
Mitochondrial DNA on Tumor-Associated Macrophages Polarization and Immunity


Abstract
:Simple Summary
Abstract
1. Introduction
2. TAMs in Tumor Progression
3. Regulation of Free mtDNA in TME
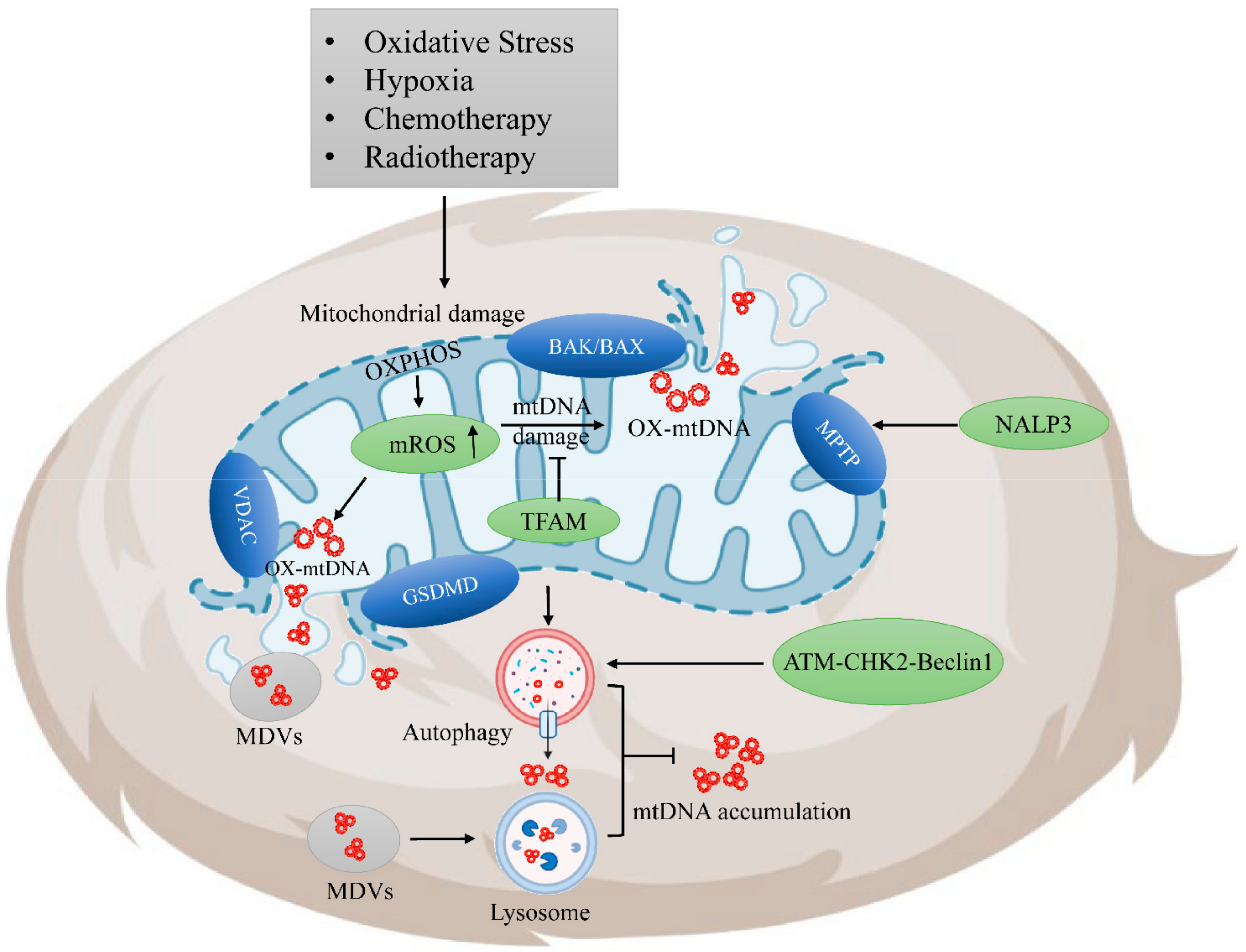
3.1. Release of Tumor-Derived mtDNA
3.2. Regulation of Tumor-Derived mtDNA Accumulation
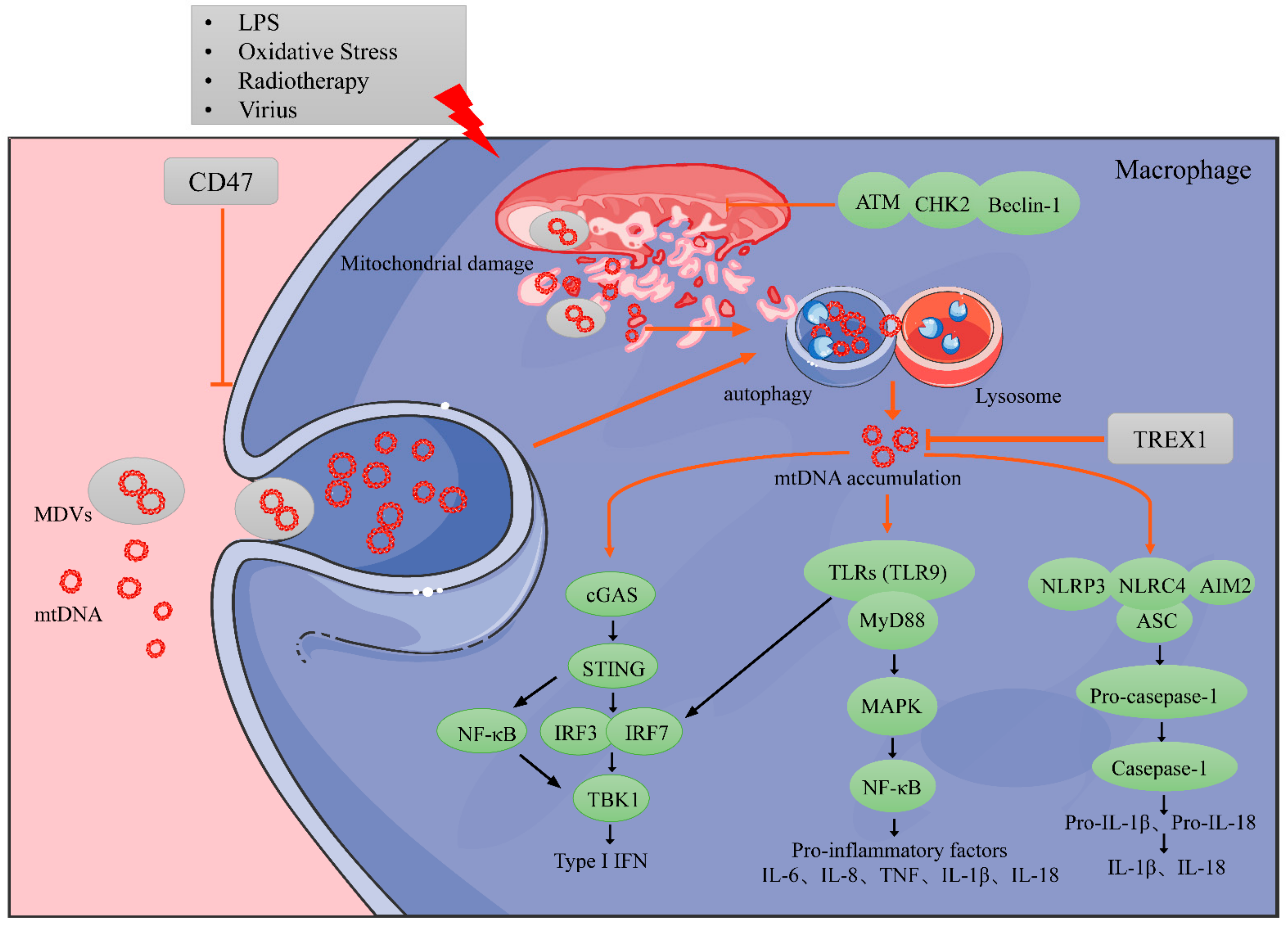
4. Regulation of mtDNA in Immune Cells
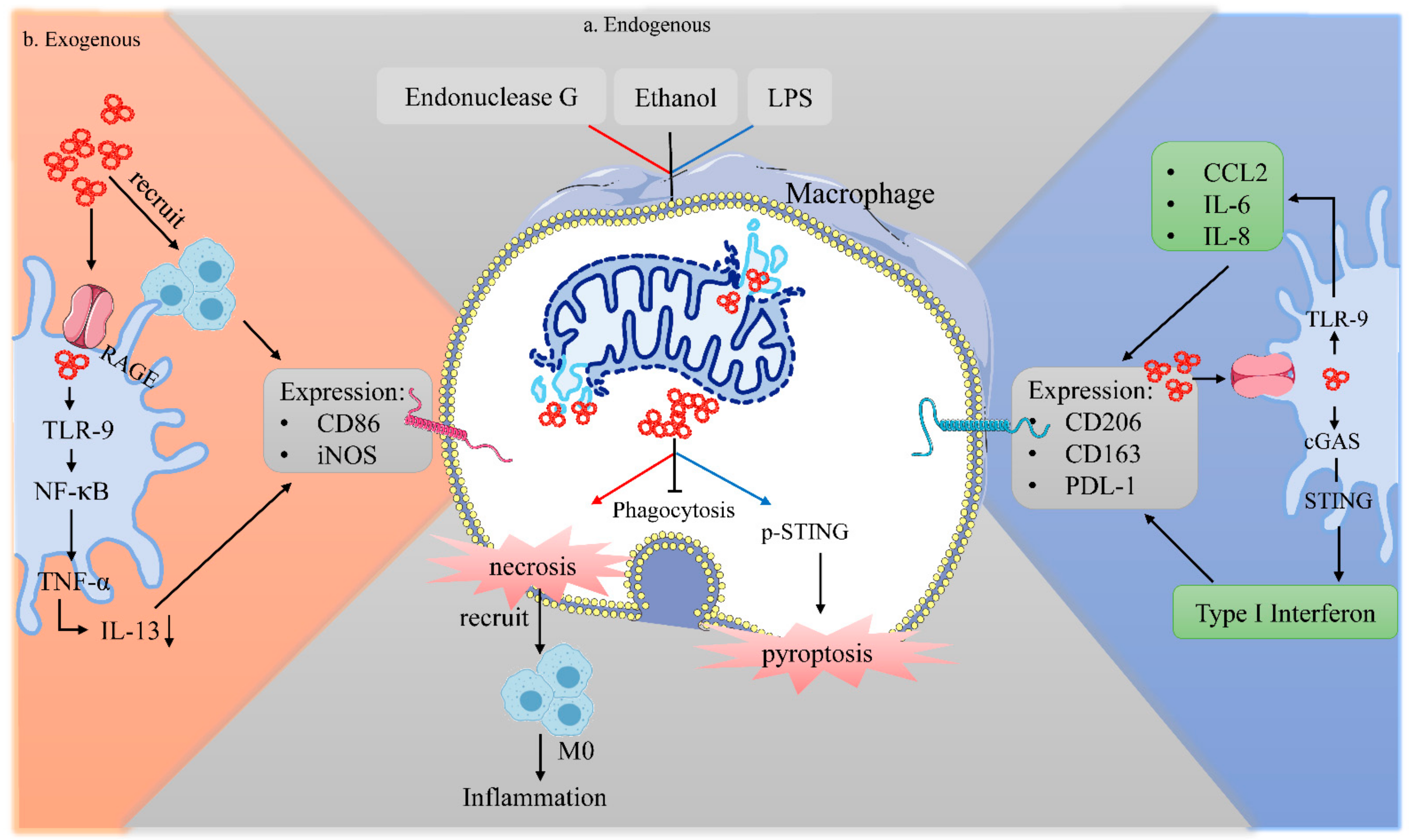
4.1. Exogenous mtDNA on Macrophages
4.2. Endogenous mtDNA on Macrophages
4.2.1. MtDNA Accumulation in Macrophages
4.2.2. Impact of Instinct Endogenous mtDNA on Macrophage Biology
4.2.3. Impact of Swallowed mtDNA on Macrophage Biology
5. Anti-Cancer Therapeutic Opportunities That Involve MtDNA
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Glossary
| Abbreviation | Meaning |
| AcAc | Acetoacetate |
| AML | Acute myeloid leukemia |
| APE1 | AP endonuclease |
| Arg-1 | Arginase-1 |
| ASC | Apoptosis-associated spot protein |
| ATM | Ataxia telangiectasia-mutated |
| BAK | Bcl-2 homologous antagonist/killer |
| BAX | Bcl-2-associated X protein |
| cCAS | cGAMP synthetase |
| CCL2 | Chemokine (C-C motif) ligand 2 |
| CCL5 | Chemokine (C-C motif) ligand 2 |
| CD47 | Antigenic surface determinant protein OA3 |
| CD206 | Mannose receptor |
| cGAMP | Cyclic GMP-AMP |
| CHK2 | Cell Cycle Checkpoint |
| CSF-1 | Colony Stimulating Factor-1 |
| CTLs | Cytotoxic T lymphocytes |
| CXCL10 | CXC chemokine ligand 10 |
| DAMPs | Damage associated molecular patterns |
| DCs | Dendritic cells |
| DDRs | DNA damage repair responses |
| DNase I | Deoxyribonuclease I |
| DNase II | Deoxyribonuclease II |
| Drp1 | Dynamin-related protein-1 |
| dsDNA | Double-stranded DNA |
| ECM | Extracellular matrix |
| ER | Endoplasmic reticulum |
| EVs | Extracellular vesicles |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GSDMD | Gasdermin-D |
| HLA | Human leukocyte antigen |
| HMGB1 | High-mobility group protein B1 |
| IFN-β | Interferon-β |
| IFN-γ | Interferon-γ |
| IL-1α | Interleukin-1 alpha |
| IL-1β | Interleukin-1 beta |
| IL-4 | Interleukin 4 |
| IL-6 | Interleukin 6 |
| IL-10 | Interleukin 10 |
| IL-12 | Interleukin 12 |
| IL-13 | Interleukin 13 |
| IL-18 | Interleukin 18 |
| IL-23IRF3 | Interleukin 23Interferon regulatory Factor 3 |
| LAP | LC3-associated phagocytosis |
| LPS | Lipopolysaccharide |
| MAPK | Mitogen-activated protein kinases |
| MDVs | Mitochondria-derived vesicles |
| MDSCs | Myeloid-derived suppressor cells |
| MHC-II | Major histocompatibility complex-II |
| MOMP | Mitochondrial outer membrane permeability |
| MPTP | Mitochondrial permeability transition pore |
| Mros | mitochondrial reactive oxygen species |
| MT1-MMP | Membrane type 1 matrix metalloproteinase |
| mtDNA | mitochondrial DNA |
| MyD88 | Myeloid differentiation factor88 |
| NALP3 | NLR family, pyrin domain containing 3 |
| nDNA | Nuclear DNA |
| NETs | Extracellular traps |
| NF-κB | Nuclear factor-κB |
| NK | Natural Killer Cell |
| NLRP3 | Pyrin domain containing protein 3 |
| NLRs | Nod-like receptors |
| OXPHOS | Oxidative phosphorylation |
| PD-L1 | Programmed death 1 ligand 1 |
| PAMPs | Pathogen-associated molecular patterns |
| POLG | Polymerase gamma |
| PRRs | Pattern recognition receptors |
| ROS | Reactive oxygen species |
| STING | Stimulator of Interferon Genes |
| TANs | Tumor-associated neutrophils |
| TAMs | Tumor-associated macrophages |
| TBK1 | TANK-binding kinase 1 |
| TDEs | Tumor-derived exosomes |
| TFAM | Transcription Factor A |
| TGF-β | Transforming Growth Factor |
| Th1 | Helper T cell |
| TLRs | Toll-like receptors |
| TME | Tumor microenvironments |
| TNBC | Triple-negative breast cancer |
| TNF-α | Tumor necrosis factor-α |
| Tregs | Regulatory T cells |
| TREX1 | Three initial repair exonuclease 1 |
| TRIF | TIR domain |
| VDAC | Voltage-dependent anion channel |
| VEGF | Vascular endothelial growth factor |
| 8-OHG | 8-hydroxyguanosine |
References
- Hitscherich, P.; Lee, E. In Vitro Crosstalk Between Cardiac Cells and Macrophages Postmyocardial Infarction: Insights from Studies. Tissue Eng. Part B Rev. 2021, 27, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Wenes, M.; Shang, M.; Di Matteo, M.; Goveia, J.; Martín-Pérez, R.; Serneels, J.; Prenen, H.; Ghesquière, B.; Carmeliet, P.; Mazzone, M. Macrophage Metabolism Controls Tumor Blood Vessel Morphogenesis and Metastasis. Cell Metab. 2016, 24, 701–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jetten, N.; Verbruggen, S.; Gijbels, M.J.; Post, M.J.; De Winther, M.P.; Donners, M.M. Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo. Angiogenesis 2014, 17, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Akalu, Y.; Rothlin, C.; Ghosh, S. TAM receptor tyrosine kinases as emerging targets of innate immune checkpoint blockade for cancer therapy. Immunol. Rev. 2017, 276, 165–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiemstra, I.; Beijer, M.; Veninga, H.; Vrijland, K.; Borg, E.; Olivier, B.; Mebius, R.; Kraal, G.; den Haan, J. The identification and developmental requirements of colonic CD169⁺ macrophages. Immunology 2014, 142, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, N.; Zhang, X.; Horng, T. Mitochondrial metabolism regulates macrophage biology. J. Biol. Chem. 2021, 297, 100904. [Google Scholar] [CrossRef]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef] [Green Version]
- Keatley, K.; Stromei-Cleroux, S.; Wiltshire, T.; Rajala, N.; Burton, G.; Holt, W.; Littlewood, D.; Briscoe, A.; Jung, J.; Ashkan, K.; et al. Integrated Approach Reveals Role of Mitochondrial Germ-Line Mutation F18L in Respiratory Chain, Oxidative Alterations, Drug Sensitivity, and Patient Prognosis in Glioblastoma. Int. J. Mol. Sci. 2019, 20, 3364. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Qu, Y.; Gao, K.; Yang, Q.; Shi, B.; Hou, P.; Ji, M. High copy number of mitochondrial DNA (mtDNA) predicts good prognosis in glioma patients. Am. J. Cancer Res. 2015, 5, 1207–1216. [Google Scholar]
