
Sequestration of Intestinal Acidic Toxins by Cationic Resin Attenuates Pancreatic Cancer Progression through Promoting Autophagic Flux for YAP Degradation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents, Plasmids, Primers, and Antibodies
2.2. Animal Experiment
2.3. Histological Analysis
2.4. Immunohistochemistry
2.5. Determining Blood Levels of Endotoxin and Total Bile Acids
2.6. Western Blotting Analysis
2.7. Cell Lines and Transfection
2.8. Generation of Lentivirus
2.9. Measurement of Autophagic Flux Using the Autophagy Tandem Sensor mRFP-GFP-LC3
2.10. Transwell Cell Migration Assay
2.11. Clone Formation Assay
2.12. Cell Viability Assay
2.13. Statistical Analysis
3. Results
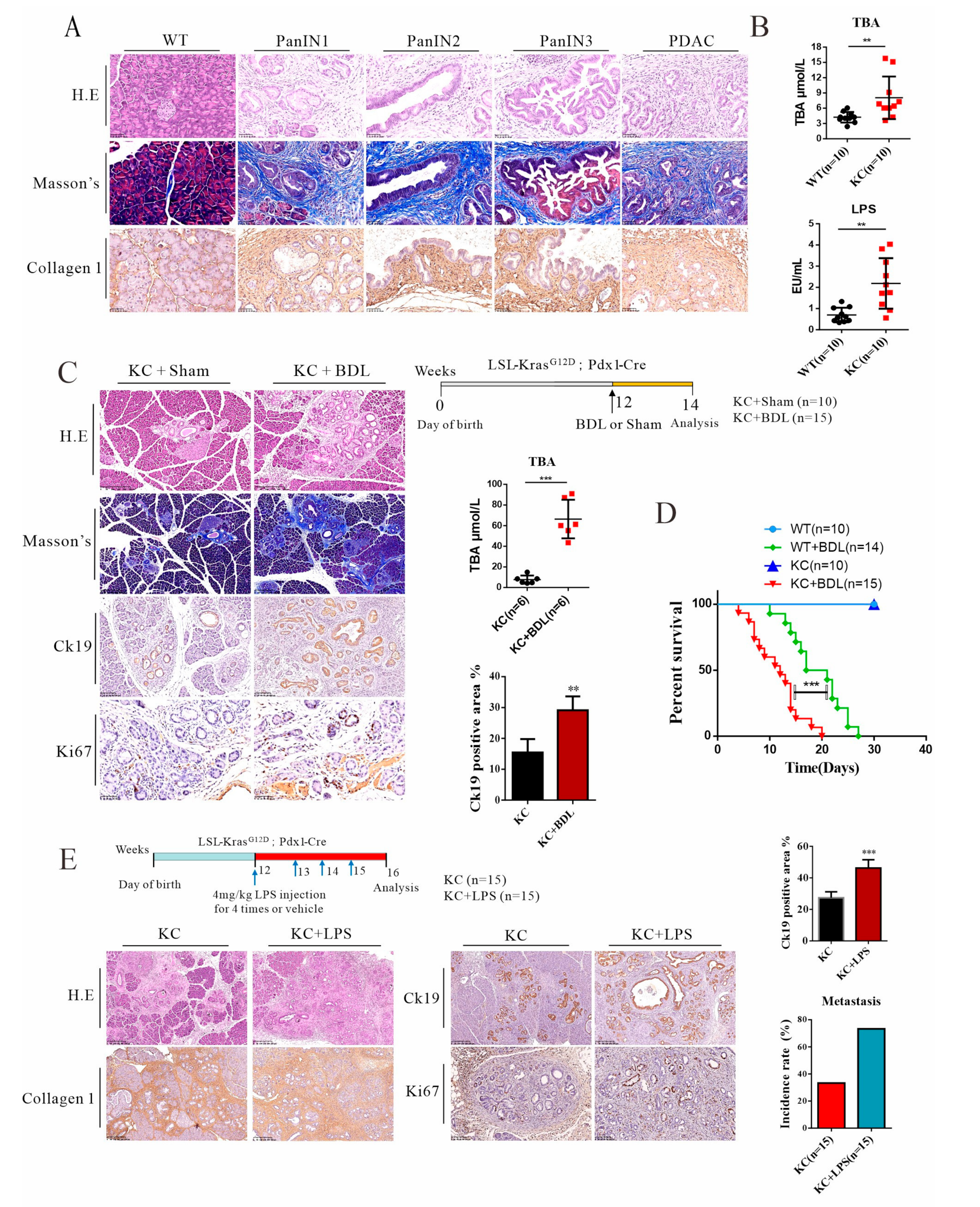
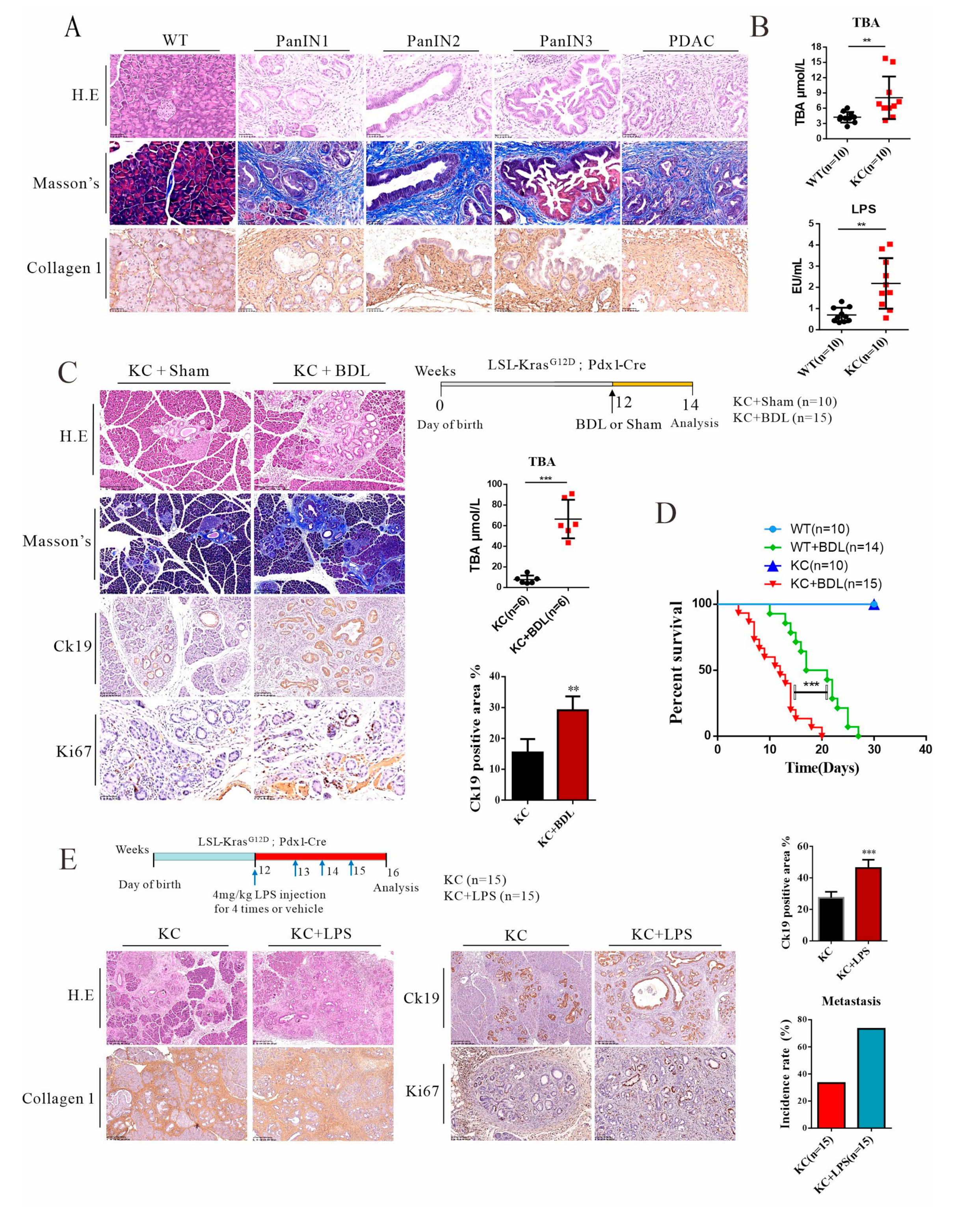
3.1. Elevated Levels of Systemic Endotoxin and BILE Acids in Pdx-1-Kras Mice Are Associated with Metastatic Progression in Mice
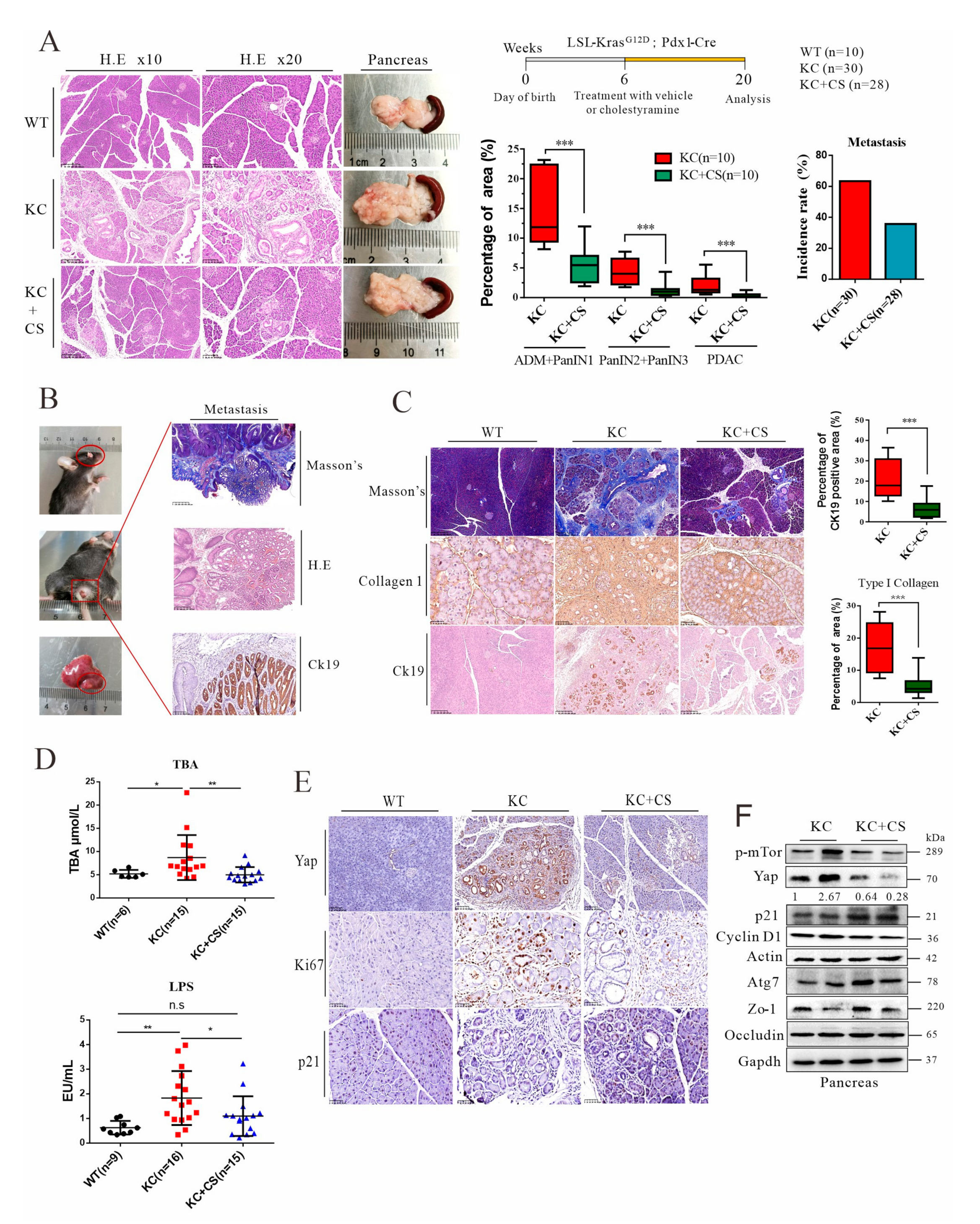
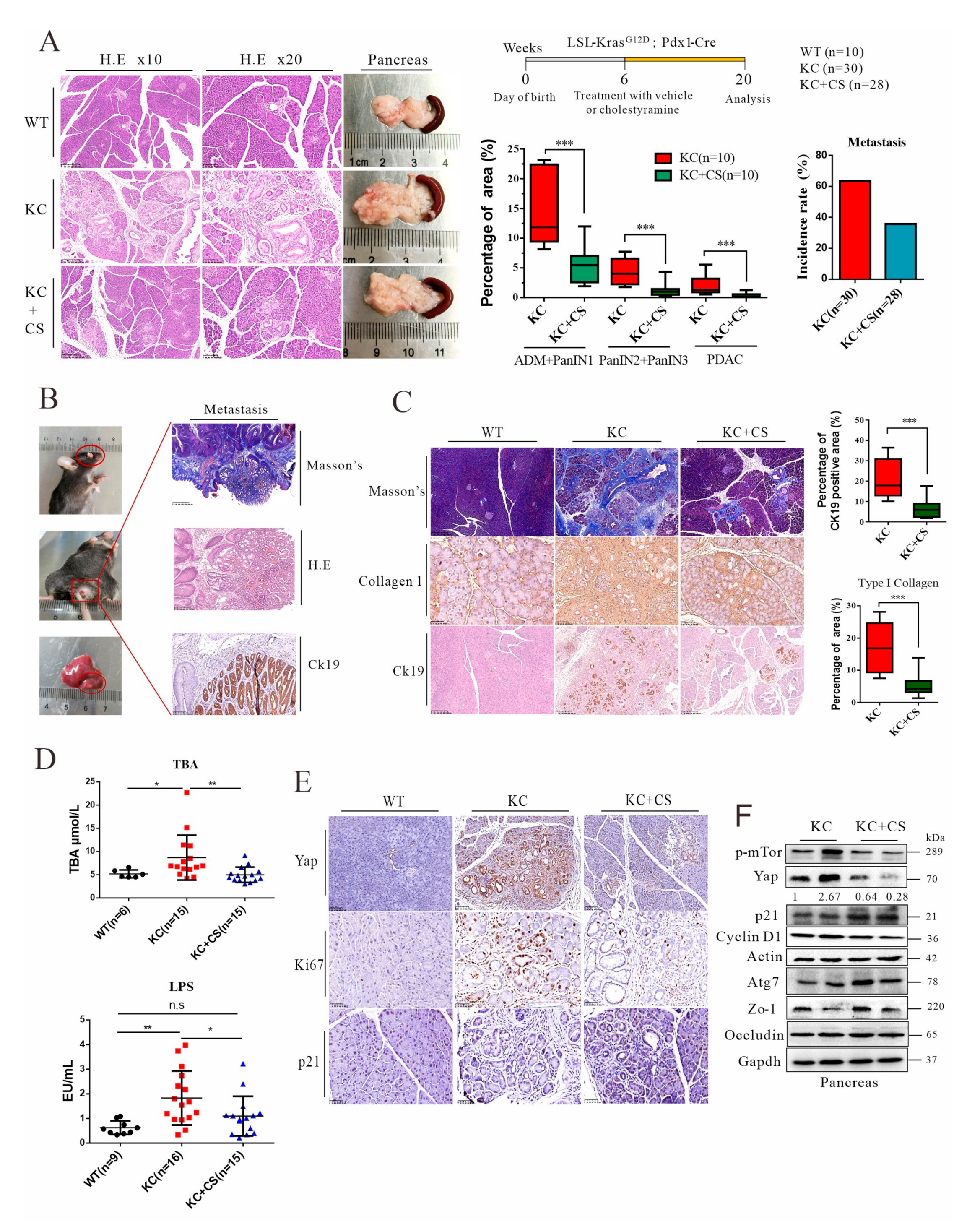
3.2. Oral Administration of Cationic Resin Attenuates Tumorigenesis and Metastasis through Sequestration of Intestinal Endotoxin and Bile Acids
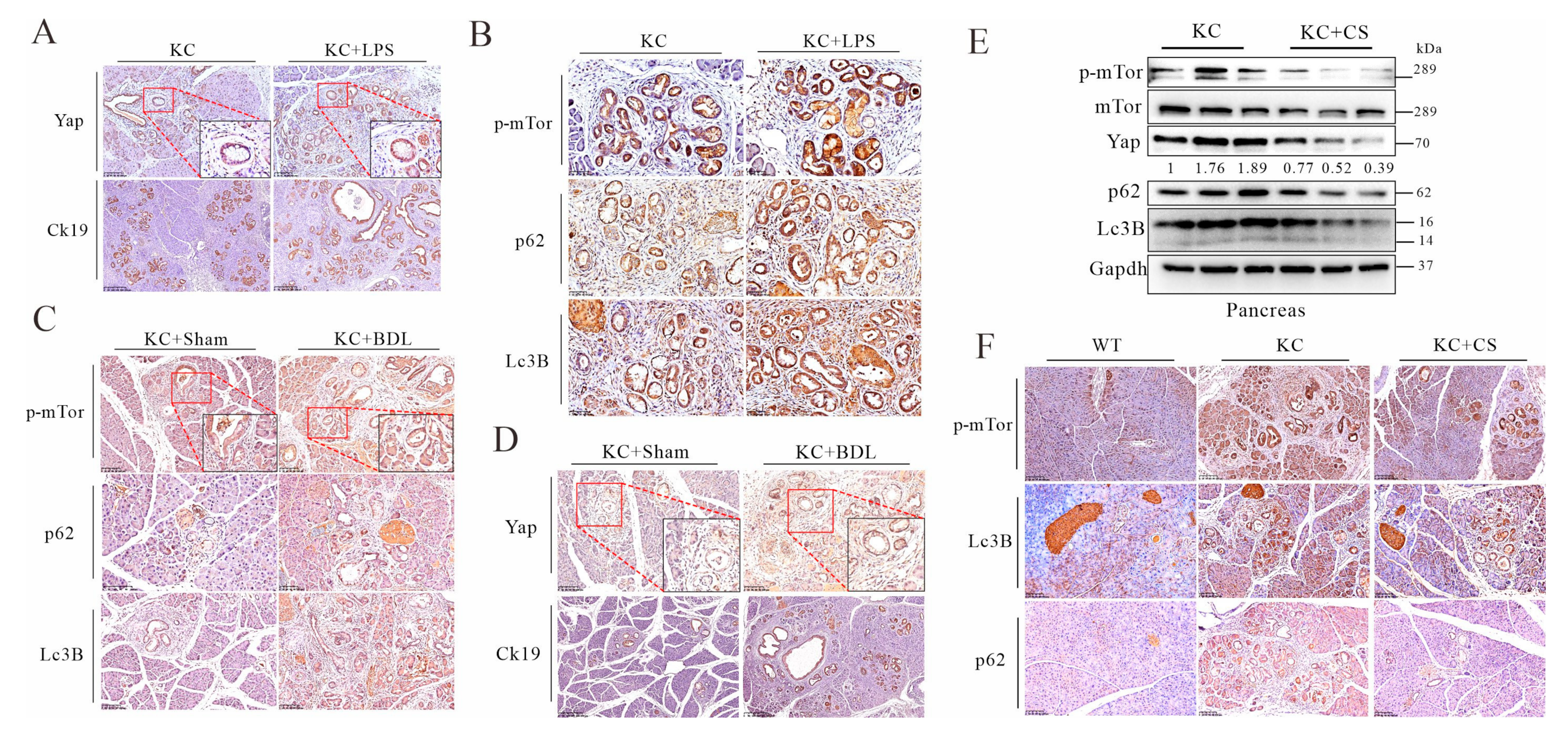
3.3. Oral Administration of Cationic Resin Leads to Mobilization of Autophagic Flux for Yap Degradation in Pancreatic Cancer Tissues
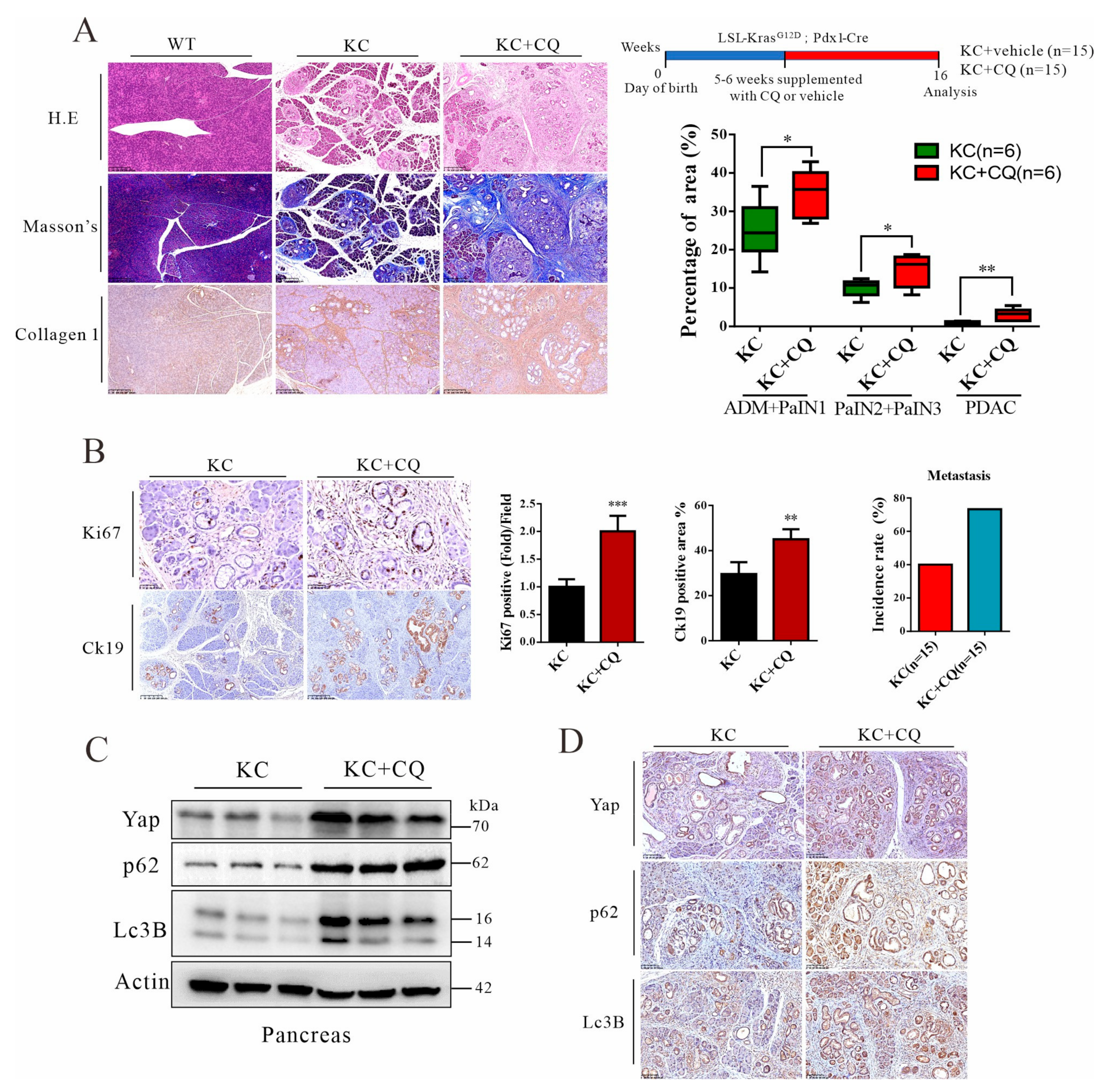
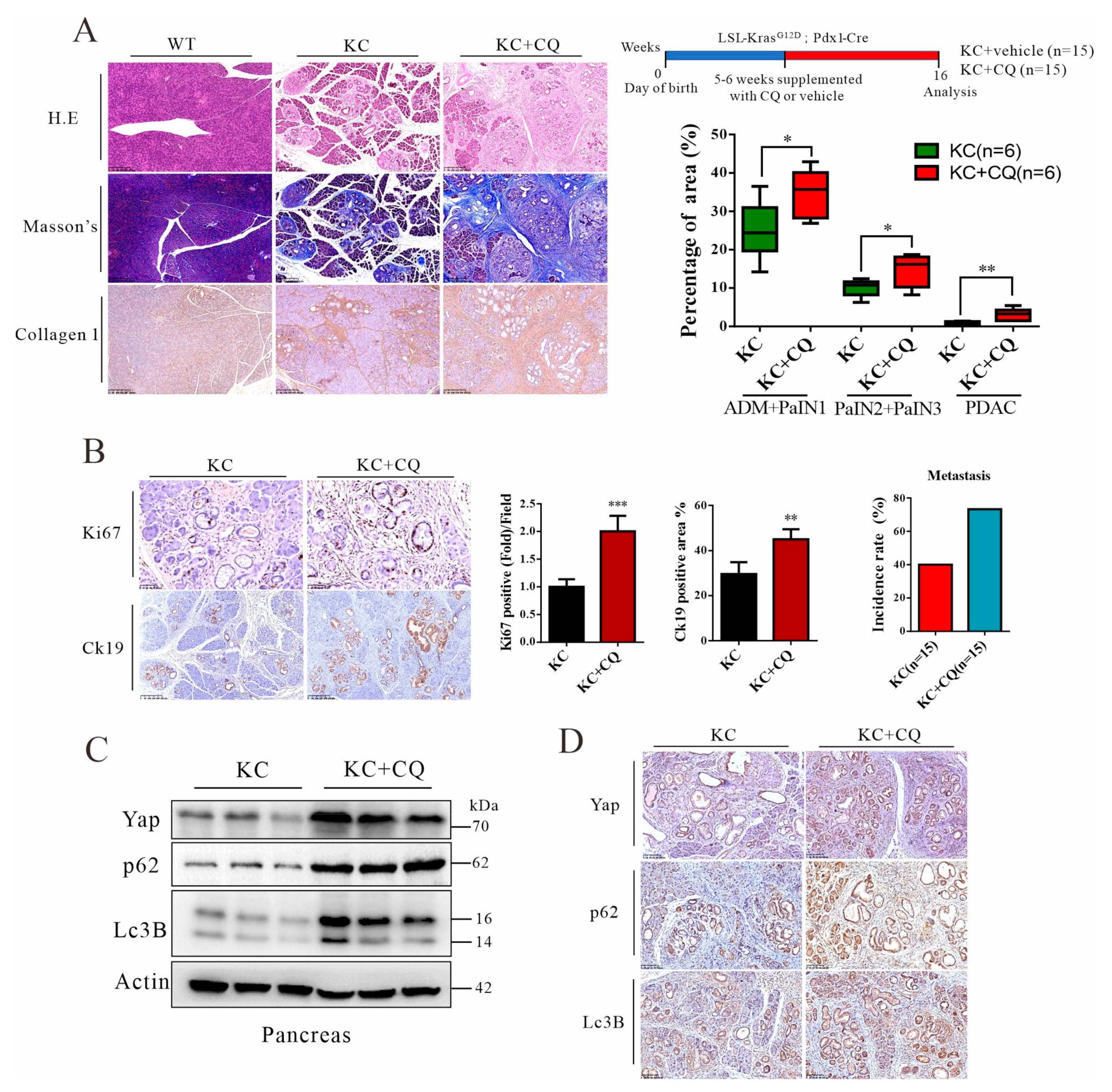
3.4. Autolysosomal Stress Induced by Chloroquine Treatment Accelerates Malignant Progression of Pancreatic Cancer in Mice
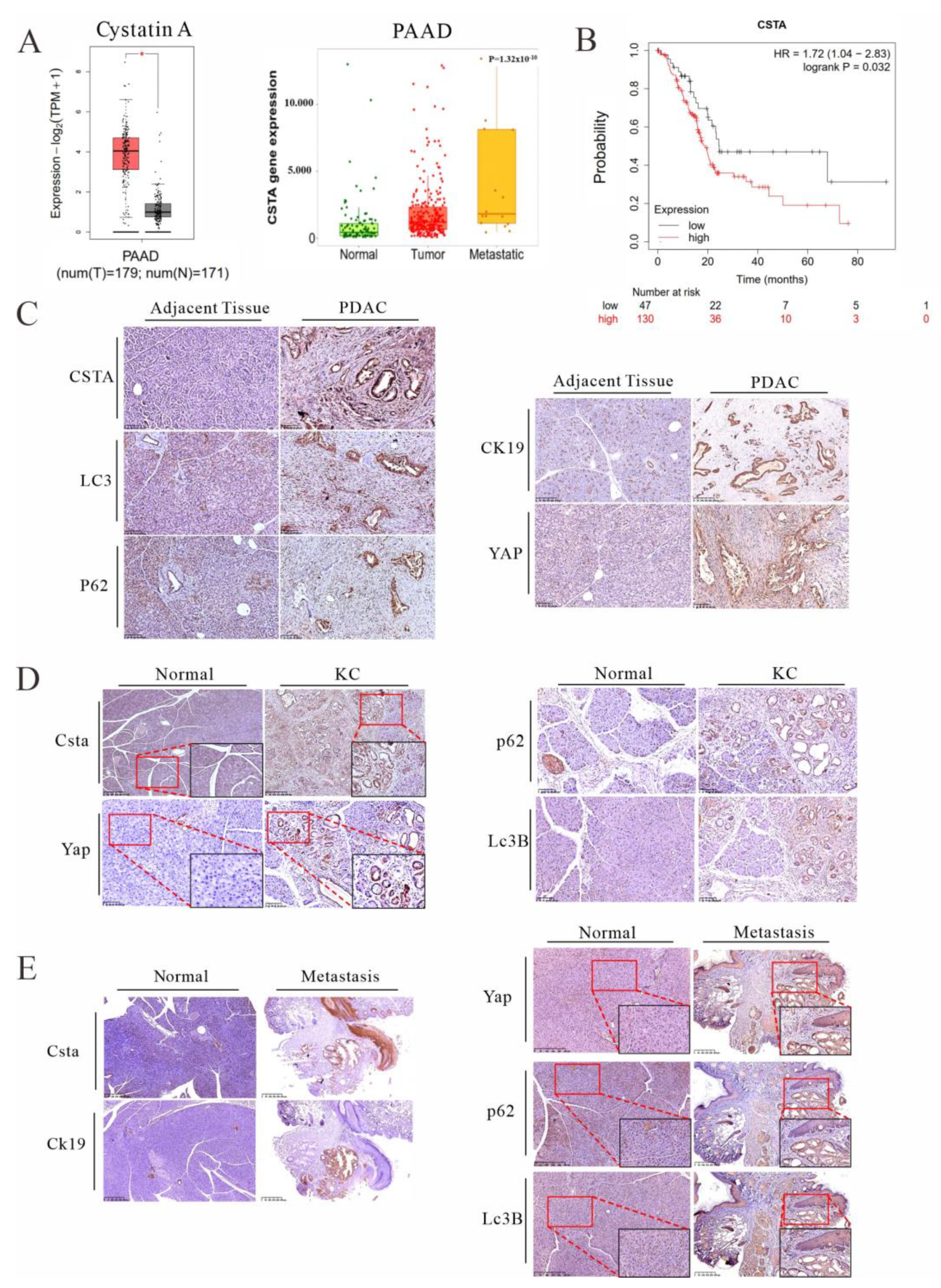
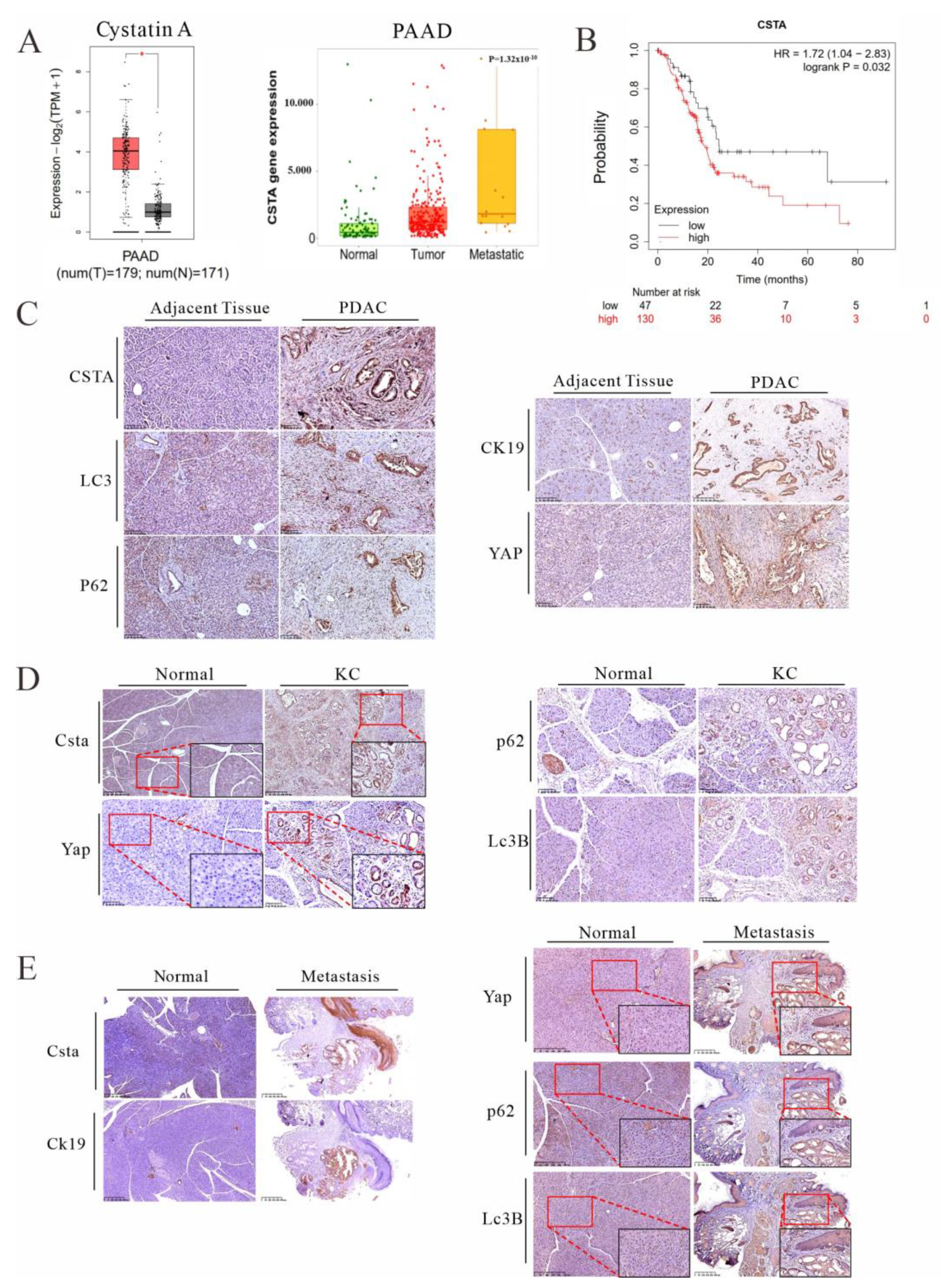
3.5. Cystatin A, an Endogenous Inhibitor of Lysosomal Acidic Proteinases, Is Upregulated in PDAC Patients and KC Mice and Is Associated with Poor Prognosis
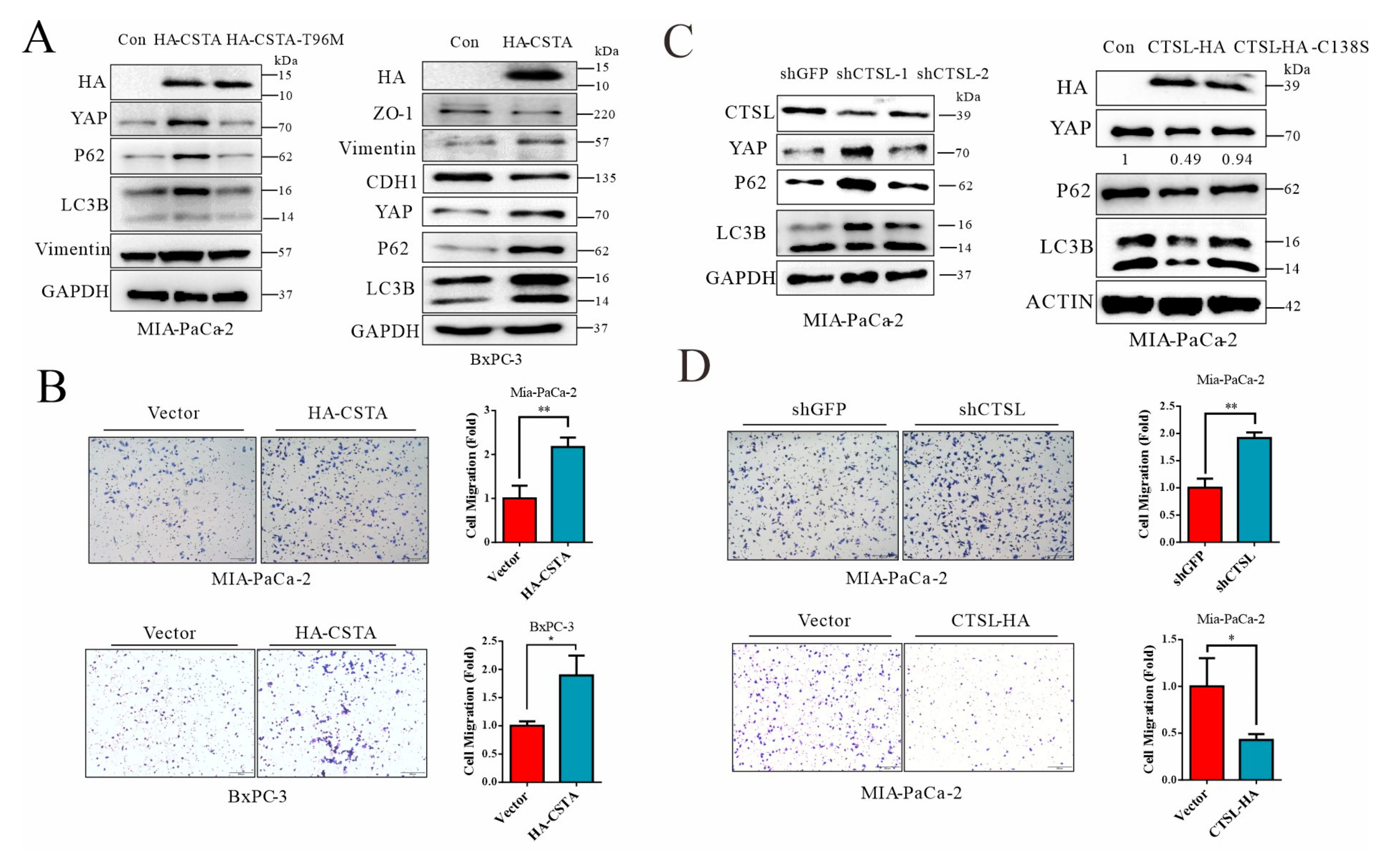
3.6. Forced Expression of Cystatin A and Lysosomal Stress Leads to YAP Accumulation in Cancer Cells
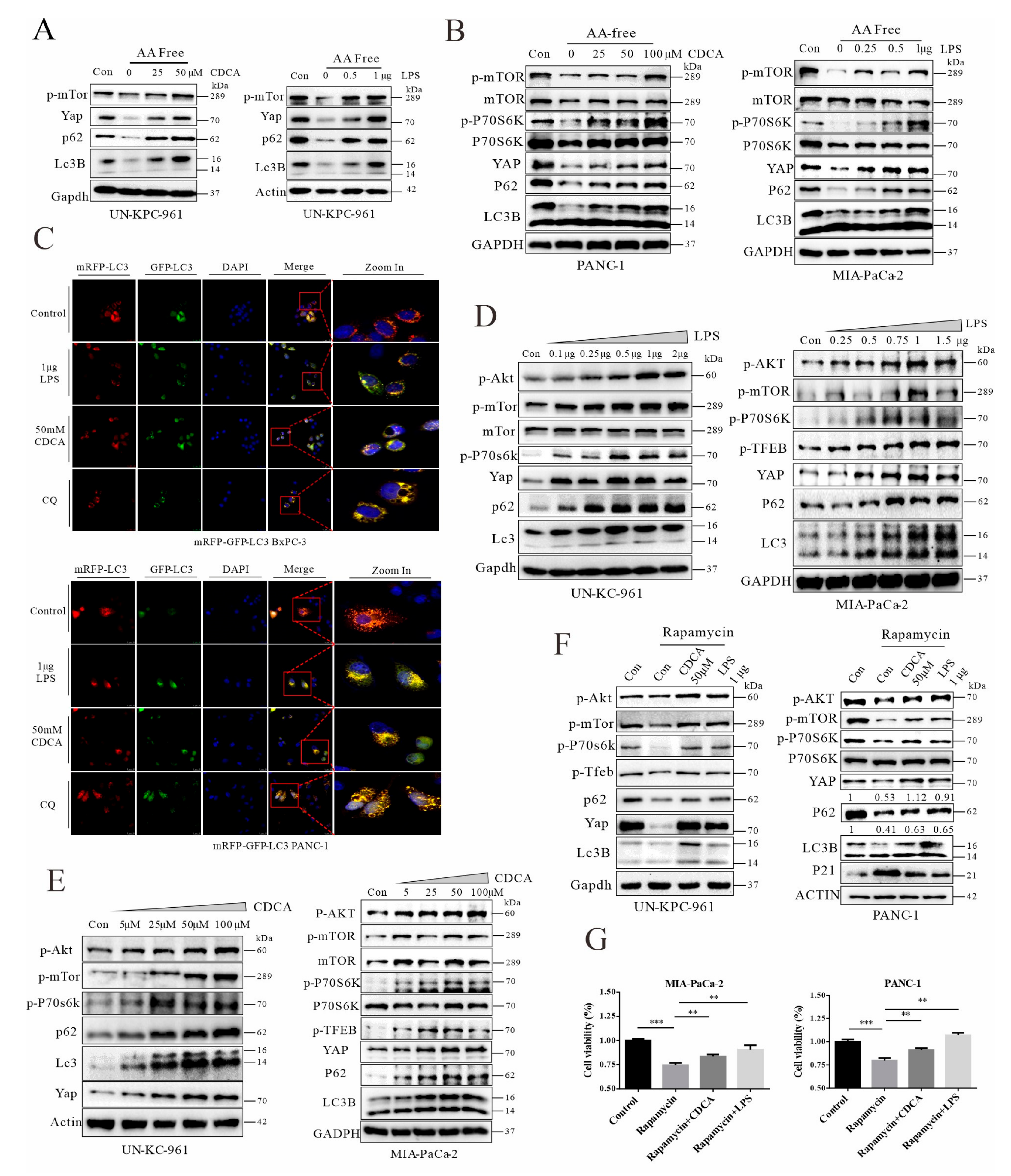
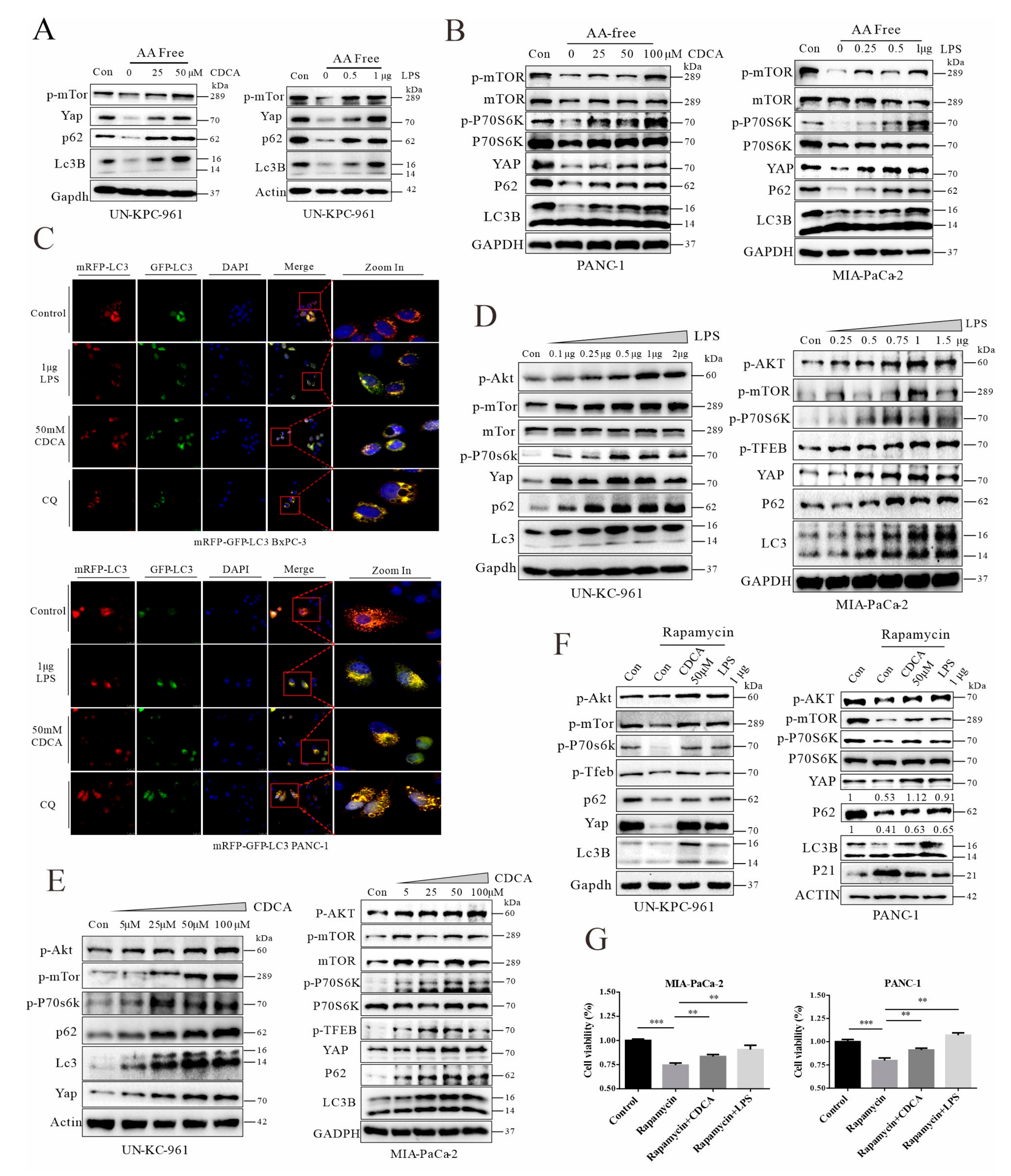
3.7. Chenodeoxycholic Acid and LPS Can Activate the AKT-mTOR Pathway, Leading to Impairment of Autophagic Flux and YAP Accumulation in Cancer Cells
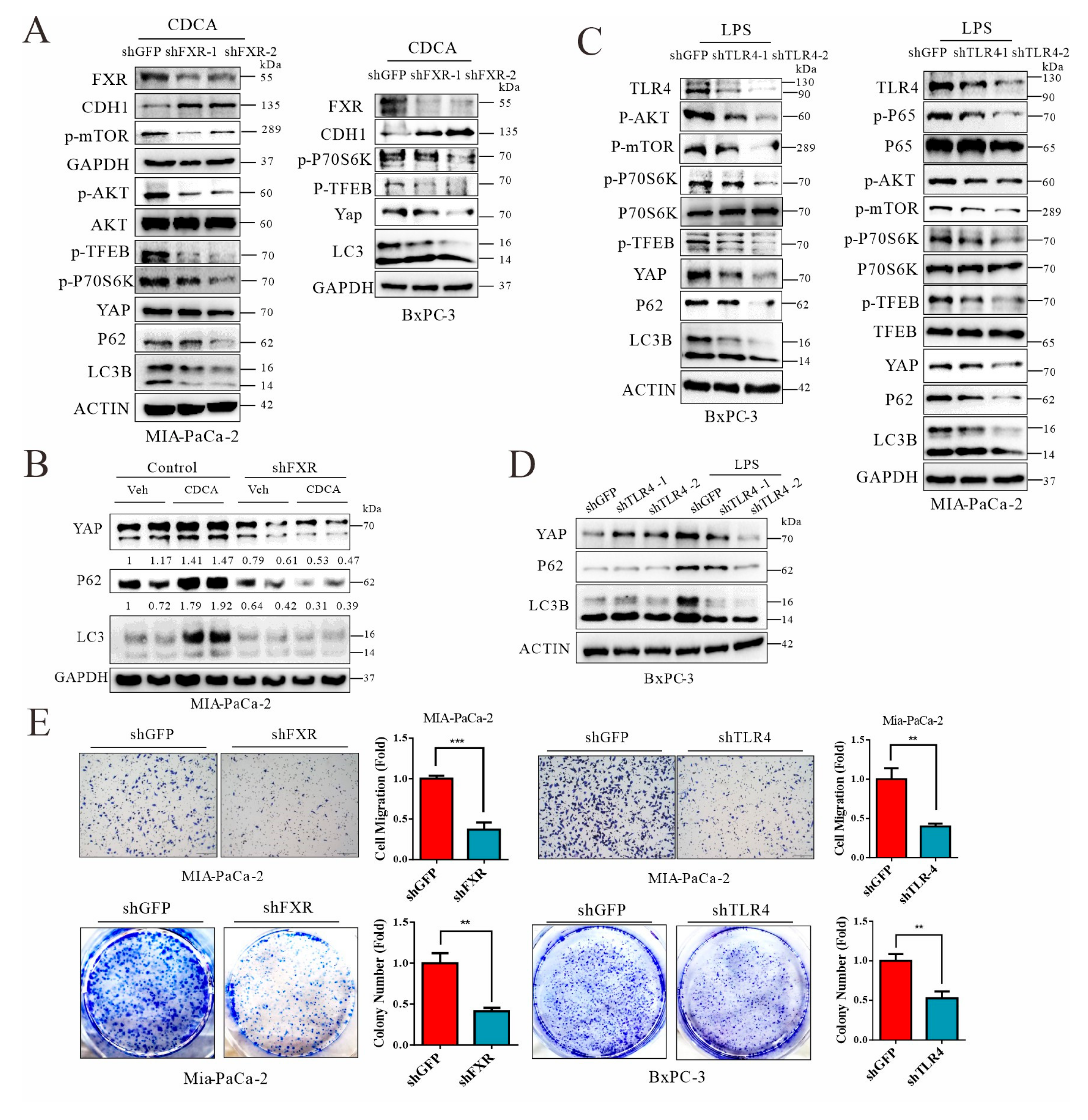
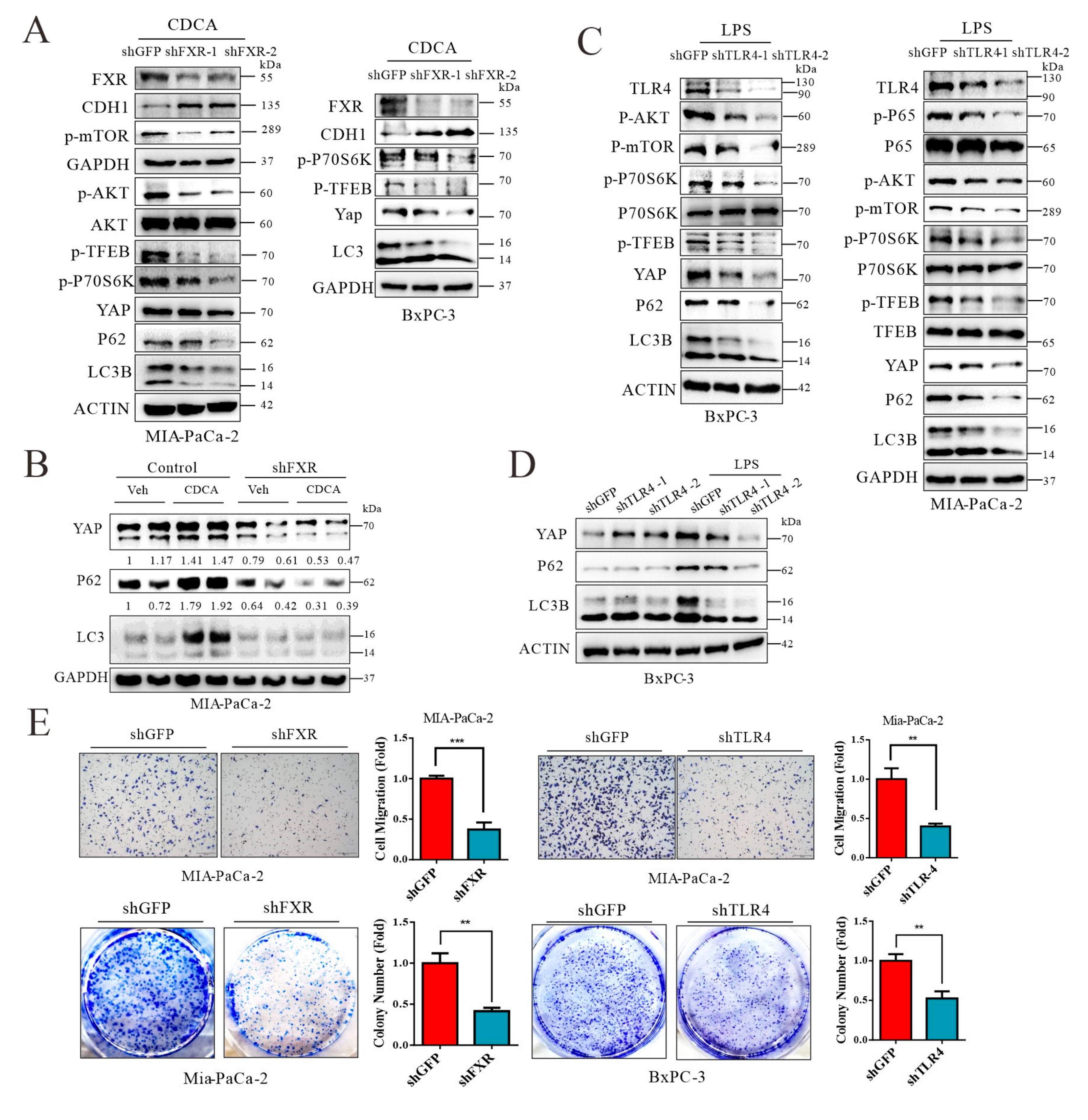
3.8. Signals from FXR and TLR4 Mediate Chenodeoxycholic Acid and LPS, Respectively, for Activation of AKT-mTOR Pathway in Pancreatic Cancer Cells
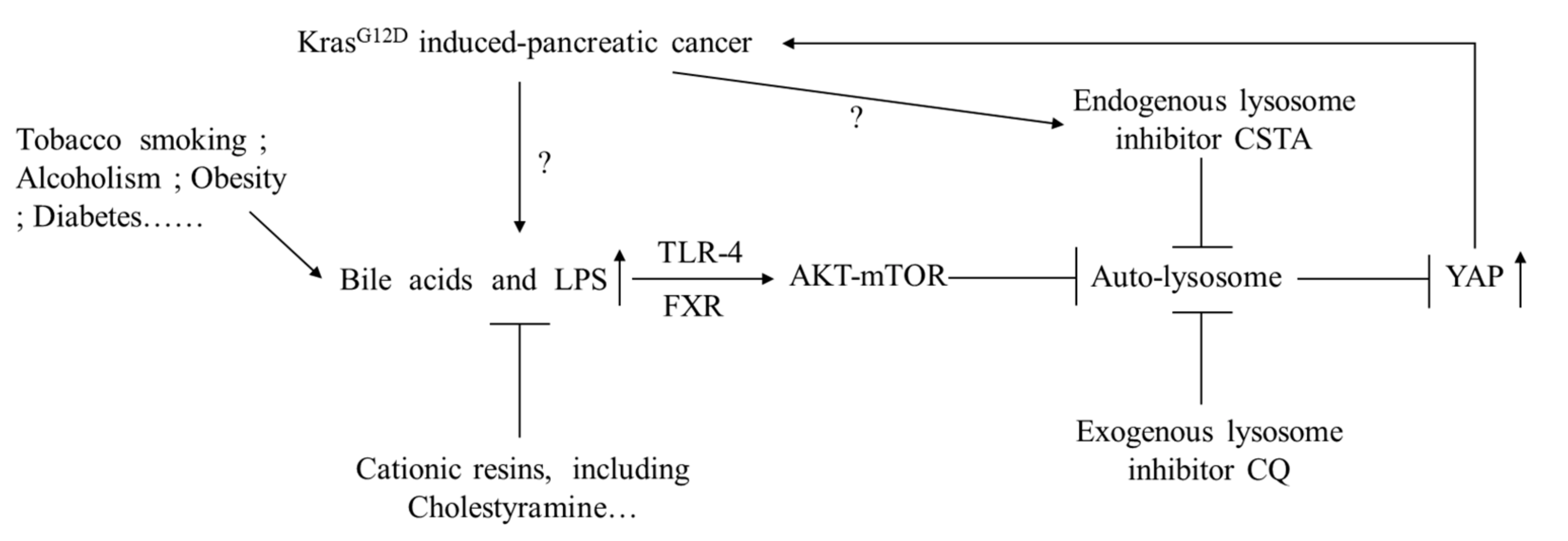
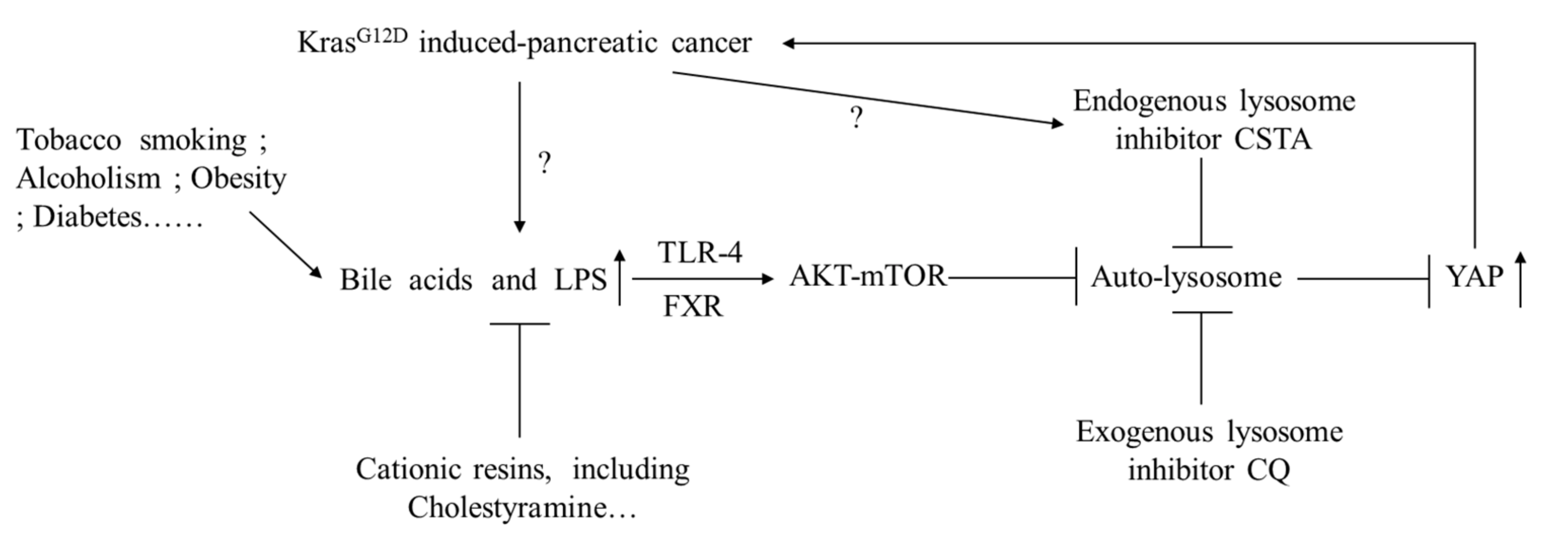
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Primers 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Grossberg, A.J.; Chu, L.C.; Deig, C.R.; Fishman, E.K.; Hwang, W.L.; Maitra, A.; Marks, D.L.; Mehta, A.; Nabavizadeh, N.; Simeone, D.M.; et al. Multidisciplinary standards of care and recent progress in pancreatic ductal adenocarcinoma. CA Cancer J. Clin. 2020, 70, 375–403. [Google Scholar] [CrossRef] [PubMed]
