
PARP Inhibitors Resistance: Mechanisms and Perspectives

 , ,
, , 
Abstract
:Simple Summary
Abstract
1. Introduction
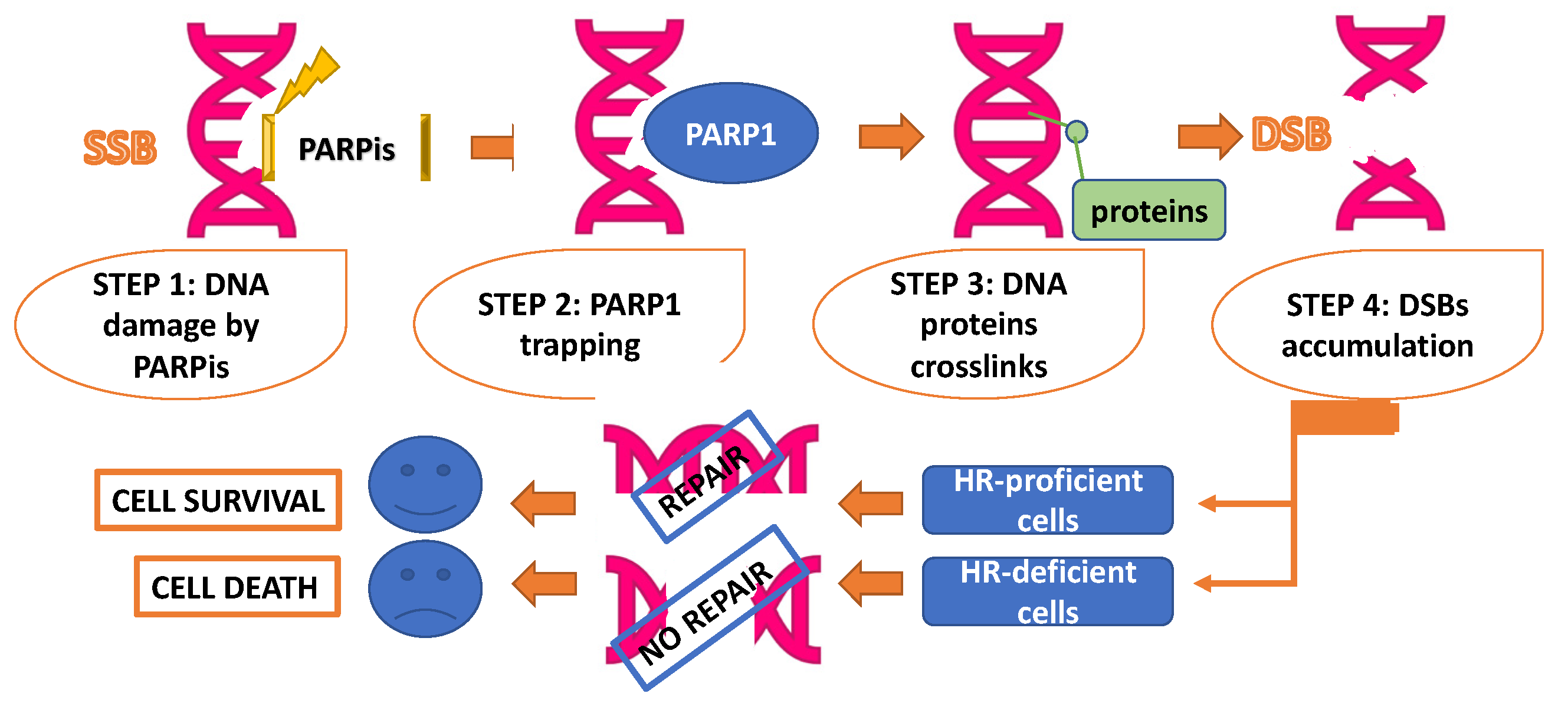
2. PARP Is Action
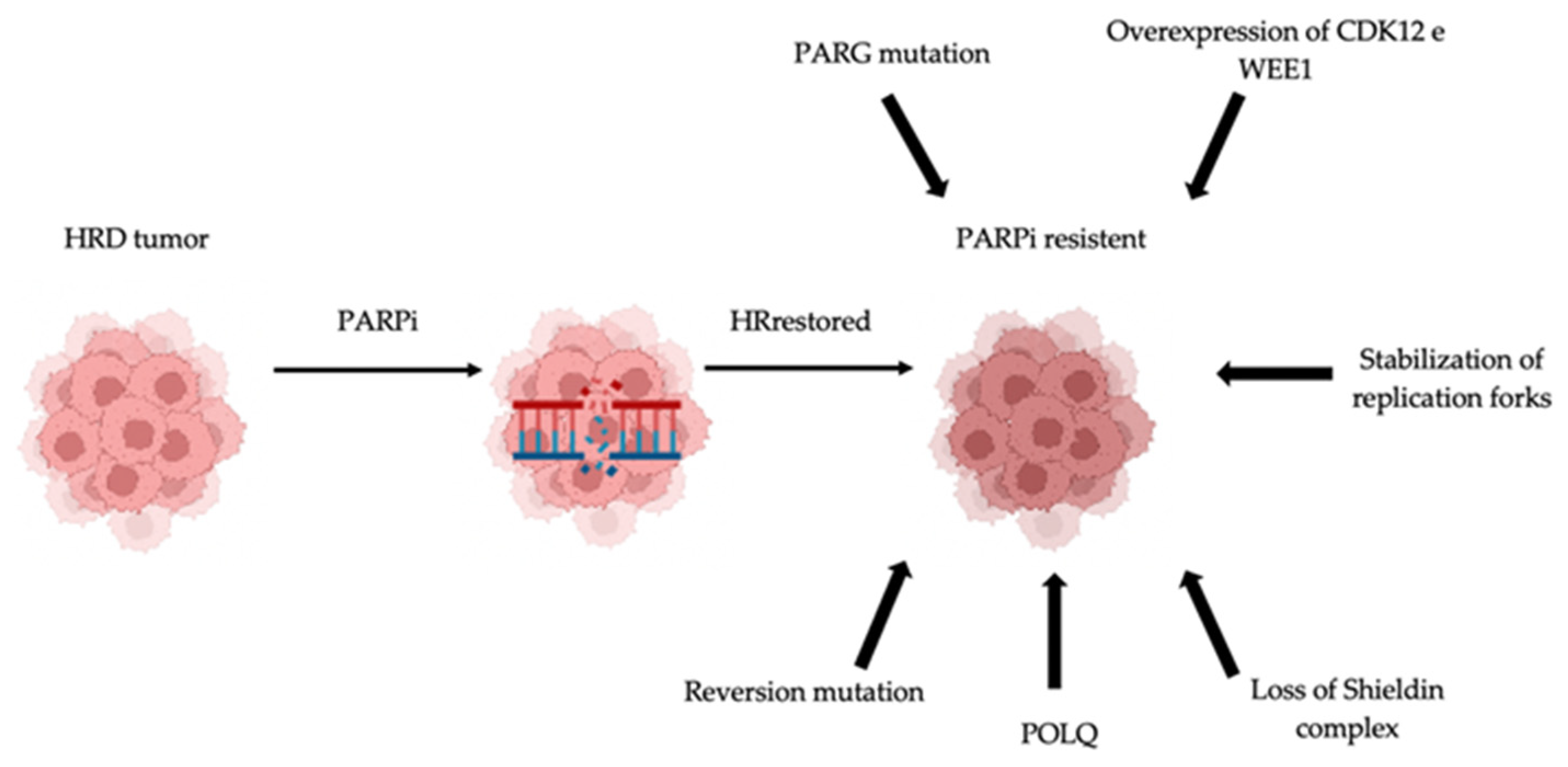
3. Mechanisms of Resistance to PARPis
3.1. Restoration of HR Activity
3.2. Reversion Mutations
3.3. DNA Polymerase θ (POLQ) (Required for MMEJ) Could Be a Driver of Resistance
3.4. Restoration of BRCA1 via Other Mechanisms
3.5. BRCA1—Independent Restoration of HR
3.6. Stabilization of Replication Forks
3.7. Increased Drug Efflux
3.8. Inhibition of PARP Trapping and PAR Glycohydrolase (PARG) Mutations Can Lead to PARP Resistance
3.9. Alterations in Cell Cycle Control
3.10. Dysregulated Signaling Pathways
4. Perspectives
4.1. Suppression of Alternative HR Pathways
4.2. Suppression of Mutator Phenotype
4.3. Indirect Inhibition of HR
4.4. Immunotherapy in HRD Deficient Cancers
4.5. Abrogation of Cell-Cycle Checkpoint Signalling
4.6. Targeting Acquired Vulnerabilities
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ait Saada, A.; Lambert, S.A.E.; Carr, A.M. Preserving Replication Fork Integrity and Competence via the Homologous Recombination Pathway. DNA Repair 2018, 71, 135–147. [Google Scholar] [CrossRef]
