
Molecular Mechanisms Linking Risk Factors to Cholangiocarcinoma Development

 ,
,  , , ,
, , ,  ,
, 
 and
and 
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. CCA Risk Factors
2.1. Risk Factors Closely Associated with CCA
2.1.1. Liver Flukes
2.1.2. Primary Sclerosing Cholangitis
2.1.3. Bile Duct Cysts
2.1.4. Hepatolithiasis
2.1.5. Thorotrast
2.2. Risk Factors with a Suggested Role in CCA Onset
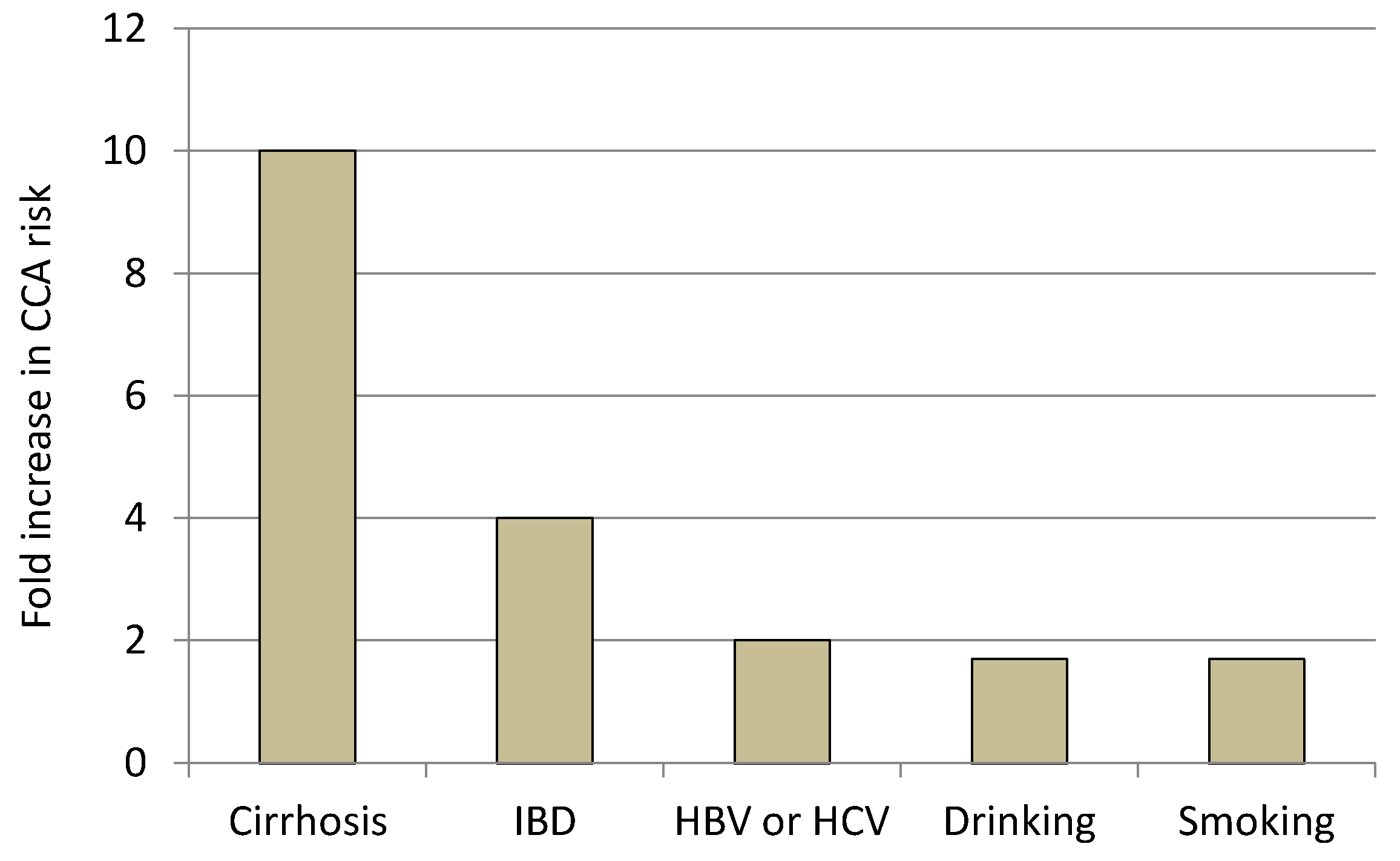
2.2.1. Liver Cirrhosis and Chronic Viral Hepatitis
2.2.2. Non-Alcoholic Fatty Liver Disease
2.2.3. Inflammatory Bowel Diseases (IBD)
2.2.4. Lifestyle Factors
3. Molecular Mechanisms Linking Risk Factors to CCA
3.1. From Liver Flukes to CCA
3.2. From PSC to CCA
3.3. From Bile Duct Cysts to CCA
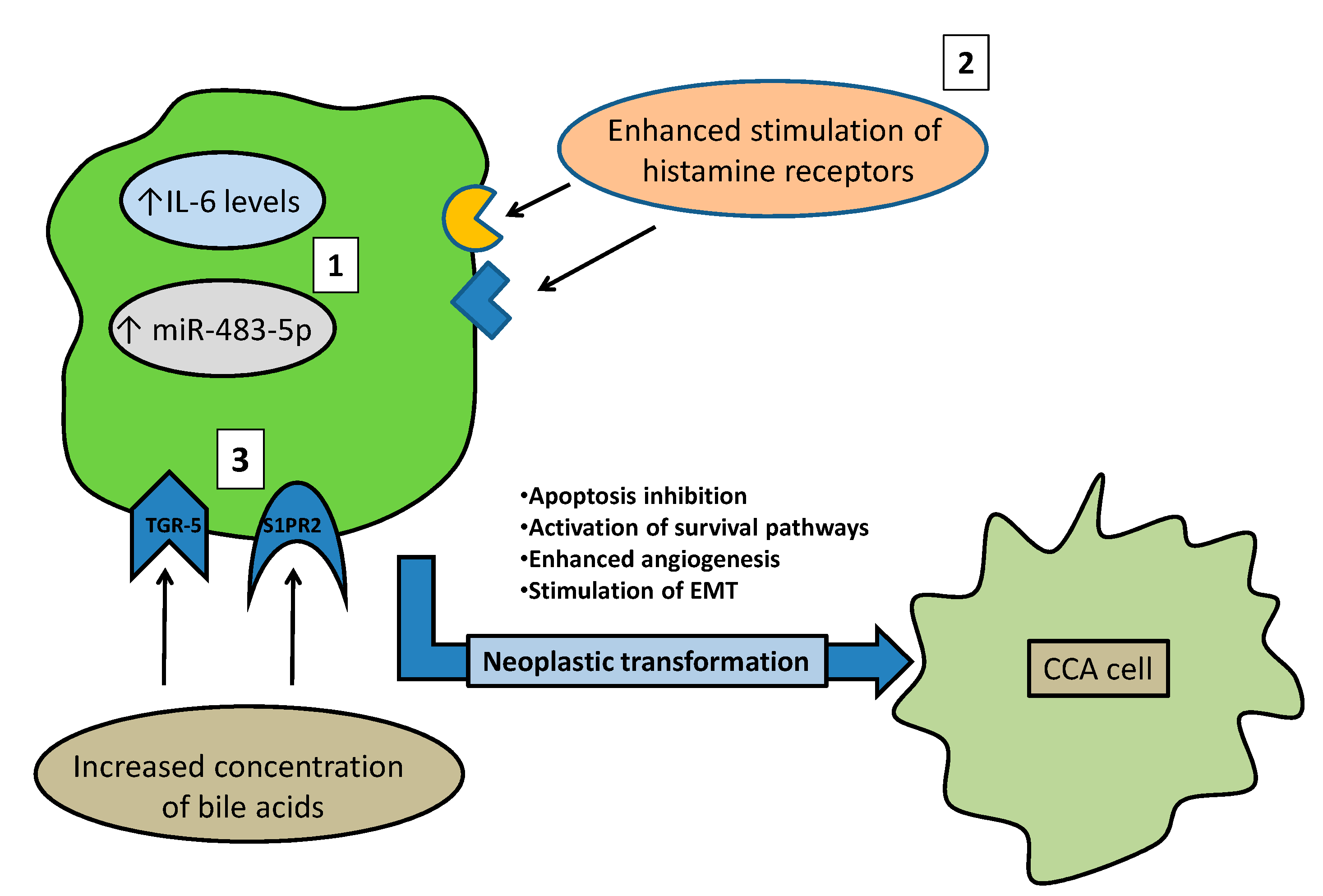
3.4. Molecular Mechanisms Linking Risk Factors with a Suggested Role in CCA Onset
4. Conclusions
Authors contribution
Funding
Conflicts of Interest
References
- Blechacz, B. Cholangiocarcinoma: Current Knowledge and New Developments. Gut Liver 2017, 11, 13–26. [Google Scholar] [CrossRef]
- Banales, J.M.; Marin, J.J.G.; Lamarca, A.; Rodrigues, P.M.; Khan, S.A.; Roberts, L.R.; Cardinale, V.; Carpino, G.; Andersen, J.B.; Braconi, C.; et al. Cholangiocarcinoma 2020: The next horizon in mechanisms and management. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 557–588. [Google Scholar] [CrossRef]
- Bertuccio, P.; Malvezzi, M.; Carioli, G.; Hashim, D.; Boffetta, P.; El-Serag, H.B.; La Vecchia, C.; Negri, E. Global trends in mortality from intrahepatic and extrahepatic cholangiocarcinoma. J. Hepatol. 2019, 71, 104–114. [Google Scholar] [CrossRef]
- Baiocchi, L.; Sato, K.; Ekser, B.; Kennedy, L.; Francis, H.; Ceci, L.; Lenci, I.; Alvaro, D.; Franchitto, A.; Onori, P.; et al. Cholangiocarcinoma: Bridging the translational gap from preclinical to clinical development and implications for future therapy. Expert Opin. Investig. Drugs 2021, 30, 365–375. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Brandi, G. Pemigatinib: Hot topics behind the first approval of a targeted therapy in cholangiocarcinoma. Cancer Treat. Res. Commun. 2021, 27, 100337. [Google Scholar] [CrossRef]
- Alsaleh, M.; Leftley, Z.; Barbera, T.A.; Koomson, L.K.; Zabron, A.; Crossey, M.M.E.; Reeves, H.L.; Cramp, M.; Ryder, S.; Greer, S.; et al. Characterisation of the Serum Metabolic Signature of Cholangiocarcinoma in a United Kingdom Cohort. J. Clin. Exp. Hepatol. 2020, 10, 17–29. [Google Scholar] [CrossRef]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39 (Suppl. 1), 19–31. [Google Scholar] [CrossRef]
- Tyson, G.L.; El-Serag, H.B. Risk factors for cholangiocarcinoma. Hepatology 2011, 54, 173–184. [Google Scholar] [CrossRef]
- Clements, O.; Eliahoo, J.; Kim, J.U.; Taylor-Robinson, S.D.; Khan, S.A. Risk factors for intrahepatic and extrahepatic cholangiocarcinoma: A systematic review and meta-analysis. J. Hepatol. 2020, 72, 95–103. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Wada, I.; Fukumoto, M. Alpha-particle carcinogenesis in Thorotrast patients: Epidemiology, dosimetry, pathology, and molecular analysis. J. Environ. Pathol. Toxicol. Oncol. 2001, 20, 311–315. [Google Scholar] [CrossRef]