- Bardeesy, N.; Aguirre, A.J.; Chu, G.C.; Cheng, K.H.; Lopez, L.V.; Hezel, A.F.; Feng, B.; Brennan, C.; Weissleder, R.; Mahmood, U.; et al. Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 5947–5952. [Google Scholar] [CrossRef] [Green Version]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniluk, J.; Liu, Y.; Deng, D.; Chu, J.; Huang, H.; Gaiser, S.; Cruz-Monserrate, Z.; Wang, H.; Ji, B.; Logsdon, C.D. An NF-κB pathway-mediated positive feedback loop amplifies Ras activity to pathological levels in mice. J. Clin. Investig. 2012, 122, 1519–1528. [Google Scholar] [CrossRef] [Green Version]
- Brat, D.J.; Lillemoe, K.D.; Yeo, C.J.; Warfield, P.B.; Hruban, R.H. Progression of pancreatic intraductal neoplasias to infiltrating adenocarcinoma of the pancreas. Am. J. Surg. Pathol. 1998, 22, 163–169. [Google Scholar] [CrossRef]
- Andea, A.; Sarkar, F.; Adsay, V.N. Clinicopathological correlates of pancreatic intraepithelial neoplasia: A comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Modern Pathol. 2003, 16, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Hruban, R.H.; Goggins, M.; Parsons, J.; Kern, S.E. Progression model for pancreatic cancer. Clin. Cancer Res. 2000, 6, 2969–2972. [Google Scholar]
- Thomas, R.M.; Jobin, C. Microbiota in pancreatic health and disease: The next frontier in microbiome research. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 53–64. [Google Scholar] [CrossRef]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Rajagopala, S.V.; Vashee, S.; Oldfield, L.M.; Suzuki, Y.; Venter, J.C.; Telenti, A.; Nelson, K.E. The Human Microbiome and Cancer. Cancer Prev. Res. 2017, 10, 226–234. [Google Scholar] [CrossRef] [Green Version]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Zhang, D.; Li, Z.; Jiang, H.; Li, J.; Ren, R.; Gao, X.; Li, J.; Wang, X.; Wang, W.; et al. The fecal microbiota of patients with pancreatic ductal adenocarcinoma and autoimmune pancreatitis characterized by metagenomic sequencing. J. Transl. Med. 2021, 19, 215. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.L.; Li, B.B.; Pan, X.H.; Sun, J. Gut microbiota in pancreatic diseases: Possible new therapeutic strategies. Acta Pharmacol. Sin. 2021, 42, 1027–1039. [Google Scholar] [CrossRef]
- Schlender, J.; Behrens, F.; McParland, V.; Müller, D.; Wilck, N.; Bartolomaeus, H.; Holle, J. Bacterial metabolites and cardiovascular risk in children with chronic kidney disease. Mol. Cell. Pediatrics 2021, 8, 17. [Google Scholar] [CrossRef]
- Vors, C.; Pineau, G.; Drai, J.; Meugnier, E.; Pesenti, S.; Laville, M.; Laugerette, F.; Malpuech-Brugère, C.; Vidal, H.; Michalski, M.C. Postprandial Endotoxemia Linked With Chylomicrons and Lipopolysaccharides Handling in Obese Versus Lean Men: A Lipid Dose-Effect Trial. J. Clin. Endocrinol. Metab. 2015, 100, 3427–3435. [Google Scholar] [CrossRef]