- Tarsounas, M.; Sung, P. The Antitumorigenic Roles of BRCA1–BARD1 in DNA Repair and Replication. Nat. Rev. Mol. Cell Biol. 2020, 21, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. BRCAness Revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Hammel, P.; Reni, M.; van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.-O.; Hochhauser, D.; Arnold, D.; Oh, D.-Y.; et al. Maintenance Olaparib for Germline BRCA—Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Helleday, T. The Underlying Mechanism for the PARP and BRCA Synthetic Lethality: Clearing up the Misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of All BRCA1 Copies Predicts Response to the PARP Inhibitor Rucaparib in Ovarian Carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [Green Version]
- Lheureux, S.; Lai, Z.; Dougherty, B.A.; Runswick, S.; Hodgson, D.R.; Timms, K.M.; Lanchbury, J.S.; Kaye, S.; Gourley, C.; Bowtell, D.; et al. Long-Term Responders on Olaparib Maintenance in High-Grade Serous Ovarian Cancer: Clinical and Molecular Characterization. Clin. Cancer Res. 2017, 23, 4086–4094. [Google Scholar] [CrossRef] [Green Version]
- Takaya, H.; Nakai, H.; Takamatsu, S.; Mandai, M.; Matsumura, N. Homologous Recombination Deficiency Status-Based Classification of High-Grade Serous Ovarian Carcinoma. Sci. Rep. 2020, 10, 2757. [Google Scholar] [CrossRef] [Green Version]
- Myriad Genetic Laboratories. BRACAnalysis® Technical Specifications. 2012. Available online: https://www.myriad.com/lib/technical-specifications/BRACAnalysis-Technical-Specifications.pdf (accessed on 30 December 2021).