- Kubo, S.; Takemura, S.; Tanaka, S.; Shinkawa, H.; Kinoshita, M.; Hamano, G.; Ito, T.; Koda, M.; Aota, T. Occupational cholangiocarcinoma caused by exposure to 1,2-dichloropropane and/or dichloromethane. Ann. Gastroenterol. Surg. 2018, 2, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Prueksapanich, P.; Piyachaturawat, P.; Aumpansub, P.; Ridtitid, W.; Chaiteerakij, R.; Rerknimitr, R. Liver Fluke-Associated Biliary Tract Cancer. Gut Liver 2018, 12, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Sripa, B.; Bethony, J.M.; Sithithaworn, P.; Kaewkes, S.; Mairiang, E.; Loukas, A.; Mulvenna, J.; Laha, T.; Hotez, P.J.; Brindley, P.J. Opisthorchiasis and Opisthorchis-associated cholangiocarcinoma in Thailand and Laos. Acta Trop. 2011, 120 (Suppl. 1), S158–S168. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.L.; Huang, Y.; Yu, X.B. Current status and perspectives of Clonorchis sinensis and clonorchiasis: Epidemiology, pathogenesis, omics, prevention and control. Infect. Dis. Poverty 2016, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Jiang, S.C.; Peng, H.J. Association between Liver Fluke Infection and Hepatobiliary Pathological Changes: A Systematic Review and Meta-Analysis. PLoS ONE 2015, 10, e0132673. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Kareemi, H.; Parab, R.; Barkema, H.W.; Quan, H.; Myers, R.P.; Kaplan, G.G. Incidence of primary sclerosing cholangitis: A systematic review and meta-analysis. Hepatology 2011, 53, 1590–1599. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Ali, A.H.; Lindor, K.D. Primary Sclerosing Cholangitis, Part 1: Epidemiology, Etiopathogenesis, Clinical Features, and Treatment. Gastroenterol. Hepatol. 2018, 14, 293–304. [Google Scholar]
- Chapman, R.; Fevery, J.; Kalloo, A.; Nagorney, D.M.; Boberg, K.M.; Shneider, B.; Gores, G.J. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010, 51, 660–678. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Baron, T.H. Endoscopic management of primary sclerosing cholangitis. Expert Rev. Gastroenterol. Hepatol. 2018, 12, 693–703. [Google Scholar] [CrossRef]
- Bowlus, C.L.; Lim, J.K.; Lindor, K.D. AGA Clinical Practice Update on Surveillance for Hepatobiliary Cancers in Patients With Primary Sclerosing Cholangitis: Expert Review. Clin. Gastroenterol. Hepatol. 2019, 17, 2416–2422. [Google Scholar] [CrossRef] [PubMed]
- Söreide, K.; Körner, H.; Havnen, J.; Söreide, J.A. Bile duct cysts in adults. Br. J. Surg. 2004, 91, 1538–1548. [Google Scholar] [CrossRef] [PubMed]
- He, X.D.; Wang, L.; Liu, W.; Liu, Q.; Qu, Q.; Li, B.L.; Hong, T. The risk of carcinogenesis in congenital choledochal cyst patients: An analysis of 214 cases. Ann. Hepatol. 2014, 13, 819–826. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, J.S.; Joo, M.K.; Lee, B.J.; Kim, J.H.; Yeon, J.E.; Park, J.J.; Byun, K.S.; Bak, Y.T. Hepatolithiasis and intrahepatic cholangiocarcinoma: A review. World J. Gastroenterol. 2015, 21, 13418–13431. [Google Scholar] [CrossRef] [PubMed]
- Tazuma, S. Gallstone disease: Epidemiology, pathogenesis, and classification of biliary stones (common bile duct and intrahepatic). Best Pract. Res. Clin. Gastroenterol. 2006, 20, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Ran, X.; Yin, B.; Ma, B. Four Major Factors Contributing to Intrahepatic Stones. Gastroenterol. Res. Pract. 2017, 2017, 7213043. [Google Scholar] [CrossRef] [PubMed]
- Lorio, E.; Patel, P.; Rosenkranz, L.; Patel, S.; Sayana, H. Management of Hepatolithiasis: Review of the Literature. Curr. Gastroenterol. Rep. 2020, 22, 30. [Google Scholar] [CrossRef]