- Torregrosa, R.; Goffart, S.; Haikonen, J.A.; Pohjoismäki, J.L.O. Low doses of ultraviolet radiation and oxidative damage induce dramatic accumulation of mitochondrial DNA replication intermediates, fork regression, and replication initiation shift. Mol. Biol. Cell 2015, 26, 4197–4208. [Google Scholar] [CrossRef]
- Reyes, A.; Kazak, L.; Wood, S.R.; Yasukawa, T.; Jacobs, H.T.; Holt, I.J. Mitochondrial DNA replication proceeds via a ‘bootlace’ mechanism involving the incorporation of processed transcripts. Nucleic Acids Res. 2013, 41, 5837–5850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, G.A.; Gahlon, H. Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res. 2020, 48, 11244–11258. [Google Scholar] [CrossRef]
- Das, P.; Chakrabarti, O. Mitochondrial DNA in innate immune responses against infectious diseases. Biochem. Soc. Trans. 2020, 48, 2823–2838. [Google Scholar] [CrossRef] [PubMed]
- Mitochondrial DNA Damage Triggers an IFN-Mediated Immune Response. Cancer Discov. 2021, 11, 1004. [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-J.; Wang, X.-H.; Gao, S.-T.; Chen, C.; Xu, X.-Y.; Sun, Q.; Zhou, Z.-H.; Wu, G.-Z.; Yu, Q.; Xu, G.; et al. Tumor-associated macrophages correlate with phenomenon of epithelial-mesenchymal transition and contribute to poor prognosis in triple-negative breast cancer patients. J. Surg. Res. 2018, 222, 93–101. [Google Scholar] [CrossRef]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef]
- Chen, P.; Zhao, D.; Li, J.; Liang, X.; Li, J.; Chang, A.; Henry, V.K.; Lan, Z.; Spring, D.J.; Rao, G.; et al. Symbiotic Macrophage-Glioma Cell Interactions Reveal Synthetic Lethality in PTEN-Null Glioma. Cancer Cell 2019, 35, 868–884.e6. [Google Scholar] [CrossRef]
- Coletta, S.; Lonardi, S.; Sensi, F.; D’Angelo, E.; Fassan, M.; Pucciarelli, S.; Valzelli, A.; Biccari, A.; Vermi, W.; Della Bella, C.; et al. Tumor Cells and the Extracellular Matrix Dictate the Pro-Tumoral Profile of Macrophages in CRC. Cancers 2021, 13, 5199. [Google Scholar] [CrossRef]
- Yuan, A.; Hsiao, Y.-J.; Chen, H.-Y.; Chen, H.-W.; Ho, C.-C.; Chen, Y.-Y.; Liu, Y.-C.; Hong, T.-H.; Yu, S.-L.; Chen, J.J.; et al. Opposite Effects of M1 and M2 Macrophage Subtypes on Lung Cancer Progression. Sci. Rep. 2015, 5, 14273. [Google Scholar] [CrossRef] [Green Version]
- Edin, S.; Wikberg, M.L.; Dahlin, A.M.; Rutegård, J.; Öberg, Å.; Oldenborg, P.-A.; Palmqvist, R. The Distribution of Macrophages with a M1 or M2 Phenotype in Relation to Prognosis and the Molecular Characteristics of Colorectal Cancer. PLoS ONE 2012, 7, e47045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colegio, O.R.; Chu, N.-Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Acebes, M.; Dalla, E.; Leader, A.M.; LeBerichel, J.; Nikolic, J.; Morales, B.M.; Brown, M.; Chang, C.; Troncoso, L.; Chen, S.T.; et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature 2021, 595, 578–584. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, T.M.; Sosnowska, A.; Matryba, P.; Rydzynska, Z.; Jasinski, M.; Nowis, D.; Golab, J. Myeloid Cell-Derived Arginase in Cancer Immune Response. Front. Immunol. 2020, 11, 938. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Fleetwood, A.J.; Lawrence, T.; Hamilton, J.A.; Cook, A. Granulocyte-Macrophage Colony-Stimulating Factor (CSF) and Macrophage CSF-Dependent Macrophage Phenotypes Display Differences in Cytokine Profiles and Transcription Factor Activities: Implications for CSF Blockade in Inflammation. J. Immunol. 2007, 178, 5245–5252. [Google Scholar] [CrossRef] [Green Version]
- de Sousa, J.R.; de Sousa, R.P.M.; Aarão, T.L.D.S.; Dias, L.B.; Carneiro, F.R.O.; Fuzii, H.T.; Quaresma, J.A.S. In situ expression of M2 macrophage subpopulation in leprosy skin lesions. Acta Trop. 2016, 157, 108–114. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef]
- Ning, C.; Li, Y.-Y.; Wang, Y.; Han, G.-C.; Wang, R.; Xiao, H.; Li, X.-Y.; Hou, C.-M.; Ma, Y.-F.; Sheng, D.-S.; et al. Complement activation promotes colitis-associated carcinogenesis through activating intestinal IL-1β/IL-17A axis. Mucosal Immunol. 2015, 8, 1275–1284. [Google Scholar] [CrossRef] [Green Version]
- Hou, Z.; Falcone, D.J.; Subbaramaiah, K.; Dannenberg, A.J. Macrophages induce COX-2 expression in breast cancer cells: Role of IL-1β autoamplification. Carcinogenesis 2011, 32, 695–702. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.-Y.; Zhang, J.-Y.; Shi, X.-Z.; Wang, Q.; Shen, W.-L.; Zhu, W.-W.; Liu, L.-K. Upregulation of CSF-1 is correlated with elevated TAM infiltration and poor prognosis in oral squamous cell carcinoma. Am. J. Transl. Res. 2020, 12, 6235–6249. [Google Scholar] [PubMed]
- Liu, M.; Zhang, L.; Zhou, Q.; Wang, Y.; Sun, Q.; Ren, X. The Distinct Impact of TAM Infiltration on the Prognosis of Patients with Cardia and Non-Cardia Gastric Cancer and Its Association with H. pylori Infection. Front. Oncol. 2021, 11, 737061. [Google Scholar] [CrossRef]
- Mulder, R.; Banete, A.; Seaver, K.; Basta, S. M(IL-4) Tissue Macrophages Support Efficient Interferon-Gamma Production in Antigen-Specific CD8+ T Cells with Reduced Proliferative Capacity. Front. Immunol. 2017, 8, 1629. [Google Scholar] [CrossRef] [Green Version]
- Philipp, D.; Suhr, L.; Wahlers, T.; Choi, Y.-H.; Paunel-Görgülü, A. Preconditioning of bone marrow-derived mesenchymal stem cells highly strengthens their potential to promote IL-6-dependent M2b polarization. Stem Cell Res. Ther. 2018, 9, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Wei, K.; Ding, Y.; Ahati, P.; Xu, H.; Fang, H.; Wang, H. M2a Macrophage-Secreted CHI3L1 Promotes Extracellular Matrix Metabolic Imbalances via Activation of IL-13Rα2/MAPK Pathway in Rat Intervertebral Disc Degeneration. Front. Immunol. 2021, 12, 666361. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wei, Y.; Liu, Y.; Wu, T.; Hu, J.; Lu, H. The Long Non-coding RNA NEAT1/miR-224-5p/IL-33 Axis Modulates Macrophage M2a Polarization and A1 Astrocyte Activation. Mol. Neurobiol. 2021, 58, 4506–4519. [Google Scholar] [CrossRef]