- Zaiss, M.M.; Jones, R.M.; Schett, G.; Pacifici, R. The gut-bone axis: How bacterial metabolites bridge the distance. J. Clin. Investig. 2019, 129, 3018–3028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenther, M.; Haas, M.; Heinemann, V.; Kruger, S.; Westphalen, C.B.; von Bergwelt-Baildon, M.; Mayerle, J.; Werner, J.; Kirchner, T.; Boeck, S.; et al. Bacterial lipopolysaccharide as negative predictor of gemcitabine efficacy in advanced pancreatic cancer-translational results from the AIO-PK0104 Phase 3 study. Br. J. Cancer 2020, 123, 1370–1376. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.X.; Huang, C.L.; Luo, S.Z.; Zhang, X.M.; Zeng, Y.; Lu, Y.Y. Characterization of the duodenal bacterial microbiota in patients with pancreatic head cancer vs. healthy controls. Pancreatology. 2018, 18, 438–445. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef] [Green Version]
- Di Ciaula, A.; Wang, D.Q.; Molina-Molina, E.; Lunardi Baccetto, R.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile Acids and Cancer: Direct and Environmental-Dependent Effects. Ann. Hepatol. 2017, 16 (Suppl. S1), S87–S105. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Wu, L.L.; Han, T.; Zhuo, M.; Lei, W.; Cui, J.J.; Jiao, F.; Wang, L.W. Correlated high expression of FXR and Sp1 in cancer cells confers a poor prognosis for pancreatic cancer: A study based on TCGA and tissue microarray. Oncotarget 2017, 8, 33265–33275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhang, S.; Zhang, W.; Yue, Y.; Qian, W.; Wang, Z. Matrix stiffness and its influence on pancreatic diseases. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188583. [Google Scholar] [CrossRef]
- Khalil, B.D.; Sanchez, R.; Rahman, T.; Rodriguez-Tirado, C.; Moritsch, S.; Martinez, A.R.; Miles, B.; Farias, E.; Mezei, M.; Nobre, A.R.; et al. An NR2F1-specific agonist suppresses metastasis by inducing cancer cell dormancy. J. Exp. Med. 2022, 219, e20210836. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Fattet, L.; Tsai, J.H.; Guo, Y.; Pai, V.H.; Majeski, H.E.; Chen, A.C.; Sah, R.L.; Taylor, S.S.; Engler, A.J.; et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol. 2015, 17, 678–688. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.J.; Cortes, E.; Lachowski, D.; Cheung, B.C.H.; Karim, S.A.; Morton, J.P.; Del Río Hernández, A. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 2017, 6, e352. [Google Scholar] [CrossRef] [Green Version]
- Vonlaufen, A.; Xu, Z.; Daniel, B.; Kumar, R.K.; Pirola, R.; Wilson, J.; Apte, M.V. Bacterial endotoxin: A trigger factor for alcoholic pancreatitis? Evidence from a novel, physiologically relevant animal model. Gastroenterology 2007, 133, 1293–1303. [Google Scholar] [CrossRef]
- Zambirinis, C.P.; Levie, E.; Nguy, S.; Avanzi, A.; Barilla, R.; Xu, Y.; Seifert, L.; Daley, D.; Greco, S.H.; Deutsch, M.; et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J. Exp. Med. 2015, 212, 2077–2094. [Google Scholar] [CrossRef]
- Zanconato, F.; Cordenonsi, M.; Piccolo, S. YAP/TAZ at the Roots of Cancer. Cancer Cell 2016, 29, 783–803. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 2014, 158, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Morvaridi, S.; Dhall, D.; Greene, M.I.; Pandol, S.J.; Wang, Q. Role of YAP and TAZ in pancreatic ductal adenocarcinoma and in stellate cells associated with cancer and chronic pancreatitis. Sci. Rep. 2015, 5, 16759. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nature 2011, 474, 179–183. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef] [Green Version]