- Lesurtel, M.; Regimbeau, J.M.; Farges, O.; Colombat, M.; Sauvanet, A.; Belghiti, J. Intrahepatic cholangiocarcinoma and hepatolithiasis: An unusual association in Western countries. Eur. J. Gastroenterol. Hepatol. 2002, 14, 1025–1027. [Google Scholar] [CrossRef]
- Guglielmi, A.; Ruzzenente, A.; Valdegamberi, A.; Bagante, F.; Conci, S.; Pinna, A.D.; Ercolani, G.; Giuliante, F.; Capussotti, L.; Aldrighetti, L.; et al. Hepatolithiasis-associated cholangiocarcinoma: Results from a multi-institutional national database on a case series of 23 patients. Eur. J. Surg. Oncol. 2014, 40, 567–575. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Mori, T.; Kato, Y.; Machinami, R.; Priest, N.D.; Kitagawa, T. Systemic deposits of thorium in thorotrast patients with particular reference to sites of minor storage. Radiat. Res. 1993, 135, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Kato, I.; Kido, C. Increased risk of death in thorotrast-exposed patients during the late follow-up period. Jpn. J. Cancer Res. 1987, 78, 1187–1192. [Google Scholar] [PubMed]
- van Kaick, G.; Bahner, M.L.; Liebermann, D.; Lührs, H.; Wesch, H. Thorotrast-induced liver cancer: Results of the German thorotrast study. Radiologe 1999, 39, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Labib, P.L.; Goodchild, G.; Pereira, S.P. Molecular Pathogenesis of Cholangiocarcinoma. BMC Cancer 2019, 19, 185. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Dixon, E. Epidemiology and risk factors: Intrahepatic cholangiocarcinoma. Hepatobiliary Surg. Nutr. 2017, 6, 101–104. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.C.; Patel, T. Are common factors involved in the pathogenesis of primary liver cancers? A meta-analysis of risk factors for intrahepatic cholangiocarcinoma. J. Hepatol. 2012, 57, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.S.; Tsai, C.R.; Chen, L.T. Medical risk factors associated with cholangiocarcinoma in Taiwan: A population-based case-control study. PLoS ONE 2013, 8, e69981. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Lee, S.S.; Jung, S.W.; Jeon, S.H.; Yun, S.C.; Oh, H.C.; Kwon, S.; Lee, S.K.; Seo, D.W.; Kim, M.H.; et al. Hepatitis B virus infection and intrahepatic cholangiocarcinoma in Korea: A case-control study. Am. J. Gastroenterol. 2008, 103, 1716–1720. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Engels, E.A.; Landgren, O.; Chiao, E.; Henderson, L.; Amaratunge, H.C.; Giordano, T.P. Risk of hepatobiliary and pancreatic cancers after hepatitis C virus infection: A population-based study of U.S. veterans. Hepatology 2009, 49, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Paik, J.M.; Golabi, P.; Younossi, Y.; Srishord, M.; Mishra, A.; Younossi, Z.M. The Growing Burden of Disability Related to Nonalcoholic Fatty Liver Disease: Data From the Global Burden of Disease 2007–2017. Hepatol. Commun. 2020, 4, 1769–1780. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Wongjarupong, N.; Assavapongpaiboon, B.; Susantitaphong, P.; Cheungpasitporn, W.; Treeprasertsuk, S.; Rerknimitr, R.; Chaiteerakij, R. Non-alcoholic fatty liver disease as a risk factor for cholangiocarcinoma: A systematic review and meta-analysis. BMC Gastroenterol. 2017, 17, 149. [Google Scholar] [CrossRef] [PubMed]
