
Innate and Adaptive Immunopathogeneses in Viral Hepatitis; Crucial Determinants of Hepatocellular Carcinoma

 ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Epidemiology
2.1. HBV Epidemiology and Comorbidity
2.2. HCV Epidemiology and Comorbidity
3. Structure, Entry and Replication of Hepatitis B and C Viruses
3.1. HBV Mode of Entry and Replication
3.2. HCV Mode of Entry and Replication
4. Host Immune Response to Hepatitis B Infection
4.1. Acute Hepatitis B Infection
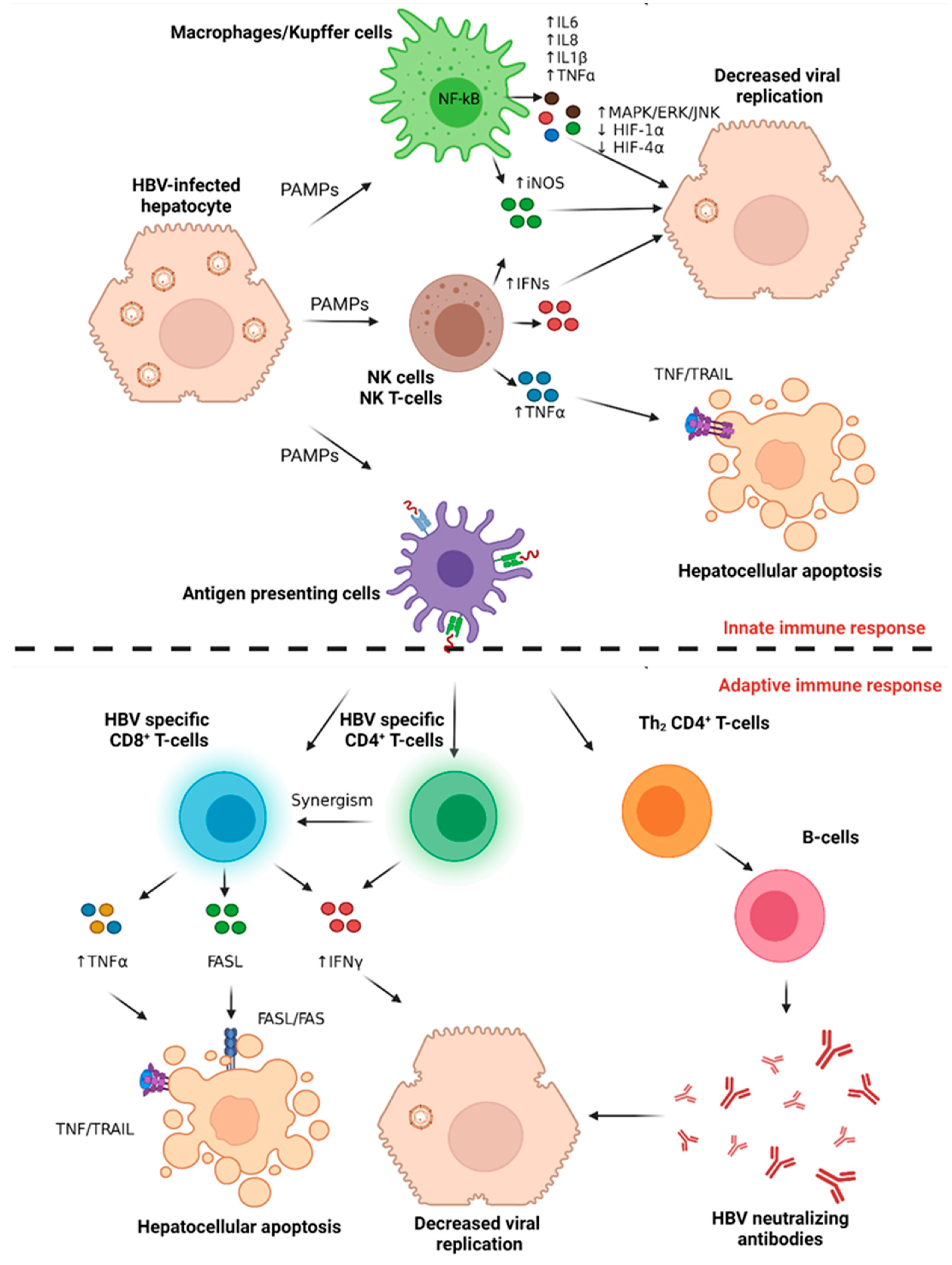
4.1.1. The Host Innate Immune Response to Acute HBV Infection
4.1.2. The Host Adaptive Immune Response to Acute HBV Infection
4.2. Chronic Hepatitis B Infection
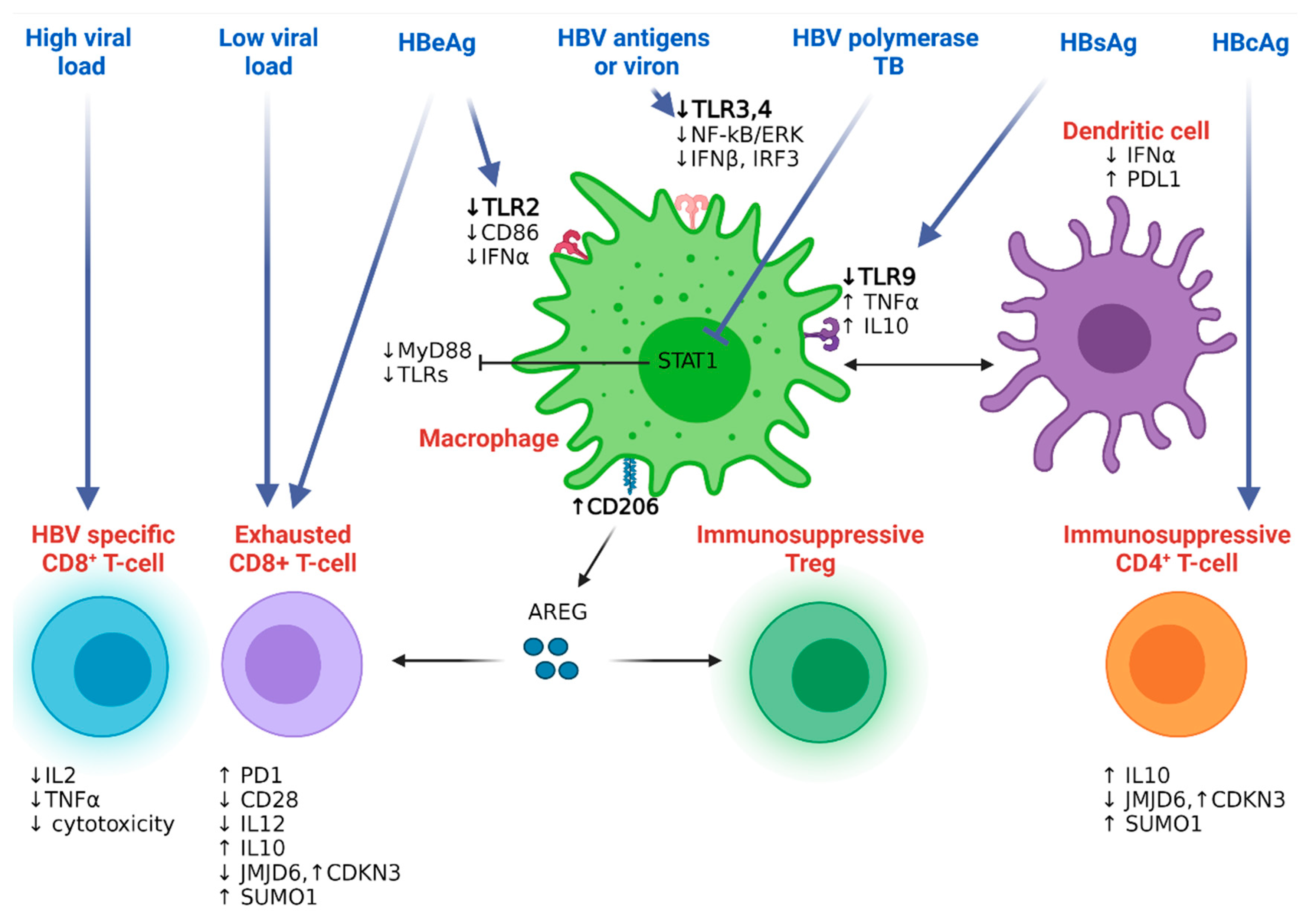
4.2.1. Hepatitis B Resistance to the Host Innate Immunity
4.2.2. Hepatitis B Resistance to the Host Adaptive Immunity
4.2.3. HBV-HDV Co-Infection Mediated Immunity
5. Host Immune Response to Hepatitis C Infection
5.1. Acute Hepatitis C Infection
5.1.1. Host Innate Immune Response to Acute HCV Infection
5.1.2. Host Adaptive Immune Response to Acute HCV Infection
5.2. Chronic Hepatitis C Infection
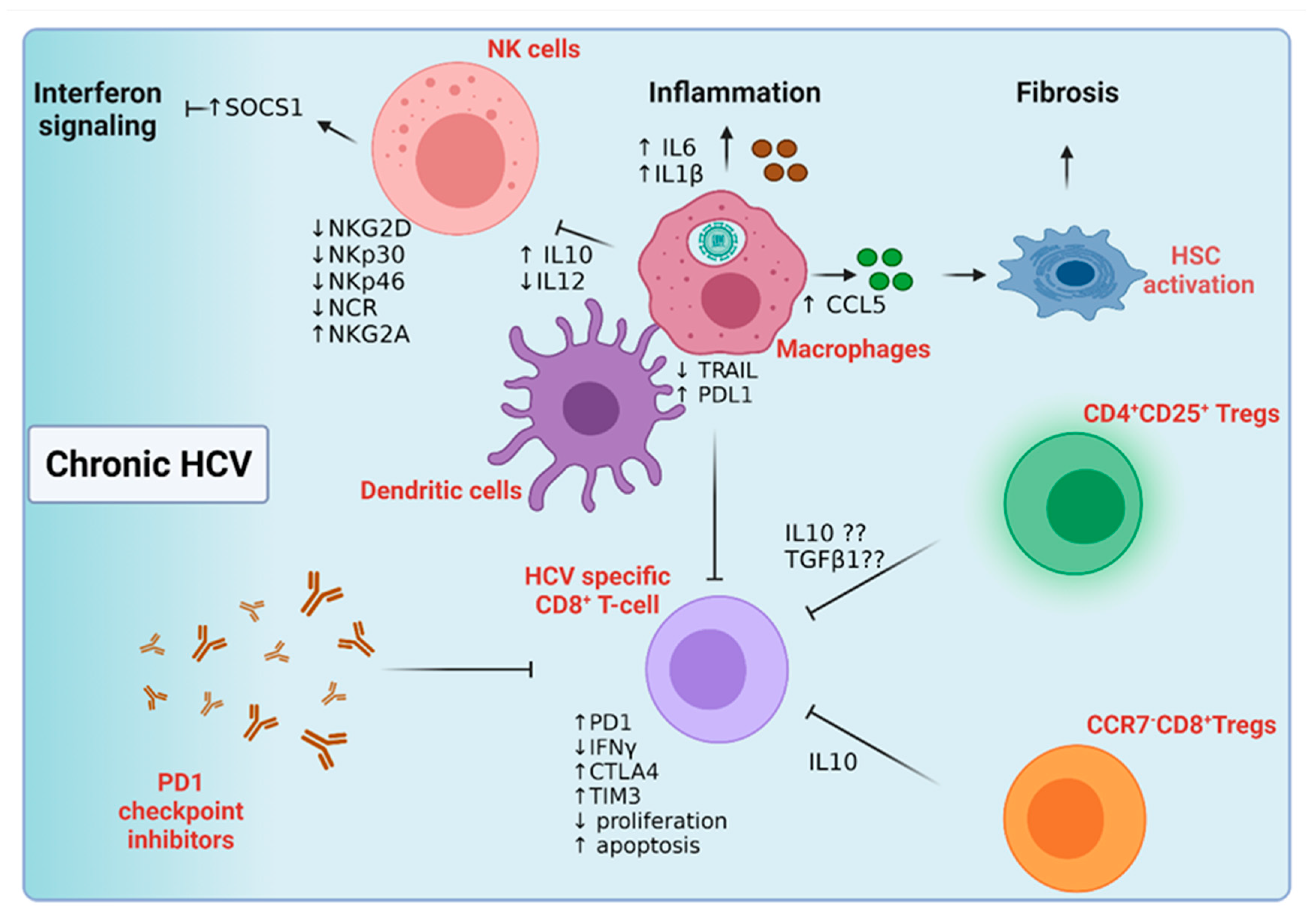
5.2.1. Host Innate Immune Response to Chronic Hepatitis C Infection
5.2.2. Host Adaptive Immune Response to Chronic Hepatitis C Infection
6. Viral Hepatitis, Immune Imbalance, and HCC Development
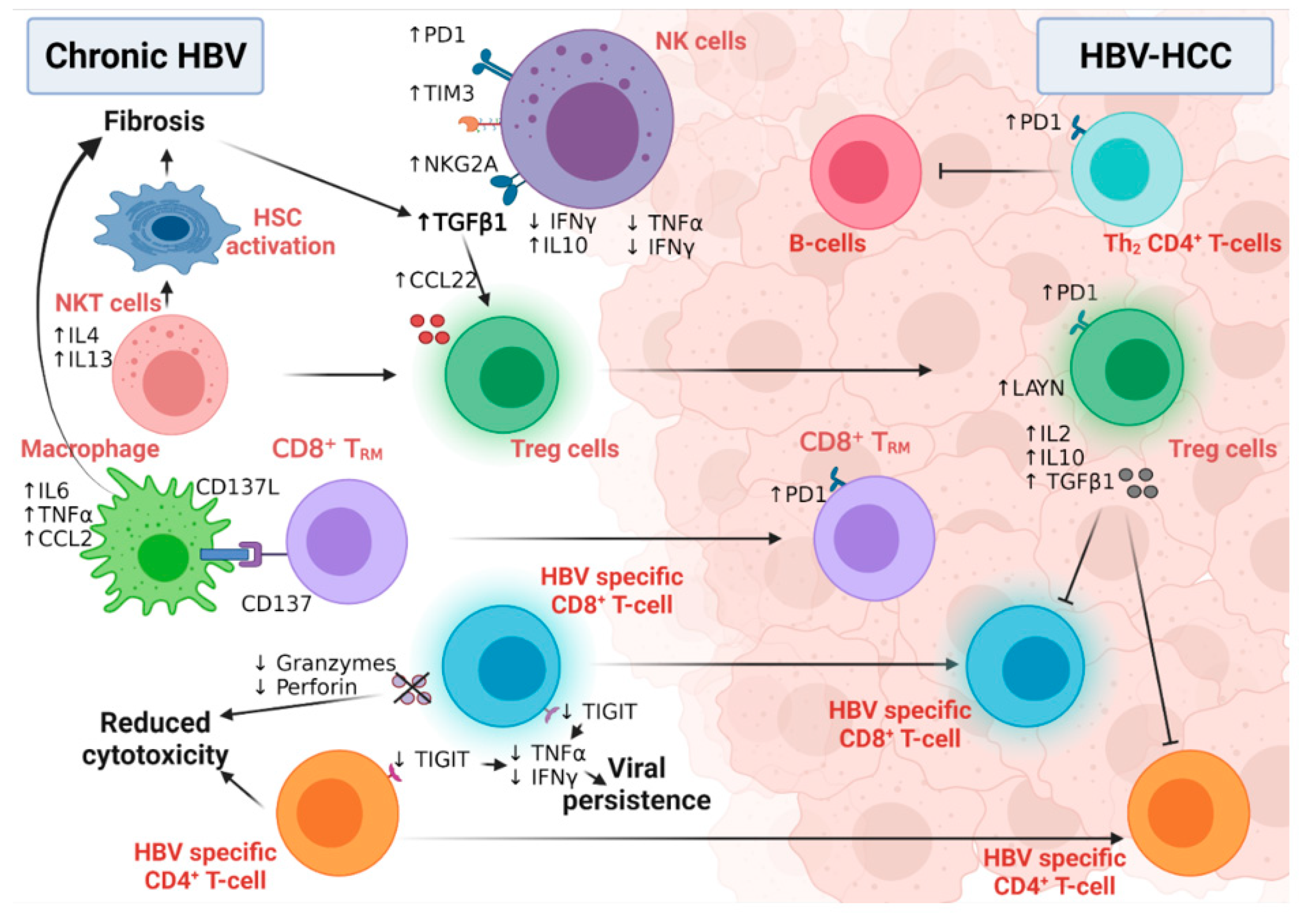
6.1. The Role of HBV-Related Immunity in the Development of HCC
6.1.1. Innate Immunity and HCC Development
6.1.2. Adaptive Immunity and HCC Development
6.2. The Role of HCV-Related Immunity in the Development of HCC
6.2.1. Innate Immunity and HCC Development
6.2.2. Adaptive Immunity and HCC Development
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424, Erratum in 2020, 70, 313. [Google Scholar] [CrossRef]
- Forner, A.; Reig, M.; Bruix, J. Hepatocellular carcinoma. Lancet 2018, 391, 1301–1314. [Google Scholar] [CrossRef]
- Arzumanyan, A.; Reis, H.M.; Feitelson, M.A. Pathogenic mechanisms in HBV- and HCV-associated hepatocellular carcinoma. Nat. Rev. Cancer 2013, 13, 123–135. [Google Scholar] [CrossRef]
- Reeves, H.L.; Zaki, M.Y.; Day, C.P. Hepatocellular Carcinoma in Obesity, Type 2 Diabetes, and NAFLD. Am. J. Dig. Dis. 2016, 61, 1234–1245. [Google Scholar] [CrossRef]
- Horton, S.; Gauvreau, C.L. Cancer in Low- and Middle-Income Countries: An Economic Overview. In Disease Control Priorities, 3rd ed.; Gelband, H., Jha, P., Sankaranarayanan, R., Horton, S., Eds.; World Bank Publications: Washington, DC, USA, 2015; Volume 3, pp. 263–280. [Google Scholar] [CrossRef]
- Hall, A.J. Liver cancer in low and middle income countries. BMJ 2003, 326, 994–995. [Google Scholar] [CrossRef]
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555. [Google Scholar] [CrossRef]
- Stanaway, J.D.; Flaxman, A.D.; Naghavi, M.; Fitzmaurice, C.; Vos, T.; Abubakar, I.; Abu-Raddad, L.J.; Assadi, R.; Bhala, N.; Cowie, B.; et al. The global burden of viral hepatitis from 1990 to 2013: Findings from the Global Burden of Disease Study 2013. Lancet 2016, 388, 1081–1088. [Google Scholar] [CrossRef]
- Morozov, V.A.; Lagaye, S. Hepatitis C virus: Morphogenesis, infection and therapy. World J. Hepatol. 2018, 10, 186–212. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- World Health Organization (WHO). Global Hepatitis Report 2017; World Health Organization: Geneva, Switzerland, 2017.
- Ott, J.; Stevens, G.; Groeger, J.; Wiersma, S. Global epidemiology of hepatitis B virus infection: New estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine 2012, 30, 2212–2219. [Google Scholar] [CrossRef]
- Mastrodomenico, M.; Muselli, M.; Provvidenti, L.; Scatigna, M.; Bianchi, S.; Fabiani, L. Long-term immune protection against HBV: Associated factors and determinants. Hum. Vaccines Immunother. 2021, 17, 2268–2272. [Google Scholar] [CrossRef] [PubMed]
- Tamandjou, C.R.; Maponga, T.G.; Chotun, N.; Preiser, W.; Andersson, M.I. Is hepatitis B birth dose vaccine needed in Africa? Pan Afr. Med. J. 2017, 27 (Suppl. S3), 18. [Google Scholar] [CrossRef] [PubMed]
- Beasley, P. Hepatitis B virus as the etiologic agent in hepatocellular carcinoma-epidemiologic consideration. Hepatology 1982, 2, 21s. [Google Scholar]
- Moraga, P.; Collaborators, G.C.o.D. Global, regional, and national age-sex specific mortality for 264 causes of death, 1980–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1151–1210. [Google Scholar]
- Gower, E.; Estes, C.; Blach, S.; Razavi-Shearer, K.; Razavi, H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 2014, 61, S45–S57. [Google Scholar] [CrossRef] [PubMed]
- Blach, S.; Zeuzem, S.; Manns, M.; Altraif, I.; Duberg, A.-S.; Muljono, D.H.; Waked, I.; Alavian, S.M.; Lee, M.-H.; Negro, F.; et al. Global prevalence and genotype distribution of hepatitis C virus infection in 2015: A modelling study. Lancet Gastroenterol. Hepatol. 2016, 2, 161–176. [Google Scholar] [CrossRef]