- Wang, Q.; Ni, H.; Lan, L.; Wei, X.; Xiang, R.; Wang, Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010, 20, 701–712. [Google Scholar] [CrossRef]
- Cho, U.; Kim, B.; Kim, S.; Han, Y.; Song, Y.S. Pro-inflammatory M1 macrophage enhances metastatic potential of ovarian cancer cells through NF-κB activation. Mol. Carcinog. 2017, 57, 235–242. [Google Scholar] [CrossRef]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS–) vs. Alternatively Activated Macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Nelson, M.P.; Christmann, B.S.; Werner, J.L.; Metz, A.E.; Trevor, J.L.; Lowell, C.A.; Steele, C. IL-33 and M2a Alveolar Macrophages Promote Lung Defense against the Atypical Fungal PathogenPneumocystis murina. J. Immunol. 2011, 186, 2372–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chizzolini, C.; Rezzonico, R.; De Luca, C.; Burger, D.; Dayer, J.-M. Th2 Cell Membrane Factors in Association with IL-4 Enhance Matrix Metalloproteinase-1 (MMP-1) While Decreasing MMP-9 Production by Granulocyte-Macrophage Colony-Stimulating Factor-Differentiated Human Monocytes. J. Immunol. 2000, 164, 5952–5960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratchev, A.; Guillot, P.; Hakiy, N.; Politz, O.; Orfanos, C.E.; Schledzewski, K.; Goerdt, S. Alternatively Activated Macrophages Differentially Express Fibronectin and Its Splice Variants and the Extracellular Matrix Protein βIG-H3. Scand. J. Immunol. 2001, 53, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Törőcsik, D.; Bárdos, H.; Nagy, L.; Ádány, R. Identification of factor XIII-A as a marker of alternative macrophage activation. Cell Mol. Life Sci. 2005, 62, 2132–2139. [Google Scholar] [CrossRef]
- Sapudom, J.; Karaman, S.; Mohamed, W.K.E.; Garcia-Sabaté, A.; Quartey, B.C.; Teo, J.C.M. 3D in vitro M2 macrophage model to mimic modulation of tissue repair. NPJ Regen. Med. 2021, 6, 1–13. [Google Scholar] [CrossRef]
- Wehling-Henricks, M.; Jordan, M.C.; Gotoh, T.; Grody, W.W.; Roos, K.P.; Tidball, J.G. Arginine Metabolism by Macrophages Promotes Cardiac and Muscle Fibrosis in mdx Muscular Dystrophy. PLoS ONE 2010, 5, e10763. [Google Scholar] [CrossRef] [Green Version]
- Anderson, C.F.; Mosser, D.M. A novel phenotype for an activated macrophage: The type 2 activated macrophage. J. Leukoc. Biol. 2002, 72, 101–106. [Google Scholar]
- Yue, Y.; Yang, X.; Feng, K.; Wang, L.; Hou, J.; Mei, B.; Qin, H.; Liang, M.; Chen, G.; Wu, Z. M2b macrophages reduce early reperfusion injury after myocardial ischemia in mice: A predominant role of inhibiting apoptosis via A20. Int. J. Cardiol. 2017, 245, 228–235. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.; Huang, W.-Y.; Niu, X.-L.; Hao, S.; Zhang, L.-N.; Hu, Y.-J. Significance of Macrophage Subtypes in the Peripheral Blood of Children with Systemic Juvenile Idiopathic Arthritis. Rheumatol. Ther. 2021, 8, 1859–1870. [Google Scholar] [CrossRef]
- Yue, Y.; Huang, S.; Li, H.; Li, W.; Hou, J.; Luo, L.; Liu, Q.; Wang, C.; Yang, S.; Lv, L.; et al. M2b macrophages protect against myocardial remodeling after ischemia/reperfusion injury by regulating kinase activation of platelet-derived growth factor receptor of cardiac fibroblast. Ann. Transl. Med. 2020, 8, 1409. [Google Scholar] [CrossRef]
- Asai, A.; Tsuchimoto, Y.; Ohama, H.; Fukunishi, S.; Tsuda, Y.; Kobayashi, M.; Higuchi, K.; Suzuki, F. Host antitumor resistance improved by the macrophage polarization in a chimera model of patients with HCC. OncoImmunology 2017, 6, e1299301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchimoto, Y.; Asai, A.; Tsuda, Y.; Ito, I.; Nishiguchi, T.; Garcia, M.C.; Suzuki, S.; Kobayashi, M.; Higuchi, K.; Suzuki, F. M2b Monocytes Provoke Bacterial Pneumonia and Gut Bacteria-Associated Sepsis in Alcoholics. J. Immunol. 2015, 195, 5169–5177. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.X.; Zhang, S.X.; Wu, H.J.; Rong, X.L.; Guo, J. M2b macrophage polarization and its roles in diseases. J. Leukoc. Biol. 2019, 106, 345–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avdic, S.; Cao, J.Z.; McSharry, B.; Clancy, L.E.; Brown, R.; Steain, M.; Gottlieb, D.; Abendroth, A.; Slobedman, B. Human Cytomegalovirus Interleukin-10 Polarizes Monocytes toward a Deactivated M2c Phenotype to Repress Host Immune Responses. J. Virol. 2013, 87, 10273–10282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapouri-Moghaddam, A.; Mohammadian, S.; Vazini, H.; Taghadosi, M.; Esmaeili, S.-A.; Mardani, F.; Seifi, B.; Mohammadi, A.; Afshari, J.T.; Sahebkar, A. Macrophage plasticity, polarization, and function in health and disease. J. Cell. Physiol. 2018, 233, 6425–6440. [Google Scholar] [CrossRef] [PubMed]
- Zizzo, G.; Hilliard, B.A.; Monestier, M.; Cohen, P.L. Efficient Clearance of Early Apoptotic Cells by Human Macrophages Requires M2c Polarization and MerTK Induction. J. Immunol. 2012, 189, 3508–3520. [Google Scholar] [CrossRef] [Green Version]
- Duluc, D.; Delneste, Y.; Tan, F.; Moles, M.-P.; Grimaud, L.; Lenoir, J.; Preisser, L.; Anegon, I.; Catala, L.; Ifrah, N.; et al. Tumor-associated leukemia inhibitory factor and IL-6 skew monocyte differentiation into tumor-associated macrophage-like cells. Blood 2007, 110, 4319–4330. [Google Scholar] [CrossRef]
- Anders, C.B.; Lawton, T.M.; Ammons, M.C.B. Metabolic immunomodulation of macrophage functional plasticity in nonhealing wounds. Curr. Opin. Infect. Dis. 2019, 32, 204–209. [Google Scholar] [CrossRef]
- Sunakawa, Y.; Stintzing, S.; Cao, S.; Heinemann, V.; Cremolini, C.; Falcone, A.; Yang, D.; Zhang, W.; Ning, Y.; Stremitzer, S.; et al. Variations in genes regulating tumor-associated macrophages (TAMs) to predict outcomes of bevacizumab-based treatment in patients with metastatic colorectal cancer: Results from TRIBE and FIRE3 trials. Ann. Oncol. 2015, 26, 2450–2456. [Google Scholar] [CrossRef]
- Wu, H.; Xu, J.-B.; He, Y.-L.; Peng, J.-J.; Zhang, X.-H.; Chen, C.-Q.; Li, W.; Cai, S.-R. Tumor-associated macrophages promote angiogenesis and lymphangiogenesis of gastric cancer. J. Surg. Oncol. 2012, 106, 462–468. [Google Scholar] [CrossRef]