- Jing, W.; Jin, G.; Zhou, X.; Zhou, Y.; Zhang, Y.; Shao, C.; Liu, R.; Hu, X. Diabetes mellitus and increased risk of cholangiocarcinoma: A meta-analysis. Eur. J. Cancer Prev. 2012, 21, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Parsi, M.A. Obesity and cholangiocarcinoma. World J. Gastroenterol. 2013, 19, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, R.; Jepsen, P.; Vilstrup, H.; Ekbom, A.; Sørensen, H.T. Incidence and prognosis of cholangiocarcinoma in Danish patients with and without inflammatory bowel disease: A national cohort study, 1978–2003. Eur. J. Epidemiol. 2009, 24, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Huai, J.P.; Ding, J.; Ye, X.H.; Chen, Y.P. Inflammatory bowel disease and risk of cholangiocarcinoma: Evidence from a meta-analysis of population-based studies. Asian Pac. J. Cancer Prev. 2014, 15, 3477–3482. [Google Scholar] [CrossRef] [PubMed]
- Mertz, A.; Nguyen, N.A.; Katsanos, K.H.; Kwok, R.M. Primary sclerosing cholangitis and inflammatory bowel disease comorbidity: An update of the evidence. Ann. Gastroenterol. 2019, 32, 124–133. [Google Scholar] [CrossRef] [PubMed]
- Erichsen, R.; Olen, O.; Sachs, M.C.; Pedersen, L.; Halfvarson, J.; Askling, J.; Ekbom, A.; Ludvigsson, J.F.; Sorensen, H.T. Hepatobiliary Cancer Risk in Patients with Inflammatory Bowel Disease: A Scandinavian Population-Based Cohort Study. Cancer Epidemiol. Biomark. Prev. 2021, 30, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Makiuchi, T.; Sobue, T.; Kitamura, T.; Sawada, N.; Iwasaki, M.; Yamaji, T.; Shimazu, T.; Inoue, M.; Tsugane, S. Smoking, Alcohol Consumption, and Risks for Biliary Tract Cancer and Intrahepatic Bile Duct Cancer. J. Epidemiol. 2019, 29, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Sripa, B.; Brindley, P.J.; Mulvenna, J.; Laha, T.; Smout, M.J.; Mairiang, E.; Bethony, J.M.; Loukas, A. The tumorigenic liver fluke Opisthorchis viverrini-multiple pathways to cancer. Trends Parasitol. 2012, 28, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Arechavaleta-Velasco, F.; Perez-Juarez, C.E.; Gerton, G.L.; Diaz-Cueto, L. Progranulin and its biological effects in cancer. Med. Oncol. 2017, 34, 194. [Google Scholar] [CrossRef] [PubMed]
- Smout, M.J.; Laha, T.; Mulvenna, J.; Sripa, B.; Suttiprapa, S.; Jones, A.; Brindley, P.J.; Loukas, A. A granulin-like growth factor secreted by the carcinogenic liver fluke, Opisthorchis viverrini, promotes proliferation of host cells. PLoS Pathog. 2009, 5, e1000611. [Google Scholar] [CrossRef] [PubMed]
- Papatpremsiri, A.; Smout, M.J.; Loukas, A.; Brindley, P.J.; Sripa, B.; Laha, T. Suppression of Ov-grn-1 encoding granulin of Opisthorchis viverrini inhibits proliferation of biliary epithelial cells. Exp. Parasitol. 2015, 148, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Arunsan, P.; Ittiprasert, W.; Smout, M.J.; Cochran, C.J.; Mann, V.H.; Chaiyadet, S.; Karinshak, S.E.; Sripa, B.; Young, N.D.; Sotillo, J.; et al. Programmed knockout mutation of liver fluke granulin attenuates virulence of infection-induced hepatobiliary morbidity. Elife 2019, 8, e41463. [Google Scholar] [CrossRef] [PubMed]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.Y.; Sneddon, J.B.; Alizadeh, A.A.; Sood, R.; West, R.B.; Montgomery, K.; Chi, J.T.; van de Rijn, M.; Botstein, D.; Brown, P.O. Gene expression signature of fibroblast serum response predicts human cancer progression: Similarities between tumors and wounds. PLoS Biol. 2004, 2, E7. [Google Scholar] [CrossRef] [PubMed]
- Smout, M.J.; Sotillo, J.; Laha, T.; Papatpremsiri, A.; Rinaldi, G.; Pimenta, R.N.; Chan, L.Y.; Johnson, M.S.; Turnbull, L.; Whitchurch, C.B.; et al. Carcinogenic Parasite Secretes Growth Factor That Accelerates Wound Healing and Potentially Promotes Neoplasia. PLoS Pathog. 2015, 11, e1005209. [Google Scholar] [CrossRef] [PubMed]