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and Validation of a Clinical Cancer Genomic Profiling Test Based on Massively Parallel DNA Sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef]
- Melinda, L.T.; Kirsten, M.T.; Julia, R.; Bryan, H.; Gordon, B.M.; Kristin, C.J.; Zoltan, S.; William, T.B.; Eric, P.W.; Nadine, M.T.; et al. Homologous Recombination Deficiency (Hrd) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [Green Version]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic Scars as Biomarkers of Homologous Recombination Deficiency and Drug Response in Breast and Ovarian Cancers. Breast Cancer Res. 2014, 16, 211. [Google Scholar] [CrossRef] [Green Version]
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.-G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef]
- Litton, J.K.; Rugo, H.S.; Ettl, J.; Hurvitz, S.A.; Gonçalves, A.; Lee, K.-H.; Fehrenbacher, L.; Yerushalmi, R.; Mina, L.A.; Martin, M.; et al. Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation. N. Engl. J. Med. 2018, 379, 753–763. [Google Scholar] [CrossRef]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP Inhibitors: Synthetic Lethality in the Clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- The DNA Damaging Revolution: PARP Inhibitors and Beyond | American Society of Clinical Oncology Educational Book. Available online: https://ascopubs.org/doi/full/10.1200/EDBK_238473 (accessed on 9 March 2022).
- Murai, J.; Pommier, Y. PARP Trapping Beyond Homologous Recombination and Platinum Sensitivity in Cancers. Annu. Rev. Cancer Biol. 2019, 3, 131–150. [Google Scholar] [CrossRef]
- Pommier, Y.; O’connor, M.J.; de Bono, J. Laying a Trap to Kill Cancer Cells: PARP Inhibitors and Their Mechanisms of Action. Sci. Transl. Med. 2016, 8, 362ps17. [Google Scholar] [CrossRef]
- LaFargue, C.J.; Dal Molin, G.Z.; Sood, A.K.; Coleman, R.L. Exploring and Comparing Adverse Events between PARP Inhibitors. Lancet Oncol. 2019, 20, e15–e28. [Google Scholar] [CrossRef]
- Rose, M.; Burgess, J.T.; O’Byrne, K.; Richard, D.J.; Bolderson, E. PARP Inhibitors: Clinical Relevance, Mechanisms of Action and Tumor Resistance. Front. Cell Dev. Biol. 2020, 8, 879. [Google Scholar] [CrossRef]
- Lin, K.K.; Harrell, M.I.; Oza, A.M.; Oaknin, A.; Ray-Coquard, I.; Tinker, A.V.; Helman, E.; Radke, M.R.; Say, C.; Vo, L.T.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar] [CrossRef] [Green Version]
- Patch, A.M.; Christie, E.L.; Etemadmoghadam, D.; Garsed, D.W.; George, J.; Fereday, S.; Nones, K.; Cowin, P.; Alsop, K.; Bailey, P.J.; et al. Whole-Genome Characterization of Chemoresistant Ovarian Cancer. Nature 2015, 521, 489–494. [Google Scholar] [CrossRef]
- Quigley, D.; Alumkal, J.J.; Wyatt, A.W.; Kothari, V.; Foye, A.; Lloyd, P.; Aggarwal, R.; Kim, W.; Lu, E.; Schwartzman, J.; et al. Analysis of Circulating Cell-Free DnA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Discov. 2017, 7, 999–1005. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [Green Version]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-Recombination-Deficient Tumours Are Dependent on Polθ-Mediated Repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Pettitt, S.J.; Krastev, D.B.; Brandsma, I.; Dréan, A.; Song, F.; Aleksandrov, R.; Harrell, M.I.; Menon, M.; Brough, R.; Campbell, J.; et al. Genome-Wide and High-Density CRISPR-Cas9 Screens Identify Point Mutations in PARP1 Causing PARP Inhibitor Resistance. Nat. Commun. 2018, 9, 1849. [Google Scholar] [CrossRef]
- Drost, R.; Bouwman, P.; Rottenberg, S.; Boon, U.; Schut, E.; Klarenbeek, S.; Klijn, C.; van der Heijden, I.; van der Gulden, H.; Wientjens, E.; et al. BRCA1 RING Function Is Essential for Tumor Suppression but Dispensable for Therapy Resistance. Cancer Cell 2011, 20, 797–809. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.; Johnson, S.F.; Yao, W.; Li, Y.C.; Choi, Y.E.; Bernhardy, A.J.; Wang, Y.; Capelletti, M.; Sarosiek, K.A.; Moreau, L.A.; et al. Stabilization of Mutant BRCA1 Protein Confers PARP Inhibitor and Platinum Resistance. Proc. Natl. Acad. Sci. USA 2013, 110, 17041–17046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Δ11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaspers, J.E.; Kersbergen, A.; Boon, U.; Sol, W.; van Deemter, L.; Zander, S.A.; Drost, R.; Wientjens, E.; Ji, J.; Aly, A.; et al. Loss of 53BP1 Causes PARP Inhibitor Resistance in BRCA1-Mutated Mouse Mammary Tumors. Cancer Discov. 