- Torres, M.P.; Rachagani, S.; Souchek, J.J.; Mallya, K.; Johansson, S.L.; Batra, S.K. Novel pancreatic cancer cell lines derived from genetically engineered mouse models of spontaneous pancreatic adenocarcinoma: Applications in diagnosis and therapy. PLoS ONE 2013, 8, e80580. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, N.; Sun, Y.; Zhu, G.; Wang, Y.; Wang, Z.; Zhang, Y.; Cheng, K.; Wang, G.; Wu, S.; et al. Screen-detected gallstone disease and risk of liver and pancreatic cancer: The Kailuan Cohort Study. Liver Int. Off. J. Int. Assoc. Study Liver 2020, 40, 1744–1755. [Google Scholar] [CrossRef] [PubMed]
- Rees, D.O.; Crick, P.J.; Jenkins, G.J.; Wang, Y.; Griffiths, W.J.; Brown, T.H.; Al-Sarireh, B. Comparison of the composition of bile acids in bile of patients with adenocarcinoma of the pancreas and benign disease. J. Steroid Biochem. Mol. Biol. 2017, 174, 290–295. [Google Scholar] [CrossRef]
- Zhu, A.; Chen, J.; Wu, P.; Luo, M.; Zeng, Y.; Liu, Y.; Zheng, H.; Zhang, L.; Chen, Z.; Sun, Q.; et al. Cationic Polystyrene Resolves Nonalcoholic Steatohepatitis, Obesity, and Metabolic Disorders by Promoting Eubiosis of Gut Microbiota and Decreasing Endotoxemia. Diabetes 2017, 66, 2137–2143. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.; Zhang, R.; Luo, M.; Zhang, T.; Pan, L.; Xu, S.; Pan, L.; Ren, F.; Ji, C.; Hu, R.; et al. Liver Injury Impaired 25-Hydroxylation of Vitamin D Suppresses Intestinal Paneth Cell defensins, leading to Gut Dysbiosis and Liver Fibrogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G685–G695. [Google Scholar] [CrossRef]
- Trobridge, P.; Knoblaugh, S.; Washington, M.K.; Munoz, N.M.; Tsuchiya, K.D.; Rojas, A.; Song, X.; Ulrich, C.M.; Sasazuki, T.; Shirasawa, S.; et al. TGF-beta receptor inactivation and mutant Kras induce intestinal neoplasms in mice via a beta-catenin-independent pathway. Gastroenterology 2009, 136, 1680–1688.e1687. [Google Scholar] [CrossRef] [Green Version]
- Xu, A.; Wang, B.; Fu, J.; Qin, W.; Yu, T.; Yang, Z.; Lu, Q.; Chen, J.; Chen, Y.; Wang, H. Diet-induced hepatic steatosis activates Ras to promote hepatocarcinogenesis via CPT1α. Cancer Lett. 2019, 442, 40–52. [Google Scholar] [CrossRef]
- Rozengurt, E.; Sinnett-Smith, J.; Eibl, G. Yes-associated protein (YAP) in pancreatic cancer: At the epicenter of a targetable signaling network associated with patient survival. Signal. Transduct. Target. Ther. 2018, 3, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Xiu, M.; Wang, S.; Brigstock, D.R.; Li, H.; Qu, L.; Gao, R. Lipopolysaccharide enhances TGF-beta1 signalling pathway and rat pancreatic fibrosis. J. Cell. Mol. Med. 2018, 22, 2346–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, C.; Zhu, H.; Han, M.; Zhuang, Y.; Wu, X.; Xu, T. Disruption of lysosome function promotes tumor growth and metastasis in Drosophila. J. Biol. Chem. 2010, 285, 21817–21823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komura, T.; Takabatake, H.; Harada, K.; Yamato, M.; Miyazawa, M.; Yoshida, K.; Honda, M.; Wada, T.; Kitagawa, H.; Ohta, T.; et al. Clinical features of cystatin A expression in patients with pancreatic ductal adenocarcinoma. Cancer Sci. 2017, 108, 2122–2129. [Google Scholar] [CrossRef] [Green Version]