- Na, B.K.; Pak, J.H.; Hong, S.J. Clonorchis sinensis and clonorchiasis. Acta Trop. 2020, 203, 105309. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.S.; Pak, J.H.; Kim, J.B.; Bahk, Y.Y. Clonorchis sinensis, an oriental liver fluke, as a human biological agent of cholangiocarcinoma: A brief review. BMB Rep. 2016, 49, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Ju, J.W.; Kim, S.M.; Shin, Y.; Chung, S.; Pak, J.H. Clonorchis sinensis infestation promotes three-dimensional aggregation and invasion of cholangiocarcinoma cells. PLoS ONE 2014, 9, e110705. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Cho, Y.; Lee, D.; Jeon, B.Y.; Ju, J.W.; Chung, S.; Pak, J.H. Clonorchis sinensis excretory-secretory products increase malignant characteristics of cholangiocarcinoma cells in three-dimensional co-culture with biliary ductal plates. PLoS Pathog. 2019, 15, e1007818. [Google Scholar] [CrossRef] [PubMed]
- Pak, J.H.; Kim, I.K.; Kim, S.M.; Maeng, S.; Song, K.J.; Na, B.K.; Kim, T.S. Induction of cancer-related microRNA expression profiling using excretory-secretory products of Clonorchis sinensis. Parasitol. Res. 2014, 113, 4447–4455. [Google Scholar] [CrossRef] [PubMed]
- Chirshev, E.; Oberg, K.C.; Ioffe, Y.J.; Unternaehrer, J.J. Let-7 as biomarker, prognostic indicator, and therapy for precision medicine in cancer. Clin. Transl. Med. 2019, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; LaRusso, N.F. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 2014, 59, 2263–2275. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Broome, U.; Ericzon, B.G.; Sumitran-Holgersson, S. High frequency of autoantibodies in patients with primary sclerosing cholangitis that bind biliary epithelial cells and induce expression of CD44 and production of interleukin 6. Gut 2002, 51, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Goydos, J.S.; Brumfield, A.M.; Frezza, E.; Booth, A.; Lotze, M.T.; Carty, S.E. Marked elevation of serum interleukin-6 in patients with cholangiocarcinoma: Validation of utility as a clinical marker. Ann. Surg. 1998, 227, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Cuenco, J.; Wehnert, N.; Blyuss, O.; Kazarian, A.; Whitwell, H.J.; Menon, U.; Dawnay, A.; Manns, M.P.; Pereira, S.P.; Timms, J.F. Identification of a serum biomarker panel for the differential diagnosis of cholangiocarcinoma and primary sclerosing cholangitis. Oncotarget 2018, 9, 17430–17442. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Han, Y.; Hughart, N.; McCarra, J.; Alpini, G.; Meng, F. Interleukin-6 and its receptor, key players in hepatobiliary inflammation and cancer. Transl. Gastrointest. Cancer 2012, 1, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Isomoto, H.; Kobayashi, S.; Werneburg, N.W.; Bronk, S.F.; Guicciardi, M.E.; Frank, D.A.; Gores, G.J. Interleukin 6 upregulates myeloid cell leukemia-1 expression through a STAT3 pathway in cholangiocarcinoma cells. Hepatology 2005, 42, 1329–1338. [Google Scholar] [CrossRef]
- Meng, F.; Yamagiwa, Y.; Ueno, Y.; Patel, T. Over-expression of interleukin-6 enhances cell survival and transformed cell growth in human malignant cholangiocytes. J. Hepatol. 2006, 44, 1055–1065. [Google Scholar] [CrossRef] [PubMed]
