
The Hodgkin Lymphoma Immune Microenvironment: Turning Bad News into Good


Abstract
:Simple Summary
Abstract
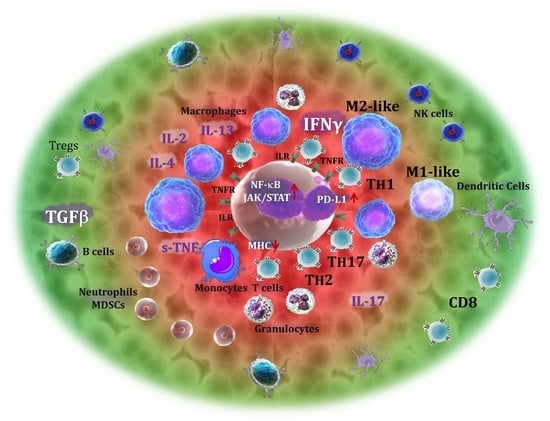
1. Introduction
2. T Cells
3. CD4+ T Cells
3.1. Th1 Cells
3.2. Th2 Cells
3.3. Th17 Cells
3.4. Regulatory T Cells
3.5. Tr1 Cells
3.6. T Follicular Helper Cells
4. CD8+ T Cells
- (1)
- (2)
- (3)
- Long distance between CD8+ T cells and tumor cells [3].
- (4)
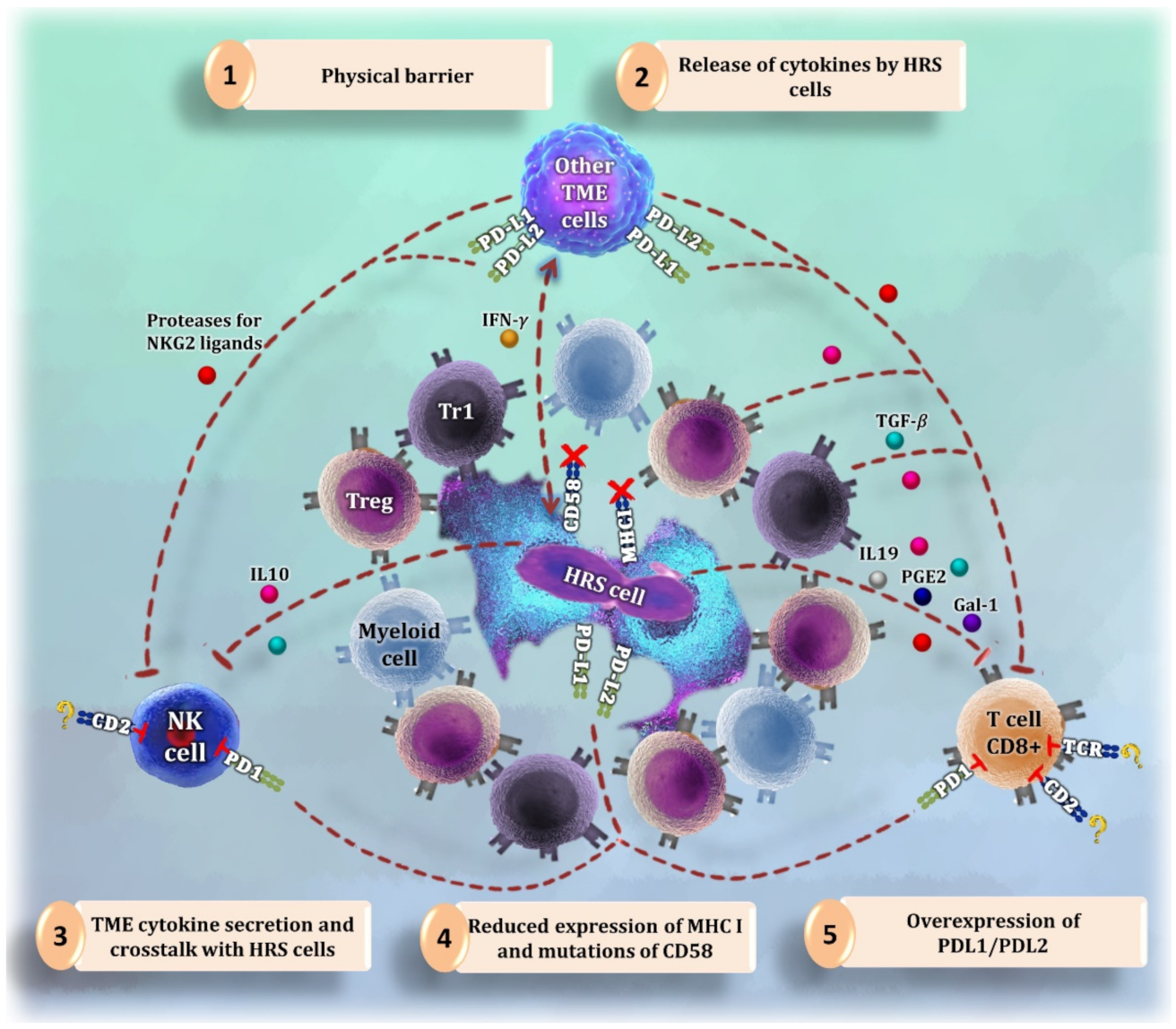
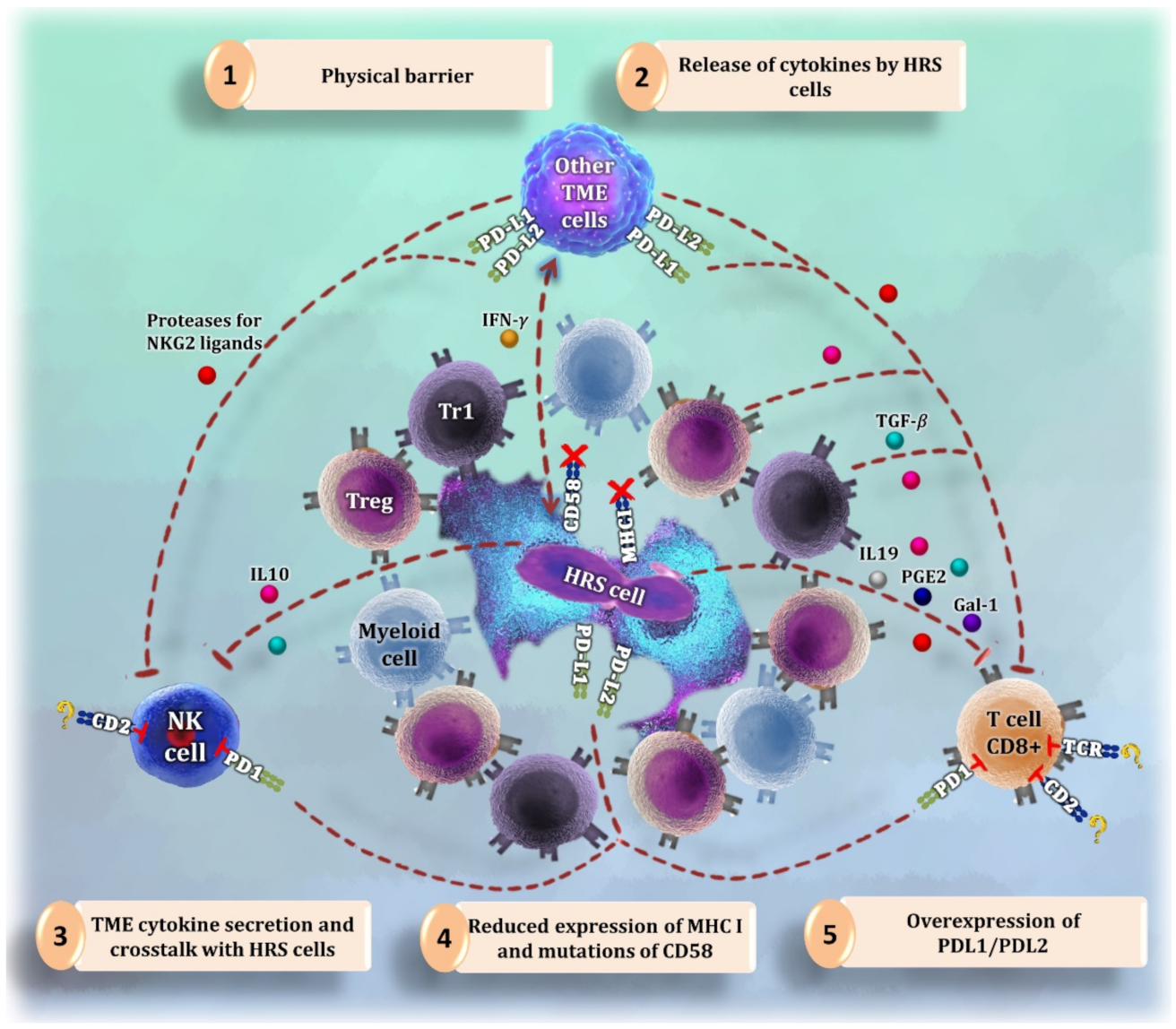
5. Natural Killer Cells
- (1)
- (2)
- A physical barrier of myeloid and T cells around HRS cells that blocks the access of NK cells to tumor cell shores [3].
