
What Do We Know about Survival in Skeletally Premature Children Aged 0 to 10 Years with Ewing Sarcoma? A Multicenter 10-Year Follow-Up Study in 60 Patients

 , , ,
, , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Patients
2.2. Statistics
3. Results
3.1. Surgery
3.2. Radiotherapy
3.3. Survival, Local Control and Distant Metastases
3.4. Secondary Malignancy
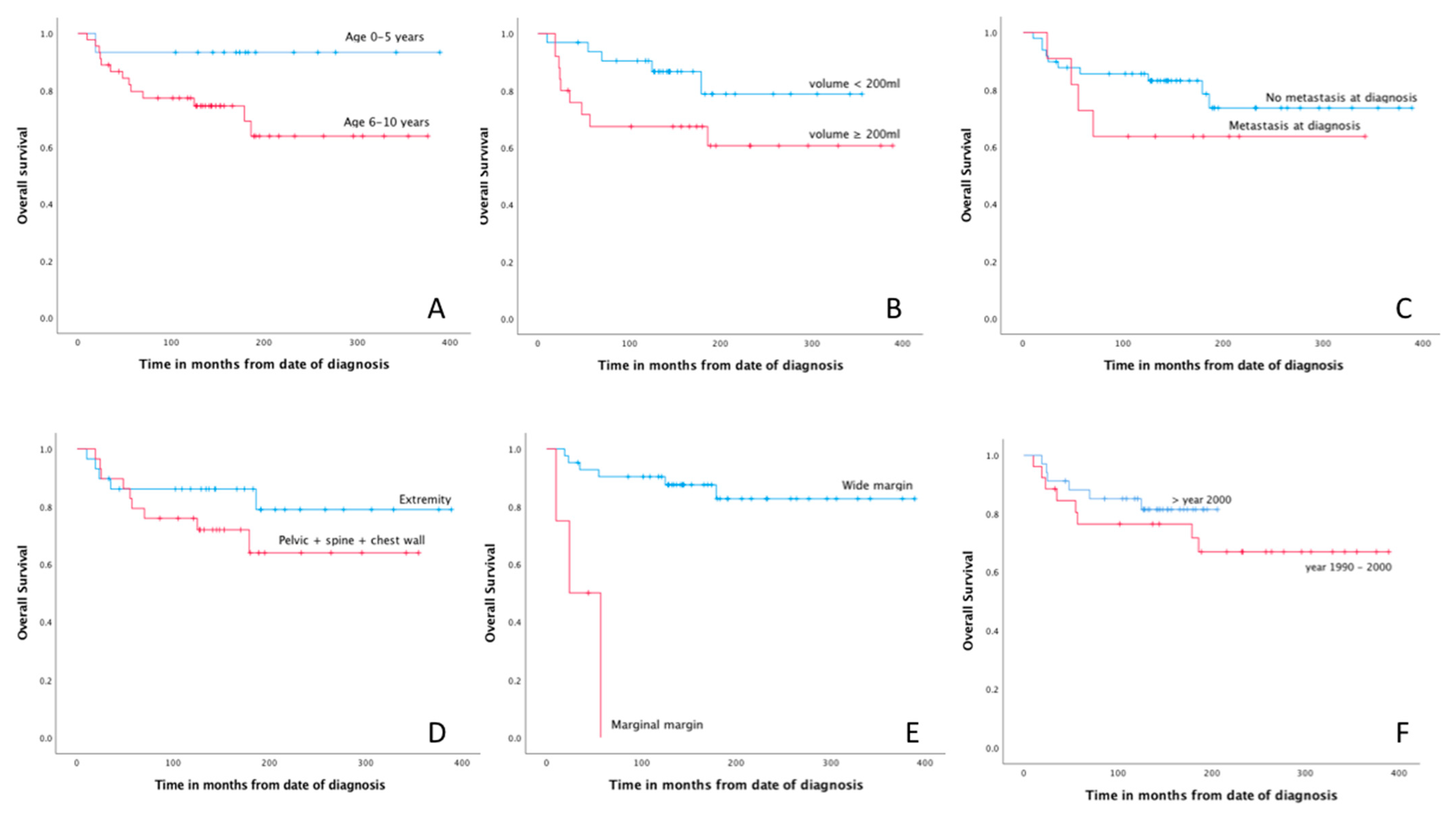
3.5. Prognostic Factors
4. Discussion
Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fletcher, C.D.M.; Hogendoorn, P.W.C.; Mertens, F. WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; IARC: Lyon, France, 2013. [Google Scholar]
- Arora, R.S.; Alston, R.D.; Eden, T.O.; Geraci, M.; Birch, J.M. The contrasting age-incidence patterns of bone tumours in teenagers and young adults: Implications for aetiology. Int. J. Cancer 2012, 131, 1678–1685. [Google Scholar] [CrossRef] [Green Version]
- Duchman, K.R.; Gao, Y.; Miller, B.J. Prognostic factors for survival in patients with Ewing’s sarcoma using the surveillance, epidemiology, and end results (SEER) program database. Cancer Epidemiol. 2015, 39, 189–195. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, H.; Kroon, H.M.; Slaar, A.; Hogendoorn, P. Incidence of biopsy-proven bone tumors in children: A report based on the Dutch pathology registration “PALGA”. J. Pediatr. Orthop. 2008, 28, 29–35. [Google Scholar] [CrossRef]
- Whelan, J.; McTiernan, A.; Cooper, N.; Wong, Y.K.; Francis, M.; Vernon, S.; Strauss, S.J. Incidence and survival of malignant bone sarcomas in England 1979–2007. Int. J. Cancer 2012, 131, E508–E517. [Google Scholar] [CrossRef]
- Worch, J.; Ranft, A.; DuBois, S.G.; Paulussen, M.; Juergens, H.; Dirksen, U. Age dependency of primary tumor sites and metastases in patients with Ewing sarcoma. Pediatr. Blood Cancer 2018, 65, e27251. [Google Scholar] [CrossRef] [PubMed]
- Stiller, C.A.; Bielack, S.S.; Jundt, G.; Steliarova-Foucher, E. Bone tumours in European children and adolescents, 1978–1997. Report from the Automated Childhood Cancer Information System project. Eur. J. Cancer 2006, 42, 2124–2135. [Google Scholar] [CrossRef] [PubMed]
- Ladenstein, R.; Potschger, U.; Le Deley, M.C.; Whelan, J.; Paulussen, M.; Oberlin, O.; van den Berg, H.; Dirksen, U.; Hjorth, L.; Michon, J.; et al. Primary disseminated multifocal Ewing sarcoma: Results of the Euro-EWING 99 trial. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2010, 28, 3284–3291. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Pappo, A.S.; Dirksen, U. Rhabdomyosarcoma, Ewing Sarcoma, and Other Round Cell Sarcomas. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.J.; Gao, Y.; Duchman, K.R. Does surgery or radiation provide the best overall survival in Ewing’s sarcoma? A review of the National Cancer Data Base. J. Surg. Oncol. 2017, 116, 384–390. [Google Scholar] [CrossRef]
