control tumorigenesis. Oncotarget 2016, 7, 19575–19588. [Google Scholar] [CrossRef] [PubMed]
- Blackiston, D.J.; McLaughlin, K.A.; Levin, M. Bioelectric controls of cell proliferation: Ion channels, membrane voltage and the cell cycle. Cell Cycle 2009, 8, 3527–3536. [Google Scholar] [CrossRef]
- Payne, S.L.; Levin, M.; Oudin, M.J. Bioelectric Control of Metastasis in Solid Tumors. Bioelectricity 2019, 1, 114–130. [Google Scholar] [CrossRef] [PubMed]
- Ruggieri, P.; Mangino, G.; Fioretti, B.; Catacuzzeno, L.; Puca, R.; Ponti, D.; Miscusi, M.; Franciolini, F.; Ragona, G.; Calogero, A. The inhibition of KCa3.1 channels activity reduces cell motility in glioblastoma derived cancer stem cells. PLoS ONE 2012, 7, e47825. [Google Scholar] [CrossRef]
- Comes, N.; Serrano-Albarras, A.; Capera, J.; Serrano-Novillo, C.; Condom, E.; Ramon, Y.C.S.; Ferreres, J.C.; Felipe, A. Involvement of potassium channels in the progression of cancer to a more malignant phenotype. Biochim. Biophys. Acta 2015, 1848 Pt B, 2477–2492. [Google Scholar] [CrossRef]
- Pollak, J.; Rai, K.G.; Funk, C.C.; Arora, S.; Lee, E.; Zhu, J.; Price, N.D.; Paddison, P.J.; Ramirez, J.M.; Rostomily, R.C. Ion channel expression patterns in glioblastoma stem cells with functional and therapeutic implications for malignancy. PLoS ONE 2017, 12, e0172884. [Google Scholar] [CrossRef]
- Jehle, J.; Schweizer, P.A.; Katus, H.A.; Thomas, D. Novel roles for hERG K(+) channels in cell proliferation and apoptosis. Cell Death Dis. 2011, 2, e193. [Google Scholar] [CrossRef]
- Breuer, E.K.; Fukushiro-Lopes, D.; Dalheim, A.; Burnette, M.; Zartman, J.; Kaja, S.; Wells, C.; Campo, L.; Curtis, K.J.; Romero-Moreno, R.; et al. Potassium channel activity controls breast cancer metastasis by affecting beta-catenin signaling. Cell Death Dis. 2019, 10, 180. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Liu, L.; Li, G.; Xia, M.; Du, C.; Zheng, Z. The role of BKCa in endometrial cancer HEC-1-B cell proliferation and migration. Gene 2018, 655, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Brandalise, F.; Ratto, D.; Leone, R.; Olivero, F.; Roda, E.; Locatelli, C.A.; Grazia Bottone, M.; Rossi, P. Deeper and Deeper on the Role of BK and Kir4.1 Channels in Glioblastoma Invasiveness: A Novel Summative Mechanism? Front. Neurosci. 2020, 14, 595664. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Qu, C.; Han, C.; Chen, M.M.; An, L.J.; Zou, W. Potassium channels and their role in glioma: A mini review. Mol. Membr. Biol. 2019, 35, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Terrasi, A.; Bertolini, I.; Martelli, C.; Gaudioso, G.; Di Cristofori, A.; Storaci, A.M.; Formica, M.; Bosari, S.; Caroli, M.; Ottobrini, L.; et al. Specific V-ATPase expression sub-classifies IDHwt lower-grade gliomas and impacts glioma growth in vivo. EBioMedicine 2019, 41, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Yekula, A.; Yekula, A.; Muralidharan, K.; Kang, K.; Carter, B.S.; Balaj, L. Extracellular Vesicles in Glioblastoma Tumor Microenvironment. Front. Immunol. 2019, 10, 3137. [Google Scholar] [CrossRef]
- Harguindey, S.; Polo Orozco, J.; Alfarouk, K.O.; Devesa, J. Hydrogen Ion Dynamics of Cancer and a New Molecular, Biochemical and Metabolic Approach to the Etiopathogenesis and Treatment of Brain Malignancies. Int. J. Mol. Sci. 2019, 20, 4278. [Google Scholar] [CrossRef]
- Hamprecht, B.; Glaser, T.; Reiser, G.; Bayer, E.; Propst, F. Culture and characteristics of hormone-responsive neuroblastoma X glioma hybrid cells. Methods Enzymol. 1985, 109, 316–341. [Google Scholar] [CrossRef] [PubMed]
- Lichtshtein, D.; Dunlop, K.; Kaback, H.R.; Blume, A.J. Mechanism of monensin-induced hyperpolarization of neuroblastoma-glioma hybrid NG108-15. Proc. Natl. Acad. Sci. USA 1979, 76, 2580–2584. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.J.; Hanson, J.C.; Quets-Nguyen, A.T.; Bergeron, M.; Smith, R.C. A quantitative method for analysis of in vitro neurite outgrowth. J. Neurosci. Methods 2007, 164, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Pancrazio, J.J.; Ma, W.; Grant, G.M.; Shaffer, K.M.; Kao, W.Y.; Liu, Q.-Y.; Manos, P.; Barker, J.L.; Stenger, D.A. A role for inwardly rectifying K+ channels in differentiation of NG108-15 neuroblastoma glioma cells. J. Neurobiol. 1999, 38, 466–474. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Ledur, P.F.; Onzi, G.R.; Zong, H.; Lenz, G. Culture conditions defining glioblastoma cells behavior: What is the impact for novel discoveries? Oncotarget 2017, 8, 69185–69197. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Chedid, K.; Kalkanis, S.N. Glioblastoma cell line-derived spheres in serumcontaining medium versus serum-free medium: A comparison of cancer stem cell properties. Int. J. Oncol. 2012, 41, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Haspels, H.N.; Rahman, M.A.; Joseph, J.V.; Gras Navarro, A.; Chekenya, M. Glioblastoma Stem-Like Cells Are More Susceptible Than Differentiated Cells to Natural Killer Cell Lysis Mediated through Killer Immunoglobulin-Like Receptors-Human Leukocyte Antigen Ligand Mismatch and Activation Receptor-Ligand Interactions. Front. Immunol. 2018, 9, 1345. [Google Scholar] [CrossRef]