- (3)
6. B Cells and Plasma Cells
7. Monocytes and Macrophages
7.1. Monocytes
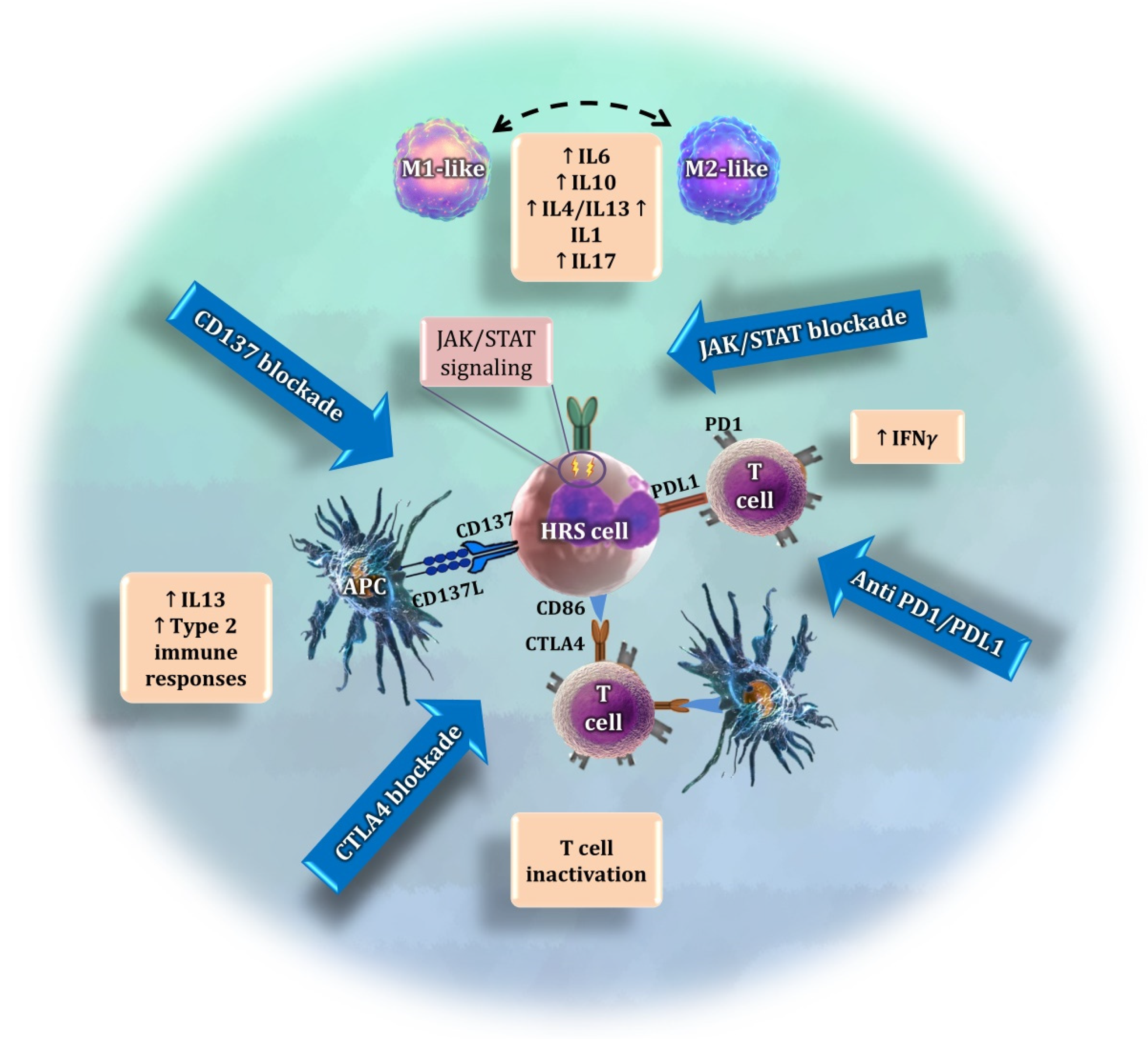
7.2. Macrophages
8. Myeloid Cells
8.1. Myeloid-Derived Suppressor Cells
8.2. Neutrophils
9. Mast Cells
10. Eosinophils
11. Dendritic Cells
11.1. Myeloid DCs
11.2. Plasmacytoid DCs
11.3. Follicular DCs (fDCs)
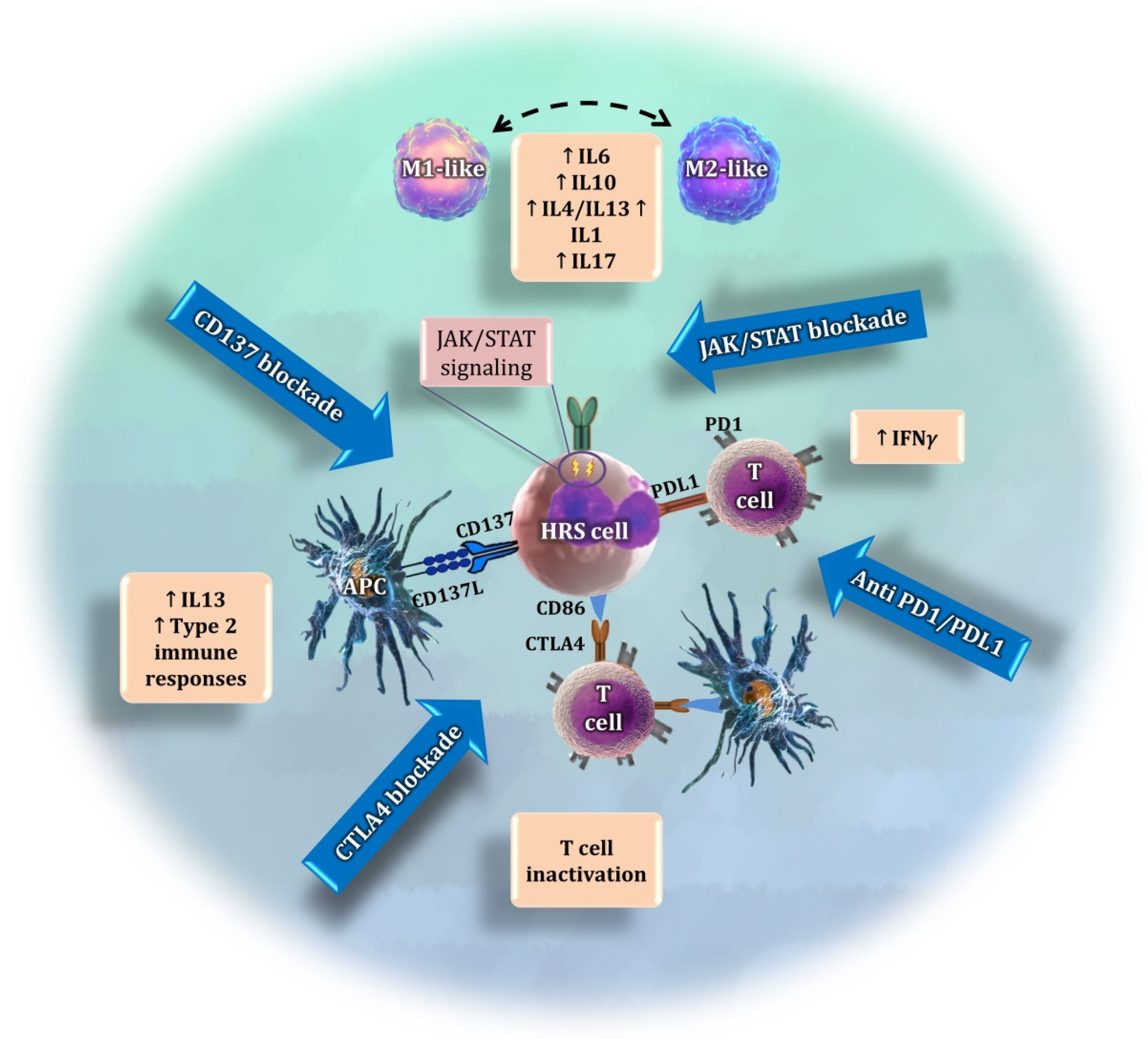