- Morton, S.D.; Cadamuro, M.; Brivio, S.; Vismara, M.; Stecca, T.; Massani, M.; Bassi, N.; Furlanetto, A.; Joplin, R.E.; Floreani, A.; et al. Leukemia inhibitory factor protects cholangiocarcinoma cells from drug-induced apoptosis via a PI3K/AKT-dependent Mcl-1 activation. Oncotarget 2015, 6, 26052–26064. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. IL-6 and related cytokines as the critical lynchpins between inflammation and cancer. Semin. Immunol. 2014, 26, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Pham, L.; Kennedy, L.; Baiocchi, L.; Meadows, V.; Ekser, B.; Kundu, D.; Zhou, T.; Sato, K.; Glaser, S.; Ceci, L.; et al. Mast Cells in Liver Disease Progression: An Update on Current Studies and Implications. Hepatology 2021. [Google Scholar] [CrossRef] [PubMed]
- Francis, H.; DeMorrow, S.; Venter, J.; Onori, P.; White, M.; Gaudio, E.; Francis, T.; Greene, J.F., Jr.; Tran, S.; Meininger, C.J.; et al. Inhibition of histidine decarboxylase ablates the autocrine tumorigenic effects of histamine in human cholangiocarcinoma. Gut 2012, 61, 753–764. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, L.; Hargrove, L.; Demieville, J.; Karstens, W.; Jones, H.; DeMorrow, S.; Meng, F.; Invernizzi, P.; Bernuzzi, F.; Alpini, G.; et al. Blocking H1/H2 histamine receptors inhibits damage/fibrosis in Mdr2(-/-) mice and human cholangiocarcinoma tumorigenesis. Hepatology 2018, 68, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- Bernuzzi, F.; Marabita, F.; Lleo, A.; Carbone, M.; Mirolo, M.; Marzioni, M.; Alpini, G.; Alvaro, D.; Boberg, K.M.; Locati, M.; et al. Serum microRNAs as novel biomarkers for primary sclerosing cholangitis and cholangiocarcinoma. Clin. Exp. Immunol. 2016, 185, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, H.; Zhu, S.; Lan, X. miR-483-5p promotes esophageal cancer progression by targeting KCNQ1. Biochem. Biophys. Res. Commun. 2020, 531, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Karakatsanis, A.; Papaconstantinou, I.; Gazouli, M.; Lyberopoulou, A.; Polymeneas, G.; Voros, D. Expression of microRNAs, miR-21, miR-31, miR-122, miR-145, miR-146a, miR-200c, miR-221, miR-222, and miR-223 in patients with hepatocellular carcinoma or intrahepatic cholangiocarcinoma and its prognostic significance. Mol. Carcinog. 2013, 52, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, J.; Suzuki, H. Role of MicroRNAs-221/222 in Digestive Systems. J. Clin. Med. 2015, 4, 1566–1577. [Google Scholar] [CrossRef] [PubMed]
- Baiocchi, L.; Zhou, T.; Liangpunsakul, S.; Lenci, I.; Santopaolo, F.; Meng, F.; Kennedy, L.; Glaser, S.; Francis, H.; Alpini, G. Dual Role of Bile Acids on the Biliary Epithelium: Friend or Foe? Int. J. Mol. Sci. 2019, 20, 1869. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Wang, H.; Dong, Y.; Zhang, Y.; Wang, J. Bile acids affect the growth of human cholangiocarcinoma via NF-kB pathway. Cancer Invest. 2013, 31, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Erice, O.; Labiano, I.; Arbelaiz, A.; Santos-Laso, A.; Munoz-Garrido, P.; Jimenez-Agüero, R.; Olaizola, P.; Caro-Maldonado, A.; Martín-Martín, N.; Carracedo, A.; et al. Differential effects of FXR or TGR5 activation in cholangiocarcinoma progression. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1335–1344. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhao, R.; Zhou, X.; Liang, X.; Campbell, D.J.; Zhang, X.; Zhang, L.; Shi, R.; Wang, G.; Pandak, W.M.; et al. Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology 2014, 60, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Soares, K.C.; Arnaoutakis, D.J.; Kamel, I.; Rastegar, N.; Anders, R.; Maithel, S.; Pawlik, T.M. Choledochal cysts: Presentation, clinical differentiation, and management. J. Am. Coll. Surg. 2014, 219, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Xie, X.; Pu, L.; Wang, Q.; Pu, S.; Ai, C.; Liu, Y.; Chen, J.; Xiang, B. Molecular Characteristics of Choledochal Cysts in Children: Transcriptome Sequencing. Front. Genet. 2021, 12, 709340. [Google Scholar] [CrossRef] [PubMed]