- Sladitschek, H.L.; Neveu, P.A. MXS-Chaining: A Highly Efficient Cloning Platform for Imaging and Flow Cytometry Approaches in Mammalian Systems. PLoS ONE 2015, 10, e0124958. [Google Scholar] [CrossRef]
- Cairns, D.M.; Chwalek, K.; Moore, Y.E.; Kelley, M.R.; Abbott, R.D.; Moss, S.; Kaplan, D.L. Expandable and Rapidly Differentiating Human Induced Neural Stem Cell Lines for Multiple Tissue Engineering Applications. Stem Cell Rep. 2016, 7, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Goedhart, J.; von Stetten, D.; Noirclerc-Savoye, M.; Lelimousin, M.; Joosen, L.; Hink, M.A.; van Weeren, L.; Gadella, T.W., Jr.; Royant, A. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93%. Nat. Commun. 2012, 3, 751. [Google Scholar] [CrossRef]
- Marh, J.; Stoytcheva, Z.; Urschitz, J.; Sugawara, A.; Yamashiro, H.; Owens, J.B.; Stoytchev, I.; Pelczar, P.; Yanagimachi, R.; Moisyadi, S. Hyperactive self-inactivating piggyBac for transposase-enhanced pronuclear microinjection transgenesis. Proc. Natl. Acad. Sci. USA 2012, 109, 19184–19189. [Google Scholar] [CrossRef] [PubMed]
- Owens, J.B.; Mathews, J.; Davy, P.; Stoytchev, I.; Moisyadi, S.; Allsopp, R. Effective Targeted Gene Knockdown in Mammalian Cells Using the piggyBac Transposase-based Delivery System. Mol. Ther. Nucleic Acids 2013, 2, e137. [Google Scholar] [CrossRef] [PubMed]
- Kamentsky, L.; Jones, T.R.; Fraser, A.; Bray, M.-A.; Logan, D.J.; Madden, K.L.; Ljosa, V.; Rueden, C.; Eliceiri, K.W.; Carpenter, A.E. Improved structure, function and compatibility for CellProfiler: Modular high-throughput image analysis software. Bioinformatics 2011, 27, 1179–1180. [Google Scholar] [CrossRef] [PubMed]
- Sero, J.E.; Bakal, C. Multiparametric Analysis of Cell Shape Demonstrates that beta-PIX Directly Couples YAP Activation to Extracellular Matrix Adhesion. Cell Syst. 2017, 4, 84–96.e6. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.Q.; Baez-Nieto, D.; Allen, A.; Wang, H.R.; Cottrell, J.R. Developing High-Throughput Assays to Analyze and Screen Electrophysiological Phenotypes. Methods Mol. Biol. 2018, 1787, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Petho, Z.; Najder, K.; Carvalho, T.; McMorrow, R.; Todesca, L.M.; Rugi, M.; Bulk, E.; Chan, A.; Lowik, C.; Reshkin, S.J.; et al. pH-Channeling in Cancer: How pH-Dependence of Cation Channels Shapes Cancer Pathophysiology. Cancers 2020, 12, 2484. [Google Scholar] [CrossRef]
- Abdul Kadir, L.; Stacey, M.; Barrett-Jolley, R. Emerging Roles of the Membrane Potential: Action beyond the Action Potential. Front. Physiol. 2018, 9, 1661. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; James, A.D.; Suman, R.; Kasprowicz, R.; Nelson, M.; O’Toole, P.J.; Brackenbury, W.J. Voltage-dependent activation of Rac1 by Nav 1.5 channels promotes cell migration. J. Cell. Physiol. 2020, 235, 3950–3972. [Google Scholar] [CrossRef] [PubMed]
- Chinnaiyan, P.; Won, M.; Wen, P.Y.; Rojiani, A.M.; Werner-Wasik, M.; Shih, H.A.; Ashby, L.S.; Michael Yu, H.H.; Stieber, V.W.; Malone, S.C.; et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: Results of NRG Oncology RTOG 0913. Neuro Oncol. 2018, 20, 666–673. [Google Scholar] [CrossRef] [PubMed]
- Fouladi, M.; Laningham, F.; Wu, J.; O’Shaughnessy, M.A.; Molina, K.; Broniscer, A.; Spunt, S.L.; Luckett, I.; Stewart, C.F.; Houghton, P.J.; et al. Phase I study of everolimus in pediatric patients with refractory solid tumors. J. Clin. Oncol. 2007, 25, 4806–4812. [Google Scholar] [CrossRef] [PubMed]
- Geeviman, K.; Babu, D.; Prakash Babu, P. Pantoprazole Induces Mitochondrial Apoptosis and Attenuates NF-kappaB Signaling in Glioma Cells. Cell. Mol. Neurobiol. 2018, 38, 1491–1504. [Google Scholar] [CrossRef] [PubMed]