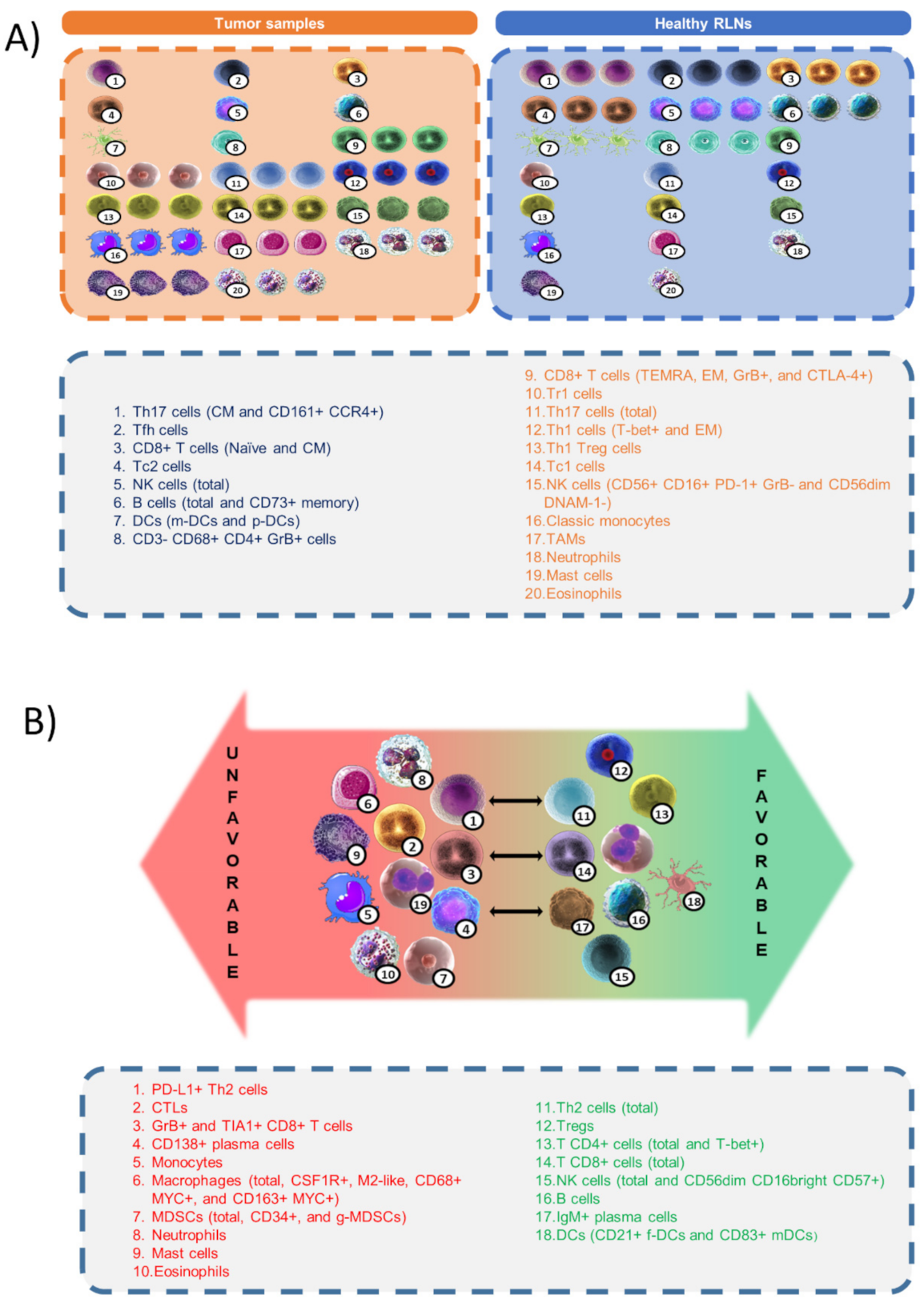
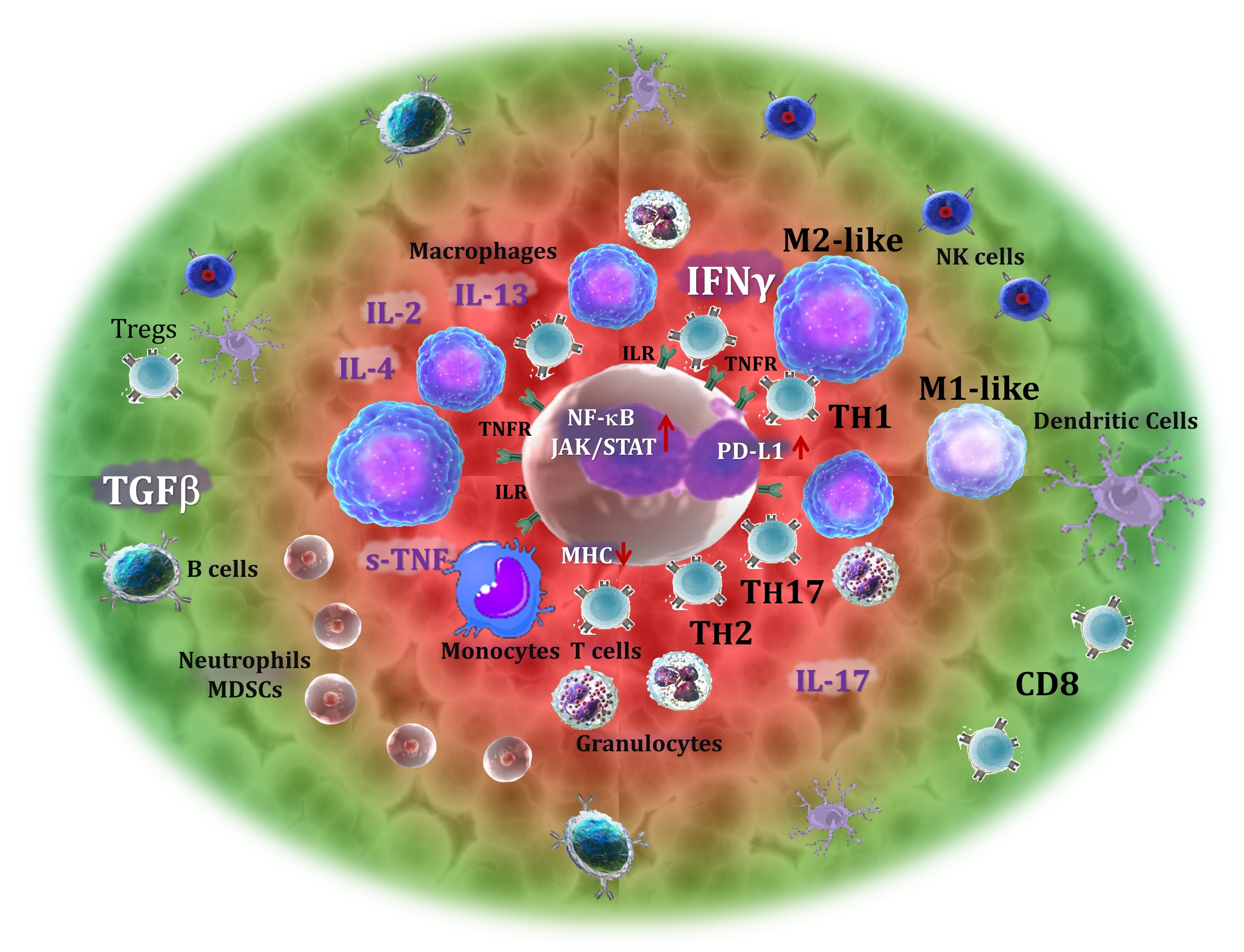
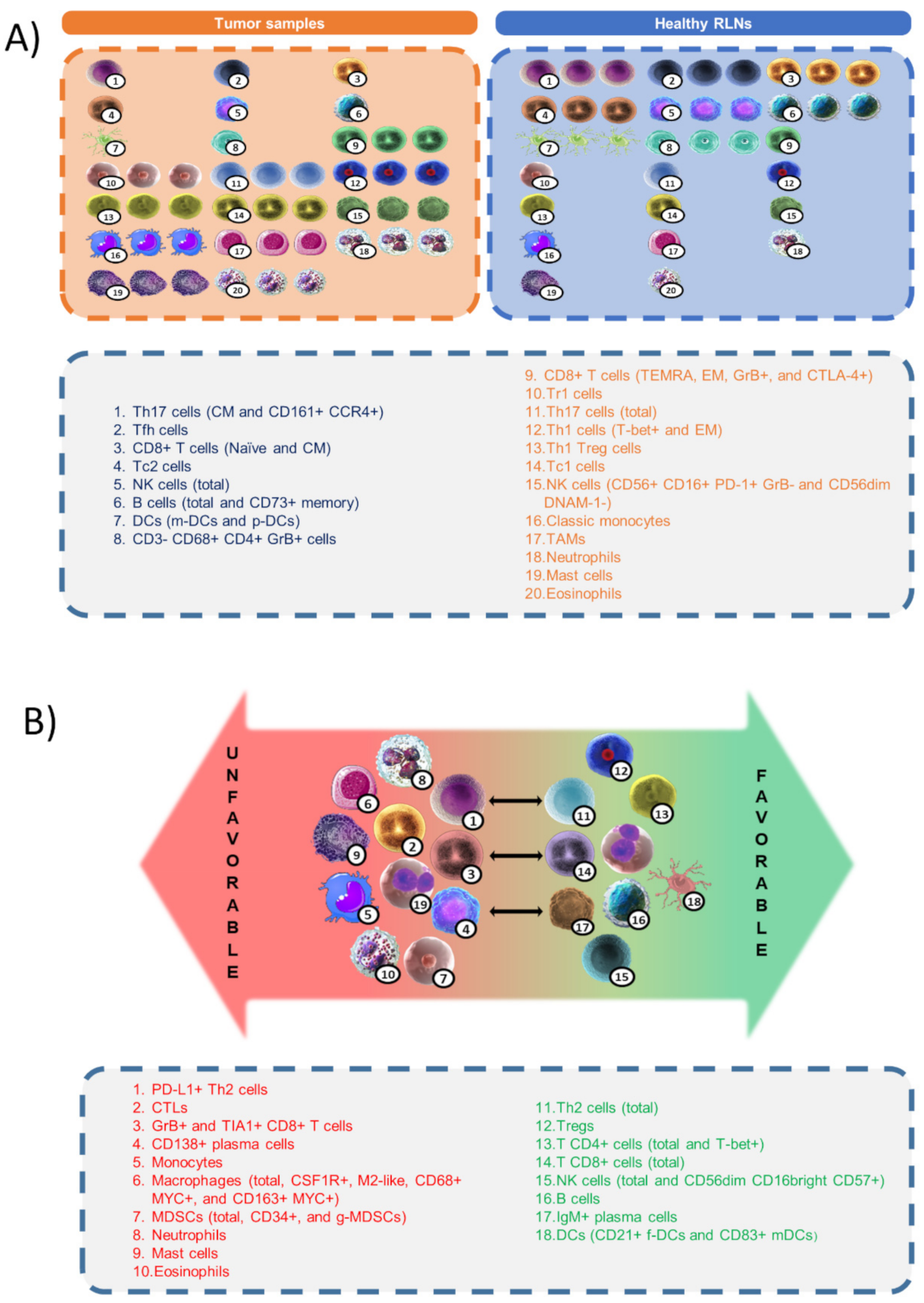
12. Discussion and Perspectives: Functional Signatures of the Tumor Microenvironment
13. Conclusions
- (1)
- (2)
- Tumor descriptors, such as gene expression analyses based on pathway-related genes, that could be upregulated or suppressed in the responsive versus non-responsive TME.