- Ohshio, G.; Manabe, T.; Tobe, T.; Yoshioka, H.; Hamashima, Y. Circulating immune complex, endotoxin, and biliary infection in patients with biliary obstruction. Am. J. Surg. 1988, 155, 343–347. [Google Scholar] [CrossRef]
- Scaldaferri, F.; Pizzoferrato, M.; Ponziani, F.R.; Gasbarrini, G.; Gasbarrini, A. Use and indications of cholestyramine and bile acid sequestrants. Intern. Emerg. Med. 2013, 8, 205–210. [Google Scholar] [CrossRef]
- Sjöberg, B.G.; Straniero, S.; Angelin, B.; Rudling, M. Cholestyramine treatment of healthy humans rapidly induces transient hypertriglyceridemia when treatment is initiated. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E167–E174. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science. 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Pandol, S.; Gukovskaya, A.; Edderkaoui, M.; Dawson, D.; Eibl, G.; Lugea, A. Epidemiology, risk factors, and the promotion of pancreatic cancer: Role of the stellate cell. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. S2), 127–134. [Google Scholar] [CrossRef] [Green Version]
- Shi, Y.; Cang, L.; Zhang, X.; Cai, X.; Wang, X.; Ji, R.; Wang, M.; Hong, Y. The use of magnetic resonance elastography in differentiating autoimmune pancreatitis from pancreatic ductal adenocarcinoma: A preliminary study. Eur. J. Radiol. 2018, 108, 13–20. [Google Scholar] [CrossRef]
- Mueller, S.; Sandrin, L. Liver stiffness: A novel parameter for the diagnosis of liver disease. Hepat. Med. 2010, 2, 49–67. [Google Scholar] [CrossRef] [Green Version]
- Apte, M.V.; Wilson, J.S.; Lugea, A.; Pandol, S.J. A starring role for stellate cells in the pancreatic cancer microenvironment. Gastroenterology 2013, 144, 1210–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Zhou, C.; Cheng, L.; Yan, B.; Chen, K.; Chen, X.; Zong, L.; Lei, J.; Duan, W.; Xu, Q.; et al. Inhibiting YAP expression suppresses pancreatic cancer progression by disrupting tumor-stromal interactions. J. Exp. Clin. Cancer Res. CR 2018, 37, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [Green Version]
- Min, K.B.; Min, J.Y. Household endotoxin exposure and increased risk of diabetes in older adults. Diabet. Med. 2015, 32, 1667–1669. [Google Scholar] [CrossRef]
- Su, D.; Nie, Y.; Zhu, A.; Chen, Z.; Wu, P.; Zhang, L.; Luo, M.; Sun, Q.; Cai, L.; Lai, Y.; et al. Vitamin D Signaling through Induction of Paneth Cell Defensins Maintains Gut Microbiota and Improves Metabolic Disorders and Hepatic Steatosis in Animal Models. Front. Physiol. 2016, 7, 498. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Enot, D.P.; Pietrocola, F.; Zitvogel, L.; Kroemer, G. Impact of Pattern Recognition Receptors on the Prognosis of Breast Cancer Patients Undergoing Adjuvant Chemotherapy. Cancer Res. 2016, 76, 3122–3126. [Google Scholar] [CrossRef] [Green Version]