- Beier, D.; Schriefer, B.; Brawanski, K.; Hau, P.; Weis, J.; Schulz, J.B.; Beier, C.P. Efficacy of clinically relevant temozolomide dosing schemes in glioblastoma cancer stem cell lines. J. Neurooncol. 2012, 109, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chen, X.; Zhang, Q.; Zheng, Y.; Jiang, H.; Yang, H.; Gao, Z. The human ether-a-go-go-related gene activator NS1643 enhances epilepsy-associated KCNQ channels. J. Pharmacol. Exp. Ther. 2014, 351, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Zou, X.; Chen, M.; Shen, Y.; Huang, S.; Guo, H.; Zhang, L.; Liu, P. Effect of pantoprazole on human gastric adenocarcinoma SGC7901 cells through regulation of phospho-LRP6 expression in Wnt/β-catenin signaling. Oncol. Rep. 2013, 30, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Gunthorpe, M.J.; Large, C.H.; Sankar, R. The mechanism of action of retigabine (ezogabine), a first-in-class K+ channel opener for the treatment of epilepsy. Epilepsia 2012, 53, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Lagrange, A. Retigabine: Bending potassium channels to our will. Epilepsy Curr. 2005, 5, 166–168. [Google Scholar] [CrossRef] [PubMed]
- Nakatani, Y.; Masuko, H.; Amano, T. Effect of lamotrigine on Na(v)1.4 voltage-gated sodium channels. J. Pharmacol. Sci. 2013, 123, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Chin, T.Y.; Kao, C.H.; Wang, H.Y.; Huang, W.P.; Ma, K.H.; Chueh, S.H. Inhibition of the mammalian target of rapamycin promotes cyclic AMP-induced differentiation of NG108-15 cells. Autophagy 2010, 6, 1139–1156. [Google Scholar] [CrossRef] [PubMed]
- Raker, V.K.; Becker, C.; Steinbrink, K. The cAMP Pathway as Therapeutic Target in Autoimmune and Inflammatory Diseases. Front. Immunol. 2016, 7, 123. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef]
- Yeh, P.S.; Wu, S.J.; Hung, T.Y.; Huang, Y.M.; Hsu, C.W.; Sze, C.I.; Hsieh, Y.J.; Huang, C.W.; Wu, S.N. Evidence for the Inhibition by Temozolomide, an Imidazotetrazine Family Alkylator, of Intermediate-Conductance Ca2+-Activated K+ Channels in Glioma Cells. Cell. Physiol. Biochem. 2016, 38, 1727–1742. [Google Scholar] [CrossRef]
- Soltani, M.H.; Pichardo, R.; Song, Z.; Sangha, N.; Camacho, F.; Satyamoorthy, K.; Sangueza, O.P.; Setaluri, V. Microtubule-associated protein 2, a marker of neuronal differentiation, induces mitotic defects, inhibits growth of melanoma cells, and predicts metastatic potential of cutaneous melanoma. Am. J. Pathol. 2005, 166, 1841–1850. [Google Scholar] [CrossRef]
- Sabelstrom, H.; Petri, R.; Shchors, K.; Jandial, R.; Schmidt, C.; Sacheva, R.; Masic, S.; Yuan, E.; Fenster, T.; Martinez, M.; et al. Driving Neuronal Differentiation through Reversal of an ERK1/2-miR-124-SOX9 Axis Abrogates Glioblastoma Aggressiveness. Cell Rep. 2019, 28, 2064–2079.e11. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Skaftnesmo, K.O.; Leiss, L.; Sleire, L.; Wang, J.; Li, X.; Enger, P.O. Neuronal markers are expressed in human gliomas and NSE knockdown sensitizes glioblastoma cells to radiotherapy and temozolomide. BMC Cancer 2011, 11, 524. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. A. Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a018309. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yi, M.; Zhou, F.; He, W.; Yang, A.; Qiu, M.; Huang, H. S100B is selectively expressed by gray matter protoplasmic astrocytes and myelinating oligodendrocytes in the developing CNS. Mol. Brain 2021, 14, 154. [Google Scholar] [CrossRef]
- Yang, Z.; Wang, K.K. Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends Neurosci. 2015, 38, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Landeira, B.S.; Santana, T.; Araujo, J.A.M.; Tabet, E.I.; Tannous, B.A.; Schroeder, T.; Costa, M.R. Activity-Independent Effects of CREB on Neuronal Survival and Differentiation during Mouse Cerebral Cortex Development. Cereb. Cortex. 2018, 28, 538–548. [Google Scholar] [CrossRef]
- Li, Y.; Yin, W.; Wang, X.; Zhu, W.; Huang, Y.; Yan, G. Cholera toxin induces malignant glioma cell differentiation via the PKA/CREB pathway. Proc. Natl. Acad. Sci. USA 2007, 104, 13438–13443. [Google Scholar] [CrossRef] [PubMed]
- Shiga, H.; Yamane, Y.; Kubo, M.; Sakurai, Y.; Asou, H.; Ito, E. Differentiation of immature oligodendrocytes is regulated by phosphorylation of cyclic AMP-response element binding protein by a protein kinase C signaling cascade. J. Neurosci. Res. 2005, 80, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Bender, R.A.; Lauterborn, J.C.; Gall, C.M.; Cariaga, W.; Baram, T.Z. Enhanced CREB phosphorylation in immature dentate gyrus granule cells precedes neurotrophin expression and indicates a specific role of CREB in granule cell differentiation. Eur. J. Neurosci. 2001, 13, 679–686. [Google Scholar] [CrossRef]
- Hitomi, M.; Deleyrolle, L.P.; Mulkearns-Hubert, E.E.; Jarrar, A.; Li, M.; Sinyuk, M.; Otvos, B.; Brunet, S.; Flavahan, W.A.; Hubert, C.G.; et al. Differential connexin function enhances self-renewal in glioblastoma. Cell Rep. 2015, 11, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.Y.; Han, J.A.; Im, J.S.; Morrone, A.; Johung, K.; Goodwin, E.C.; Kleijer, W.J.; DiMaio, D.; Hwang, E.S. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 2006, 5, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.M.; Beesley, J.S.; Grinspan, J.B.; Seth, P.; Kamholz, J.; Cambi, F. Cell Cycle Arrest Induced by Ectopic Expression of p27 Is Not Sufficient to Promote Oligodendrocyte Differentiation. J. Cell. Biochem. 1999, 76, 270–279. [Google Scholar] [CrossRef]