- (3)
- Phenotype-specific dysregulated descriptors of the chemokine milieu.
- (4)
- Relevant cell populations and functionalities (such as T cell and monocyte signatures).
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Opinto, G.; Agostinelli, C.; Ciavarella, S.; Guarini, A.; Maiorano, E.; Ingravallo, G. Hodgkin Lymphoma: A Special Micro-environment. J. Clin. Med. 2021, 10, 4665. [Google Scholar] [CrossRef] [PubMed]
- Greaves, P.; Clear, A.; Coutinho, R.; Wilson, A.; Matthews, J.; Owen, A.; Shanyinde, M.; Lister, T.A.; Calaminici, M.; Gribben, J.G. Expression of FOXP3, CD68, and CD20 at Diagnosis in the Microenvironment of Classical Hodgkin Lymphoma Is Predictive of Outcome. J. Clin. Oncol. 2013, 31, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrarini, I.; Rigo, A.; Visco, C.; Krampera, M.; Vinante, F. The Evolving Knowledge on T and NK Cells in Classic Hodgkin Lymphoma: Insights into Novel Subsets Populating the Immune Microenvironment. Cancers 2020, 12, 3757. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Aguilera, A.; Montalbán, C.; de la Cueva, P.; Sánchez-Verde, L.; Morente, M.M.; García-Cosío, M.; García-Laraña, J.; Bellas, C.; Provencio, M.; Romagosa, V.; et al. Tumor Microenvironment and Mitotic Checkpoint Are Key Factors in the Outcome of Classic Hodgkin Lymphoma. Blood 2006, 108, 662–668. [Google Scholar] [CrossRef]
- Steidl, C.; Connors, J.M.; Gascoyne, R.D. Molecular Pathogenesis of Hodgkin’s Lymphoma: Increasing Evidence of the Importance of the Microenvironment. J. Clin. Oncol. 2011, 29, 1812–1826. [Google Scholar] [CrossRef]
- Küppers, R.; Rajewsky, K.; Zhao, M.; Simons, G.; Laumann, R.; Fischer, R.; Hansmann, M.L. Hodgkin Disease: Hodgkin and Reed-Sternberg Cells Picked from Histological Sections Show Clonal Immunoglobulin Gene Rearrangements and Appear to Be Derived from B Cells at Various Stages of Development. Proc. Natl. Acad. Sci. USA 1994, 91, 10962–10966. [Google Scholar] [CrossRef] [Green Version]
- Bosch Príncep, R.; Lejeune, M.; Salvadó Usach, M.T.; Jaén Martínez, J.; Pons Ferré, L.E.; Alvaro Naranjo, T. Decreased Number of Granzyme B+ Activated CD8+ Cytotoxic T Lymphocytes in the Inflammatory Background of HIV-Associated Hodgkin’s Lymphoma. Ann. Hematol. 2005, 84, 661–666. [Google Scholar] [CrossRef]
- Ciavarella, S.; Vegliante, M.C.; Fabbri, M.; De Summa, S.; Melle, F.; Motta, G.; De Iuliis, V.; Opinto, G.; Enjuanes, A.; Rega, S.; et al. Dissection of DLBCL Microenvironment Provides a Gene Expression-Based Predictor of Survival Applicable to Forma-lin-Fixed Paraffin-Embedded Tissue. Ann. Oncol. 2018, 29, 2363–2370. [Google Scholar] [CrossRef] [Green Version]
- Henry, M.; Buck, S.; Savaşan, S. Flow Cytometry for Assessment of the Tumor Microenvironment in Pediatric Hodgkin Lymphoma. Pediatr. Blood Cancer 2018, 65, e27307. [Google Scholar] [CrossRef]
- Chiu, J.; Ernst, D.M.; Keating, A. Acquired Natural Killer Cell Dysfunction in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Front. Immunol. 2018, 9, 267. [Google Scholar] [CrossRef]
- Hess, J.L.; Bodis, S.; Pinkus, G.; Silver, B.; Mauch, P. Histopathologic Grading of Nodular Sclerosis Hodgkin’s Disease. Lack of Prognostic Significance in 254 Surgically Staged Patients. Cancer 1994, 74, 708–714. [Google Scholar] [CrossRef]
- Aldinucci, D.; Celegato, M.; Casagrande, N. Microenvironmental Interactions in Classical Hodgkin Lymphoma and Their Role in Promoting Tumor Growth Immune Escape and Drug Resistance. Cancer Lett. 2016, 380, 243–252. [Google Scholar] [CrossRef]
- Lin, Y.; Gustafson, M.P.; Bulur, P.A.; Gastineau, D.A.; Witzig, T.E.; Dietz, A.B. Immunosuppressive CD14+HLA-DRlow/− Monocytes in B-Cell Non-Hodgkin Lymphoma. Blood 2011, 117, 872–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Álvaro, T.; Lejeune, M.; Salvadó, M.T.; Bosch, R.; García, J.F.; Jaén, J.; Banham, A.H.; Roncador, G.; Montalbán, C.; Piris, M.A. Outcome in Hodgkin’s Lymphoma Can Be Predicted from the Presence of Accompanying Cytotoxic and Regulatory T Cells. Clin. Cancer. Res. 2005, 11, 1467–1473. [Google Scholar] [CrossRef] [Green Version]
- Oshi, M.; Tokumaru, Y.; Asaoka, M.; Yan, L.; Satyananda, V.; Matsuyama, R.; Matsuhashi, N.; Futamura, M.; Ishikawa, T.; Yoshida, K.; et al. M1 Macrophage and M1/M2 Ratio Defined by Transcriptomic Signatures Resemble Only Part of Their Conventional Clinical Characteristics in Breast Cancer. Sci. Rep. 2020, 10, 16554. [Google Scholar] [CrossRef] [PubMed]
- Honkanen, T.J.; Tikkanen, A.; Karihtala, P.; Mäkinen, M.; Väyrynen, J.P.; Koivunen, J.P. Prognostic and Predictive Role of Tumour-Associated Macrophages in HER2 Positive Breast Cancer. Sci. Rep. 2019, 9, 10961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minoda, Y.; Virshup, I.; Leal Rojas, I.; Haigh, O.; Wong, Y.; Miles, J.J.; Wells, C.A.; Radford, K.J. Human CD141+ Dendritic Cell and CD1c+ Dendritic Cell Undergo Concordant Early Genetic Programming after Activation in Humanized Mice In Vivo. Front. Immunol. 2017, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Mulder, T.A.; Wahlin, B.E.; Österborg, A.; Palma, M. Targeting the Immune Microenvironment in Lymphomas of B-Cell Origin: From Biology to Clinical Application. Cancers 2019, 11, 915. [Google Scholar] [CrossRef] [Green Version]
- Kline, J.; Godfrey, J.; Ansell, S.M. The Immune Landscape and Response to Immune Checkpoint Blockade Therapy in Lymphoma. Blood 2020, 135, 523–533. [Google Scholar] [CrossRef]
- Liu, Y.; Sattarzadeh, A.; Diepstra, A.; Visser, L.; van den Berg, A. The Microenvironment in Classical Hodgkin Lymphoma: An Actively Shaped and Essential Tumor Component. Semin. Cancer Biol. 2014, 24, 15–22. [Google Scholar] [CrossRef]
- Calabretta, E.; d’Amore, F.; Carlo-Stella, C. Immune and Inflammatory Cells of the Tumor Microenvironment Represent Novel Therapeutic Targets in Classical Hodgkin Lymphoma. Int. J. Mol. Sci. 2019, 20, 5503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, C.S.; Stuart, A.E. Reed-Sternberg/Lymphocyte Rosette: Lymphocyte Subpopulations as Defined by Monoclonal Antibodies. J. Clin. Pathol. 1984, 37, 767–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, P.; Clear, A.; Owen, A.; Iqbal, S.; Lee, A.; Matthews, J.; Wilson, A.; Calaminici, M.; Gribben, J.G. Defining Characteristics of Classical Hodgkin Lymphoma Microenvironment T-Helper Cells. Blood 2013, 122, 2856–2863. [Google Scholar] [CrossRef] [PubMed]
- Okoye, I.S.; Wilson, M.S. CD4+ T Helper 2 Cells-Microbial Triggers Differentiation Requirements and Effector Functions. Immunology 2011, 134, 368–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynn, T. A Type 2 Cytokines: Mechanisms and Therapeutic Strategies. Nat. Rev. Immunol. 2015, 15, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Oliphant, C.J.; Barlow, J.L.; McKenzie, A.N.J. Insights into the Initiation of Type 2 Immune Responses. Immunology 2011, 134, 378–385. [Google Scholar] [CrossRef]