- Castano-Rodriguez, N.; Kaakoush, N.O.; Mitchell, H.M. Pattern-recognition receptors and gastric cancer. Front. Immunol. 2014, 5, 1–23. [Google Scholar]
- Ren, Z.; Jiang, J.; Xie, H.; Li, A.; Lu, H.; Xu, S.; Zhou, L.; Zhang, H.; Cui, G.; Chen, X.; et al. Gut microbial profile analysis by MiSeq sequencing of pancreatic carcinoma patients in China. Oncotarget 2017, 8, 95176–95191. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, T.; Fukaya, R.; Takamatsu, S.; Itoyama, S.; Fukuoka, T.; Yamada, M.; Hata, T.; Nagaoka, S.; Kawamoto, K.; Eguchi, H.; et al. Possible involvement of Enterococcus infection in the pathogenesis of chronic pancreatitis and cancer. Biochem. Biophys. Res. Commun. 2018, 506, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Song, X.; Peng, Y.M.; Wang, W.N.; Song, Z. Association of disease condition with changes in intestinal flora, and plasma endotoxin and vascular endothelial growth factor levels in patients with liver cancer. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 3605–3613. [Google Scholar]
- Ikebe, M.; Kitaura, Y.; Nakamura, M.; Tanaka, H.; Yamasaki, A.; Nagai, S.; Wada, J.; Yanai, K.; Koga, K.; Sato, N.; et al. Lipopolysaccharide (LPS) increases the invasive ability of pancreatic cancer cells through the TLR4/MyD88 signaling pathway. J. Surg. Oncol. 2009, 100, 725–731. [Google Scholar] [CrossRef]
- Thomas, R.M.; Gharaibeh, R.Z.; Gauthier, J.; Beveridge, M.; Pope, J.L.; Guijarro, M.V.; Yu, Q.; He, Z.; Ohland, C.; Newsome, R.; et al. Intestinal microbiota enhances pancreatic carcinogenesis in preclinical models. Carcinogenesis 2018, 39, 1068–1078. [Google Scholar] [CrossRef]
- Adachi, T.; Tajima, Y.; Kuroki, T.; Mishima, T.; Kitasato, A.; Fukuda, K.; Tsutsumi, R.; Kanematsu, T. Bile-reflux into the pancreatic ducts is associated with the development of intraductal papillary carcinoma in hamsters. J. Surg. Res. 2006, 136, 106–111. [Google Scholar] [CrossRef]
- Joshi, S.; Cruz, E.; Rachagani, S.; Guha, S.; Brand, R.E.; Ponnusamy, M.P.; Kumar, S.; Batra, S.K. Bile acids-mediated overexpression of MUC4 via FAK-dependent c-Jun activation in pancreatic cancer. Mol. Oncol. 2016, 10, 1063–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, G.; Wang, X.; Huang, F.; Zhao, A.; Chen, W.; Yan, J.; Zhang, Y.; Lei, S.; Ge, K.; Zheng, X.; et al. Dysregulated hepatic bile acids collaboratively promote liver carcinogenesis. Int. J. Cancer 2016, 139, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; He, S.J.; Ma, J.J.; Hu, H.; Gong, Y.P.; Wang, Y.L.; Hu, B.J.; Xie, J.Z.; Tu, W.Z.; Huang, Q.; et al. High expression of TGR5 predicts a poor prognosis in patients with pancreatic cancer. Int. J. Clin. Exp. Pathol. 2018, 11, 3567–3574. [Google Scholar]