- Schiappacassi, M.; Lovat, F.; Canzonieri, V.; Belletti, B.; Berton, S.; Di Stefano, D.; Vecchione, A.; Colombatti, A.; Baldassarre, G. p27Kip1 expression inhibits glioblastoma growth, invasion, and tumor-induced neoangiogenesis. Mol. Cancer Ther. 2008, 7, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, R.V.; Erickson, L.A.; Jin, L.; Kulig, E.; Qian, X.; Cheville, J.C.; Scheithauer, B.W. p27kip1: A multifunctional cyclin-dependent kinase inhibitor with prognostic significance in human cancers. Am. J. Pathol. 1999, 154, 313–323. [Google Scholar] [CrossRef]
- McKay, L.K.; White, J.P. The AMPK/p27(Kip1) Pathway as a Novel Target to Promote Autophagy and Resilience in Aged Cells. Cells 2021, 10, 1430. [Google Scholar] [CrossRef] [PubMed]
- Love, R. Cyclin D1 and P27KIP1: The Gatekeepers of Dysplasia. J. Immunol. Sci. 2018, 2, 30–39. [Google Scholar] [CrossRef]
- Collado, M.; Medema, R.H.; Garcia-Cao, I.; Dubuisson, M.L.; Barradas, M.; Glassford, J.; Rivas, C.; Burgering, B.M.; Serrano, M.; Lam, E.W. Inhibition of the phosphoinositide 3-kinase pathway induces a senescence-like arrest mediated by p27Kip1. J. Biol. Chem. 2000, 275, 21960–21968. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.H.; Chang, C.Y.; Lin, B.Z.; Wu, Y.L.; Wu, M.H.; Lin, L.T.; Huang, W.C.; Holz, J.D.; Sheu, T.J.; Lee, J.S.; et al. Up-regulation of cofilin-1 in cell senescence associates with morphological change and p27(kip1) -mediated growth delay. Aging Cell 2021, 20, e13288. [Google Scholar] [CrossRef] [PubMed]
- Kawamata, S.; Sakaida, H.; Hori, T.; Maeda, M.; Uchiyama, T. The upregulation of p27Kip1 by rapamycin results in G1 arrest in exponentially growing T-cell lines. Blood 1998, 91, 561–569. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, L.A.; Davoli, M.A.; Belliveau, C.; Tanti, A.; Ma, J.C.; Farmer, W.T.; Turecki, G.; Murai, K.K.; Mechawar, N. Characterization of Vimentin-Immunoreactive Astrocytes in the Human Brain. Front. Neuroanat. 2020, 14, 31. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Du, W.W.; Fang, L.; Shan, S.W.; Qian, J.; Lin, J.; Qian, W.; Ma, J.; Rutnam, Z.J.; Yang, B.B. The intermediate filament vimentin mediates microRNA miR-378 function in cellular self-renewal by regulating the expression of the Sox2 transcription factor. J. Biol. Chem. 2013, 288, 319–331. [Google Scholar] [CrossRef]
- Adams, D.S.; Levin, M. Measuring resting membrane potential using the fluorescent voltage reporters DiBAC4(3) and CC2-DMPE. Cold Spring Harb. Protoc. 2012, 2012, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.S.; Levin, M. General principles for measuring resting membrane potential and ion concentration using fluorescent bioelectricity reporters. Cold Spring Harb. Protoc. 2012, 2012, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.N.; Shi, Z.Y.; Dang, Y.F.; Cheng, Y.N.; Guan, Y.H.; Hao, Z.J.; Tian, B.; He, H.W.; Guo, X.L. Pantoprazole pretreatment elevates sensitivity to vincristine in drug-resistant oral epidermoid carcinoma in vitro and in vivo. Biomed. Pharmacother. 2019, 120, 109478. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Baker, S.S.; Trinidad, J.; Burlingame, A.L.; Baker, R.D.; Forte, J.G.; Virtuoso, L.P.; Egilmez, N.K.; Zhu, L. Inhibition of lysosomal enzyme activities by proton pump inhibitors. J. Gastroenterol. 2013, 48, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Chen, M.; Tang, D.H.; Yan, H.L.; Ding, X.W.; Zhou, F.; Zhang, M.M.; Xu, G.F.; Zhang, W.J.; Zhang, S.; et al. The proton pump inhibitor pantoprazole disrupts protein degradation systems and sensitizes cancer cells to death under various stresses. Cell Death Dis. 2018, 9, 604. [Google Scholar] [CrossRef]
- Lu, Z.N.; Niu, W.X.; Zhang, N.; Ge, M.X.; Bao, Y.Y.; Ren, Y.; Guo, X.L.; He, H.W. Pantoprazole ameliorates liver fibrosis and suppresses hepatic stellate cell activation in bile duct ligation rats by promoting YAP degradation. Acta Pharmacol. Sin. 2021, 42, 1808–1820. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Shi, X.Y.; Li, Z.M.; Pan, X.H.; Li, Z.L.; Chen, Y.; Yan, S.J.; Xiao, L. Proton pump inhibitors can reverse the YAP mediated paclitaxel resistance in epithelial ovarian cancer. BMC Mol. Cell Biol. 2019, 20, 49. [Google Scholar] [CrossRef] [PubMed]
- Lian, I.; Kim, J.; Okazawa, H.; Zhao, J.; Zhao, B.; Yu, J.; Chinnaiyan, A.; Israel, M.A.; Goldstein, L.S.; Abujarour, R.; et al. The role of YAP transcription coactivator in regulating stem cell self-renewal and differentiation. Genes Dev. 2010, 24, 1106–1118. [Google Scholar] [CrossRef] [PubMed]