- Arafa, N.; El Hoseiny, M.; Rekacewicz, C.; Bakr, I.; El-Kafrawy, S.; El Daly, M.; Aoun, S.; Marzouk, D.; Mohamed, M.K.; Fontanet, A. Changing pattern of hepatitis C virus spread in rural areas of Egypt. J. Hepatol. 2005, 43, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.R.; Barnes, E.; Cox, A.L. Approaches, Progress, and Challenges to Hepatitis C Vaccine Development. Gastroenterology 2019, 156, 418–430. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.; Asch, S.M.; Chayanupatkul, M.; Cao, Y.; El-Serag, H.B. Risk of Hepatocellular Cancer in HCV Patients Treated With Direct-Acting Antiviral Agents. Gastroenterology 2017, 153, 996–1005.e1. [Google Scholar] [CrossRef]
- Seitz, S.; Urban, S.; Antoni, C.; Böttcher, B. Cryo-electron microscopy of hepatitis B virions reveals variability in envelope capsid interactions. EMBO J. 2007, 26, 4160–4167. [Google Scholar] [CrossRef]
- Sureau, C.; Salisse, J. A conformational heparan sulfate binding site essential to infectivity overlaps with the conserved hepatitis B virus A-determinant. Hepatology 2012, 57, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, S.; Nassal, M. A role for the host DNA damage response in hepatitis B virus cccDNA formation—And beyond? Viruses 2017, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Glebe, D.; Bremer, C.M. The molecular virology of hepatitis B virus. Semin. Liver Dis. 2013, 33, 103–112. [Google Scholar]
- Dubuisson, J.; Rey, F.; Moradpour, D.; Pawlotsky, J.-M. Structural biology of hepatitis C virus. Hepatology 2004, 39, 5–19. [Google Scholar] [CrossRef]
- Zeisel, M.B.; Felmlee, D.J.; Baumert, T.F. Hepatitis C virus entry. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Springer: Amsterdam, The Netherlands, 2013; pp. 87–112. [Google Scholar]
- André, P.; Komurian-Pradel, F.; Deforges, S.; Perret, M.; Berland, J.L.; Sodoyer, M.; Pol, S.; Bréchot, C.; Paranhos-Baccalà, G.; Lotteau, V. Characterization of Low- and Very-Low-Density Hepatitis C Virus RNA-Containing Particles. J. Virol. 2002, 76, 7040–7048. [Google Scholar] [CrossRef]
- Honda, M.; Beard, M.R.; Ping, L.-H.; Lemon, S.M. A Phylogenetically Conserved Stem-Loop Structure at the 5′ Border of the Internal Ribosome Entry Site of Hepatitis C Virus Is Required for Cap-Independent Viral Translation. J. Virol. 1999, 73, 1165–1174. [Google Scholar] [CrossRef]
- Niepmann, M. Hepatitis C virus RNA translation. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Springer: Amsterdam, The Netherlands, 2013; pp. 143–166. [Google Scholar]
- Moradpour, D.; Penin, F. Hepatitis C virus proteins: From structure to function. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Springer: Amsterdam, The Netherlands, 2013; pp. 113–142. [Google Scholar]
- Lohmann, V. Hepatitis C virus RNA replication. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Springer: Amsterdam, The Netherlands, 2013; pp. 167–198. [Google Scholar]
- Lindenbach, B.D. Virion assembly and release. In Hepatitis C Virus: From Molecular Virology to Antiviral Therapy; Springer: Amsterdam, The Netherlands, 2013; pp. 199–218. [Google Scholar]
- Fattovich, G. Natural History and Prognosis of Hepatitis B. Semin. Liver Dis. 2003, 23, 047–058. [Google Scholar] [CrossRef]
- Rehermann, B.; Nascimbeni, M. Immunology of hepatitis B virus and hepatitis C virus infection. Nat. Rev. Immunol. 2005, 5, 215–229. [Google Scholar] [CrossRef]
- Chen, M.S.; Billaud, J.-N.; Sällberg, M.; Guidotti, L.G.; Chisari, F.; Jones, J.; Hughes, J.; Milich, D.R. A function of the hepatitis B virus precore protein is to regulate the immune response to the core antigen. Proc. Natl. Acad. Sci. USA 2004, 101, 14913–14918. [Google Scholar] [CrossRef]
- Whalley, S.A.; Brown, D.; Webster, G.J.; Jacobs, R.; Reignat, S.; Bertoletti, A.; Teo, C.-G.; Emery, V.; Dusheiko, G.M. Evolution of hepatitis B virus during primary infection in humans: Transient generation of cytotoxic T-cell mutants. Gastroenterology 2004, 127, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Hösel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782. [Google Scholar] [CrossRef]
- Lebossé, F.; Testoni, B.; Fresquet, J.; Facchetti, F.; Galmozzi, E.; Fournier, M.; Hervieu, V.; Berthillon, P.; Berby, F.; Bordes, I.; et al. Intrahepatic innate immune response pathways are downregulated in untreated chronic hepatitis B. J. Hepatol. 2016, 66, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, L.G.; McClary, H.; Loudis, J.M.; Chisari, F. Nitric Oxide Inhibits Hepatitis B Virus Replication in the Livers of Transgenic Mice. J. Exp. Med. 2000, 191, 1247–1252. [Google Scholar] [CrossRef] [PubMed]