- Cui, X.; Morales, R.-T.T.; Qian, W.; Wang, H.; Gagner, J.-P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M.; et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials 2018, 161, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Raes, G.; De Baetselier, P.; Noël, W.; Beschin, A.; Brombacher, F.; Gh, G.H. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J. Leukoc. Biol. 2002, 71, 597–602. [Google Scholar] [PubMed]
- Ju, X.; Zhang, H.; Zhou, Z.; Chen, M.; Wang, Q. Tumor-associated macrophages induce PD-L1 expression in gastric cancer cells through IL-6 and TNF-ɑ signaling. Exp. Cell Res. 2020, 396, 112315. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Wahyuningtyas, R.; Aui, S.; Chang, K. AutocrineVEGFsignalling on M2 macrophages regulatesPD-L1 expression for immunomodulation of T cells. J. Cell. Mol. Med. 2018, 23, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Daley, D.; Mani, V.R.; Mohan, N.; Akkad, N.; Pandian, G.S.B.; Savadkar, S.; Lee, K.B.; Torres-Hernandez, A.; Aykut, B.; Diskin, B.; et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J. Exp. Med. 2017, 214, 1711–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plitas, G.; Konopacki, C.; Wu, K.; Bos, P.D.; Morrow, M.; Putintseva, E.V.; Chudakov, D.M.; Rudensky, A.Y. Regulatory T Cells Exhibit Distinct Features in Human Breast Cancer. Immunity 2016, 45, 1122–1134. [Google Scholar] [CrossRef] [Green Version]
- Curiel, T.J.; Coukos, G.; Zou, L.; Alvarez, X.; Cheng, P.; Mottram, P.; Evdemon-Hogan, M.; Conejo-Garcia, J.R.; Zhang, L.; Burow, M.; et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat. Med. 2004, 10, 942–949. [Google Scholar] [CrossRef]
- Fu, Q.; Xu, L.; Wang, Y.; Jiang, Q.; Liu, Z.; Zhang, J.; Zhou, Q.; Zeng, H.; Tong, S.; Wang, T.; et al. Tumor-associated Macrophage-derived Interleukin-23 Interlinks Kidney Cancer Glutamine Addiction with Immune Evasion. Eur. Urol. 2019, 75, 752–763. [Google Scholar] [CrossRef]
- Del Barrio, I.M.; Penski, C.; Schlahsa, L.; Stein, R.G.; Diessner, J.; Wöckel, A.; Dietl, J.; Lutz, M.B.; Mittelbronn, M.; Wischhusen, J.; et al. Adenosine-generating ovarian cancer cells attract myeloid cells which differentiate into adenosine-generating tumor associated macrophages—A self-amplifying, CD39- and CD73-dependent mechanism for tumor immune escape. J. Immunother. Cancer 2016, 4, 49. [Google Scholar] [CrossRef] [Green Version]
- Kersten, K.; Coffelt, S.; Hoogstraat, M.; Verstegen, N.; Vrijland, K.; Ciampricotti, M.; Doornebal, C.W.; Hau, C.-S.; Wellenstein, M.D.; Salvagno, C.; et al. Mammary tumor-derived CCL2 enhances pro-metastatic systemic inflammation through upregulation of IL1β in tumor-associated macrophages. OncoImmunology 2017, 6, e1334744. [Google Scholar] [CrossRef]
- Shieh, Y.-S.; Hung, Y.-J.; Hsieh, C.-B.; Chen, J.-S.; Chou, K.-C.; Liu, S.-Y. Tumor-Associated Macrophage Correlated with Angiogenesis and Progression of Mucoepidermoid Carcinoma of Salivary Glands. Ann. Surg. Oncol. 2008, 16, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Xuhui, M.; Zhang, S.; Jia, L.; Wang, S.; Liu, J.; Ma, X.; Wang, C.; Fu, Y.; Luo, Y. E-M, an Engineered Endostatin with High ATPase Activity, Inhibits the Recruitment and Alternative Activation of Macrophages in Non-small Cell Lung Cancer. Front. Pharmacol. 2017, 8, 532. [Google Scholar] [CrossRef] [Green Version]
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG Inhibits Tumor Growth and Metastasis by Inducing Macrophage Polarization and Vessel Normalization through Downregulation of PlGF. Cancer Cell 2011, 19, 31–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adam, C.; Paolini, L.; Gueguen, N.; Mabilleau, G.; Preisser, L.; Blanchard, S.; Pignon, P.; Manero, F.; Le Mao, M.; Morel, A.; et al. Acetoacetate protects macrophages from lactic acidosis-induced mitochondrial dysfunction by metabolic reprograming. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.-J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Fang, C.; Mo, F.; Liu, L.; Du, J.; Luo, M.; Men, K.; Na, F.; Wang, W.; Yang, H.; Wei, X. Oxidized mitochondrial DNA sensing by STING signaling promotes the antitumor effect of an irradiated immunogenic cancer cell vaccine. Cell. Mol. Immunol. 2021, 18, 2211–2223. [Google Scholar] [CrossRef]
- Yang, Y.; Wu, M.; Cao, D.; Yang, C.; Jin, J.; Wu, L.; Hong, X.; Li, W.; Lu, L.; Li, J.; et al. ZBP1-MLKL necroptotic signaling potentiates radiation-induced antitumor immunity via intratumoral STING pathway activation. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef]
- Patrushev, M.; Kasymov, V.; Patrusheva, V.; Ushakova, T.; Gogvadze, V.; Gaziev, A. Mitochondrial permeability transition triggers the release of mtDNA fragments. Cell. Mol. Life Sci. 2004, 61, 3100–3103. [Google Scholar] [CrossRef]
- Kim, J.; Gupta, R.; Blanco, L.P.; Yang, S.; Shteinfer-Kuzmine, A.; Wang, K.; Zhu, J.; Yoon, H.E.; Wang, X.; Kerkhofs, M.; et al. VDAC oligomers form mitochondrial pores to release mtDNA fragments and promote lupus-like disease. Science 2019, 366, 1531–1536. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.-J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; Chin, H.S.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torre-Minguela, C.; Gómez, A.I.; Couillin, I.; Pelegrín, P. Gasdermins mediate cellular release of mitochondrial DNA during pyroptosis and apoptosis. FASEB J. 2021, 35, e21757. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Rauf, W.; Khan, K.; Khan, S.; Bell, K.M.; de Oliveira, V.C.; Tariq, M.; Bakhshalizadeh, S.; Touraine, P.; Katsanis, N.; et al. A recessive variant in TFAM causes mtDNA depletion associated with primary ovarian insufficiency, seizures, intellectual disability and hearing loss. Qual. Life Res. 2021, 140, 1733–1751. [Google Scholar] [CrossRef]
- Long, Q.; Zhou, Y.; Wu, H.; Du, S.; Hu, M.; Qi, J.; Li, W.; Guo, J.; Wu, Y.; Yang, L.; et al. Phase separation drives the self-assembly of mitochondrial nucleoids for transcriptional modulation. Nat. Struct. Mol. Biol. 2021, 28, 900–908. [Google Scholar] [CrossRef]