- Verma, V.; Denniston, K.A.; Lin, C.J.; Lin, C. A Comparison of Pediatric vs. Adult Patients with the Ewing Sarcoma Family of Tumors. Front. Oncol. 2017, 7, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fizazi, K.; Dohollou, N.; Blay, J.Y.; Guerin, S.; Le Cesne, A.; Andre, F.; Pouillart, P.; Tursz, T.; Nguyen, B.B. Ewing’s family of tumors in adults: Multivariate analysis of survival and long-term results of multimodality therapy in 182 patients. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 1998, 16, 3736–3743. [Google Scholar] [CrossRef] [PubMed]
- Albergo, J.I.; Gaston, C.L.; Laitinen, M.; Darbyshire, A.; Jeys, L.M.; Sumathi, V.; Parry, M.; Peake, D.; Carter, S.R.; Tillman, R.; et al. Ewing’s sarcoma: Only patients with 100% of necrosis after chemotherapy should be classified as having a good response. Bone Jt. J. 2016, 98, 1138–1144. [Google Scholar] [CrossRef] [PubMed]
- Biswas, B.; Rastogi, S.; Khan, S.A.; Shukla, N.K.; Deo, S.V.; Agarwala, S.; Mohanti, B.K.; Sharma, M.C.; Vishnubhatla, S.; Bakhshi, S. Developing a prognostic model for localized Ewing sarcoma family of tumors: A single institutional experience of 224 cases treated with uniform chemotherapy protocol. J. Surg. Oncol. 2015, 111, 683–689. [Google Scholar] [CrossRef]
- Provost, B.; Missenard, G.; Pricopi, C.; Mercier, O.; Mussot, S.; Fabre, D.; Langer, N.; Mir, O.; Le Pechoux, C.; Dartevelle, P.; et al. Ewing Sarcoma of the Chest Wall: Prognostic Factors of Multimodal Therapy Including En Bloc Resection. Ann. Thorac. Surg. 2018, 106, 207–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedetti, B.; Wiebe, K.; Ranft, A.; Aebert, H.; Schmidt, J.; Jurgens, H.; Dirksen, U. Local control in Ewing sarcoma of the chest wall: Results of the EURO-EWING 99 trial. Ann. Surg. Oncol. 2015, 22, 2853–2859. [Google Scholar] [CrossRef] [PubMed]
- Bacci, G.; Longhi, A.; Briccoli, A.; Bertoni, F.; Versari, M.; Picci, P. The role of surgical margins in treatment of Ewing’s sarcoma family tumors: Experience of a single institution with 512 patients treated with adjuvant and neoadjuvant chemotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Cotterill, S.J.; Ahrens, S.; Paulussen, M.; Jurgens, H.F.; Voute, P.A.; Gadner, H.; Craft, A.W. Prognostic factors in Ewing’s tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2000, 18, 3108–3114. [Google Scholar] [CrossRef]
- Gaspar, N.; Rey, A.; Berard, P.M.; Michon, J.; Gentet, J.C.; Tabone, M.D.; Roche, H.; Defachelles, A.S.; Lejars, O.; Plouvier, E.; et al. Risk adapted chemotherapy for localised Ewing’s sarcoma of bone: The French EW93 study. Eur. J. Cancer 2012, 48, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Foulon, S.; Brennan, B.; Gaspar, N.; Dirksen, U.; Jeys, L.; Cassoni, A.; Claude, L.; Seddon, B.; Marec-Berard, P.; Whelan, J.; et al. Can postoperative radiotherapy be omitted in localised standard-risk Ewing sarcoma? An observational study of the Euro-E.W.I.N.G group. Eur. J. Cancer 2016, 61, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hoang, B.H.; Ziogas, A.; Zell, J.A. Analysis of prognostic factors in Ewing sarcoma using a population-based cancer registry. Cancer 2010, 116, 1964–1973. [Google Scholar] [CrossRef] [PubMed]