- Thomson, A.W.; Knolle, P.A. Antigen-presenting cell function in the tolerogenic liver environment. Nat. Rev. Immunol. 2010, 10, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Stadler, D.; Lucifora, J.; Reisinger, F.; Webb, D.; Hösel, M.; Michler, T.; Wisskirchen, K.; Cheng, X.; Zhang, K. Interferon-γ and tumor necrosis factor-α produced by T cells reduce the HBV persistence form, cccDNA, without cytolysis. Gastroenterology 2016, 150, 194–205. [Google Scholar] [CrossRef] [PubMed]
- Isorce, N.; Testoni, B.; Locatelli, M.; Fresquet, J.; Rivoire, M.; Luangsay, S.; Zoulim, F.; Durantel, D. Antiviral activity of various interferons and pro-inflammatory cytokines in non-transformed cultured hepatocytes infected with hepatitis B virus. Antivir. Res. 2016, 130, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, G.A.; Scisciani, C.; Pediconi, N.; Lupacchini, L.; Alfalate, D.; Guerrieri, F.; Calvo, L.; Salerno, D.; Di Cocco, S.; Levrero, M.; et al. IL6 Inhibits HBV Transcription by Targeting the Epigenetic Control of the Nuclear cccDNA Minichromosome. PLoS ONE 2015, 10, e0142599. [Google Scholar] [CrossRef]
- Belloni, L.; Allweiss, L.; Guerrieri, F.; Pediconi, N.; Volz, T.; Pollicino, T.; Petersen, J.; Raimondo, G.; Dandri, M.; Levrero, M. IFN-α inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Investig. 2012, 122, 529–537. [Google Scholar] [CrossRef]
- Maini, M.K.; Pallett, L.J. Defective T-cell immunity in hepatitis B virus infection: Why therapeutic vaccination needs a helping hand. Lancet Gastroenterol. Hepatol. 2018, 3, 192–202. [Google Scholar] [CrossRef]
- Thimme, R.; Wieland, S.; Steiger, C.; Ghrayeb, J.; Reimann, K.A.; Purcell, R.H.; Chisari, F.V. CD8 + T Cells Mediate Viral Clearance and Disease Pathogenesis during Acute Hepatitis B Virus Infection. J. Virol. 2003, 77, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Asabe, S.; Wieland, S.F.; Chattopadhyay, P.; Roederer, M.; Engle, R.; Purcell, R.H.; Chisari, F.V. The Size of the Viral Inoculum Contributes to the Outcome of Hepatitis B Virus Infection. J. Virol. 2009, 83, 9652–9662. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.; Peppa, D.; Khanna, P.; Nebbia, G.; Jones, M.; Brendish, N.; Lascar, R.M.; Brown, D.; Gilson, R.J.; Tedder, R.; et al. Temporal Analysis of Early Immune Responses in Patients With Acute Hepatitis B Virus Infection. Gastroenterology 2009, 137, 1289–1300. [Google Scholar] [CrossRef]
- Shin, E.-C.; Sung, P.S.; Park, S.-H. Immune responses and immunopathology in acute and chronic viral hepatitis. Nat. Rev. Immunol. 2016, 16, 509–523. [Google Scholar] [CrossRef]
- Shi, B.; Ren, G.; Hu, Y.; Wang, S.; Zhang, Z.; Yuan, Z. HBsAg Inhibits IFN-α Production in Plasmacytoid Dendritic Cells through TNF-α and IL-10 Induction in Monocytes. PLoS ONE 2012, 7, e44900. [Google Scholar] [CrossRef]
- Wu, J.; Meng, Z.; Jiang, M.; Pei, R.; Trippler, M.; Broering, R.; Bucchi, A.; Sowa, J.-P.; Dittmer, U.; Yang, D.; et al. Hepatitis B virus suppresses toll-like receptor-mediated innate immune responses in murine parenchymal and nonparenchymal liver cells. Hepatology 2008, 49, 1132–1140. [Google Scholar] [CrossRef]
- Wu, M.; Xu, Y.; Lin, S.; Zhang, X.; Xiang, L.; Yuan, Z. Hepatitis B virus polymerase inhibits the interferon-inducible MyD88 promoter by blocking nuclear translocation of Stat1. J. Gen. Virol. 2007, 88, 3260–3269. [Google Scholar] [CrossRef]
- Visvanathan, K.; Skinner, N.A.; Thompson, A.J.; Riordan, S.M.; Sozzi, V.; Edwards, R.; Rodgers, S.; Kurtovic, J.; Chang, J.; Lewin, S.R.; et al. Regulation of Toll-like receptor-2 expression in chronic hepatitis B by the precore protein. Hepatology 2006, 45, 102–110. [Google Scholar] [CrossRef]
- Dunn, C.; Brunetto, M.; Reynolds, G.; Christophides, T.; Kennedy, P.T.; Lampertico, P.; Das, A.; Lopes, A.R.; Borrow, P.; Williams, K.; et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell–mediated liver damage. J. Exp. Med. 2007, 204, 667–680. [Google Scholar] [CrossRef]
- Peppa, D.; Gill, U.S.; Reynolds, G.; Easom, N.J.; Pallett, L.J.; Schurich, A.; Micco, L.; Nebbia, G.; Singh, H.D.; Adams, D.; et al. Up-regulation of a death receptor renders antiviral T cells susceptible to NK cell–mediated deletion. J. Exp. Med. 2012, 210, 99–114. [Google Scholar] [CrossRef]
- Li, J.; Liu, F.-W.; Wu, D.-B.; Chen, E.-Q.; Chen, X.-J.; Chen, S.-C.; Liu, C.; Zhao, L.-S.; Tang, H.; Zhou, T.-Y. TRAIL inhibits HBV replication and expression by down-regulating liver-enriched transcription factors. Arab J. Gastroenterol. 2020, 21, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.-C.; Easom, N.J.; Tang, X.-Z.; Gill, U.S.; Singh, H.; Robertson, F.; Chang, C.; Trowsdale, J.; Davidson, B.R.; Rosenberg, W.M.; et al. T Cells Infiltrating Diseased Liver Express Ligands for the NKG2D Stress Surveillance System. J. Immunol. 2016, 198, 1172–1182. [Google Scholar] [CrossRef] [PubMed]