2013, 3, 68–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunting, S.F.; Callén, E.; Wong, N.; Chen, H.T.; Polato, F.; Gunn, A.; Bothmer, A.; Feldhahn, N.; Fernandez-Capetillo, O.; Cao, L.; et al. 53BP1 Inhibits Homologous Recombination in Brca1-Deficient Cells by Blocking Resection of DNA Breaks. Cell 2010, 141, 243–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dev, H.; Chiang, T.W.W.; Lescale, C.; de Krijger, I.; Martin, A.G.; Pilger, D.; Coates, J.; Sczaniecka-Clift, M.; Wei, W.; Ostermaier, M.; et al. Shieldin Complex Promotes DNA End-Joining and Counters Homologous Recombination in BRCA1-Null Cells. Nat. Cell Biol. 2018, 20, 954–965. [Google Scholar] [CrossRef]
- Mirman, Z.; Lottersberger, F.; Takai, H.; Kibe, T.; Gong, Y.; Takai, K.; Bianchi, A.; Zimmermann, M.; Durocher, D.; de Lange, T. 53BP1–RIF1–Shieldin Counteracts DSB Resection through CST- and Polα-Dependent Fill-In. Nature 2018, 560, 112–116. [Google Scholar] [CrossRef]
- Liao, H.; Ji, F.; Helleday, T.; Ying, S. Mechanisms for Stalled Replication Fork Stabilization: New Targets for Synthetic Lethality Strategies in Cancer Treatments. EMBO Rep. 2018, 19, e46263. [Google Scholar] [CrossRef]
- Rottenberg, S.; Jaspers, J.E.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.H.; Zander, S.A.L.; Derksen, P.W.B.; de Bruin, M.; Zevenhoven, J.; Lau, A.; et al. High Sensitivity of BRCA1-Deficient Mammary Tumors to the PARP Inhibitor AZD2281 Alone and in Combination with Platinum Drugs. Proc. Natl. Acad. Sci. USA 2008, 105, 17079–17084. [Google Scholar] [CrossRef] [Green Version]
- Leitner, I.; Nemeth, J.; Feurstein, T.; Abrahim, A.; Matzneller, P.; Lagler, H.; Erker, T.; Langer, O.; Zeitlinger, M. The Third-Generation P-Glycoprotein Inhibitor Tariquidar May Overcome Bacterial Multidrug Resistance by Increasing Intracellular Drug Concentration. J. Antimicrob. Chemother. 2011, 66, 834–839. [Google Scholar] [CrossRef] [Green Version]
- Christie, E.L.; Pattnaik, S.; Beach, J.; Copeland, A.; Rashoo, N.; Fereday, S.; Hendley, J.; Alsop, K.; Brady, S.L.; Lamb, G.; et al. Multiple ABCB1 Transcriptional Fusions in Drug Resistant High-Grade Serous Ovarian and Breast Cancer. Nat. Commun. 2019, 10, 1295. [Google Scholar] [CrossRef] [Green Version]
- Vaidyanathan, A.; Sawers, L.; Gannon, A.L.; Chakravarty, P.; Scott, A.L.; Bray, S.E.; Ferguson, M.J.; Smith, G. ABCB1 (MDR1) Induction Defines a Common Resistance Mechanism in Paclitaxel- and Olaparib-Resistant Ovarian Cancer Cells. Br. J. Cancer 2016, 115, 431–441. [Google Scholar] [CrossRef] [Green Version]
- Bitler, B.G.; Watson, Z.L.; Wheeler, L.J.; Behbakht, K. PARP Inhibitors: Clinical Utility and Possibilities of Overcoming Resistance. Gynecol. Oncol. 2017, 147, 695–704. [Google Scholar] [CrossRef] [Green Version]
- Gogola, E.; Duarte, A.A.; de Ruiter, J.R.; Wiegant, W.W.; Schmid, J.A.; de Bruijn, R.; James, D.I.; Guerrero Llobet, S.; Vis, D.J.; Annunziato, S.; et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell 2018, 33, 1078–1093. [Google Scholar] [CrossRef] [Green Version]
- Bajrami, I.; Frankum, J.R.; Konde, A.; Miller, R.E.; Rehman, F.L.; Brough, R.; Campbell, J.; Sims, D.; Rafiq, R.; Hooper, S.; et al. Genome-Wide Profiling of Genetic Synthetic Lethality Identifies CDK12 as a Novel Determinant of PARP1/2 Inhibitor Sensitivity. Cancer Res. 2014, 74, 287–297. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Yamaguchi, H.; Wei, Y.; Hsu, J.L.; Wang, H.L.; Hsu, Y.H.; Lin, W.C.; Yu, W.H.; Leonard, P.G.; Lee, G.R.; et al. Blocking C-Met-Mediated PARP1 Phosphorylation Enhances Anti-Tumor Effects of PARP Inhibitors. Nat. Med. 2016, 22, 194–201. [Google Scholar] [CrossRef]
- Yazinski, S.A.; Comaills, V.; Buisson, R.; Genois, M.M.; Nguyen, H.D.; Ho, C.K.; Kwan, T.T.; Morris, R.; Lauffer, S.; Nussenzweig, A.; et al. ATR Inhibition Disrupts Rewired Homologous Recombination and Fork Protection Pathways in PARP Inhibitor-Resistant BRCA-Deficient Cancer Cells. Genes Dev. 2017, 31, 318–332. [Google Scholar] [CrossRef]