- Jawad, M.U.; Cheung, M.C.; Min, E.S.; Schneiderbauer, M.M.; Koniaris, L.G.; Scully, S.P. Ewing sarcoma demonstrates racial disparities in incidence-related and sex-related differences in outcome: An analysis of 1631 cases from the SEER database, 1973-2005. Cancer 2009, 115, 3526–3536. [Google Scholar] [CrossRef]
- Bosma, S.E.; Ayu, O.; Fiocco, M.; Gelderblom, H.; Dijkstra, P.D.S. Prognostic factors for survival in Ewing sarcoma: A systematic review. Surg. Oncol. 2018, 27, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Marina, N.; Granowetter, L.; Grier, H.E.; Womer, R.B.; Randall, R.L.; Marcus, K.J.; McIlvaine, E.; Krailo, M. Age, Tumor Characteristics, and Treatment Regimen as Event Predictors in Ewing: A Children’s Oncology Group Report. Sarcoma 2015, 2015, 927123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gobel, V.; Jurgens, H.; Etspuler, G.; Kemperdick, H.; Jungblut, R.M.; Stienen, U.; Gobel, U. Prognostic significance of tumor volume in localized Ewing’s sarcoma of bone in children and adolescents. J. Cancer Res. Clin. Oncol. 1987, 113, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Hense, H.W.; Ahrens, S.; Paulussen, M.; Lehnert, M.; Jurgens, H. Factors associated with tumor volume and primary metastases in Ewing tumors: Results from the (EI)CESS studies. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 1999, 10, 1073–1077. [Google Scholar] [CrossRef] [PubMed]
- Schuck, A.; Ahrens, S.; Paulussen, M.; Kuhlen, M.; Konemann, S.; Rube, C.; Winkelmann, W.; Kotz, R.; Dunst, J.; Willich, N.; et al. Local therapy in localized Ewing tumors: Results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int. J. Radiat. Oncol. Biol. Phys. 2003, 55, 168–177. [Google Scholar] [CrossRef]
- Bacci, G.; Ferrari, S.; Barbieri, E.; Longhi, A.; Forni, C.; Cesari, M.; Sarti, M.; Gasbarrini, A.; Rosito, P.; Delprever, A. Long-term follow-up for patients with Ewing’s sarcoma of bone treated with adjuvant and neoadjuvant chemotherapy. Oncol. Rep. 1997, 4, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, J.P.; Goodman, P.; Leisenring, W.; Ness, K.K.; Meyers, P.A.; Wolden, S.L.; Smith, S.M.; Stovall, M.; Hammond, S.; Robison, L.L.; et al. Long-term survivors of childhood Ewing sarcoma: Report from the childhood cancer survivor study. J. Natl. Cancer Inst. 2010, 102, 1272–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacci, G.; Forni, C.; Longhi, A.; Ferrari, S.; Donati, D.; De Paolis, M.; Barbieri, E.; Pignotti, E.; Rosito, P.; Versari, M. Long-term outcome for patients with non-metastatic Ewing’s sarcoma treated with adjuvant and neoadjuvant chemotherapies. 402 patients treated at Rizzoli between 1972 and 1992. Eur. J. Cancer 2004, 40, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.A.; Pappo, A.; Saunders, N.; Hopyan, S.; Ferguson, P.; Wunder, J.; O’Sullivan, B.; Catton, C.; Greenberg, M.; Blackstein, M. Clinical outcome of children and adults with localized Ewing sarcoma: Impact of chemotherapy dose and timing of local therapy. Cancer 2010, 116, 3189–3194. [Google Scholar] [CrossRef]