- Crouse, J.; Bedenikovic, G.; Wiesel, M.; Ibberson, M.; Xenarios, I.; Von Laer, D.; Kalinke, U.; Vivier, E.; Jonjic, S.; Oxenius, A. Type I Interferons Protect T Cells against NK Cell Attack Mediated by the Activating Receptor NCR1. Immunity 2014, 40, 961–973. [Google Scholar] [CrossRef]
- Xu, H.C.; Grusdat, M.; Pandyra, A.A.; Polz, R.; Huang, J.; Sharma, P.; Deenen, R.; Köhrer, K.; Rahbar, R.; Diefenbach, A.; et al. Type I Interferon Protects Antiviral CD8+ T Cells from NK Cell Cytotoxicity. Immunity 2014, 40, 949–960. [Google Scholar] [CrossRef]
- Gill, U.S.; Peppa, D.; Micco, L.; Singh, H.D.; Carey, I.; Foster, G.R.; Maini, M.K.; Kennedy, P.T.F. Interferon Alpha Induces Sustained Changes in NK Cell Responsiveness to Hepatitis B Viral Load Suppression In Vivo. PLoS Pathog. 2016, 12, e1005788. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000, 64, 51–68. [Google Scholar] [CrossRef]
- Bowen, D.G.; Zen, M.; Holz, L.; Davis, T.; Mccaughan, G.; Bertolino, P. The site of primary T cell activation is a determinant of the balance between intrahepatic tolerance and immunity. J. Clin. Investig. 2004, 114, 701–712. [Google Scholar] [CrossRef]
- Wherry, E.J.; Blattman, J.N.; Murali-Krishna, K.; van der Most, R.; Ahmed, R. Viral Persistence Alters CD8 T-Cell Immunodominance and Tissue Distribution and Results in Distinct Stages of Functional Impairment. J. Virol. 2003, 77, 4911–4927. [Google Scholar] [CrossRef]
- Boni, C.; Fisicaro, P.; Valdatta, C.; Amadei, B.; Di Vincenzo, P.; Giuberti, T.; Laccabue, D.; Zerbini, A.; Cavalli, A.; Missale, G.; et al. Characterization of Hepatitis B Virus (HBV)-Specific T-Cell Dysfunction in Chronic HBV Infection. J. Virol. 2007, 81, 4215–4225. [Google Scholar] [CrossRef]
- Gane, E.; Verdon, D.J.; Brooks, A.E.; Gaggar, A.; Nguyen, A.H.; Subramanian, G.M.; Schwabe, C.; Dunbar, R. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: A pilot study. J. Hepatol. 2019, 71, 900–907. [Google Scholar] [CrossRef]
- Li, B.; Yan, C.; Zhu, J.; Chen, X.; Fu, Q.; Zhang, H.; Tong, Z.; Liu, L.; Zheng, Y.; Zhao, P.; et al. Anti–PD-1/PD-L1 Blockade Immunotherapy Employed in Treating Hepatitis B Virus Infection–Related Advanced Hepatocellular Carcinoma: A Literature Review. Front. Immunol. 2020, 11, 1037. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Z.; Chen, W.; Zhang, Z.; Li, Y.; Shi, M.; Zhang, J.; Chen, L.; Wang, S.; Wang, F.-S. B7-H1 up-regulation on myeloid dendritic cells significantly suppresses T cell immune function in patients with chronic hepatitis B. J. Immunol. 2007, 178, 6634–6641. [Google Scholar] [CrossRef]
- Schurich, A.; Pallett, L.J.; Lubowiecki, M.; Singh, H.D.; Gill, U.S.; Kennedy, P.T.; Nastouli, E.; Tanwar, S.; Rosenberg, W.; Maini, M.K. The Third Signal Cytokine IL-12 Rescues the Anti-Viral Function of Exhausted HBV-Specific CD8 T Cells. PLoS Pathog. 2013, 9, e1003208. [Google Scholar] [CrossRef]
- Chen, C.-F.; Feng, X.; Liao, H.-Y.; Jin, W.-J.; Zhang, J.; Wang, Y.; Gong, L.-L.; Liu, J.-J.; Yuan, X.-H.; Zhao, B.-B.; et al. Regulation of T cell proliferation by JMJD6 and PDGF-BB during chronic hepatitis B infection. Sci. Rep. 2014, 4, 6359. [Google Scholar] [CrossRef]
- Li, X.; Kong, H.; Tian, L.; Zhu, Q.; Wang, Y.; Dong, Y.; Ni, Q.; Chen, Y. Changes of Costimulatory Molecule CD28 on Circulating CD8+T Cells Correlate with Disease Pathogenesis of Chronic Hepatitis B. BioMed Res. Int. 2014, 2014, 423181. [Google Scholar] [CrossRef] [PubMed]
- Barboza, L.; Salmen, S.; Peterson, D.L.; Montes, H.; Colmenares, M.; Hernández, M.; Berrueta-Carrillo, L.E.; Berrueta, L. Altered T cell costimulation during chronic hepatitis B infection. Cell. Immunol. 2009, 257, 61–68. [Google Scholar] [CrossRef]
- Dai, K.; Huang, L.; Sun, X.; Yang, L.; Gong, Z. Hepatic CD206-positive macrophages express amphiregulin to promote the immunosuppressive activity of regulatory T cells in HBV infection. J. Leukoc. Biol. 2015, 98, 1071–1080. [Google Scholar] [CrossRef]
- Fisicaro, P.; Barili, V.; Montanini, B.; Acerbi, G.; Ferracin, M.; Guerrieri, F.; Salerno, D.; Boni, C.; Massari, M.; Cavallo, M.C.; et al. Targeting mitochondrial dysfunction can restore antiviral activity of exhausted HBV-specific CD8 T cells in chronic hepatitis B. Nat. Med. 2017, 23, 327–336. [Google Scholar] [CrossRef]
- Schurich, A.; Pallett, L.J.; Jajbhay, D.; Wijngaarden, J.; Otano, I.; Gill, U.S.; Hansi, N.; Kennedy, P.T.; Nastouli, E.; Gilson, R.; et al. Distinct Metabolic Requirements of Exhausted and Functional Virus-Specific CD8 T Cells in the Same Host. Cell Rep. 2016, 16, 1243–1252. [Google Scholar] [CrossRef]
- Pallett, L.J.; Gill, U.S.; Quaglia, A.; Sinclair, L.V.; Jover-Cobos, M.; Schurich, A.; Singh, K.P.; Thomas, N.; Das, A.; Chen, A.; et al. Metabolic regulation of hepatitis B immunopathology by myeloid-derived suppressor cells. Nat. Med. 2015, 21, 591–600. [Google Scholar] [CrossRef]