- Nacson, J.; Krais, J.J.; Bernhardy, A.J.; Clausen, E.; Feng, W.; Wang, Y.; Nicolas, E.; Cai, K.Q.; Tricarico, R.; Hua, X.; et al. BRCA1 Mutation-Specific Responses to 53BP1 Loss-Induced Homologous Recombination and PARP Inhibitor Resistance. Cell Rep. 2018, 24, 3513–3527. [Google Scholar] [CrossRef] [Green Version]
- Callen, E.; Zong, D.; Wu, W.; Wong, N.; Stanlie, A.; Ishikawa, M.; Pavani, R.; Dumitrache, L.C.; Byrum, A.K.; Mendez-Dorantes, C.; et al. 53BP1 Enforces Distinct Pre- and Post-Resection Blocks on Homologous Recombination. Mol. Cell 2020, 77, 26–38. [Google Scholar] [CrossRef]
- Zong, D.; Adam, S.; Wang, Y.; Sasanuma, H.; Callén, E.; Murga, M.; Day, A.; Kruhlak, M.J.; Wong, N.; Munro, M.; et al. BRCA1 Haploinsufficiency Is Masked by RNF168-Mediated Chromatin Ubiquitylation. Mol. Cell 2019, 73, 1267–1281. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yücel, H.; Davis, R.E.; Färkkilä, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A First-in-Class Polymerase Theta Inhibitor Selectively Targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef]
- Pilié, P.G.; Tang, C.; Mills, G.B.; Yap, T.A. State-of-the-Art Strategies for Targeting the DNA Damage Response in Cancer. Nat. Rev. Clin. Oncol. 2019, 16, 81–104. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Liu, J.F.; Brady, M.F.; Matulonis, U.A.; Miller, A.; Kohn, E.C.; Swisher, E.M.; Tew, W.P.; Cloven, N.G.; Muller, C.; Bender, D.; et al. A Phase III Study Comparing Single-Agent Olaparib or the Combination of Cediranib and Olaparib to Standard Platinum-Based Chemotherapy in Recurrent Platinum-Sensitive Ovarian Cancer. J. Clin. Oncol. 2020, 38, 6003. [Google Scholar] [CrossRef]
- Lee, J.; Moore, R.G.; Ghamande, S.A.; Park, M.S.; Diaz, J.P.; Chapman, J.A.; Kendrick, J.E.; Slomovitz, B.M.; Tewari, K.S.; Lowe, E.S.; et al. Cediranib in Combination with Olaparib in Patients without a Germline BRCA1/2 Mutation with Recurrent Platinum-Resistant Ovarian Cancer: Phase IIb CONCERTO Trial. J. Clin. Oncol. 2020, 38, 6056. [Google Scholar] [CrossRef]
- Lheureux, S.; Oaknin, A.; Garg, S.; Bruce, J.P.; Madariaga, A.; Dhani, N.C.; Bowering, V.; White, J.; Accardi, S.; Tan, Q.; et al. EVOLVE: A Multicenter Open-Label Single-Arm Clinical and Translational Phase II Trial of Cediranib plus Olaparib for Ovarian Cancer after PARP Inhibition Progression. Clin. Cancer Res. 2020, 26, 4206–4215. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [Green Version]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.J.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase i Trial of the Parp Inhibitor Olaparib and Akt Inhibitor Capivasertib in Patients with Brca1/2-and Non–Brca1/2-Mutant Cancers. Cancer Discov. 2020, 10, 1528–1543. [Google Scholar] [CrossRef]
- Mahdi, H.; Hafez, N.; Doroshow, D.; Sohal, D.; Keedy, V.; Do, K.T.; Lorusso, P.; Urgensmeier, J.J.; Avedissian, M.; Sklar, J.; et al. Ceralasertib-Mediated ATR Inhibition Combined with Olaparib in Advanced Cancers Harboring DNA Damage Response and Repair Alterations (Olaparib Combinations). JCO Precis. Oncol. 2021, 5, 1432–1442. [Google Scholar] [CrossRef]
- Shah, P.D.; Wethington, S.L.; Pagan, C.; Latif, N.; Tanyi, J.; Martin, L.P.; Morgan, M.; Burger, R.A.; Haggerty, A.; Zarrin, H.; et al. Combination ATR and PARP Inhibitor (CAPRI): A Phase 2 Study of Ceralasertib plus Olaparib in Patients with Recurrent, Platinum-Resistant Epithelial Ovarian Cancer. Gynecol. Oncol. 2021, 163, 246–253. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.; Cadoo, K.A.; Shapiro, G.I.; Mayer, E.L.; O’Cearbhaill, R.E.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and α-Specific PI3K Inhibitor Alpelisib for Patients with Epithelial Ovarian Cancer: A Dose-Escalation and Dose-Expansion Phase 1b Trial. Lancet Oncol. 2019, 20, 570. [Google Scholar] [CrossRef]
- Alpelisib Plus Olaparib in Platinum-Resistant/Refractory, High-Grade Serous Ovarian Cancer, with No Germline BRCA Mutation Detected—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04729387 (accessed on 12 January 2022).