- Yang, S.; He, X.; Zhao, J.; Wang, D.; Guo, S.; Gao, T.; Wang, G.; Jin, C.; Yan, Z.; Wang, N.; et al. Mitochondrial transcription factor A plays opposite roles in the initiation and progression of colitis-associated cancer. Cancer Commun. 2021, 41, 695–714. [Google Scholar] [CrossRef]
- Correia, R.; Oba-Shinjo, S.; Uno, M.; Huang, N.; Marie, S. Mitochondrial DNA depletion and its correlation with TFAM, TFB1M, TFB2M and POLG in human diffusely infiltrating astrocytomas. Mitochondrion 2011, 11, 48–53. [Google Scholar] [CrossRef]
- Bazzani, V.; Barchiesi, A.; Radecka, D.; Pravisani, R.; Guadagno, A.; Di Loreto, C.; Baccarani, U.; Vascotto, C. Mitochondrial apurinic/apyrimidinic endonuclease 1 enhances mtDNA repair contributing to cell proliferation and mitochondrial integrity in early stages of hepatocellular carcinoma. BMC Cancer 2020, 20, 1–13. [Google Scholar] [CrossRef]
- Rongvaux, A.; Jackson, R.; Harman, C.C.; Li, T.; West, A.P.; De Zoete, M.R.; Wu, Y.; Yordy, B.; Lakhani, S.; Kuan, C.-Y.; et al. Apoptotic Caspases Prevent the Induction of Type I Interferons by Mitochondrial DNA. Cell 2014, 159, 1563–1577. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, T.; Kirchmair, A.; Sato, A.; Buqué, A.; Rybstein, M.; Petroni, G.; Bloy, N.; Finotello, F.; Stafford, L.; Manzano, E.N.; et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat. Immunol. 2020, 21, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Wang, S.; Xu, H.; Li, X.; Guan, Y.; Yi, F.; Zhou, T.; Jiang, B.; Bai, N.; Ma, M.; et al. ATM—CHK 2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020, 39, e103111. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 axis promotes cGAS/STING signaling in ARID1A-deficient tumors. J. Clin. Investig. 2020, 130, 5951–5966. [Google Scholar] [CrossRef] [PubMed]
- Todkar, K.; Chikhi, L.; Desjardins, V.; El-Mortada, F.; Pépin, G.; Germain, M. Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 2021, 12, 1971. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.M.; Pu, Y.; Han, D.; Shi, Y.; Cao, X.; Liang, H.; Chen, X.; Li, X.-D.; Deng, L.; Chen, Z.; et al. Dendritic Cells but Not Macrophages Sense Tumor Mitochondrial DNA for Cross-priming through Signal Regulatory Protein α Signaling. Immunity 2017, 47, 363–373.e5. [Google Scholar] [CrossRef] [PubMed]
- Pazmandi, K.; Agod, Z.; Kumar, B.V.; Szabo, A.; Fekete, T.; Sogor, V.; Veres, A.; Boldogh, I.; Rajnavolgyi, E.; Lanyi, A.; et al. Oxidative modification enhances the immunostimulatory effects of extracellular mitochondrial DNA on plasmacytoid dendritic cells. Free Radic. Biol. Med. 2014, 77, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Scott Worthen, G.; Albelda, S.M. Polarization of tumor-associated neutrophil pheno-type by TGF-beta: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Pylaeva, E.; Lang, S.; Jablonska, J. The Essential Role of Type I Interferons in Differentiation and Activation of Tumor-Associated Neutrophils. Front. Immunol. 2016, 7, 629. [Google Scholar] [CrossRef] [Green Version]
- Keshari, R.S.; Jyoti, A.; Kumar, S.; Dubey, M.; Verma, A.; Srinag, B.S.; Krishnamurthy, H.; Barthwal, M.K.; Dikshit, M. Neutrophil extracellular traps contain mitochondrial as well as nuclear DNA and exhibit inflammatory potential. Cytom. Part A J. Int. Soc. Anal. Cytol. 2011, 81, 238–247. [Google Scholar] [CrossRef]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wang, Z.; Li, L.; Zhang, Z.; Jin, X.; Wu, P.; Sun, S.; Pan, J.; Su, K.; Jia, F.; et al. Aged neutrophils form mitochondria-dependent vital NETs to promote breast cancer lung metastasis. J. Immunother. Cancer 2021, 9, e002875. [Google Scholar] [CrossRef] [PubMed]
- Itagaki, K.; Kaczmarek, E.; Lee, Y.T.; Tang, I.T.; Isal, B.; Adibnia, Y.; Sandler, N.; Grimm, M.J.; Segal, B.H.; Otterbein, L.E.; et al. Mitochondrial DNA Released by Trauma Induces Neutrophil Extracellular Traps. PLoS ONE 2015, 10, e0120549. [Google Scholar] [CrossRef] [PubMed]
- Singel, K.; Grzankowski, K.S.; Khan, A.N.M.N.H.; Grimm, M.J.; D’Auria, A.C.; Morrell, K.; Eng, K.H.; Hylander, B.; Mayor, P.C.; Emmons, T.R.; et al. Mitochondrial DNA in the tumour microenvironment activates neutrophils and is associated with worse outcomes in patients with advanced epithelial ovarian cancer. Br. J. Cancer 2019, 120, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caielli, S.; Veiga, D.F.T.; Balasubramanian, P.; Athale, S.; Domic, B.; Murat, E.; Banchereau, R.; Xu, Z.; Chandra, M.; Chung, C.-H.; et al. A CD4+ T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat. Med. 2019, 25, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Schuster, P.; Thomann, S.; Donhauser, N.; Vollmer, J.; Schmidt, B. Identification of novel oligonucleotides from mitochondrial DNA that spontaneously induce plasmacytoid dendritic cell activation. J. Leukoc. Biol. 2013, 94, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Ingelsson, B.; Söderberg, D.; Strid, T.; Söderberg, A.; Bergh, A.-C.; Loitto, V.; Lotfi, K.; Segelmark, M.; Spyrou, G.; Rosén, A. Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non-CpG oligodeoxynucleotides of class C. Proc. Natl. Acad. Sci. USA 2018, 115, E478–E487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pisetsky, D.S. Unexpected link between mitochondrial DNA and T cell help in systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, e59. [Google Scholar] [CrossRef]
- Grasso, C.; Eccles, D.A.; Boukalova, S.; Fabre, M.-S.; Dawson, R.H.; Neuzil, J.; Herst, P.M.; Berridge, M.V. Mitochondrial DNA Affects the Expression of Nuclear Genes Involved in Immune and Stress Responses in a Breast Cancer Model. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef]
- Jiang, H.; Guo, Y.; Wei, C.; Hu, P.; Shi, J. Nanocatalytic Innate Immunity Activation by Mitochondrial DNA Oxidative Damage for Tumor-Specific Therapy. Adv. Mater. 2021, 33, 2008065. [Google Scholar] [CrossRef]
- Jiang, G.; Yang, X.; Zhou, H.; Long, J.; Zhang, L.; Lu, D. cGAS-STING signaling regulates control of microglia polarization in cerebral ischemic stroke. Res. Sq. 2020. preprint. [Google Scholar] [CrossRef] [Green Version]