- Pallett, L.J.; Davies, J.; Colbeck, E.J.; Robertson, F.; Hansi, N.; Easom, N.J.; Burton, A.R.; Stegmann, K.A.; Schurich, A.; Swadling, L.; et al. IL-2high tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med. 2017, 214, 1567–1580. [Google Scholar] [CrossRef]
- Gill, U.S.; Pallett, L.J.; Thomas, N.; Burton, A.R.; Patel, A.; Yona, S.; Kennedy, P.T.F.; Maini, M.K. Fine needle aspirates comprehensively sample intrahepatic immunity. Gut 2018, 68, 1493–1503. [Google Scholar] [CrossRef]
- Gerlich, W.H. Medical Virology of Hepatitis B: How it began and where we are now. Virol. J. 2013, 10, 239–259. [Google Scholar] [CrossRef]
- Cai, Y.; Yin, W. The Multiple Functions of B Cells in Chronic HBV Infection. Front. Immunol. 2020, 11, 3272. [Google Scholar] [CrossRef]
- Puigvehí, M.; Moctezuma-Velázquez, C.; Villanueva, A.; Llovet, J.M. The oncogenic role of hepatitis delta virus in hepatocellular carcinoma. JHEP Rep. 2019, 1, 120–130. [Google Scholar] [CrossRef]
- Pugnale, P.; Pazienza, V.; Guilloux, K.; Negro, F. Hepatitis delta virus inhibits alpha interferon signaling. Hepatology 2009, 49, 398–406. [Google Scholar] [CrossRef]
- Wu, J.C.; Chen, P.-J.; Kuo, M.Y.; Lee, S.D.; Chen, D.S.; Ting, L.P. Production of hepatitis delta virus and suppression of helper hepatitis B virus in a human hepatoma cell line. J. Virol. 1991, 65, 1099–1104. [Google Scholar] [CrossRef]
- Kefalakes, H.; Horgan, X.J.; Jung, M.K.; Amanakis, G.; Kapuria, D.; Bolte, F.J.; Kleiner, D.E.; Koh, C.; Heller, T.; Rehermann, B. Liver-Resident Bystander CD8+ T Cells Contribute to Liver Disease Pathogenesis in Chronic Hepatitis D Virus Infection. Gastroenterology 2021, 161, 1567–1583.e9. [Google Scholar] [CrossRef]
- Lunemann, S.; Malone, D.; Grabowski, J.; Port, K.; Béziat, V.; Bremer, B.; Malmberg, K.-J.; Manns, M.P.; Sandberg, J.; Cornberg, M.; et al. Effects of HDV infection and pegylated interferon α treatment on the natural killer cell compartment in chronically infected individuals. Gut 2014, 64, 469–482. [Google Scholar] [CrossRef]
- Lunemann, S.; Malone, D.F.G.; Hengst, J.; Port, K.; Grabowski, J.; Deterding, K.; Markova, A.; Bremer, B.; Schlaphoff, V.; Cornberg, M.; et al. Compromised Function of Natural Killer Cells in Acute and Chronic Viral Hepatitis. J. Infect. Dis. 2013, 209, 1362–1373. [Google Scholar] [CrossRef]
- Thimme, R.; Oldach, D.; Chang, K.-M.; Steiger, C.; Ray, S.; Chisari, F.V. Determinants of Viral Clearance and Persistence during Acute Hepatitis C Virus Infection. J. Exp. Med. 2001, 194, 1395–1406. [Google Scholar] [CrossRef]
- Major, M.E.; Dahari, H.; Mihalik, K.; Puig, M.; Rice, C.M.; Neumann, A.U.; Feinstone, S.M. Hepatitis C virus kinetics and host responses associated with disease and outcome of infection in chimpanzees. Hepatology 2004, 39, 1709–1720. [Google Scholar] [CrossRef]
- Marukian, S.; Andrus, L.; Sheahan, T.P.; Jones, C.T.; Charles, E.D.; PLoSs, A.; Rice, C.M.; Dustin, L.B. Hepatitis C virus induces interferon-λ and interferon-stimulated genes in primary liver cultures. Hepatology 2011, 54, 1913–1923. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Takahashi, K.; Asabe, S.; Wieland, S.; Garaigorta, U.; Gastaminza, P.; Isogawa, M.; Chisari, F.V. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. USA 2010, 107, 7431–7436. [Google Scholar] [CrossRef]
- Suzuki, R.; Matsuda, M.; Shimoike, T.; Watashi, K.; Aizaki, H.; Kato, T.; Suzuki, T.; Muramatsu, M.; Wakita, T. Activation of protein kinase R by hepatitis C virus RNA-dependent RNA polymerase. Virology 2019, 529, 226–233. [Google Scholar] [CrossRef]
- Rosen, H.R. Emerging concepts in immunity to hepatitis C virus infection. J. Clin. Investig. 2013, 123, 4121–4130. [Google Scholar] [CrossRef]
- Thoens, C.; Berger, C.; Trippler, M.; Siemann, H.; Lutterbeck, M.; Broering, R.; Schlaak, J.; Heinemann, F.M.; Heinold, A.; Nattermann, J.; et al. KIR2DL3+NKG2A− natural killer cells are associated with protection from productive hepatitis C virus infection in people who inject drugs. J. Hepatol. 2014, 61, 475–481. [Google Scholar] [CrossRef]
- Amadei, B.; Urbani, S.; Cazaly, A.; Fisicaro, P.; Zerbini, A.; Ahmed, P.; Missale, G.; Ferrari, C.; Khakoo, S.I. Activation of Natural Killer Cells During Acute Infection With Hepatitis C Virus. Gastroenterology 2010, 138, 1536–1545. [Google Scholar] [CrossRef]
- Werner, J.; Heller, T.; Gordon, A.M.; Sheets, A.; Sherker, A.H.; Kessler, E.; Bean, K.S.; Stevens, M.; Schmitt, J.; Rehermann, B. Innate immune responses in hepatitis C virus-exposed healthcare workers who do not develop acute infection. Hepatology 2013, 58, 1621–1631. [Google Scholar] [CrossRef]