- Strickland, K.C.; Howitt, B.E.; Shukla, S.A.; Rodig, S.; Ritterhouse, L.L.; Liu, J.F.; Garber, J.E.; Chowdhury, D.; Wu, C.J.; D’andrea, A.D.; et al. Association and Prognostic Significance of BRCA1/2-Mutation Status with Neoantigen Load, Number of Tumor-Infiltrating Lymphocytes and Expression of PD-1/PD-L1 in High Grade Serous Ovarian Cancer. Oncotarget 2016, 7, 13587. [Google Scholar] [CrossRef] [Green Version]
- Shen, J.; Zhao, W.; Ju, Z.; Wang, L.; Peng, Y.; Labrie, M.; Yap, T.A.; Mills, G.B.; Peng, G. PARPI Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCANEss. Cancer Res. 2019, 79, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Pantelidou, C.; Sonzogni, O.; Taveira, M.D.O.; Mehta, A.K.; Kothari, A.; Wang, D.; Visal, T.; Li, M.K.; Pinto, J.; Castrillon, J.A.; et al. Parp Inhibitor Efficacy Depends on CD8+ T-Cell Recruitment via Intratumoral Sting Pathway Activation in Brca-Deficient Models of Triple-Negative Breast Cancer. Cancer Discov. 2019, 9, 722–737. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Yi, M.; Qin, S.; Chu, Q.; Luo, S.; Wu, K. Prospects for Combining Immune Checkpoint Blockade with PARP Inhibition. J. Hematol. Oncol. 2019, 12, 98. [Google Scholar] [CrossRef]
- Domchek, S.M.; Postel-Vinay, S.; Im, S.A.; Park, Y.H.; Delord, J.P.; Italiano, A.; Alexandre, J.; You, B.; Bastian, S.; Krebs, M.G.; et al. Olaparib and Durvalumab in Patients with Germline BRCA-Mutated Metastatic Breast Cancer (MEDIOLA): An Open-Label, Multicentre, Phase 1/2, Basket Study. Lancet Oncol. 2020, 21, 1155–1164. [Google Scholar] [CrossRef]
- Durvalumab Treatment in Combination with Chemotherapy and Bevacizumab, Followed by Maintenance Durvalumab, Bevacizumab and Olaparib Treatment in Advanced Ovarian Cancer Patients—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03737643 (accessed on 12 January 2022).
- Study of Chemotherapy with Pembrolizumab (MK-3475) Followed by Maintenance with Olaparib (MK-7339) for the First-Line Treatment of Women with BRCA Non-Mutated Advanced Epithelial Ovarian Cancer (EOC) (MK-7339-001/KEYLYNK-001/ENGOT-Ov43/GOG-3036)—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03740165 (accessed on 13 January 2022).