- Alonso-Álvarez, S.; Vidriales, M.B.; Caballero, M.D.; Blanco, O.; Puig, N.; Martin, A.; Peñarrubia, M.J.; Zato, E.; Galende, J.; Bárez, A.; et al. The Number of Tumor Infiltrating T-Cell Subsets in Lymph Nodes from Patients with Hodgkin Lymphoma Is As-sociated with the Outcome after First Line ABVD Therapy. Leuk. Lymphoma 2017, 58, 1144–1152. [Google Scholar] [CrossRef]
- Aoki, T.; Chong, L.C.; Takata, K.; Milne, K.; Hav, M.; Colombo, A.; Chavez, E.A.; Nissen, M.; Wang, X.; Miyata-Takata, T.; et al. Single-Cell Transcriptome Analysis Reveals Disease-Defining T-Cell Subsets in the Tumor Microenvironment of Classic Hodgkin Lymphoma. Cancer Discov. 2020, 10, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Cader, F.Z.; Hu, X.; Goh, W.L.; Wienand, K.; Ouyang, J.; Mandato, E.; Redd, R.; Lawton, L.N.; Chen, P.H.; Weirather, J.L.; et al. A Peripheral Immune Signature of Responsiveness to PD-1 Blockade in Patients with Classical Hodgkin Lymphoma. Nat. Med. 2020, 26, 1468–1479. [Google Scholar] [CrossRef]
- Cader, F.Z.; Schackmann, R.C.J.; Hu, X.; Wienand, K.; Redd, R.; Chapuy, B.; Ouyang, J.; Paul, N.; Gjini, E.; Lipschitz, M.; et al. Mass Cytometry of Hodgkin Lymphoma Reveals a CD4+ Regulatory T-Cell–Rich and Exhausted T-Effector Microenvironment. Blood 2018, 132, 825–836. [Google Scholar] [CrossRef]
- Takeuchi, A.; Saito, T. CD4 CTL a Cytotoxic Subset of CD4+ T Cells Their Differentiation and Function. Front. Immunol. 2017, 8, 194. [Google Scholar] [CrossRef] [Green Version]
- Mowen, K.A.; Glimcher, L.H. Signaling Pathways in Th2 Development. Immunol. Rev. 2004, 202, 203–222. [Google Scholar] [CrossRef] [PubMed]
- Dehghani, M.; Ramzi, M.; Kalani, M.; Golmoghaddam, H.; Arandi, N. Higher Peripheral Blood IFN-γ-/IL-4+ Th2 Lym-phocytes Are Associated with Lower Rate of Relapse in Patients with Lymphoma. Immunol. Investig. 2020, 51, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, A.; Wali, S.; Nurieva, R. T Helper 2 and T Follicular Helper Cells: Regulation and Function of Interleukin-4. Cytokine Growth Factor Rev. 2016, 30, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schreck, S.; Friebel, D.; Buettner, M.; Distel, L.; Grabenbauer, G.; Young, L.S.; Niedobitek, G. Prognostic Impact of Tu-mour-Infiltrating Th2 and Regulatory T Cells in Classical Hodgkin Lymphoma. Hematol. Oncol. 2009, 27, 31–39. [Google Scholar] [CrossRef]
- Martín-Moreno, A.M.; Roncador, G.; Maestre, L.; Mata, E.; Jiménez, S.; Martínez-Torrecuadrada, J.L.; Reyes-García, A.I.; Rubio, C.; Tomás, J.F.; Estévez, M.; et al. CSF1R Protein Expression in Reactive Lymphoid Tissues and Lymphoma: Its Relevance in Classical Hodgkin Lymphoma. PLoS ONE 2015, 10, e0125203. [Google Scholar] [CrossRef]
- Dehghani, M.; Kalani, M.; Golmoghaddam, H.; Ramzi, M.; Arandi, N. Aberrant Peripheral Blood CD4+ CD25+ FOXP3+ Regulatory T Cells/T Helper-17 Number Is Associated with the Outcome of Patients with Lymphoma. Cancer Immunol. Immunother. 2020, 69, 1917–1928. [Google Scholar] [CrossRef]
- Asadzadeh, Z.; Mohammadi, H.; Safarzadeh, E.; Hemmatzadeh, M.; Mahdian-Shakib, A.; Jadidi-Niaragh, F.; Azizi, G.; Baradaran, B. The Paradox of Th17 Cell Functions in Tumor Immunity. Cell Immunol. 2017, 322, 15–25. [Google Scholar] [CrossRef]
- Sun, R.; Zheng, Z.; Wang, L.; Cheng, S.; Shi, Q.; Qu, B.; Fu, D.; Leboeuf, C.; Zhao, Y.; Ye, J.; et al. A Novel Prognostic Model Based on Four Circulating MiRNA in Diffuse Large B-Cell Lymphoma: Implications for the Roles of MDSC and Th17 Cells in Lymphoma Progression. Mol. Oncol. 2021, 15, 246–261. [Google Scholar] [CrossRef]
- Ferrarini, I.; Rigo, A.; Zamò, A.; Vinante, F. Classical Hodgkin Lymphoma Cells May Promote an IL-17-Enriched Micro-environment. Leuk. Lymphoma 2019, 60, 3395–3405. [Google Scholar] [CrossRef]
- Du, R.; Zhao, H.; Yan, F.; Li, H. IL-17+Foxp3+ T Cells: An Intermediate Differentiation Stage between Th17 Cells and Regulatory T Cells. J. Leukoc. Biol. 2014, 96, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Grygorowicz, M.A.; Biernacka, M.; Bujko, M.; Nowak, E.; Rymkiewicz, G.; Paszkiewicz-Kozik, E.; Borycka, I.S.; Bystydzienski, Z.; Walewski, J.; Markowicz, S. Human Regulatory T Cells Suppress Proliferation of B Lymphoma Cells. Leuk. Lymphoma 2016, 57, 1903–1920. [Google Scholar] [CrossRef] [PubMed]
- Veldman, J.; Visser, L.; Huberts-Kregel, M.; Muller, N.; Hepkema, B.; van den Berg, A.; Diepstra, A. Rosetting T Cells in Hodgkin Lymphoma Are Activated by Immunological Synapse Components HLA Class II and CD. Blood 2020, 136, 2437–2441. [Google Scholar] [CrossRef] [PubMed]
- Roemer, M.G.M.; Redd, R.A.; Cader, F.Z.; Pak, C.J.; Abdelrahman, S.; Ouyang, J.; Sasse, S.; Younes, A.; Fanale, M.; Santoro, A.; et al. Major Histocompatibility Complex Class II and Programmed Death Ligand 1 Expression Predict Outcome After Pro-grammed Death 1 Blockade in Classic Hodgkin Lymphoma. J. Clin. Oncol. 2016, 36, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. T Follicular Helper Cell Differentiation Function and Roles in Disease. Immunity 2014, 41, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Nurieva, R.I.; Chung, Y. Understanding the Development and Function of T Follicular Helper Cells. Cell Mol. Immunol. 2010, 7, 190–197. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.M.; Xu, Y.X.; Zhang, L.Y.; Sun, Y.; Wang, Z.Y.; Yuan, Y.Q.; Fu, J.X. The Role of Follicular T Helper Cells in Patients with Malignant Lymphoid Disease. Hematol. Amst. Neth. 2017, 22, 412–418. [Google Scholar] [CrossRef] [Green Version]
- Dzangué-Tchoupou, G.; Mariampillai, K.; Bolko, L.; Amelin, D.; Mauhin, W.; Corneau, A.; Blanc, C.; Allenbach, Y.; Benveniste, O. CD8+T-Bet+ Cells as a Predominant Biomarker for Inclusion Body Myositis. Autoimmun. Rev. 2019, 18, 325–333. [Google Scholar] [CrossRef]
- Frisullo, G.; Iorio, R.; Plantone, D.; Marti, A.; Nociti, V.; Patanella, A.K.; Batocchi, A.P. CD4+T-Bet+ CD4+pSTAT3+ and CD8+T-Bet+ T Cells Accumulate in Peripheral Blood during NZB Treatment. Mult. Scler. J. 2011, 17, 556–566. [Google Scholar] [CrossRef]
- Vari, F.; Arpon, D.; Keane, C.; Hertzberg, M.S.; Talaulikar, D.; Jain, S.; Cui, Q.; Han, E.; Tobin, J.; Bird, R.; et al. Immune Evasion via PD-1/PD-L1 on NK Cells and Monocyte/Macrophages Is More Prominent in Hodgkin Lymphoma than DLBCL. Blood 2018, 131, 1809–1819. [Google Scholar] [CrossRef] [Green Version]
- Upshaw, J.L.; Leibson, P.J. NKG2D-Mediated Activation of Cytotoxic Lymphocytes: Unique Signaling Pathways and Distinct Functional Outcomes. Semin. Immunol. 2006, 18, 167–175. [Google Scholar] [CrossRef] [PubMed]
- Lückel, C.; Picard, F.S.R.; Huber, M. Tc17 Biology and Function: Novel Concepts. Eur. J. Immunol. 2020, 50, 1257–1267. [Google Scholar] [CrossRef] [PubMed]