- Diao, M.; Li, L.; Cheng, W. Congenital biliary dilatation may consist of 2 disease entities. J. Pediatr. Surg. 2011, 46, 1503–1509. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Tang, S. WNT/β-catenin signaling in the development of liver cancers. Biomed. Pharmacother. 2020, 132, 110851. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Oyesanya, R.A.; Campbell, D.J.; Almenara, J.A.; Dewitt, J.L.; Sirica, A.E. Preclinical assessment of simultaneous targeting of epidermal growth factor receptor (ErbB1) and ErbB2 as a strategy for cholangiocarcinoma therapy. Hepatology 2010, 52, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Strazzabosco, M.; Somlo, S. Polycystic liver diseases: Congenital disorders of cholangiocyte signaling. Gastroenterology 2011, 140, 1855–1859.e1. [Google Scholar] [CrossRef]
- Gallagher, A.R.; Esquivel, E.L.; Briere, T.S.; Tian, X.; Mitobe, M.; Menezes, L.F.; Markowitz, G.S.; Jain, D.; Onuchic, L.F.; Somlo, S. Biliary and pancreatic dysgenesis in mice harboring a mutation in Pkhd1. Am. J. Pathol. 2008, 172, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Fava, G.; Marzioni, M.; Benedetti, A.; Glaser, S.; DeMorrow, S.; Francis, H.; Alpini, G. Molecular pathology of biliary tract cancers. Cancer Lett. 2007, 250, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Yasoshima, M.; Sato, Y.; Furubo, S.; Kizawa, K.; Sanzen, T.; Ozaki, S.; Harada, K.; Nakanuma, Y. Matrix proteins of basement membrane of intrahepatic bile ducts are degraded in congenital hepatic fibrosis and Caroli’s disease. J. Pathol. 2009, 217, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Shaib, Y.H.; El-Serag, H.B.; Davila, J.A.; Morgan, R.; McGlynn, K.A. Risk factors of intrahepatic cholangiocarcinoma in the United States: A case-control study. Gastroenterology 2005, 128, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishna, G.; Rastogi, A.; Trehanpati, N.; Sen, B.; Khosla, R.; Sarin, S.K. From cirrhosis to hepatocellular carcinoma: New molecular insights on inflammation and cellular senescence. Liver Cancer 2013, 2, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Rogler, L.E.; Teperman, L.; Morgan, G.; Rogler, C.E. Identification of hepatocytic and bile ductular cell lineages and candidate stem cells in bipolar ductular reactions in cirrhotic human liver. Hepatology 2007, 45, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Gigante, E.; Paradis, V.; Ronot, M.; Cauchy, F.; Soubrane, O.; Ganne-Carrié, N.; Nault, J.C. New insights into the pathophysiology and clinical care of rare primary liver cancers. JHEP Rep. 2021, 3, 100174. [Google Scholar] [CrossRef] [PubMed]
- Minouchi, K.; Kaneko, S.; Kobayashi, K. Mutation of p53 gene in regenerative nodules in cirrhotic liver. J. Hepatol. 2002, 37, 231–239. [Google Scholar] [CrossRef][Green Version]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. TP53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166–2176. [Google Scholar] [CrossRef]