- Hardy-Bessard, A.-C.; Moore, K.N.; Mirza, M.R.; Asselain, B.; Redondo, A.; Pfisterer, J.; Pignata, S.; Provencher, D.M.; Cibula, D.; Reyners, A.K.L.; et al. ENGOT-OV44/FIRST Study: A Randomized, Double-Blind, Adaptive, Phase III Study of Standard of Care (SOC) Platinum-Based Therapy ± Dostarlimab Followed by Niraparib ± Dostarlimab Maintenance as First-Line (1L) Treatment of Stage 3 or 4 Ovarian Cancer (OC). J. Clin. Oncol. 2020, 38, TPS6101. [Google Scholar] [CrossRef]
- Monk, B.J.; Coleman, R.L.; Fujiwara, K.; Wilson, M.K.; Oza, A.M.; Oaknin, A.; O’Malley, D.M.; Lorusso, D.; Westin, S.N.; Safra, T.; et al. ATHENA (GOG-3020/ENGOT-Ov45): A Randomized, Phase III Trial to Evaluate Rucaparib as Monotherapy (ATHENA–MONO) and Rucaparib in Combination with Nivolumab (ATHENA–COMBO) as Maintenance Treatment Following Frontline Platinum-Based Chemotherapy in Ovarian Cancer. Int. J. Gynecol. Cancer 2021, 31, 1589. [Google Scholar] [CrossRef]
- Musacchio, L.; Salutari, V.; Pignata, S.; Braicu, E.; Cibula, D.; Colombo, N.; Frenel, J.S.; Zagouri, F.; Carbone, V.; Ghizzoni, V.; et al. Randomized Phase III Trial on Niraparib-TSR-042 (Dostarlimab) versus Physician’s Choice Chemotherapy in Recurrent Ovarian, Fallopian Tube, or Primary Peritoneal Cancer Patients Not Candidate for Platinum Retreatment: NItCHE Trial (MITO 33). Int. J. Gynecol. Cancer 2021, 31, 1369–1373. [Google Scholar] [CrossRef]
- Blank, S.; Mahdi, H.; Tehrani, O.; Ghamande, S.; Jain, S.; Nicacio, L.; Soumaoro, I.; O’malley, D.M.; Randall, L.M.; Monk, B.J.; et al. 883TiP MOONSTONE/GOG-3032: A Phase II, Open-Label, Single-Arm Study to Evaluate the Efficacy and Safety of Niraparib + Dostarlimab in Patients with Platinum-Resistant Ovarian Cancer. Ann. Oncol. 2020, 31, S646–S647. [Google Scholar] [CrossRef]
- Gaillard, S.; Berg, M.; Harrison, J.; Huang, P.; Leatherman, J.M.; Doucet, M.; Sen, R.; Suru, A.; Cai, H.; Durham, J.N.; et al. A Clinical Study of Tremelimumab Alone or in Combination with Olaparib in Patients with Advanced Epithelial Ovarian Cancer. J. Clin. Oncol. 2020, 38, 6045. [Google Scholar] [CrossRef]
- Tri Association in Patient with Advanced Epithelial Ovarian Cancer in Relapse—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04015739 (accessed on 22 January 2022).
- A Study to Evaluate the Efficacy and Safety of Niraparib Novel Treatment Combinations in Participants with Recurrent Ovarian Cancer—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03574779 (accessed on 2 February 2022).
- Comparison of Standard of Care Treatment with a Triplet Combination of Targeted Immunotherapeutic Agents—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT04739800 (accessed on 2 February 2022).
- Zimmer, A.S.; Nichols, E.; Cimino-Mathews, A.; Peer, C.; Cao, L.; Lee, M.J.; Kohn, E.C.; Annunziata, C.M.; Lipkowitz, S.; Trepel, J.B.; et al. A Phase I Study of the PD-L1 Inhibitor, Durvalumab, in Combination with a PARP Inhibitor, Olaparib, and a VEGFR1–3 Inhibitor, Cediranib, in Recurrent Women’s Cancers with Biomarker Analyses. J. Immunother. Cancer 2019, 7, 197. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Waggoner, S.; Vidal, G.A.; Mita, M.; Moroney, J.W.; Holloway, R.; van Le, L.; Sachdev, J.C.; Chapman-Davis, E.; Colon-Otero, G.; et al. Single-Arm Phases 1 and 2 Trial of Niraparib in Combination with Pembrolizumab in Patients with Recurrent Platinum-Resistant Ovarian Carcinoma. JAMA Oncol. 2019, 5, 1141–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez Martin, A.; Sanchez Lorenzo, L.; Colombo, N.; Depont Christensen, R.; Heitz, F.; Meirovitz, M.; Selle, F.; van Gorp, T.; Alvarez, N.; Sanchez, J.; et al. A Phase III, Randomized, Double Blinded Trial of Platinum Based Chemotherapy with or without Atezolizumab Followed by Niraparib Maintenance with or without Atezolizumab in Patients with Recurrent Ovarian, Tubal, or Peritoneal Cancer and Platinum Treatment Free Interval of More than 6 Months: ENGOT-Ov41/GEICO 69-O/ANITA Trial. Int. J. Gynecol. Cancer 2021, 31, 617–622. [Google Scholar] [CrossRef]
- Ray-Coquard, I.L.; Leary, A.; Bigot, F.; Montane, L.; Fabbro, M.; Hardy-Bessard, A.-C.; Selle, F.; Chakiba, C.; Lortholary, A.; Berton, D.; et al. ROCSAN Trial (GINECO-EN203b/ENGOT-EN8): A Multicentric Randomized Phase II/III Evaluating Dostarlimab in Combination with Niraparib versus Niraparib Alone Compared to Chemotherapy in the Treatment of Endometrial/Ovarian Carcinosarcoma after at Least One Line of Platinum Based Chemotherapy. J. Clin. Oncol. 2021, 39, TPS5604. [Google Scholar] [CrossRef]
- Kim, H.; Xu, H.; George, E.; Hallberg, D.; Kumar, S.; Jagannathan, V.; Medvedev, S.; Kinose, Y.; Devins, K.; Verma, P.; et al. Combining PARP with ATR Inhibition Overcomes PARP Inhibitor and Platinum Resistance in Ovarian Cancer Models. Nat. Commun. 2020, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Peng, Y.; Wei, L.; Zhang, W.; Yang, L.; Lan, L.; Kapoor, P.; Ju, Z.; Mo, Q.; Shih, I.M.; et al. ARID1A Deficiency Impairs the DNA Damage Checkpoint and Sensitizes Cells to PARP Inhibitors. Cancer Discov. 2015, 5, 752–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Ross Chapman, J.; Brandsma, I.; Yuan, J.; Mistrik, M.; Bouwman, P.; Bartkova, J.; Gogola, E.; Warmerdam, D.; Barazas, M.; et al. REV7 Counteracts DNA Double-Strand Break Resection and Affects PARP Inhibition. Nature 2015, 521, 541–544. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.; McGrail, D.J.; Sun, C.; Labrie, M.; Chen, X.; Zhang, D.; Ju, Z.; Vellano, C.P.; Lu, Y.; Li, Y.; et al. Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity While Maintaining Efficacy. Cancer Cell 2019, 35, 851–867.e7. [Google Scholar] [CrossRef]
- Westin, S.N.; Coleman, R.L.; Fellman, B.M.; Yuan, Y.; Sood, A.K.; Soliman, P.T.; Wright, A.A.; Horowitz, N.S.; Campos, S.M.; Konstantinopoulos, P.A.; et al. EFFORT: EFFicacy of Adavosertib in Parp ResisTance: A Randomized Two-Arm Non-Comparative Phase II Study of Adavosertib with or without Olaparib in Women with PARP-Resistant Ovarian Cancer. J. Clin. Oncol. 2021, 39, 5505. [Google Scholar] [CrossRef]
- Selumetinib and Olaparib in Solid Tumors—Full Text View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/NCT03162627 (accessed on 29 December 2021).