- Tseng, C.-T.K.; Klimpel, G.R. Binding of the Hepatitis C Virus Envelope Protein E2 to CD81 Inhibits Natural Killer Cell Functions. J. Exp. Med. 2001, 195, 43–50. [Google Scholar] [CrossRef]
- Jinushi, M.; Takehara, T.; Tatsumi, T.; Kanto, T.; Miyagi, T.; Suzuki, T.; Kanazawa, Y.; Hiramatsu, N.; Hayashi, N. Negative Regulation of NK Cell Activities by Inhibitory Receptor CD94/NKG2A Leads to Altered NK Cell-Induced Modulation of Dendritic Cell Functions in Chronic Hepatitis C Virus Infection. J. Immunol. 2004, 173, 6072–6081. [Google Scholar] [CrossRef]
- Shin, E.; Park, S.; DeMino, M.; Nascimbeni, M.; Mihalik, K.; Major, M.; Veerapu, N.S.; Heller, T.; Feinstone, S.M.; Rice, C.M.; et al. Delayed Induction, Not Impaired Recruitment, of Specific CD8+ T Cells Causes the Late Onset of Acute Hepatitis C. Gastroenterology 2011, 141, 686–695.e1. [Google Scholar] [CrossRef]
- Thimme, R.; Bukh, J.; Spangenberg, H.C.; Wieland, S.; Pemberton, J.; Steiger, C.; Govindarajan, S.; Purcell, R.H.; Chisari, F.V. Viral and immunological determinants of hepatitis C virus clearance, persistence, and disease. Proc. Natl. Acad. Sci. USA 2002, 99, 15661–15668. [Google Scholar] [CrossRef]
- Klenerman, P.; Thimme, R. T cell responses in hepatitis C: The good, the bad and the unconventional. Gut 2011, 61, 1226–1234. [Google Scholar] [CrossRef]
- Shoukry, N.H.; Grakoui, A.; Houghton, M.; Chien, D.Y.; Ghrayeb, J.; Reimann, K.A.; Walker, C.M. Memory CD8+ T Cells Are Required for Protection from Persistent Hepatitis C Virus Infection. J. Exp. Med. 2003, 197, 1645–1655. [Google Scholar] [CrossRef]
- Grakoui, A.; Shoukry, N.H.; Woollard, D.J.; Han, J.-H.; Hanson, H.L.; Ghrayeb, J.; Murthy, K.K.; Rice, C.M.; Walker, C.M. HCV Persistence and Immune Evasion in the Absence of Memory T Cell Help. Science 2003, 302, 659–662. [Google Scholar] [CrossRef]
- Kared, H.; Fabre, T.; Bédard, N.; Bruneau, J.; Shoukry, N.H. Galectin-9 and IL-21 Mediate Cross-regulation between Th17 and Treg Cells during Acute Hepatitis C. PLoS Pathog. 2013, 9, e1003422. [Google Scholar] [CrossRef]
- Logvinoff, C.; Major, M.E.; Oldach, D.; Heyward, S.; Talal, A.; Balfe, P.; Feinstone, S.M.; Alter, H.; Rice, C.M.; McKeating, J. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 10149–10154. [Google Scholar] [CrossRef]
- Pestka, J.M.; Zeisel, M.B.; Blaser, E.; Schurmann, P.; Bartosch, B.; Cosset, F.-L.; Patel, A.H.; Meisel, H.; Baumert, J.; Viazov, S.; et al. Rapid induction of virus-neutralizing antibodies and viral clearance in a single-source outbreak of hepatitis C. Proc. Natl. Acad. Sci. USA 2007, 104, 6025–6030. [Google Scholar] [CrossRef]
- Nattermann, J.; Feldmann, G.; Ahlenstiel, G.; Langhans, B.; Sauerbruch, T.; Spengler, U. Surface expression and cytolytic function of natural killer cell receptors is altered in chronic hepatitis C. Gut 2006, 55, 869–877. [Google Scholar] [CrossRef][Green Version]
- Miyagi, T.; Takehara, T.; Nishio, K.; Shimizu, S.; Kohga, K.; Li, W.; Tatsumi, T.; Hiramatsu, N.; Kanto, T.; Hayashi, N. Altered interferon-α-signaling in natural killer cells from patients with chronic hepatitis C virus infection. J. Hepatol. 2010, 53, 424–430. [Google Scholar] [CrossRef]
- Oliviero, B.; Varchetta, S.; Paudice, E.; Michelone, G.; Zaramella, M.; Mavilio, D.; De Filippi, F.; Bruno, S.; Mondelli, M. Natural Killer Cell Functional Dichotomy in Chronic Hepatitis B and Chronic Hepatitis C Virus Infections. Gastroenterology 2009, 137, 1151–1160.e7. [Google Scholar] [CrossRef]
- Ahlenstiel, G.; Titerence, R.H.; Koh, C.; Edlich, B.; Feld, J.J.; Rotman, Y.; Ghany, M.G.; Hoofnagle, J.H.; Liang, T.J.; Heller, T.; et al. Natural Killer Cells Are Polarized Toward Cytotoxicity in Chronic Hepatitis C in an Interferon-Alfa–Dependent Manner. Gastroenterology 2010, 138, 325–335.e2. [Google Scholar] [CrossRef]
- Yoon, J.C.; Lim, J.-B.; Park, J.H.; Lee, J.M. Cell-to-Cell Contact with Hepatitis C Virus-Infected Cells Reduces Functional Capacity of Natural Killer Cells. J. Virol. 2011, 85, 12557–12569. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, W.; Zou, Z.; Hu, Z.; Fan, Q.; Xiong, J. Hepatitis C Virus Entry into Macrophages/Monocytes Mainly Depends on the Phagocytosis of Macrophages. Am. J. Dig. Dis. 2018, 64, 1226–1237. [Google Scholar] [CrossRef]
- Sasaki, R.; Devhare, P.B.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus-induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology 2017, 66, 746–757. [Google Scholar] [CrossRef]
- Zhang, S.; Saha, B.; Kodys, K.; Szabo, G. IFN-γ production by human natural killer cells in response to HCV-infected hepatoma cells is dependent on accessory cells. J. Hepatol. 2013, 59, 442–449. [Google Scholar] [CrossRef][Green Version]