- Lu, G.-F.; Chen, S.-C.; Xia, Y.-P.; Ye, Z.-M.; Cao, F.; Hu, B. Synergistic inflammatory signaling by cGAS may be involved in the development of atherosclerosis. Aging 2021, 13, 5650–5673. [Google Scholar] [CrossRef] [PubMed]
- Kratochvill, F.; Neale, G.; Haverkamp, J.M.; Van De Velde, L.-A.; Smith, A.M.; Kawauchi, D.; McEvoy, J.; Roussel, M.F.; Dyer, M.A.; Qualls, J.E.; et al. TNF Counterbalances the Emergence of M2 Tumor Macrophages. Cell Rep. 2015, 12, 1902–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhang, X.; Wu, F.; Zhou, Y.; Bao, Z.; Li, H.; Zheng, P.; Zhao, S. Gastric cancer-derived mesenchymal stromal cells trigger M2 macrophage polarization that promotes metastasis and EMT in gastric cancer. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, B.; Zhu, S.-J.; Xiao, S.-S.; Xue, M. MiR-217 Inhibits M2-Like Macrophage Polarization by Suppressing Secretion of Interleukin-6 in Ovarian Cancer. Inflammation 2019, 42, 1517–1529. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; He, Z.; Huang, M.; Liu, T.; Wang, Y.; Xu, H.; Duan, H.; Ma, P.; Zhang, L.; Zamvil, S.S.; et al. Vascular niche IL-6 induces alternative macrophage activation in glioblastoma through HIF-2α. Nat. Commun. 2018, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Dubey, A.; Izakelian, L.; Ayaub, E.A.; Ho, L.; Stephenson, K.; Wong, S.; Kwofie, K.; Austin, R.C.; Botelho, F.; Ask, K.; et al. Separate roles of IL-6 and oncostatin M in mouse macrophage polarizationin vitroandin vivo. Immunol. Cell Biol. 2018, 96, 257–272. [Google Scholar] [CrossRef]
- David, J.M.; Dominguez, C.; Hamilton, D.H.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines 2016, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Bénard, A.; Sakwa, I.; Schierloh, P.; Colom, A.; Mercier, I.; Tailleux, L.; Jouneau, L.; Boudinot, P.; Al-Saati, T.; Lang, R.; et al. B Cells Producing Type I IFN Modulate Macrophage Polarization in Tuberculosis. Am. J. Respir. Crit. Care Med. 2018, 197, 801–813. [Google Scholar] [CrossRef]
- Shen, Y.J.; Le Bert, N.; Chitre, A.A.; Koo, C.X.; Nga, X.H.; Ho, S.S.W.; Khatoo, M.; Tan, N.Y.; Ishii, K.J.; Gasser, S. Genome-Derived Cytosolic DNA Mediates Type I Interferon-Dependent Rejection of B Cell Lymphoma Cells. Cell Rep. 2015, 11, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, K.J.; Carroll, P.; Martin, C.-A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Hao, C.; Liu, X.; Han, G.; Yin, J.; Zou, Z.; Zhou, J.; Xu, C. MitoQ protects against liver injury induced by severe burn plus delayed resuscitation by suppressing the mtDNA-NLRP3 axis. Int. Immunopharmacol. 2020, 80, 106189. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Hikita, H.; Nozaki, Y.; Kai, Y.; Makino, Y.; Nakabori, T.; Tanaka, S.; Yamada, R.; Shigekawa, M.; Kodama, T.; et al. DNase II activated by the mitochondrial apoptotic pathway regulates RIP1-dependent non-apoptotic hepatocyte death via the TLR9/IFN-β signaling pathway. Cell Death Differ. 2019, 26, 470–486. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Liu, S.; Wang, X.; Khaidakov, M.; Dai, Y.; Mehta, J.L. Oxidant stress in mitochondrial DNA damage, autophagy and inflammation in atherosclerosis. Sci. Rep. 2013, 3, srep01077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokolenko, I.N.; Alexeyev, M.F.; Robertson, F.M.; LeDoux, S.P.; Wilson, G.L. The expression of Exonuclease III from E. coli in mitochondria of breast cancer cells diminishes mitochondrial DNA repair capacity and cell survival after oxidative stress. DNA Repair 2003, 2, 471–482. [Google Scholar] [CrossRef]
- Ning, L.; Wei, W.; Wenyang, J.; Rui, X.; Qing, G. Cytosolic DNA-STING-NLRP3 axis is involved in murine acute lung injury induced by lipopolysaccharide. Clin. Transl. Med. 2020, 10, e228. [Google Scholar] [CrossRef]
- Li, Z.; Fan, E.K.; Liu, J.; Scott, M.J.; Li, Y.; Li, S.; Xie, W.; Billiar, T.R.; Wilson, M.A.; Jiang, Y.; et al. Cold-inducible RNA-binding protein through TLR4 signaling induces mitochondrial DNA fragmentation and regulates macrophage cell death after trauma. Cell Death Dis. 2017, 8, e2775. [Google Scholar] [CrossRef]
- Moore, J.A.; Mistry, J.J.; Hellmich, C.; Horton, R.H.; Wojtowicz, E.E.; Jibril, A.; Jefferson, M.; Wileman, T.; Beraza, N.; Bowles, K.M.; et al. LC3-associated phagocytosis in bone marrow macrophages suppresses acute myeloid leukemia progression through STING activation. J. Clin. Investig. 2022. [Google Scholar] [CrossRef]
- Sadikot, R.T.; Bedi, B.; Li, J.; Yeligar, S.M. Alcohol-induced mitochondrial DNA damage promotes injurious crosstalk between alveolar epithelial cells and alveolar macrophages. Alcohol 2019, 80, 65–72. [Google Scholar] [CrossRef]
- Bellizzi, D.; D’Aquila, P.; Scafone, T.; Giordano, M.; Riso, V.; Riccio, A.; Passarino, G. The Control Region of Mitochondrial DNA Shows an Unusual CpG and Non-CpG Methylation Pattern. DNA Res. 2013, 20, 537–547. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Li, T.; Li, X.-D.; Chen, X.; Li, Q.-Z.; Wight-Carter, M.; Chen, Z.J. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. USA 2015, 112, E5699–E5705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695, Erratum in Nature 2015, 525, 144. [Google Scholar] [CrossRef] [PubMed]
- Motwani, M.; Pesiridis, S.; Fitzgerald, K.A. DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 2019, 20, 657–674. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Liang, H.; Xu, M.; Yang, X.; Burnette, B.; Arina, A.; Li, X.-D.; Mauceri, H.; Beckett, M.; Darga, T.; et al. STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 2014, 41, 843–852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schadt, L.; Sparano, C.; Schweiger, N.A.; Silina, K.; Cecconi, V.; Lucchiari, G.; Yagita, H.; Guggisberg, E.; Saba, S.; Nascakova, Z.; et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep. 2019, 29, 1236–1248.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, Z.; Liu, J.; Ren, J.; Sun, T. Mitochondrial DNA induces inflammation and increases TLR9/NF-κB expression in lung tissue. Int. J. Mol. Med. 2014, 33, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Desmet, C.J.; Ishii, K. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat. Rev. Immunol. 2012, 12, 479–491. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–107. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yan, W.; Tohme, S.; Chen, M.; Fu, Y.; Tian, D.; Lotze, M.; Tang, D.; Tsung, A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J. Hepatol. 2015, 63, 114–121. [Google Scholar] [CrossRef] [Green Version]