- Khan, S.A.; Taylor-Robinson, S.D.; Carmichael, P.L.; Habib, N.; Lemoine, N.R.; Thomas, H.C. Analysis of p53 mutations for a mutational signature in human intrahepatic cholangiocarcinoma. Int. J. Oncol. 2006, 28, 1269–1277. [Google Scholar] [CrossRef][Green Version]
- Katz, S.F.; Lechel, A.; Obenauf, A.C.; Begus-Nahrmann, Y.; Kraus, J.M.; Hoffmann, E.M.; Duda, J.; Eshraghi, P.; Hartmann, D.; Liss, B.; et al. Disruption of Trp53 in livers of mice induces formation of carcinomas with bilineal differentiation. Gastroenterology 2012, 142, 1229–1239.e3. [Google Scholar] [CrossRef] [PubMed]
- Tschaharganeh, D.F.; Xue, W.; Calvisi, D.F.; Evert, M.; Michurina, T.V.; Dow, L.E.; Banito, A.; Katz, S.F.; Kastenhuber, E.R.; Weissmueller, S.; et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 2014, 158, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Machado, M.V.; Diehl, A.M. NAFLD, NASH and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 656–665. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, S.; Tovoli, F.; Mazzotta, A.; Vasuri, F.; Edeline, J.; Malvi, D.; Boudjema, K.; Renzulli, M.; Jeddou, H.; D’Errico, A.; et al. Non-Alcoholic Steatohepatitis as a Risk Factor for Intrahepatic Cholangiocarcinoma and Its Prognostic Role. Cancers 2020, 12, 3182. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, V.; Renzi, A.; Carpino, G.; Torrice, A.; Bragazzi, M.C.; Giuliante, F.; DeRose, A.M.; Fraveto, A.; Onori, P.; Napoletano, C.; et al. Profiles of cancer stem cell subpopulations in cholangiocarcinomas. Am. J. Pathol. 2015, 185, 1724–1739. [Google Scholar] [CrossRef]
- Loeuillard, E.; Fischbach, S.R.; Gores, G.J.; Rizvi, S. Animal models of cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 982–992. [Google Scholar] [CrossRef]
- Sato, K.; Zhang, W.; Safarikia, S.; Isidan, A.; Chen, A.M.; Li, P.; Francis, H.; Kennedy, L.; Baiocchi, L.; Alvaro, D.; et al. Organoids and Spheroids as Models for Studying Cholestatic Liver Injury and Cholangiocarcinoma. Hepatology 2021, 74, 491–502. [Google Scholar] [CrossRef]
- Liu, C.H.; Huang, Q.; Jin, Z.Y.; Xie, F.; Zhu, C.L.; Liu, Z.; Wang, C. Circulating microRNA-21 as a prognostic, biological marker in cholangiocarcinoma. J. Cancer Res. Ther. 2018, 14, 220–225. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceci, L.; Zhou, T.; Lenci, I.; Meadows, V.; Kennedy, L.; Li, P.; Ekser, B.; Milana, M.; Zhang, W.; Wu, C.; et al. Molecular Mechanisms Linking Risk Factors to Cholangiocarcinoma Development. Cancers 2022, 14, 1442. https://doi.org/10.3390/cancers14061442
Ceci L, Zhou T, Lenci I, Meadows V, Kennedy L, Li P, Ekser B, Milana M, Zhang W, Wu C, et al. Molecular Mechanisms Linking Risk Factors to Cholangiocarcinoma Development. Cancers. 2022; 14(6):1442. https://doi.org/10.3390/cancers14061442
Chicago/Turabian StyleCeci, Ludovica, Tianhao Zhou, Ilaria Lenci, Vik Meadows, Lindsey Kennedy, Ping Li, Burcin Ekser, Martina Milana, Wenjun Zhang, Chaodong Wu, and et al. 2022. "Molecular Mechanisms Linking Risk Factors to Cholangiocarcinoma Development" Cancers 14, no. 6: 1442. https://doi.org/10.3390/cancers14061442
APA StyleCeci, L., Zhou, T., Lenci, I., Meadows, V., Kennedy, L., Li, P., Ekser, B., Milana, M., Zhang, W., Wu, C., Sato, K., Chakraborty, S., Glaser, S. S., Francis, H., Alpini, G., & Baiocchi, L. (2022). Molecular Mechanisms Linking Risk Factors to Cholangiocarcinoma Development. Cancers, 14(6), 1442. https://doi.org/10.3390/cancers14061442