- Novosad, O.; Gorbach, O.; Skachkova, O.; Khranovska, N.; Kryachok, I. Role of Circulating Myeloid-Derived Suppressor Cell (MDSC) in Hodgkin Lymphoma (HL) Progression: Updated Prospective Study. Blood 2020, 136, 31. [Google Scholar] [CrossRef]
- Challa-Malladi, M.; Lieu, Y.K.; Califano, O.; Holmes, A.; Bhagat, G.; Murty, V.V.; Dominguez-Sola, D.; Pasqualucci, L.; Dalla-Favera, R. Combined Genetic Inactivation of Beta2-Microglobulin and CD58 Reveals Frequent Escape from Immune Recognition in Diffuse Large B-Cell Lymphoma. Cancer Cell. 2011, 20, 728–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.; Schneider, S.; Zühlke-Jenisch, R.; Klapper, W.; Sundström, C.; Hartmann, S.; Hansmann, M.L.; Siebert, R.; Küppers, R.; Giefing, M. Alterations of the CD58 Gene in Classical Hodgkin Lymphoma. Genes Chromosomes Cancer 2015, 54, 638–645. [Google Scholar] [CrossRef]
- Abdul Razak, F.R.; Diepstra, A.; Visser, L.; van den Berg, A. CD58 Mutations Are Common in Hodgkin Lymphoma Cell Lines and Loss of CD58 Expression in Tumor Cells Occurs in Hodgkin Lymphoma Patients Who Relapse. Genes Immun. 2016, 17, 363–366. [Google Scholar] [CrossRef]
- Gholiha, A.R.; Hollander, P.; Hedstrom, G.; Sundstrom, C.; Molin, D.; Smedby, K.E.; Hjalgrim, H.; Glimelius, I.; Amini, R.M.; Enblad, G. High Tumour Plasma Cell Infiltration Reflects an Important Microenvironmental Component in Classic Hodgkin Lymphoma Linked to Presence of B-Symptoms. Br. J. Haematol. 2019, 184, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Steidl, C.; Lee, T.; Shah, S.P.; Farinha, P.; Han, G.; Nayar, T.; Delaney, A.; Jones, S.J.; Iqbal, J.; Weisenburger, D.D.; et al. Tu-mor-Associated Macrophages and Survival in Classic Hodgkin’s Lymphoma. N. Engl. J. Med. 2010, 362, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Chetaille, B.; Bertucci, F.; Finetti, P.; Esterni, B.; Stamatoullas, A.; Picquenot, J.M.; Copin, M.C.; Morschhauser, F.; Casasnovas, O.; Petrella, T.; et al. Molecular Profiling of Classical Hodgkin Lymphoma Tissues Uncovers Variations in the Tumor Microenvi-ronment and Correlations with EBV Infection and Outcome. Blood 2009, 113, 2765–3775. [Google Scholar] [CrossRef] [Green Version]
- Calame, K.L.; Lin, K.I.; Tunyaplin, C. Regulatory Mechanisms That Determine the Development and Function of Plasma Cells. Annu. Rev. Immunol. 2003, 21, 205–230. [Google Scholar] [CrossRef]
- Tudor, C.S.; Distel, L.V.; Eckhardt, J.; Hartmann, A.; Niedobitek, G.; Buettner, M. B Cells in Classical Hodgkin Lymphoma Are Important Actors Rather than Bystanders in the Local Immune Reaction. Hum. Pathol. 2013, 44, 2475–2486. [Google Scholar] [CrossRef] [PubMed]
- McKee, S.J.; Tuong, Z.K.; Kobayashi, T.; Doff, B.L.; Soon, M.S.; Nissen, M.; Lam, P.Y.; Keane, C.; Vari, F.; Moi, D.; et al. B Cell Lymphoma Progression Promotes the Accumulation of Circulating Ly6Clo Monocytes with Immunosuppressive Activity. Oncoimmunology 2017, 7, e1393599. [Google Scholar] [CrossRef]
- Marzaioli, V.; Canavan, M.; Floudas, A.; Wade, S.C.; Low, C.; Veale, D.J.; Fearon, U. Monocyte-Derived Dendritic Cell Dif-ferentiation in Inflammatory Arthritis Is Regulated by the JAK/STAT Axis via NADPH Oxidase Regulation. Front. Immunol. 2020, 11, 1406. [Google Scholar] [CrossRef] [PubMed]
- Tudor, C.S.; Bruns, H.; Daniel, C.; Distel, L.V.; Hartmann, A.; Gerbitz, A.; Buettner, M.J. Macrophages and Dendritic Cells as Actors in the Immune Reaction of Classical Hodgkin Lymphoma. PLoS ONE 2014, 9, e114345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parisi, L.; Gini, E.; Baci, D.; Tremolati, M.; Fanuli, M.; Bassani, B.; Farronato, G.; Bruno, A.; Mortara, L. Macrophage Polarization in Chronic Inflammatory Diseases: Killers or Builders? J. Immunol. Res. 2018, 2018, e8917804. [Google Scholar] [CrossRef]
- Gotti, M.; Nicola, M.; Lucioni, M.; Fiaccadori, V.; Ferretti, V.; Sciarra, R.; Costanza, M.; Bono, E.; Molo, S.; Maffi, A.; et al. Independent Prognostic Impact of Tumour-Infiltrating Macrophages in Early-Stage Hodgkin’s Lymphoma. Hematol. Oncol. 2017, 35, 296–302. [Google Scholar] [CrossRef]
- Azambuja, D.; Natkunam, Y.; Biasoli, I.; Lossos, I.S.; Anderson, M.W.; Morais, J.C.; Spector, N. Lack of Association of Tu-mor-Associated Macrophages with Clinical Outcome in Patients with Classical Hodgkin’s Lymphoma. Ann. Oncol. 2012, 23, 736–742. [Google Scholar] [CrossRef]
- Cencini, E.; Fabbri, A.; Rigacci, L.; Lazzi, S.; Gini, G.; Cox, M.C.; Mancuso, S.; Abruzzese, E.; Kovalchuk, S.; Goteri, G.; et al. Evaluation of the Prognostic Role of Tumour-Associated Macrophages in Newly Diagnosed Classical Hodgkin Lymphoma and Cor-relation with Early FDG-PET Assessment. Hematol. Oncol. 2017, 35, 69–78. [Google Scholar] [CrossRef]
- Werner, L.; Dreyer, J.H.; Hartmann, D.; Barros, M.H.M.; Büttner-Herold, M.; Grittner, U.; Niedobitek, G. Tumor-Associated Macrophages in Classical Hodgkin Lymphoma: Hormetic Relationship to Outcome. Sci. Rep. 2020, 10, 9410. [Google Scholar] [CrossRef]
- Ino, Y.; Yamazaki-Itoh, R.; Shimada, K.; Iwasaki, M.; Kosuge, T.; Kanai, Y.; Hiraoka, N. Immune Cell Infiltration as an Indicator of the Immune Microenvironment of Pancreatic Cancer. Br. J. Cancer 2013, 108, 914–923. [Google Scholar] [CrossRef]
- Balermpas, P.; Rödel, F.; Liberz, R.; Oppermann, J.; Wagenblast, J.; Ghanaati, S.; Harter, P.N.; Mittelbronn, M.; Weiss, C.; Rödel, C.; et al. Head and Neck Cancer Relapse after Chemoradiotherapy Correlates with CD163+ Macrophages in Primary Tumour and CD11b+ Myeloid Cells in Recurrences. Br. J. Cancer 2014, 111, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Elliott, L.A.; Doherty, G.A.; Sheahan, K.; Ryan, E.J. Human Tumor-Infiltrating Myeloid Cells: Phenotypic and Functional Diversity. Front. Immunol. 2017, 8, 86. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Arlt, A.; von Bonin, F.; Rehberg, T.; Perez-Rubio, P.; Engelmann, J.C.; Limm, K.; Reinke, S.; Dullin, C.; Sun, X.; Specht, R.; et al. High CD206 Levels in Hodgkin Lymphoma-Educated Macrophages Are Linked to Matrix-Remodeling and Lymphoma Dissem-ination. Mol. Oncol. 2020, 14, 571–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, H.; Hang, J.J.; Han, T.; Zhuo, M.; Jiao, F.; Wang, L.W. The M2 Phenotype of Tumor-Associated Macrophages in the Stroma Confers a Poor Prognosis in Pancreatic Cancer. Tumour Biol. J. Int. Soc. Oncodev. Biol. Med. 2016, 37, 8657–8664. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000 Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- De Kleer, I.; Willems, F.; Lambrecht, B.; Goriely, S. Ontogeny of Myeloid Cells. Front. Immunol. 2014, 5, 423. [Google Scholar] [CrossRef] [Green Version]