- Marques, P.E.; Amaral, S.S.; Pires, D.A.; Nogueira, L.L.; Soriani, F.M.; Lima, B.H.; Lopes, G.A.; Russo, R.C.; Ávila, T.V.; Melgaço, J.G.; et al. Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 2012, 56, 1971–1982. [Google Scholar] [CrossRef] [PubMed]
- Compan, V.; Martín-Sánchez, F.; Baroja-Mazo, A.; Lopez-Castejon, G.; Gomez, A.I.; Verkhratsky, A.; Brough, D.; Pelegrín, P. Apoptosis-Associated Speck-like Protein Containing a CARD Forms Specks but Does Not Activate Caspase-1 in the Absence of NLRP3 during Macrophage Swelling. J. Immunol. 2015, 194, 1261–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houtman, J.; Freitag, K.; Gimber, N.; Schmoranzer, J.; Heppner, F.; Jendrach, M. Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP 3. EMBO J. 2019, 38, e99430. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elliott, E.; Sutterwala, F.S. Initiation and perpetuation of NLRP3 inflammasome activation and assembly. Immunol. Rev. 2015, 265, 35–52. [Google Scholar] [CrossRef] [Green Version]
- Bao, D.; Zhao, J.; Zhou, X.; Yang, Q.; Chen, Y.; Zhu, J.; Yuan, P.; Yang, J.; Qin, T.; Wan, S.; et al. Mitochondrial fission-induced mtDNA stress promotes tumor-associated macrophage infiltration and HCC progression. Oncogene 2019, 38, 5007–5020. [Google Scholar] [CrossRef] [Green Version]
- Fousek, K.; Horn, L.A.; Palena, C. Interleukin-8: A chemokine at the intersection of cancer plasticity, angiogenesis, and immune suppression. Pharmacol. Ther. 2021, 219, 107692. [Google Scholar] [CrossRef]
- Guescini, M.; Genedani, S.; Stocchi, V.; Agnati, L.F. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J. Neural Transm. 2010, 117, 1–4. [Google Scholar] [CrossRef]
- Puhm, F.; Afonyushkin, T.; Resch, U.; Obermayer, G.; Rohde, M.; Penz, T.; Schuster, M.; Wagner, G.; Rendeiro, A.; Melki, I.; et al. Mitochondria Are a Subset of Extracellular Vesicles Released by Activated Monocytes and Induce Type I IFN and TNF Responses in Endothelial Cells. Circ. Res. 2019, 125, 43–52. [Google Scholar] [CrossRef]
- Bernimoulin, M.; Waters, E.K.; Foy, M.; Steele, B.M.; Sullivan, M.; Falet, H.; Walsh, M.T.; Barteneva, N.; Geng, J.-G.; Hartwig, J.H.; et al. Differential stimulation of monocytic cells results in distinct populations of microparticles. J. Thromb. Haemost. 2009, 7, 1019–1028. [Google Scholar] [CrossRef] [Green Version]
- Rabas, N.; Palmer, S.; Mitchell, L.; Ismail, S.; Gohlke, A.; Riley, J.S.; Tait, S.W.; Gammage, P.; Soares, L.L.; Macpherson, I.R.; et al. PINK1 drives production of mtDNA-containing extracellular vesicles to promote invasiveness. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef] [PubMed]
- Abad, E.; Lyakhovich, A. Movement of Mitochondria with Mutant DNA through Extracellular Vesicles Helps Cancer Cells Acquire Chemoresistance. ChemMedChem 2021, 17. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.N.; Cheng, L.-C.; Kuo, C.-L.; Lo, Y.K.; Chou, H.-Y.; Chen, C.-H.; Wang, Y.-H.; Chuang, T.-H.; Cheng, S.-J.; Lee, A.Y.-L. Mitochondrial Lon-induced mtDNA leakage contributes to PD-L1–mediated immunoescape via STING-IFN signaling and extracellular vesicles. J. Immunother. Cancer 2020, 8, e001372. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015, 212, 435–445. [Google Scholar] [CrossRef]
- Morrissey, S.M.; Zhang, F.; Ding, C.; Montoya-Durango, D.E.; Hu, X.; Yang, C.; Wang, Z.; Yuan, F.; Fox, M.; Zhang, H.-G.; et al. Tumor-derived exosomes drive immunosuppressive macrophages in a pre-metastatic niche through glycolytic dominant metabolic reprogramming. Cell Metab. 2021, 33, 2040–2058.e10. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, Y.; Zhang, Y.; Jia, Z.; Zhang, Z.; Yang, J. Cancer derived exosomes induce macrophages immunosuppressive polarization to promote bladder cancer progression. Cell Commun. Signal. 2021, 19, 1–13. [Google Scholar] [CrossRef]
- Ohashi, T.; Aoki, M.; Tomita, H.; Akazawa, T.; Sato, K.; Kuze, B.; Mizuta, K.; Hara, A.; Nagaoka, H.; Inoue, N.; et al. M2-like macrophage polarization in high lactic acid-producing head and neck cancer. Cancer Sci. 2017, 108, 1128–1134. [Google Scholar] [CrossRef] [Green Version]
- Bonekamp, N.A.; Peter, B.; Hillen, H.S.; Felser, A.; Bergbrede, T.; Choidas, A.; Horn, M.; Unger, A.; Di Lucrezia, R.; Atanassov, I.; et al. Small-molecule inhibitors of human mitochondrial DNA transcription. Nature 2020, 588, 712–716. [Google Scholar] [CrossRef]
- Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol. Cancer Ther. 2015, 14, 1794–1804. [Google Scholar] [CrossRef] [Green Version]
- Riley, J.S.; Quarato, G.; Cloix, C.; Lopez, J.; O’Prey, J.; Pearson, M.; Chapman, J.; Sesaki, H.; Carlin, L.M.; Passos, J.F.; et al. Mitochondrial inner membrane permeabilisation enables mt DNA release during apoptosis. EMBO J. 2018, 37, e99238. [Google Scholar] [CrossRef]
- White, M.J.; McArthur, K.; Metcalf, D.; Lane, R.M.; Cambier, J.; Herold, M.; van Delft, M.; Bedoui, S.; Lessene, G.; Ritchie, M.; et al. Apoptotic Caspases Suppress mtDNA-Induced STING-Mediated Type I IFN Production. Cell 2014, 159, 1549–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, W.-T.; Lo, Y.-C.; Wu, M.-S.; Li, C.-Y.; Kuo, Y.-P.; Lai, Y.-H.; Tsai, Y.; Chen, K.-C.; Chuang, T.-H.; Yao, C.-H.; et al. Mycotoxin Patulin Suppresses Innate Immune Responses by Mitochondrial Dysfunction and p62/Sequestosome-1-dependent Mitophagy. J. Biol. Chem. 2016, 291, 19299–19311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Feng, L.; Hu, W.; Chen, J.; Wang, X.; Wang, L.; He, Y. Optineurin inhibits NLRP3 inflammasome activation by enhancing mitophagy of renal tubular cells in diabetic nephropathy. FASEB J. 2019, 33, 4571–4585. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Ma, H.; Liu, H.; Ye, W.; Li, Z.; Cheng, L.; Zhang, L.; Lei, Y.; Shen, L.; Zhang, F. The Glycoprotein and Nucleocapsid Protein of Hantaviruses Manipulate Autophagy Flux to Restrain Host Innate Immune Responses. Cell Rep. 2019, 27, 2075–2091.e5. [Google Scholar] [CrossRef] [Green Version]