- Idoate, M.Á.; Aquerreta, J.D.; Lamo-Espinosa, J.M.; San-Julian, M. A Reassessment of the Barrier Effect of the Physis against Metaphyseal Osteosarcoma: A Comprehensive Pathological Study with Its Radiological and Clinical Follow-Up Correlations. Diagnostics 2022, 12, 450. [Google Scholar] [CrossRef] [PubMed]
- Simpson, P.M.; Reid, R.; Porter, D. Ewing’s Sarcoma of the Upper Extremity: Presenting Symptoms, Diagnostic Delay and Outcome. Sarcoma 2005, 9, 15–20. [Google Scholar] [CrossRef]
- Sneppen, O.; Hansen, L.M. Presenting symptoms and treatment delay in osteosarcoma and Ewing’s sarcoma. Acta Radiol. Oncol. 1984, 23, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Daugaard, S.; Sunde, L.M.; Kamby, C.; Schiodt, T.; Jensen, O.M. Ewing’s sarcoma. A retrospective study of prognostic factors and treatment results. Acta Oncol. 1987, 26, 281–287. [Google Scholar] [CrossRef] [PubMed]
- De Ioris, M.A.; Prete, A.; Cozza, R.; Podda, M.; Manzitti, C.; Pession, A.; Schiavello, E.; Contoli, B.; Balter, R.; Fagioli, F.; et al. Ewing sarcoma of the bone in children under 6 years of age. PLoS ONE 2013, 8, e53223. [Google Scholar] [CrossRef] [PubMed]
- Huh, W.W.; Daw, N.C.; Herzog, C.E.; Munsell, M.F.; McAleer, M.F.; Lewis, V.O. Ewing sarcoma family of tumors in children younger than 10 years of age. Pediatr. Blood Cancer 2017, 64, e26275. [Google Scholar] [CrossRef]
- Koshy, M.; Paulino, A.C.; Mai, W.Y.; Teh, B.S. Radiation-induced osteosarcomas in the pediatric population. Int. J. Radiat. Oncol. Biol. Phys. 2005, 63, 1169–1174. [Google Scholar] [CrossRef] [PubMed]
- Kuttesch, J.F.; Wexler, L.H.; Marcus, R.B.; Fairclough, D.; WeaverMcClure, L.; White, M.; Mao, L.; Delaney, T.F.; Pratt, C.B.; Horowitz, M.E.; et al. Second malignancies after Ewing’s sarcoma: Radiation dose-dependency of secondary sarcomas. J. Clin. Oncol. 1996, 14, 2818–2825. [Google Scholar] [CrossRef]
- Nguyen, F.; Rubino, C.; Guerin, S.; Diallo, I.; Samand, A.; Hawkins, M.; Oberlin, O.; Lefkopoulos, D.; De Vathaire, F. Risk of a second malignant neoplasm after cancer in childhood treated with radiotherapy: Correlation with the integral dose restricted to the irradiated fields. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 908–915. [Google Scholar] [CrossRef] [PubMed]
- Strauss, S.J.; Frezza, A.M.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Blay, J.Y.; Bolle, S.; Bonvalot, S.; et al. Bone sarcomas: ESMO-EURACAN-GENTURIS-ERN PaedCan Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 1520–1536. [Google Scholar] [CrossRef] [PubMed]