- Perides, G.; Laukkarinen, J.M.; Vassileva, G.; Steer, M.L. Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology 2010, 138, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Xie, L.; Bergmann, F.; Endris, V.; Strobel, O.; Büchler, M.W.; Kroemer, G.; Hackert, T.; Fortunato, F. The bile acid receptor FXR attenuates acinar cell autophagy in chronic pancreatitis. Cell Death Discov. 2017, 3, 17027. [Google Scholar] [CrossRef]
- Mancias, J.D.; Kimmelman, A.C. Targeting autophagy addiction in cancer. Oncotarget 2011, 2, 1302–1306. [Google Scholar] [CrossRef] [Green Version]
- Gugnoni, M.; Sancisi, V.; Manzotti, G.; Gandolfi, G.; Ciarrocchi, A. Autophagy and epithelial-mesenchymal transition: An intricate interplay in cancer. Cell Death Dis. 2016, 7, e2520. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.Y.; Teng, X.; Laddha, S.V.; Ma, S.; van Nostrand, S.C.; Yang, Y.; Khor, S.; Chan, C.S.; Rabinowitz, J.D.; White, E. Autophagy provides metabolic substrates to maintain energy charge and nucleotide pools in Ras-driven lung cancer cells. Genes Dev. 2016, 30, 1704–1717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lock, R.; Roy, S.; Kenific, C.M.; Su, J.S.; Salas, E.; Ronen, S.M.; Debnath, J. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Mol. Biol. Cell 2011, 22, 165–178. [Google Scholar] [CrossRef]
- Guo, J.Y.; Chen, H.Y.; Mathew, R.; Fan, J.; Strohecker, A.M.; Karsli-Uzunbas, G.; Kamphorst, J.J.; Chen, G.; Lemons, J.M.; Karantza, V.; et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011, 25, 460–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgendy, M.; Sheridan, C.; Brumatti, G.; Martin, S.J. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Mol. Cell 2011, 42, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Karasic, T.B.; O’Hara, M.H.; Loaiza-Bonilla, A.; Reiss, K.A.; Teitelbaum, U.R.; Borazanci, E.; de Jesus-Acosta, A.; Redlinger, C.; Burrell, J.A.; Laheru, D.A.; et al. Effect of Gemcitabine and nab-Paclitaxel With or Without Hydroxychloroquine on Patients With Advanced Pancreatic Cancer: A Phase 2 Randomized Clinical Trial. JAMA Oncol. 2019, 5, 993–998. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Rubinson, D.A.; Wang, X.; Chan, J.A.; Cleary, J.M.; Enzinger, P.C.; Fuchs, C.S.; McCleary, N.J.; Meyerhardt, J.A.; Ng, K.; et al. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist 2014, 19, 637–638. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Yelamanchi, R. Pancreatic adenocarcinoma: A review of recent paradigms and advances in epidemiology, clinical diagnosis and management. World J. Gastroenterol. 2021, 27, 3158–3181. [Google Scholar] [CrossRef]
- Nassour, J.; Radford, R.; Correia, A.; Fusté, J.M.; Schoell, B.; Jauch, A.; Shaw, R.J.; Karlseder, J. Autophagic cell death restricts chromosomal instability during replicative crisis. Nature 2019, 565, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Vander Heiden, M.G. Critical Functions of the Lysosome in Cancer Biology. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 481–507. [Google Scholar] [CrossRef] [PubMed]