- Zou, R.; Xu, Y.H.; Feng, Y.F.; Shen, M.Q.; Yuan, F.; Yuan, Y.Z. YAP nuclear-cytoplasmic translocation is regulated by mechanical signaling, protein modification, and metabolism. Cell Biol. Int. 2020, 44, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [PubMed]
- Markowska, A.; Kaysiewicz, J.; Markowska, J.; Huczynski, A. Doxycycline, salinomycin, monensin and ivermectin repositioned as cancer drugs. Bioorg. Med. Chem. Lett. 2019, 29, 1549–1554. [Google Scholar] [CrossRef]
- Jiang, L.; Wang, P.; Sun, Y.J.; Wu, Y.J. Ivermectin reverses the drug resistance in cancer cells through EGFR/ERK/Akt/NF-kappaB pathway. J. Exp. Clin. Cancer Res. 2019, 38, 265. [Google Scholar] [CrossRef] [PubMed]
- Intuyod, K.; Hahnvajanawong, C.; Pinlaor, P.; Pinlaor, S. Anti-parasitic Drug Ivermectin Exhibits Potent Anticancer Activity against Gemcitabine-resistant Cholangiocarcinoma In Vitro. Anticancer Res. 2019, 39, 4837–4843. [Google Scholar] [CrossRef]
- Juarez, M.; Schcolnik-Cabrera, A.; Duenas-Gonzalez, A. The multitargeted drug ivermectin: From an antiparasitic agent to a repositioned cancer drug. Am. J. Cancer Res. 2018, 8, 317–331. [Google Scholar]
- Gupta, P.B.; Onder, T.T.; Jiang, G.; Tao, K.; Kuperwasser, C.; Weinberg, R.A.; Lander, E.S. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell 2009, 138, 645–659. [Google Scholar] [CrossRef] [PubMed]
- Fukushiro-Lopes, D.F.; Hegel, A.D.; Rao, V.; Wyatt, D.; Baker, A.; Breuer, E.K.; Osipo, C.; Zartman, J.J.; Burnette, M.; Kaja, S.; et al. Preclinical study of a Kv11.1 potassium channel activator as antineoplastic approach for breast cancer. Oncotarget 2018, 9, 3321–3337. [Google Scholar] [CrossRef] [PubMed]
- Gentile, S. hERG1 potassium channel in cancer cells: A tool to reprogram immortality. Eur. Biophys. J. 2016, 45, 649–655. [Google Scholar] [CrossRef]
- Perez-Neut, M.; Shum, A.; Cuevas, B.D.; Miller, R.; Gentile, S. Stimulation of hERG1 channel activity promotes a calcium-dependent degradation of cyclin E2, but not cyclin E1, in breast cancer cells. Oncotarget 2015, 6, 1631–1639. [Google Scholar] [CrossRef]
- Perez-Neut, M.; Rao, V.R.; Gentile, S. hERG1/Kv11.1 activation stimulates transcription of p21waf/cip in breast cancer cells via a calcineurin-dependent mechanism. Oncotarget 2015, 7, 58893–58902. [Google Scholar] [CrossRef] [PubMed]
- Spugnini, E.P.; Citro, G.; Fais, S. Proton pump inhibitors as anti vacuolar-ATPases drugs: A novel anticancer strategy. J. Exp. Clin. Cancer Res. 2010, 29, 44. [Google Scholar] [CrossRef] [PubMed]
- Kale, V.P.; Amin, S.G.; Pandey, M.K. Targeting ion channels for cancer therapy by repurposing the approved drugs. Biochim. Biophys. Acta 2015, 1848 Pt B, 2747–2755. [Google Scholar] [CrossRef]
- Villalonga, N.; Ferreres, J.C.; Argiles, J.M.; Condom, E.; Felipe, A. Potassium channels are a new target field in anticancer drug design. Recent Pat. Anti-Cancer Drug Discov. 2007, 2, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Becchetti, A. Ion channels and transporters in cancer. 1. Ion channels and cell proliferation in cancer. Am. J. Physiol. Cell Physiol. 2011, 301, C255–C265. [Google Scholar] [CrossRef]
- Arcangeli, A.; Crociani, O.; Lastraioli, E.; Masi, A.; Pillozzi, S.; Becchetti, A. Targeting ion channels in cancer: A novel frontier in antineoplastic therapy. Curr. Med. Chem. 2009, 16, 66–93. [Google Scholar] [CrossRef]
- Churchill, C.D.M.; Winter, P.; Tuszynski, J.A.; Levin, M. EDEn—Electroceutical Design Environment: An Ion Channel Database with Small Molecule Modulators and Tissue Expression Information. iScience 2018, 11, 42–56. [Google Scholar] [CrossRef]
- Tuszynski, J.; Tilli, T.M.; Levin, M. Ion Channel and Neurotransmitter Modulators as Electroceutical Approaches to the Control of Cancer. Curr. Pharm. Des. 2017, 23, 4827–4841. [Google Scholar] [CrossRef] [PubMed]
- Pai, V.P.; Cervera, J.; Mafe, S.; Willocq, V.; Lederer, E.K.; Levin, M. HCN2 Channel-Induced Rescue of Brain Teratogenesis via Local and Long-Range Bioelectric Repair. Front. Cell. Neurosci. 2020, 14, 136. [Google Scholar] [CrossRef] [PubMed]
- Pai, V.P.; Pietak, A.; Willocq, V.; Ye, B.; Shi, N.Q.; Levin, M. HCN2 Rescues brain defects by enforcing endogenous voltage pre-patterns. Nat. Commun. 2018, 9, 998. [Google Scholar] [CrossRef]
- Levin, M. Bioelectrical approaches to cancer as a problem of the scaling of the cellular self. Prog. Biophys. Mol. Biol. 2021, 165, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Tojima, T.; Yamane, Y.; Takahashi, M.; Ito, E. Acquisition of neuronal proteins during differentiation of NG108-15 cells. Neurosci. Res. 2000, 37, 153–161. [Google Scholar] [CrossRef]