- Uyttendaele, D.; De Schryver, A.; Claessens, H.; Roels, H.; Berkvens, P.; Mondelaers, W. Limb conservation in primary bone tumours by resection, extracorporeal irradiation and re-implantation. J. Bone Jt. Surg. Br. Vol. 1988, 70, 348–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, A.; Byregowda, S.; Gulia, A.; Patil, V.; Crasto, S.; Laskar, S. Reconstructing diaphyseal tumors using radiated (50 Gy) autogenous tumor bone graft. J. Surg. Oncol. 2018, 118, 138–143. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, L.; Farfalli, G.L.; Balacó, I.; Alves, C.; Salom, M.; Lamo-Espinosa, J.M.; San-Julián, M.; van de Sande, M.A.J. Biology and technology in the surgical treatment of malignant bone tumours in children and adolescents, with a special note on the very young. J. Child. Orthop. 2021, 15, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Raciborska, A.; Bilska, K.; Koziński, T.; Rodriguez-Galindo, C. Subsequent Malignant Neoplasm of Bone in Children and Adolescent—Possibility of Multimodal Treatment. Curr. Oncol. 2022, 29, 1001–1007. [Google Scholar] [CrossRef]
- Inskip, P.D.; Curtis, R.E. New malignancies following childhood cancer in the United States, 1973–2002. Int. J. Cancer 2007, 121, 2233–2240. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.; Valenzuela, R.G.; Petersen, I.A.; Arndt, C.A.; Sim, F.H. Ewing’s sarcoma and the development of secondary malignancies. Clin. Orthop. Relat. Res. 2003, 415, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, L.M.; Liu, Q.; Yasui, Y.; Henderson, T.O.; Gibson, T.M.; Leisenring, W.; Arnold, M.A.; Howell, R.M.; Green, D.M.; Armstrong, G.T.; et al. Chemotherapy and Risk of Subsequent Malignant Neoplasms in the Childhood Cancer Survivor Study Cohort. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 3310–3319. [Google Scholar] [CrossRef] [PubMed]
- Caruso, J.; Shulman, D.S.; DuBois, S.G. Second malignancies in patients treated for Ewing sarcoma: A systematic review. Pediatr. Blood Cancer 2019, 66, e27938. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
| All Patients | Deceased Patients | |
|---|---|---|
| N (%) | N (%) | |
| Total | 60 | 14 (23) |
| Gender | ||
| Male | 37 (62) | 6 (43) |
| Female | 23 (38) | 8 (57) |
| Age (year) (mean, SD) | 7 (2.7) | 8.4 (1.9) |
| Location primary tumor | ||
| Extremity | 29 (48) | 5 (36) |
| Upper extremity | 5 (8) | 3 (22) |
| Lower extremity | 24 (40) | 2 (14) |
| Axial: | 22 (37) | 9 (63) |
| Pelvic | 10 (17) | 3 (22) |
| Spine | 1 (2) | 1 (7) |
| Chest wall (costa/sternum/scapula) | 11 (18) | 5 (36) |
| Other | ||
| Retroperitoneal, soft tissue | 6 (10) | |
| Skull/cranial | 3 (5) | |
| Volume | ||
| <200 mL | 32 (53) | 5 (36) |
| ≥200 mL | 25 (42) | 9 (63) |
| Missing | 3 (5) | |
| Size | ||
| <8 cm | 21 (35) | 3 (21) |
| ≥8 cm | 36 (60) | 11 (79) |
| Missing | 3 (5) | |
| Metastasis at diagnosis | ||
| No | 49 (82) | 10 (71) |
| Lung metastasis | 8 (13) | 1 (7) |
| Bone metastasis | 3 (5) | 3 (21) |
| Treatment | ||
| Radiotherapy | 32 (53) | 11 (79) |
| Definitive | 12 (20) | 6 (43) |
| Preoperative | 12 (20) | 4 (29) |
| Postoperative | 6 (10) | 1 (7) |
| Extracorporeal RT | 2 (3) | |