- Romano, A.; Parrinello, N.L.; Vetro, C.; Forte, S.; Chiarenza, A.; Figuera, A.; Motta, G.; Palumbo, G.A.; Ippolito, M.; Consoli, U.; et al. Circulating Myeloid-Derived Suppressor Cells Correlate with Clinical Outcome in Hodgkin Lymphoma Patients Treated up-Front with a Risk-Adapted Strategy. Br. J. Haematol. 2015, 168, 689–700. [Google Scholar] [CrossRef]
- Marini, O.; Spina, C.; Mimiola, E.; Cassaro, A.; Malerba, G.; Todeschini, G.; Perbellini, O.; Scupoli, M.; Carli, G.; Facchinelli, D.; et al. Identification of Granulocytic Myeloid-Derived Suppressor Cells (G-MDSCs) in the Peripheral Blood of Hodgkin and Non-Hodgkin Lymphoma Patients. Oncotarget 2016, 7, 27676–27688. [Google Scholar] [CrossRef] [Green Version]
- Vardhana, S.; Younes, A. The Immune Microenvironment in Hodgkin Lymphoma: T Cells B Cells and Immune Checkpoints. Haematologica 2016, 101, 794–802. [Google Scholar] [CrossRef] [Green Version]
- Au, Q.; Fang, J.; Juncker-Jensen, A.; Kuo, J.; Leones, E.; Sahafi, F.; Padmanabhan, R.; Hoe, N.; William, J. Characterization of Myeloid-Derived Suppressor Cells and Tumor Associated Macrophages Using MultiOmyxTM Hyperplexed Immunofluo-rescence Assay in Hodgkin Lymphoma. Blood 2018, 132, 4135. [Google Scholar] [CrossRef]
- Koh, Y.W.; Kang, H.J.; Park, C.; Yoon, D.H.; Kim, S.; Suh, C.; Kim, J.E.; Kim, C.W.; Huh, J. Prognostic Significance of the Ratio of Absolute Neutrophil Count to Absolute Lymphocyte Count in Classic Hodgkin Lymphoma. Am. J. Clin. Pathol. 2012, 138, 846–854. [Google Scholar] [CrossRef] [Green Version]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Marone, G.; Iannone, R.; Marone, G.; Granata, F. Are Mast Cells MASTers in Cancer? Front. Immunol. 2017, 8, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, M.D.; Kamper, P.; Nielsen, P.S.; Bendix, K.; Riber-Hansen, R.; Steiniche, T.; Hamilton-Dutoit, S.; Clausen, M.; d’Amore, F. Tumour-Associated Mast Cells in Classical Hodgkin’s Lymphoma: Correlation with Histological Subtype Other Tu-mour-Infiltrating Inflammatory Cell Subsets and Outcome. Eur. J. Haematol. 2016, 96, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Keresztes, K.; Szollosi, Z.; Simon, Z.; Tarkanyi, I.; Nemes, Z.; Illes, A. Retrospective Analysis of the Prognostic Role of Tissue Eosinophil and Mast Cells in Hodgkin’s Lymphoma. Pathol. Oncol. Res. 2007, 13, 237–242. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Lucarini, V.; Marone, G.; Mattei, F.; Marone, G.; Schiavoni, G. Eosinophils: The Unsung Heroes in Cancer? OncoImmunology 2018, 7, e1393134. [Google Scholar] [CrossRef] [Green Version]
- Von Wasielewski, R.; Seth, S.; Franklin, J.; Fischer, R.; Hübner, K.; Hansmann, M.L.; Diehl, V.; Georgii, A. Tissue Eosinophilia Correlates Strongly with Poor Prognosis in Nodular Sclerosing Hodgkin’s Disease Allowing for Known Prognostic Factors. Blood 2000, 95, 1207–1213. [Google Scholar] [CrossRef]
- Colby, T.V.; Hoppe, R.T.; Warnke, R.A. Hodgkin’s Disease: A Clinicopathologic Study of 659 Cases. Cancer 1982, 49, 1848–1858. [Google Scholar] [CrossRef]
- Hart, D.N.J. Dendritic Cells: Unique Leukocyte Populations Which Control the Primary Immune Response. Blood 1997, 90, 3245–3287. [Google Scholar] [CrossRef]
- Galati, D.; Zanotta, S.; Corazzelli, G.; Bruzzese, D.; Capobianco, G.; Morelli, E.; Arcamone, M.; De Filippi, R.; Pinto, A. Circulating Dendritic Cells Deficiencies as a New Biomarker in Classical Hodgkin Lymphoma. Br. J. Haematol. 2019, 184, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jongbloed, S.L.; Kassianos, A.J.; McDonald, K.J.; Clark, G.J.; Ju, X.; Angel, C.E.; Chen, C.J.J.; Dunbar, P.R.; Wadley, R.B.; Jeet, V.; et al. Human CD141+ (BDCA-3)+ Dendritic Cells (DCs) Represent a Unique Myeloid DC Subset That Cross-Presents Necrotic Cell Antigens. J. Exp. Med. 2010, 207, 1247–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shodell, M.; Kempin, S.; Siegal, F.P. Plasmacytoid Dendritic Cell and CD4 + T Cell Deficiencies in Untreated Hodgkin Disease: Implications for Susceptibility to Opportunistic Infections. Leuk. Lymphoma 2014, 55, 2656–2657. [Google Scholar] [CrossRef] [PubMed]
- Aldinucci, D.; Poletto, D.; Gloghini, A.; Nanni, P.; Degan, M.; Perin, T.; Ceolin, P.; Rossi, F.M.; Gattei, V.; Carbone, A.; et al. Ex-pression of Functional Interleukin-3 Receptors on Hodgkin and Reed-Sternberg Cells. Am. J. Pathol. 2002, 160, 585–596. [Google Scholar] [CrossRef] [Green Version]
- Diefenbach, C.S.; Sabado, R.; Brooks, C.L.; Baquero-Buitrago, J.; Cruz, C.; Vengco, I.; Montanari, F.; Marchi, E.; Scotto, L.; Cirrito, T.P.; et al. Hodgkin’s Lymphoma Cell Lines Have up-Regulated IL-3 Receptor α (IL-3Rα) Expression and Are Sensitive to SL-401 An IL-3Rα Targeted Drug. Blood 2011, 118, 3737. [Google Scholar] [CrossRef]
- Ruella, M.; Klichinsky, M.; Kenderian, S.S.; Shestova, O.; Ziober, A.; Kraft, D.O.; Feldman, M.; Wasik, M.A.; June, C.H.; Gill, S. Overcoming the Immunosuppressive Tumor Microenvironment of Hodgkin Lymphoma Using Chimeric Antigen Receptor T Cells. Cancer Discov. 2017, 7, 1154–1167. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 Blockade with Nivolumab in Relapsed or Refractory Hodgkin’s Lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.S.; Weirather, J.L.; Lipschitz, M.; Lako, A.; Chen, P.H.; Griffin, G.K.; Armand, P.; Shipp, M.A.; Rodig, S.J. The Microenvi-ronmental Niche in Classic Hodgkin Lymphoma Is Enriched for CTLA-4-Positive T Cells That Are PD-1-Negative. Blood 2019, 134, 2059–2069. [Google Scholar] [CrossRef]
- Kim, H.; Lee, J.E.; Hong, S.H.; Lee MAh Kang, J.H.; Kim, I.H. The Effect of Antibiotics on the Clinical Outcomes of Patients with Solid Cancers Undergoing Immune Checkpoint Inhibitor Treatment: A Retrospective Study. BMC Cancer 2019, 19, 1100. [Google Scholar] [CrossRef]
- Neste, E.V.D.; André, M.; Gastinne, T.; Stamatoullas, A.; Haioun, C.; Belhabri, A.; Reman, O.; Casasnovas, O.; Ghesquieres, H.; Verhoef, G.; et al. A Phase II Study of the Oral JAK1/JAK2 Inhibitor Ruxolitinib in Advanced Relapsed/Refractory Hodgkin Lymphoma. Haematologica 2018, 103, 840–848. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative Analysis Reveals Selective 9p24.1 Amplification Increased PD-1 Ligand Expression and Further Induction via JAK2 in Nodular Sclerosing Hodgkin Lymphoma and Primary Mediastinal Large B-Cell Lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiacci, E.; Ladewig, E.; Schiavoni, G.; Penson, A.; Fortini, E.; Pettirossi, V.; Wang, Y.; Rosseto, A.; Venanzi, A.; Vlasevska, S.; et al. Pervasive Mutations of JAK-STAT Pathway Genes in Classical Hodgkin Lymphoma. Blood 2018, 131, 2454–2465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mata, E.; Fernández, S.; Astudillo, A.; Fernández, R.; García-Cosío, M.; Sánchez-Beato, M.; Provencio, M.; Estévez, M.; Montalbán, C.; Piris, M.A.; et al. Genomic Analyses of Microdissected Hodgkin and Reed-Sternberg Cells: Mutations in Epigenetic Regulators and P53 Are Frequent in Refractory Classic Hodgkin Lymphoma. Blood Cancer J. 2019, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Pardoll, D.; Jove, R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-García, L.; Higueras, M.Á.; Herranz, S.; Hernández-López, M.; Luque, A.; de las Heras, B.; Hortelano, S. A Hispanolone-Derived Diterpenoid Inhibits M2-Macrophage Polarization in Vitro via JAK/STAT and Attenuates Chitin Induced Inflammation in Vivo. Biochem. Pharmacol. 2018, 154, 373–383. [Google Scholar] [CrossRef]