- Noordermeer, S.M.; Adam, S.; Setiaputra, D.; Barazas, M.; Pettitt, S.J.; Ling, A.K.; Olivieri, M.; Álvarez-Quilón, A.; Moatti, N.; Zimmermann, M.; et al. The Shieldin Complex Mediates 53BP1-Dependent DNA Repair. Nature 2018, 560, 117–121. [Google Scholar] [CrossRef]
- Barcellini, A.; Loap, P.; Murata, K.; Villa, R.; Kirova, Y.; Okonogi, N.; Orlandi, E. Parp Inhibitors in Combination with Radiotherapy: To Do or Not to Do? Cancers 2021, 13, 5380. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Perspectives for Eluding PARPI Resistance | |
|---|---|
| Suppression of Alternative HR Pathways | |
| Suppression of mutator phenotype | Loss of RNF168 |
| POLQ inhibition by novobiocin kills HR deficient tumors in vitro/in vivo | |
| Indirect inhibition of HR | CONCERTO trial: cediranib + olaparib |
| AKT inhibitor: capivasertib + olaparib | |
| ceralasertib + olaparib | |
| Immunotherapy in HRD deficient cancers | durvalumab + olaparib + bevacizumab (AGO-DUO) (BOLD) |
| durvalumab + olaparib + cediranib | |
| pembrolizumab + olaparib (ENGOT ov43) | |
| pembrolizumab + niraparib (TOPACIO) | |
| atezolizumab + niraparib (ANITA) | |
| nivolumab + rucaparib (ATHENA combo) | |
| dostarlimab + niraparib (MITO33) (MOONSTONE) (ROCSAN) | |
| dostarlimab + niraparib + bevacizumab (OPAL) tremelimumab + olaparib | |
| tremelimumab + durvalumab + olaparib | |
| Abrogation of cell-cycle checkpoint signalling | EFFORT trial: adavosertib (WEE1 inhibitor) + olaparib |
| SOLAR trial: selumetinib (MEK inhibitor) + olaparib | |
| Targeting acquired vulnerabilities | Ioniziting radiation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giudice, E.; Gentile, M.; Salutari, V.; Ricci, C.; Musacchio, L.; Carbone, M.V.; Ghizzoni, V.; Camarda, F.; Tronconi, F.; Nero, C.; et al. PARP Inhibitors Resistance: Mechanisms and Perspectives. Cancers 2022, 14, 1420. https://doi.org/10.3390/cancers14061420
Giudice E, Gentile M, Salutari V, Ricci C, Musacchio L, Carbone MV, Ghizzoni V, Camarda F, Tronconi F, Nero C, et al. PARP Inhibitors Resistance: Mechanisms and Perspectives. Cancers. 2022; 14(6):1420. https://doi.org/10.3390/cancers14061420
Chicago/Turabian StyleGiudice, Elena, Marica Gentile, Vanda Salutari, Caterina Ricci, Lucia Musacchio, Maria Vittoria Carbone, Viola Ghizzoni, Floriana Camarda, Francesca Tronconi, Camilla Nero, and et al. 2022. "PARP Inhibitors Resistance: Mechanisms and Perspectives" Cancers 14, no. 6: 1420. https://doi.org/10.3390/cancers14061420
APA StyleGiudice, E., Gentile, M., Salutari, V., Ricci, C., Musacchio, L., Carbone, M. V., Ghizzoni, V., Camarda, F., Tronconi, F., Nero, C., Ciccarone, F., Scambia, G., & Lorusso, D. (2022). PARP Inhibitors Resistance: Mechanisms and Perspectives. Cancers, 14(6), 1420. https://doi.org/10.3390/cancers14061420