- Dennemarker, J.; Lohmuller, T.; Mayerle, J.; Tacke, M.; Lerch, M.M.; Coussens, L.M.; Peters, C.; Reinheckel, T. Deficiency for the cysteine protease cathepsin L promotes tumor progression in mouse epidermis. Oncogene 2010, 29, 1611–1621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebert, E.; Werle, B.; Julke, B.; Kopitar-Jerala, N.; Kos, J.; Lah, T.; Abrahamson, M.; Spiess, E.; Ebert, W. Expression of cysteine protease inhibitors stefin A, stefin B, and cystatin C in human lung tumor tissue. Adv. Exp. Med. Biol. 1997, 421, 259–265. [Google Scholar]
- Kuopio, T.; Kankaanranta, A.; Jalava, P.; Kronqvist, P.; Kotkansalo, T.; Weber, E.; Collan, Y. Cysteine proteinase inhibitor cystatin A in breast cancer. Cancer Res. 1998, 58, 432–436. [Google Scholar]
- Liang, X.H.; Jackson, S.; Seaman, M.; Brown, K.; Kempkes, B.; Hibshoosh, H.; Levine, B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999, 402, 672–676. [Google Scholar] [CrossRef]
- Görgülü, K.; Diakopoulos, K.N.; Ai, J.; Schoeps, B.; Kabacaoglu, D.; Karpathaki, A.F.; Ciecielski, K.J.; Kaya-Aksoy, E.; Ruess, D.A.; Berninger, A.; et al. Levels of the Autophagy-Related 5 Protein Affect Progression and Metastasis of Pancreatic Tumors in Mice. Gastroenterology 2019, 156, 203–217.e220. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.A.; Noon, L.A.; Akat, K.M.; Ybanez, M.D.; Lee, T.F.; Berres, M.L.; Fujiwara, N.; Goossens, N.; Chou, H.I.; Parvin-Nejad, F.P.; et al. Autophagy is a gatekeeper of hepatic differentiation and carcinogenesis by controlling the degradation of Yap. Nature Commun. 2018, 9, 4962. [Google Scholar] [CrossRef]
- Wang, P.; Gong, Y.; Guo, T.; Li, M.; Fang, L.; Yin, S.; Kamran, M.; Liu, Y.; Xu, J.; Xu, L.; et al. Activation of Aurora A kinase increases YAP stability via blockage of autophagy. Cell Death Dis. 2019, 10, 432. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, G.; Zhang, T.; Liu, W.; Edderkaoui, M.; Hu, R.; Li, J.; Pandol, S.J.; Fu, X.; Han, Y.-P. Sequestration of Intestinal Acidic Toxins by Cationic Resin Attenuates Pancreatic Cancer Progression through Promoting Autophagic Flux for YAP Degradation. Cancers 2022, 14, 1407. https://doi.org/10.3390/cancers14061407
Zhao G, Zhang T, Liu W, Edderkaoui M, Hu R, Li J, Pandol SJ, Fu X, Han Y-P. Sequestration of Intestinal Acidic Toxins by Cationic Resin Attenuates Pancreatic Cancer Progression through Promoting Autophagic Flux for YAP Degradation. Cancers. 2022; 14(6):1407. https://doi.org/10.3390/cancers14061407
Chicago/Turabian StyleZhao, Guangfu, Tianci Zhang, Wei Liu, Mouad Edderkaoui, Richard Hu, Jun Li, Stephen J. Pandol, Xiangsheng Fu, and Yuan-Ping Han. 2022. "Sequestration of Intestinal Acidic Toxins by Cationic Resin Attenuates Pancreatic Cancer Progression through Promoting Autophagic Flux for YAP Degradation" Cancers 14, no. 6: 1407. https://doi.org/10.3390/cancers14061407
APA StyleZhao, G., Zhang, T., Liu, W., Edderkaoui, M., Hu, R., Li, J., Pandol, S. J., Fu, X., & Han, Y.-P. (2022). Sequestration of Intestinal Acidic Toxins by Cationic Resin Attenuates Pancreatic Cancer Progression through Promoting Autophagic Flux for YAP Degradation. Cancers, 14(6), 1407. https://doi.org/10.3390/cancers14061407