- Irey, E.A.; Lassiter, C.M.; Brady, N.J.; Chuntova, P.; Wang, Y.; Knutson, T.P.; Henzler, C.; Chaffee, T.S.; Vogel, R.I.; Nelson, A.C.; et al. JAK/STAT Inhibition in Macrophages Promotes Therapeutic Resistance by Inducing Expression of Protumorigenic Factors. Proc. Natl. Acad. Sci. USA 2019, 116, 12442–12451. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Huang, C.; Lin, Z.; Zhan, S.; Kong, L.; Fang, C.; Li, J. Macrophage Polarization and Function with Emphasis on the Evolving Roles of Coordinated Regulation of Cellular Signaling Pathways. Cell. Signal. 2014, 26, 192–197. [Google Scholar] [CrossRef]
- Ho, W.T.; Pang, W.L.; Chong, S.M.; Castella, A.; Al-Salam, S.; Tan, T.E.; Moh, M.C.; Koh, L.K.; Gan, S.U.; Cheng, C.K.; et al. Expression of CD137 on Hodgkin and Reed–Sternberg Cells Inhibits T-Cell Activation by Eliminating CD137 Ligand Expression. Cancer Res. 2013, 73, 652–661. [Google Scholar] [CrossRef] [Green Version]
- Anderson, M.W.; Zhao, S.; Freud, A.G.; Czerwinski, D.K.; Kohrt, H.; Alizadeh, A.A.; Houot, R.; Azambuja, D.; Biasoli, I.; Morais, J.C.; et al. CD137 Is Expressed in Follicular Dendritic Cell Tumors and in Classical Hodgkin and T-Cell Lymphomas: Diagnostic and Therapeutic Implications. Am. J. Pathol. 2012, 181, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Rajendran, S.; Ho, W.T.; Schwarz, H. CD137 Signaling in Hodgkin and Reed-Sternberg Cell Lines Induces IL-13 Secretion Immune Deviation and Enhanced Growth. Oncoimmunology 2016, 5, e1160188. [Google Scholar] [CrossRef] [Green Version]
- Cabannes, E.; Khan, G.; Aillet, F.; Jarrett, R.F.; Hay, R.T. Mutations in the IkBa Gene in Hodgkin’s Disease Suggest a Tumour Suppressor Role for IκBα. Oncogene 1999, 18, 3063–3070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jungnickel, B.; Staratschek-Jox, A.; Bräuninger, A.; Spieker, T.; Wolf, J.; Diehl, V.; Hansmann, M.L.; Rajewsky, K.; Küppers, R. Clonal Deleterious Mutations in the IkappaBalpha Gene in the Malignant Cells in Hodgkin’s Lymphoma. J. Exp. Med. 2000, 191, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Hansmann, M.L.; Bohle, V.; Martin-Subero, J.I.; Hartmann, S.; Mechtersheimer, G.; Klapper, W.; Vater, I.; Giefing, M.; Gesk, S.; et al. TNFAIP3 (A20) Is a Tumor Suppressor Gene in Hodgkin Lymphoma and Primary Mediastinal B Cell Lymphoma. J. Exp. Med. 2009, 206, 981–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Sanada, M.; Kato, I.; Sato, Y.; Takita, J.; Takeuchi, K.; Niwa, A.; Chen, Y.; Nakazaki, K.; Nomoto, J.; et al. Frequent Inac-tivation of A20 in B-Cell Lymphomas. Nature 2009, 459, 712–716. [Google Scholar] [CrossRef]
- Weniger, M.A.; Melzner, I.; Menz, C.K.; Wegener, S.; Bucur, A.J.; Dorsch, K.; Mattfeldt, T.; Barth, T.F.E.; Möller, P. Mutations of the Tumor Suppressor Gene SOCS-1 in Classical Hodgkin Lymphoma Are Frequent and Associated with Nuclear Phos-pho-STAT5 Accumulation. Oncogene 2006, 25, 2679–2684. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Population | Outcome Association | Details | References | ||
|---|---|---|---|---|---|
| T CD4+ cells | General | Favorable | Better outcome | [23,27,29] | |
| T-bet+ | Favorable | Better outcome and response to anti-PD-L1 immunotherapy | [23,27,29] | ||
| Th2 | General | Favorable | Improved disease-free and event-free survival | [35] | |
| PD-L1+ | Unfavorable | Worse outcome and shorter survival | [36] | ||
| Tregs | CD25+ and FOXP3+ | Favorable | Positively associated with survival rate | [2,5,23,37,42] | |
| FOXP3+ GrB+ | Favorable | Improved survival | [5,14] | ||
| T CD8+ cells | General | Favorable | Better outcome, particularly in the advanced-disease group | [9,27] | |
| Cytotoxic (CTLs) | Unfavorable | Worse clinical outcome | [5,7,14,36] | ||
| GrB+ TIA1+ | Unfavorable | Worse prognosis | [5,7,14,36] | ||
| NK cells | General | Favorable | Infiltration and activation confer better prognosis | [10] | |
| CD56dim CD16bright CD57+ | Favorable | Better prognostic factors | [10] | ||
| B cells | General | Favorable | Better outcome | [2,5,21,58] | |
| CD138+ Plasma cells | Unfavorable | Associated with advanced stage and poor survival | [57] | ||
| IgM+ Plasma cells | Favorable | Better survival | [2,61] | ||
| Monocytes | General | Unfavorable | Poor prognosis for frequency, gene signature, and markers | [5,12,28,58,62] | |
| Macrophages | General | Unfavorable | Poor outcomes Intermediate numbers show best outcomes | [2,5,9,27,58,66] [69] | |
| CSF1R+ | Unfavorable | Shorter survival | [36] | ||
| M2-like (CD68+ CD163+) | Unfavorable | Poor clinical outcomes | [20,21,36,50] | ||
| CD68+/CD163+ MYC+ | Unfavorable | Worse outcome | [69] | ||
| MDSCs (CD11b+ CD33+ HL-DR−) | General | Unfavorable | Correlated with disease aggressiveness and poor prognosis | [21] | |
| CD34+ | Unfavorable | Poor outcomes | [79] | ||
| g-MDSCs (CD14− or CD66+ CD33dim, or CD14− CD15+) | Unfavorable | Worse prognosis | [53,79,82] | ||
| Neutrophils | General | Unfavorable | Poor prognosis and tumor recurrence | [21,72,83] | |
| Mast cells | General | Unfavorable | Poor prognosis and fibrosis promotion | [5,12,20,21] | |
| Eosinophils | General | Unfavorable | Inferior prognosis | [5,12,21,88] | |
| Dendritic cells | CD21+ Follicular DCs | Favorable | Better outcome | [5,61] | |
| CD83+ mDCs | Favorable | Improved outcome | [64] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Menéndez, V.; Solórzano, J.L.; Fernández, S.; Montalbán, C.; García, J.F. The Hodgkin Lymphoma Immune Microenvironment: Turning Bad News into Good. Cancers 2022, 14, 1360. https://doi.org/10.3390/cancers14051360
Menéndez V, Solórzano JL, Fernández S, Montalbán C, García JF. The Hodgkin Lymphoma Immune Microenvironment: Turning Bad News into Good. Cancers. 2022; 14(5):1360. https://doi.org/10.3390/cancers14051360
Chicago/Turabian StyleMenéndez, Victoria, José L. Solórzano, Sara Fernández, Carlos Montalbán, and Juan F. García. 2022. "The Hodgkin Lymphoma Immune Microenvironment: Turning Bad News into Good" Cancers 14, no. 5: 1360. https://doi.org/10.3390/cancers14051360
APA StyleMenéndez, V., Solórzano, J. L., Fernández, S., Montalbán, C., & García, J. F. (2022). The Hodgkin Lymphoma Immune Microenvironment: Turning Bad News into Good. Cancers, 14(5), 1360. https://doi.org/10.3390/cancers14051360