- Sène, D.; Levasseur, F.; Abel, M.; Lambert, M.; Camous, X.; Hernandez, C.; Pène, V.; Rosenberg, A.R.; Jouvin-Marche, E.; Marche, P.; et al. Hepatitis C Virus (HCV) Evades NKG2D-Dependent NK Cell Responses through NS5A-Mediated Imbalance of Inflammatory Cytokines. PLoS Pathog. 2010, 6, e1001184. [Google Scholar] [CrossRef]
- Gerlach, J.; Diepolder, H.; Grüner, N.; Jung, M.; Zachoval, R.; Hoffmann, R.; Schirren, C.; Schraut, W.; Santantonio, T.; Houghton, M.; et al. Recurrence of hepatitis C virus after loss of virus specific CD4+ T cell response in acute hepatitis C. J. Hepatol. 1998, 28, 44. [Google Scholar] [CrossRef]
- Neumann-Haefelin, C.; Thimme, R. Adaptive Immune Responses in Hepatitis C Virus Infection. Curr. Top. Microbiol. Immunol. 2013, 369, 243–262. [Google Scholar] [CrossRef] [PubMed]
- Golden-Mason, L.; Palmer, B.; Klarquist, J.; Mengshol, J.A.; Castelblanco, N.; Rosen, H.R. Upregulation of PD-1 Expression on Circulating and Intrahepatic Hepatitis C Virus-Specific CD8 + T Cells Associated with Reversible Immune Dysfunction. J. Virol. 2007, 81, 9249–9258. [Google Scholar] [CrossRef] [PubMed]
- Rehermann, B. Hepatitis C virus versus innate and adaptive immune responses: A tale of coevolution and coexistence. J. Clin. Investig. 2009, 119, 1745–1754. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, N.; Kaplan, D.E.; Coleclough, J.; Li, Y.; Valiga, M.E.; Kaminski, M.; Shaked, A.; Olthoff, K.; Gostick, E.; Price, D.; et al. Functional Restoration of HCV-Specific CD8 T Cells by PD-1 Blockade Is Defined by PD-1 Expression and Compartmentalization. Gastroenterology 2008, 134, 1927–1937.e2. [Google Scholar] [CrossRef]
- Radziewicz, H.; Ibegbu, C.C.; Fernandez, M.L.; Workowski, K.A.; Obideen, K.; Wehbi, M.; Hanson, H.L.; Steinberg, J.P.; Masopust, D.; Wherry, E.J.; et al. Liver-Infiltrating Lymphocytes in Chronic Human Hepatitis C Virus Infection Display an Exhausted Phenotype with High Levels of PD-1 and Low Levels of CD127 Expression. J. Virol. 2007, 81, 2545–2553. [Google Scholar] [CrossRef]
- Kroy, D.C.; Ciuffreda, D.; Cooperrider, J.H.; Tomlinson, M.; Hauck, G.D.; Aneja, J.; Berger, C.; Wolski, D.; Carrington, M.; Wherry, E.J.; et al. Liver Environment and HCV Replication Affect Human T-Cell Phenotype and Expression of Inhibitory Receptors. Gastroenterology 2014, 146, 550–561. [Google Scholar] [CrossRef]
- Bengsch, B.; Seigel, B.; Ruhl, M.; Timm, J.; Kuntz, M.; Blum, H.E.; Pircher, H.; Thimme, R. Coexpression of PD-1, 2B4, CD160 and KLRG1 on Exhausted HCV-Specific CD8+ T Cells Is Linked to Antigen Recognition and T Cell Differentiation. PLoS Pathog. 2010, 6, e1000947. [Google Scholar] [CrossRef]
- Fuller, M.J.; Callendret, B.; Zhu, B.; Freeman, G.J.; Hasselschwert, D.L.; Satterfield, W.; Sharpe, A.H.; Dustin, L.; Rice, C.M.; Grakoui, A.; et al. Immunotherapy of chronic hepatitis C virus infection with antibodies against programmed cell death-1 (PD-1). Proc. Natl. Acad. Sci. USA 2013, 110, 15001–15006. [Google Scholar] [CrossRef]
- Semmo, N.; Day, C.L.; Ward, S.M.; Lucas, M.; Harcourt, G.; Loughry, A.; Klenerman, P. Preferential loss of IL-2-secreting CD4+ T helper cells in chronic HCV infection. Hepatology 2005, 41, 1019–1028. [Google Scholar] [CrossRef]
- Boettler, T.; Spangenberg, H.C.; Neumann-Haefelin, C.; Panther, E.; Urbani, S.; Ferrari, C.; Blum, H.E.; von Weizsäcker, F.; Thimme, R. T Cells with a CD4 + CD25 + Regulatory Phenotype Suppress In Vitro Proliferation of Virus-Specific CD8 + T Cells during Chronic Hepatitis C Virus Infection. J. Virol. 2005, 79, 7860–7867. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, R.; Tu, Z.; Xu, Y.; Firpi, R.J.; Rosen, H.R.; Liu, C.; Nelson, D.R. An immunomodulatory role for CD4+CD25+ regulatory T lymphocytes in hepatitis C virus infection. Hepatology 2004, 40, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Rushbrook, S.M.; Ward, S.M.; Unitt, E.; Vowler, S.L.; Lucas, M.; Klenerman, P.; Alexander, G.J.M. Regulatory T Cells Suppress In Vitro Proliferation of Virus-Specific CD8 + T Cells during Persistent Hepatitis C Virus Infection. J. Virol. 2005, 79, 7852–7859. [Google Scholar] [CrossRef]
- Accapezzato, D.; Francavilla, V.; Paroli, M.; Casciaro, M.; Chircu, L.V.; Cividini, A.; Abrignani, S.; Mondelli, M.; Barnaba, V. Hepatic expansion of a virus-specific regulatory CD8+ T cell population in chronic hepatitis C virus infection. J. Clin. Investig. 2004, 113, 963–972. [Google Scholar] [CrossRef]
- Brimacombe, C.L.; Grove, J.; Meredith, L.W.; Hu, K.; Syder, A.J.; Flores, M.V.; Timpe, J.M.; Krieger, S.E.; Baumert, T.F.; Tellinghuisen, T.L.; et al. Neutralizing Antibody-Resistant Hepatitis C Virus Cell-to-Cell Transmission. J. Virol. 2011, 85, 596–605. [Google Scholar] [CrossRef] [PubMed]
- von Hahn, T.; Yoon, J.C.; Alter, H.; Rice, C.M.; Rehermann, B.; Balfe, P.; McKeating, J. Hepatitis C Virus Continuously Escapes From Neutralizing Antibody and T-Cell Responses During Chronic Infection In Vivo. Gastroenterology 2007, 132, 667–678. [Google Scholar] [CrossRef]