- Seidman, K.J.; Barsuk, J.H.; Johnson, R.F.; Weyhenmeyer, J.A. Differentiation of NG108-15 neuroblastoma cells by serum starvation or dimethyl sulfoxide results in marked differences in angiotensin II receptor subtype expression. J. Neurochem. 1996, 66, 1011–1018. [Google Scholar] [CrossRef]
- Ma, W.; Pancrazio, J.J.; Coulombe, M.; Dumm, J.; Sathanoori, R.; Barker, J.L.; Kowtha, V.C.; Stenger, D.A.; Hickman, J.J. Neuronal and glial epitopes and transmitter-synthesizing enzymes appear in parallel with membrane excitability during neuroblastoma×glioma hybrid differentiation. Dev. Brain Res. 1998, 106, 155–163. [Google Scholar] [CrossRef]
- Nurse, P. A long twentieth century of the cell cycle and beyond. Cell 2000, 100, 71–78. [Google Scholar] [CrossRef]
- Rieske, P.; Golanska, E.; Zakrzewska, M.; Piaskowski, S.; Hulas-Bigoszewska, K.; Wolanczyk, M.; Szybka, M.; Witusik-Perkowska, M.; Jaskolski, D.J.; Zakrzewski, K.; et al. Arrested neural and advanced mesenchymal differentiation of glioblastoma cells-comparative study with neural progenitors. BMC Cancer 2009, 9, 54. [Google Scholar] [CrossRef]
- Wolanczyk, M.; Hulas-Bigoszewska, K.; Witusik-Perkowska, M.; Papierz, W.; Jaskolski, D.; Liberski, P.P.; Rieske, P. Imperfect oligodendrocytic and neuronal differentiation of glioblastoma cells. Folia Neuropathol. 2010, 48, 27–34. [Google Scholar] [PubMed]
- Zhou, X.; Song, M.; Chen, D.; Wei, L.; Yu, S.P. Potential role of KCNQ/M-channels in regulating neuronal differentiation in mouse hippocampal and embryonic stem cell-derived neuronal cultures. Exp. Neurol. 2011, 229, 471–483. [Google Scholar] [CrossRef]
- Bilet, A.; Bauer, C.K. Effects of the small molecule HERG activator NS1643 on Kv11.3 channels. PLoS ONE 2012, 7, e50886. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP and TAZ: A signalling hub of the tumour microenvironment. Nat. Rev. Cancer 2019, 19, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Chen, J.; Feng, H.; Peng, S.; Adams, U.; Bai, Y.; Huang, L.; Li, J.; Huang, J.; Meng, S.; et al. YAP/TEAD-mediated transcription controls cellular senescence. Cancer Res. 2013, 73, 3615–3624. [Google Scholar] [CrossRef]
- Wolff, C.; Fuks, B.; Chatelain, P. Comparative study of membrane potential-sensitive fluorescent probes and their use in ion channel screening assays. J. Biomol. Screen. 2003, 8, 533–543. [Google Scholar] [CrossRef]
- Yamada, A.; Gaja, N.; Ohya, S.; Muraki, K.; Narita, H.; Ohwada, T.; Imaizumi, Y. Usefulness and limitation of DiBAC4(3), a voltage-sensitive fluorescent dye, for the measurement of membrane potentials regulated by recombinant large conductance Ca2+-activated K+ channels in HEK293 cells. Jpn. J. Pharmacol. 2001, 86, 342–350. [Google Scholar] [CrossRef]
- Perez-Neut, M.; Haar, L.; Rao, V.; Santha, S.; Lansu, K.; Rana, B.; Jones, W.K.; Gentile, S. Activation of hERG3 channel stimulates autophagy and promotes cellular senescence in melanoma. Oncotarget 2016, 7, 21991–22004. [Google Scholar] [CrossRef]
- Santini, M.P.; Talora, C.; Seki, T.; Bolgan, L.; Dotto, G.P. Cross talk among calcineurin, Sp1/Sp3, and NFAT in control of p21(WAF1/CIP1) expression in keratinocyte differentiation. Proc. Natl. Acad. Sci. USA 2001, 98, 9575–9580. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.H.; Hong, J.M.; Park, S.Y. Calcineurin Activation by Prion Protein Induces Neurotoxicity via Mitochondrial Reactive Oxygen Species. Oxidative Med. Cell. Longev. 2021, 2021, 5572129. [Google Scholar] [CrossRef] [PubMed]
- Livnat-Levanon, N.; Kevei, E.; Kleifeld, O.; Krutauz, D.; Segref, A.; Rinaldi, T.; Erpapazoglou, Z.; Cohen, M.; Reis, N.; Hoppe, T.; et al. Reversible 26S proteasome disassembly upon mitochondrial stress. Cell Rep. 2014, 7, 1371–1380. [Google Scholar] [CrossRef]
- Kudo, Y.; Takata, T.; Ogawa, I.; Kaneda, T.; Sato, S.; Takekoshi, T.; Zhao, M.; Miyauchi, M.; Nikai, H. p27Kip1 accumulation by inhibition of proteasome function induces apoptosis in oral squamous cell carcinoma cells. Clin. Cancer Res. 2000, 6, 916–923. [Google Scholar] [PubMed]
- Kossatz, U.; Dietrich, N.; Zender, L.; Buer, J.; Manns, M.P.; Malek, N.P. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Gene Dev. 2004, 18, 2602–2607. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Kim, B.C.; Park, M.J.; Lee, Y.S.; Kim, Y.N.; Lee, B.L.; Lee, J.S. PTEN status switches cell fate between premature senescence and apoptosis in glioma exposed to ionizing radiation. Cell Death Differ. 2011, 18, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Tompson, D.J.; Crean, C.S.; Reeve, R.; Berry, N.S. Efficacy and tolerability exposure-response relationship of retigabine (ezogabine) immediate-release tablets in patients with partial-onset seizures. Clin. Ther. 2013, 35, 1174–1185.e4. [Google Scholar] [CrossRef] [PubMed]