- Vanpouille-Box, C.; Alard, A.; Aryankalayil, M.J.; Sarfraz, Y.; Diamond, J.M.; Schneider, R.J.; Inghirami, G.; Coleman, C.N.; Formenti, S.C.; DeMaria, S. DNA exonuclease Trex1 regulates radiotherapy-induced tumour immunogenicity. Nat. Commun. 2017, 8, 15618. [Google Scholar] [CrossRef]
- Han, C.; Liu, Z.; Zhang, Y.; Shen, A.; Dong, C.; Zhang, A.; Moore, C.; Ren, Z.; Lu, C.; Cao, X.; et al. Tumor cells suppress radiation-induced immunity by hijacking caspase 9 signaling. Nat. Immunol. 2020, 21, 546–554. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Stimuli | Markers | Secret | Function | Mechanisms | Ref | |
|---|---|---|---|---|---|---|
| M1 | IFNγ LPS GM-CSF | CD40 CD86 CD80 CD68 MHC-II IL1R TLR2 TLR4 SOCS3 | TNF-α IL-1α IL-1β IL-6 IL-12 IL-18 IL-23 CCL4 CCL5 CCL8 CCL10 CCL11 CXCL9 | Inflammation, tissue damage, pathogen clearance, inhibit cancer invasion and metastasis, enhance the metastatic potential of ovarian cancer cells | Enhances antigen presentation, activates Th1 cells, and secretes pro-inflammatory cytokines, release TNF-α to active NF-κB signaling pathway | [38,39,40] |
| M2a | IL-4 IL-13 Fungal/helminth infection LPS IFN-ɣ | Ym-1 CD163 MHC-II SRs CD206 YM1a FIZZ1a ARG1a CD86 iNOS Fizz | CCL17 CCL22 CCL24 RELM-α TGF-β IGF NO IL-10 | Inflammation, tissue damage, pathogen clearance, tissue proliferation and repair and fibrogenesis, involved in parasitic infections | TGF-β1 promotes matrix synthesis and remodeling, arginase-mediated hydrolysis of arginine that drives the production of ornithine to promote fibrogenesis, support efficient IFN-ɣ production in CD8+ T cells | [28,33,41,42,43,44,45,46] |
| M2b | IL-1R LPS IFN-ɣ IL-4 | CD86 MHC-II IL-10 TNF IL-1β IL-6 MerTK SPHK1 LIGHT | IL-10 IL-1 IL-6 IL-10 TNF-α CCL1 | Anti-inflammation, Minor tissue damage, Ameliorated myocardial ischemia/reperfusion injury, tissue remodeling, promote tumor development and infections | inhibited IFN-ɣ expression in CD4+T, active kinase of platelet-derived growth factor receptor of cardiac fibroblast, inhibit the immune and inflammatory response | [47,48,49,50,51,52,53] |
| M2c | IL-10 TGF-β Glucocorticoids IFN-ɣ IL-6 | CD163 TLR-1 TLR-8 IL-10 TGF-β CD206 SLAM MerTK | IL-10 TGF-β CCR2 | Anti-inflammation, tissue remodeling, phagocyte apoptotic cells | Reduce CD4+T cell activation and proliferation, TGF-β1 promotes matrix synthesis and remodeling, high expression of MerTK | [45,54,55,56] |
| M2d | IL-6 TLR ligands A2R Adenosine | VEGF IL-10 TGF-β | IL-10 IL-12 TNF-α TGF-β CCL5 CXCL10 CXCL16 | Tissue repair, Angiogenesis, promote cancer growth and metastasis | Regulate integrin (avb3) receptors and Src-PI3K-YAP signaling to promote angiogenic activity, secrete anti-inflammatory cytokines and suppress T-cell immunity | [37,57,58,59,60,61] |
| Cell Populations | Protein Expression | Function | Anti-Tumor Effects | Ref |
|---|---|---|---|---|
| Dendritic Cells | (↑) CD86 (↑) CD83 (↑) HLA-DQ (↑) TNF-α (↑) IFN-β (↑) IL-3 (↓) CXCR4 (↓) CXCR3 (↑) CCR7 | (↑) activation (↑) maturation (↑) migration | Anti-tumor immunity | [78,95,96,104,105] |
| Tumor-associated neutrophil | (↑) myeloperoxidase (MPO) (↑) type I IFN | (↑) recruitment (↑) activation induces NETs | accelerated progression, facilitated metastatic seeding and progression, poor prognosis | [99,100,102,103] |
| B cell | (↑) type I IFN | - | - | [106] |
| CD4+T cell | (↑) IL-10 | (↑) activation | Anti-tumor immunity | [104,107] |
| CD8+T cell | - | (↑) proliferation | Anti-tumor immunity | [78] |
| mtDNA Source | Mechanisms | Effects | Polarization | Anti-Tumor Effects | Ref |
|---|---|---|---|---|---|
| Endogenous | (↑) P-STING (↑) TNF-α | Inflammation, oxidative stress, pyroptosis | (↑) M1 | - | [110,126] |
| mtDNA accumulation | Necroptosis, inflammatory in surrounding naive macrophages | (↑) M1 | - | [127] | |
| Ethanol-induced mtDNA exosome release | (↓) Phagocytosis | - | [129] | ||
| cGAS-STING, TLRs-MyD88, NLRs-ASC | Inflammation, (↑) Type I IFN, IL-6, IL-8, TNF, IL-1β, IL-18 | (↑) M1 | Remodeling the TME | [77,132,133,137,144] | |
| Exogenous | - | TAM recruitment, (↑) CD86, (↓) CD206 | (↑) M1 | Inhibit progression and growth of distant tumors in pancreatic cancer | [108,109] |
| TLR-9 induce CCL2, IL-6, IL-8 production | TAM infiltration, MDSCs recruitment | (↑) M2 | Promote progression in HCC, Promote epithelial-mesenchymal transition and metastasis in gastric cancer | [113,146,147] | |
| STING induce IFN-I production | (↑) PD-L1,IL-10; (↓) IL-1b | (↑) M2 | - | [118] | |
| TLR9- NF-κB and cGAS-STING induce TNF-α production | Inflammation, (↓) IL-13 | (↑) M1 | Modify the inflammatory microenvironment, anti-tumor | [110,111,112] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Tsai, H.-i.; Zhang, L.; Zhu, H. Mitochondrial DNA on Tumor-Associated Macrophages Polarization and Immunity. Cancers 2022, 14, 1452. https://doi.org/10.3390/cancers14061452
Guo Y, Tsai H-i, Zhang L, Zhu H. Mitochondrial DNA on Tumor-Associated Macrophages Polarization and Immunity. Cancers. 2022; 14(6):1452. https://doi.org/10.3390/cancers14061452
Chicago/Turabian StyleGuo, Yaxin, Hsiang-i Tsai, Lirong Zhang, and Haitao Zhu. 2022. "Mitochondrial DNA on Tumor-Associated Macrophages Polarization and Immunity" Cancers 14, no. 6: 1452. https://doi.org/10.3390/cancers14061452
APA StyleGuo, Y., Tsai, H.-i., Zhang, L., & Zhu, H. (2022). Mitochondrial DNA on Tumor-Associated Macrophages Polarization and Immunity. Cancers, 14(6), 1452. https://doi.org/10.3390/cancers14061452