| Dose (Gy) (mean, SD) | 49 (17.9) | |
| Surgery | 48 (80) | 8 (57) |
| Rotationplasty | 2 (3) | 1 (7) |
| Allograft | 14 (23) | 2 (14) |
| Autograft | 6 (10) | |
| Prosthesis | 5 (8) | 1 (7) |
| Amputation | 2 (3) | |
| Resection without reconstruction | 19 (32) | 4 (29) |
| Surgical margin | ||
| R0 | 42 (70) | 5 (36) |
| R1 | 4 (7) | 3 (21) |
| Other * | 2 (3) | |
| Histological response | ||
| 100% | 27 (45) | 5 (36) |
| 90–99% | 13 (22) | 2 (14) |
| <90% | 2 (3) | 1 (7) |
| Missing | 6 (10) |
| Surgery Alone | Radiotherapy Alone | Surgery Combined with Radiotherapy | |
|---|---|---|---|
| All patients | 30 | 12 | 18 |
| Deceased patients | 3 (10%) | 6 (50%) | 5 (28%) |
| Variables | Univariate Analysis | |
|---|---|---|
| HR (95% CI) | p-Value | |
| Gender 1 | ||
| Male | 1 | |
| Female | 2.51 (0.87–7.24) | 0.088 |
| Age 1 | ||
| 0–5 years | 1 | |
| 6–10 years | 5.06 (0.66–38.69) | 0.119 |
| Volume 1 | ||
| <200 mL | 1 | |
| ≥200 mL | 2.55 (0.85–7.64) | 0.095 |
| Location 1 | ||
| Extremity | 1 | |
| Axial (spine + chest wall) | 1.84 (0.44–7.75) | 0.404 |
| Pelvic | 1.88 (0.57–6.17) | 0.300 |
| Metastasis at diagnosis 1 | ||
| No | 1 | |
| Yes | 1.84 (0.58–5.86) | 0.306 |
| Histological response 2 | ||
| 100% | 1 | |
| 90–99% | 0.90 (0.18–4.65) | 0.901 |
| <90% | 2.15 (0.25–18.65) | 0.489 |
| Surgical margin 2 | ||
| R0 | 1 | |
| R1 | 13.57 (2.92–63.04) | <0.001 |
| Treatment era | ||
| >2000 | 1 | |
| 1990–2000 | 1.56 (0.53–4.60) | 0.423 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosma, S.E.; van der Heijden, L.; Sierrasesúmaga, L.; Merks, H.J.H.M.; Haveman, L.M.; van de Sande, M.A.J.; San-Julián, M. What Do We Know about Survival in Skeletally Premature Children Aged 0 to 10 Years with Ewing Sarcoma? A Multicenter 10-Year Follow-Up Study in 60 Patients. Cancers 2022, 14, 1456. https://doi.org/10.3390/cancers14061456
Bosma SE, van der Heijden L, Sierrasesúmaga L, Merks HJHM, Haveman LM, van de Sande MAJ, San-Julián M. What Do We Know about Survival in Skeletally Premature Children Aged 0 to 10 Years with Ewing Sarcoma? A Multicenter 10-Year Follow-Up Study in 60 Patients. Cancers. 2022; 14(6):1456. https://doi.org/10.3390/cancers14061456
Chicago/Turabian StyleBosma, Sarah E., Lizz van der Heijden, Luis Sierrasesúmaga, Hans J. H. M. Merks, Lianne M. Haveman, Michiel A. J. van de Sande, and Mikel San-Julián. 2022. "What Do We Know about Survival in Skeletally Premature Children Aged 0 to 10 Years with Ewing Sarcoma? A Multicenter 10-Year Follow-Up Study in 60 Patients" Cancers 14, no. 6: 1456. https://doi.org/10.3390/cancers14061456
APA StyleBosma, S. E., van der Heijden, L., Sierrasesúmaga, L., Merks, H. J. H. M., Haveman, L. M., van de Sande, M. A. J., & San-Julián, M. (2022). What Do We Know about Survival in Skeletally Premature Children Aged 0 to 10 Years with Ewing Sarcoma? A Multicenter 10-Year Follow-Up Study in 60 Patients. Cancers, 14(6), 1456. https://doi.org/10.3390/cancers14061456