- Large, C.H.; Sokal, D.M.; Nehlig, A.; Gunthorpe, M.J.; Sankar, R.; Crean, C.S.; Vanlandingham, K.E.; White, H.S. The spectrum of anticonvulsant efficacy of retigabine (ezogabine) in animal models: Implications for clinical use. Epilepsia 2012, 53, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Myatt, D.P. The correlation of plasma proteins binding capacity and flavopiridol cellular and clinical trial studies. Biomed. Spectrosc. Imaging 2017, 6, 59–73. [Google Scholar] [CrossRef]
- Basa-Denes, O.; Angi, R.; Karpati, B.; Jordan, T.; Otvos, Z.; Erdosi, N.; Ujhelyi, A.; Ordasi, B.; Molnar, L.; McDermott, J.; et al. Dose Escalation Study to Assess the Pharmacokinetic Parameters of a Nano-amorphous Oral Sirolimus Formulation in Healthy Volunteers. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Cheer, S.M.; Prakash, A.; Faulds, D.; Lamb, H.M. Pantoprazole: An update of its pharmacological properties and therapeutic use in the management of acid-related disorders. Drugs 2003, 63, 101–133. [Google Scholar] [CrossRef] [PubMed]
- Sigaroudi, A.; Stelzer, C.; Braun, T.; Frechen, S.; Huttner, S.; Schroter, M.; Kinzig, M.; Fuhr, U.; Holzgrabe, U.; Sorgel, F. Comparison of Pantoprazole Concentrations in Simultaneous Cerebrospinal Fluid and Serum Samples. Pharmacology 2016, 98, 70–72. [Google Scholar] [CrossRef] [PubMed]
- Terstappen, G.C.; Meyer, A.H.; Bell, R.D.; Zhang, W. Strategies for delivering therapeutics across the blood-brain barrier. Nat. Rev. Drug Discov. 2021, 20, 362–383. [Google Scholar] [CrossRef] [PubMed]
- Lugini, L.; Federici, C.; Borghi, M.; Azzarito, T.; Marino, M.L.; Cesolini, A.; Spugnini, E.P.; Fais, S. Proton pump inhibitors while belonging to the same family of generic drugs show different anti-tumor effect. J. Enzym. Inhib. Med. Chem. 2016, 31, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Urrego, D.; Tomczak, A.P.; Zahed, F.; Stuhmer, W.; Pardo, L.A. Potassium channels in cell cycle and cell proliferation. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130094. [Google Scholar] [CrossRef]
- Ghiani, C.A.; Yuan, X.; Eisen, A.M.; Knutson, P.L.; DePinho, R.A.; McBain, C.J.; Gallo, V. Voltage-Activated K+ Channels and Membrane Depolarization Regulate Accumulation of the Cyclin-Dependent Kinase Inhibitors p27Kip1and p21CIP1in Glial Progenitor Cells. J. Neurosci. 1999, 19, 5380–5392. [Google Scholar] [CrossRef] [PubMed]
- Chittajallu, R.; Chen, Y.; Wang, H.; Yuan, X.; Ghiani, C.A.; Heckman, T.; McBain, C.J.; Gallo, V. Regulation of Kv1 subunit expression in oligodendrocyte progenitor cells and their role in G1/S phase progression of the cell cycle. Proc. Natl. Acad. Sci. USA 2002, 99, 2350–2355. [Google Scholar] [CrossRef] [PubMed]
- Harley, W.; Floyd, C.; Dunn, T.; Zhang, X.D.; Chen, T.Y.; Hegde, M.; Palandoken, H.; Nantz, M.H.; Leon, L.; Carraway, K.L., 3rd; et al. Dual inhibition of sodium-mediated proton and calcium efflux triggers non-apoptotic cell death in malignant gliomas. Brain Res. 2010, 1363, 159–169. [Google Scholar] [CrossRef]
- Marathe, K.; McVicar, N.; Li, A.; Bellyou, M.; Meakin, S.; Bartha, R. Topiramate induces acute intracellular acidification in glioblastoma. J. Neurooncol. 2016, 130, 465–472. [Google Scholar] [CrossRef]
- Gatti, M.; Pinato, S.; Maspero, E.; Soffientini, P.; Polo, S.; Penengo, L. A novel ubiquitin mark at the N-terminal tail of histone H2As targeted by RNF168 ubiquitin ligase. Cell Cycle 2012, 11, 2538–2544. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Bernal, G.M.; Cahill, K.E.; Pytel, P.; Fitzpatrick, C.A.; Mashek, H.; Weichselbaum, R.R.; Yamini, B. BCL3 expression promotes resistance to alkylating chemotherapy in gliomas. Sci. Transl. Med. 2018, 10, eaar2238. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tang, Z.G.; Yang, J.Q.; Zhou, Y.; Meng, L.H.; Wang, H.; Li, C.L. Low concentration of quercetin antagonizes the invasion and angiogenesis of human glioblastoma U251 cells. Onco. Targets Ther. 2017, 10, 4023–4028. [Google Scholar] [CrossRef] [PubMed]
- Vengoji, R.; Macha, M.A.; Batra, S.K.; Shonka, N.A. Natural products: A hope for glioblastoma patients. Oncotarget 2018, 9, 22194–22219. [Google Scholar] [CrossRef] [PubMed]
- Cenciarini, M.; Valentino, M.; Belia, S.; Sforna, L.; Rosa, P.; Ronchetti, S.; D’Adamo, M.C.; Pessia, M. Dexamethasone in Glioblastoma Multiforme Therapy: Mechanisms and Controversies. Front. Mol. Neurosci. 2019, 12, 65. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Ye, D.; Gessler, F.A.; Lee, Y.J.; Peruzzotti-Jametti, L.; Baumgarten, P.; Johnson, K.R.; Maric, D.; Yang, W.; Kogel, D.; et al. Topotecan is a potent inhibitor of SUMOylation in glioblastoma multiforme and alters both cellular replication and metabolic programming. Sci. Rep. 2017, 7, 7425. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.J.; Synold, T.; Mamelak, A.; Lim, D.; Al-Kadhimi, Z.; Twardowski, P.; Leong, L.; Chow, W.; Margolin, K.; Shibata, S.; et al. Plasma and cerebrospinal fluid pharmacokinetics of topotecan in a phase I trial of topotecan, tamoxifen, and carboplatin, in the treatment of recurrent or refractory brain or spinal cord tumors. Cancer Chemother. Pharmacol. 2010, 66, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, D.; Cairncross, G.; Stewart, D.; Forsyth, P.; Sawka, C.; Wainman, N.; Eisenhauer, E. Phase II study of topotecan in patients with recurrent malignant glioma. National Clinical Institute of Canada Clinical Trials Group. Ann. Oncol. 1996, 7, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.Y.; Park, C.K.; Kim, S.K.; Phi, J.H.; Kim, J.H.; Kim, C.Y.; Wang, K.C.; Cho, B.K. CKD-602, a camptothecin derivative, inhibits proliferation and induces apoptosis in glioma cell lines. Oncol. Rep. 2009, 21, 1413–1419. [Google Scholar] [CrossRef]
- Graham, C.D.; Kaza, N.; Klocke, B.J.; Gillespie, G.Y.; Shevde, L.A.; Carroll, S.L.; Roth, K.A. Tamoxifen Induces Cytotoxic Autophagy in Glioblastoma. J. Neuropathol. Exp. Neurol. 2016, 75, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Wan, W.; Zhang, X.; Huang, C.; Chen, L.; Yang, X.; Bao, K.; Peng, T. Monensin inhibits glioblastoma angiogenesis via targeting multiple growth factor receptor signaling. Biochem. Biophys. Res. Commun. 2020, 530, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Kang, Y.J.; Kim, I.Y.; Kim, E.H.; Lee, J.A.; Lim, J.H.; Kwon, T.K.; Choi, K.S. Monensin, a polyether ionophore antibiotic, overcomes TRAIL resistance in glioma cells via endoplasmic reticulum stress, DR5 upregulation and c-FLIP downregulation. Carcinogenesis 2013, 34, 1918–1928. [Google Scholar] [CrossRef] [PubMed]
- Enriquez Perez, J.; Fritzell, S.; Kopecky, J.; Visse, E.; Darabi, A.; Siesjo, P. The effect of locally delivered cisplatin is dependent on an intact immune function in an experimental glioma model. Sci. Rep. 2019, 9, 5632. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Sasagawa, S.; Itoh, K. Sodium butyrate induces senescence and inhibits the invasiveness of glioblastoma cells. Oncol. Lett. 2018, 15, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Engelhard, H.H.; Duncan, H.A.; Kim, S.; Criswell, P.S.; Van Eldik, L. Therapeutic effects of sodium butyrate on glioma cells in vitro and in the rat C6 glioma model. Neurosurgery 2001, 48, 616–624, discussion 624–625. [Google Scholar] [CrossRef] [PubMed]



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Mechanism | Status |
|---|---|---|
| cAMP | Activates variety of ion channels and protein kinases | Cannot be used clinically for GBM [16] |
| Rapamycin | Inhibits mTOR and induces autophagy | Clinical trial for GBM [81,82] |
| Retigabine | Opens KCNQ2-5/Kv7.2–7.5 channels | Novel application for GBM |
| Minoxidil | Opens K(ATP) channels | Novel application for GBM |
| NS1643 | Opens hERG and potentiates KCNQ2-4 channels | Novel application for GBM |
| Lamotrigine | Blocks voltage gated sodium channels | Novel application for GBM |
| Pantoprazole | Proton pump inhibitor | Published for GBM [83] |
| Temozolomide (TMZ) | Alkylates/methylates DNA, induces autophagy, used in GBM treatment | Current standard treatment for GBM [84] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mathews, J.; Kuchling, F.; Baez-Nieto, D.; Diberardinis, M.; Pan, J.Q.; Levin, M. Ion Channel Drugs Suppress Cancer Phenotype in NG108-15 and U87 Cells: Toward Novel Electroceuticals for Glioblastoma. Cancers 2022, 14, 1499. https://doi.org/10.3390/cancers14061499
Mathews J, Kuchling F, Baez-Nieto D, Diberardinis M, Pan JQ, Levin M. Ion Channel Drugs Suppress Cancer Phenotype in NG108-15 and U87 Cells: Toward Novel Electroceuticals for Glioblastoma. Cancers. 2022; 14(6):1499. https://doi.org/10.3390/cancers14061499
Chicago/Turabian StyleMathews, Juanita, Franz Kuchling, David Baez-Nieto, Miranda Diberardinis, Jen Q. Pan, and Michael Levin. 2022. "Ion Channel Drugs Suppress Cancer Phenotype in NG108-15 and U87 Cells: Toward Novel Electroceuticals for Glioblastoma" Cancers 14, no. 6: 1499. https://doi.org/10.3390/cancers14061499
APA StyleMathews, J., Kuchling, F., Baez-Nieto, D., Diberardinis, M., Pan, J. Q., & Levin, M. (2022). Ion Channel Drugs Suppress Cancer Phenotype in NG108-15 and U87 Cells: Toward Novel Electroceuticals for Glioblastoma. Cancers, 14(6), 1499. https://doi.org/10.3390/cancers14061499