- Falkowska, E.; Kajumo, F.; Garcia, E.; Reinus, J.; Dragic, T. Hepatitis C Virus Envelope Glycoprotein E2 Glycans Modulate Entry, CD81 Binding, and Neutralization. J. Virol. 2007, 81, 8072–8079. [Google Scholar] [CrossRef]
- Helle, F.; Goffard, A.; Morel, V.; Duverlie, G.; McKeating, J.; Keck, Z.-Y.; Foung, S.; Penin, F.; Dubuisson, J.; Voisset, C. The Neutralizing Activity of Anti-Hepatitis C Virus Antibodies Is Modulated by Specific Glycans on the E2 Envelope Protein. J. Virol. 2007, 81, 8101–8111. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The Hallmarks of Cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.Y.; Reeves, H.L. The genetic heterogeneity of hepatocellular carcinoma and the implications for personalised medicine. Transl. Cancer Res. 2016, 5, S1–S4. [Google Scholar] [CrossRef]
- Xue, R.; Li, R.; Guo, H.; Guo, L.; Su, Z.; Ni, X.; Qi, L.; Zhang, T.; Li, Q.; Zhang, Z.; et al. Variable Intra-Tumor Genomic Heterogeneity of Multiple Lesions in Patients with Hepatocellular Carcinoma. Gastroenterology 2016, 150, 998–1008. [Google Scholar] [CrossRef]
- Ahmad, J.; Eng, F.J.; Branch, A.D. HCV and HCC: Clinical Update and a Review of HCC-Associated Viral Mutations in theCoreGene. Semin. Liver Dis. 2011, 31, 347–355. [Google Scholar] [CrossRef]
- Pinyol, R.; Torrecilla, S.; Wang, H.; Montironi, C.; Pique-Gili, M.; Torres-Martin, M.; Wei-Qiang, L.; Willoughby, C.E.; Ramadori, P.; Andreu-Oller, C.; et al. Molecular characterization of hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Zucman-Rossi, J.; Villanueva, A.; Nault, J.C.; Llovet, J.M. Genetic Landscape and Biomarkers of Hepatocellular Carcinoma. Gastroenterology 2015, 149, 1226–1239.e4. [Google Scholar] [CrossRef]
- Levrero, M.; Zucman-Rossi, J. Mechanisms of HBV-induced hepatocellular carcinoma. J. Hepatol. 2016, 64, S84–S101. [Google Scholar] [CrossRef]
- He, Y.; Tian, Z. NK cell education via nonclassical MHC and non-MHC ligands. Cell. Mol. Immunol. 2016, 14, 321–330. [Google Scholar] [CrossRef]
- Peppa, D.; Micco, L.; Javaid, A.; Kennedy, P.T.F.; Schurich, A.; Dunn, C.; Pallant, C.; Ellis, G.; Khanna, P.; Dusheiko, G.; et al. Blockade of Immunosuppressive Cytokines Restores NK Cell Antiviral Function in Chronic Hepatitis B Virus Infection. PLoS Pathog. 2010, 6, e1001227. [Google Scholar] [CrossRef]
- Xu, D.; Han, Q.; Hou, Z.; Zhang, C.; Zhang, J. miR-146a negatively regulates NK cell functions via STAT1 signaling. Cell. Mol. Immunol. 2016, 14, 712–720. [Google Scholar] [CrossRef]
- Marraco, S.A.F.; Neubert, N.; Verdeil, G.; Speiser, D. Inhibitory Receptors Beyond T Cell Exhaustion. Front. Immunol. 2015, 6, 310. [Google Scholar] [CrossRef]
- Chen, Y.; Hao, X.; Sun, R.; Wei, H.; Tian, Z. Natural Killer Cell–Derived Interferon-Gamma Promotes Hepatocellular Carcinoma Through the Epithelial Cell Adhesion Molecule–Epithelial-to-Mesenchymal Transition Axis in Hepatitis B Virus Transgenic Mice. Hepatology 2019, 69, 1735–1750. [Google Scholar] [CrossRef]
- Maini, M.K.; Peppa, D. NK Cells: A Double-Edged Sword in Chronic Hepatitis B Virus Infection. Front. Immunol. 2013, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Sun, R.; Wei, H.; Gao, X.; Chen, Y.; Tian, Z. Accelerated liver fibrosis in hepatitis B virus transgenic mice: Involvement of natural killer T cells. Hepatology 2010, 53, 219–229. [Google Scholar] [CrossRef]
- Wynn, T.A.; Vannella, K.M. Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhao, W.; Cheng, L.; Guo, M.; Li, D.; Li, X.; Tan, Y.; Ma, S.; Li, S.; Yang, Y.; et al. CD137-Mediated Pathogenesis from Chronic Hepatitis to Hepatocellular Carcinoma in Hepatitis B Virus-Transgenic Mice. J. Immunol. 2010, 185, 7654–7662. [Google Scholar] [CrossRef]
- Sun, C.; Lan, P.; Han, Q.; Huang, M.; Zhang, Z.; Xu, G.; Song, J.; Wang, J.; Wei, H.; Zhang, J.; et al. Oncofetal gene SALL4 reactivation by hepatitis B virus counteracts miR-200c in PD-L1-induced T cell exhaustion. Nat. Commun. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Lim, C.J.; Lee, Y.H.; Pan, L.; Lai, L.; Chua, C.; Wasser, M.; Lim, T.K.H.; Yeong, J.; Toh, H.C.; Lee, S.Y.; et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 2018, 68, 916–927. [Google Scholar] [CrossRef]
- Kim, G.-A.; Lim, Y.-S.; Han, S.; Choi, J.; Shim, J.H.; Kim, K.M.; Lee, H.C.; Lee, Y.S. High risk of hepatocellular carcinoma and death in patients with immune-tolerant-phase chronic hepatitis B. Gut 2017, 67, 945–952. [Google Scholar] [CrossRef]
- Mason, W.S.; Gill, U.S.; Litwin, S.; Zhou, Y.; Peri, S.; Pop, O.; Hong, M.; Naik, S.; Quaglia, A.; Bertoletti, A.; et al. HBV DNA Integration and Clonal Hepatocyte Expansion in Chronic Hepatitis B Patients Considered Immune Tolerant. Gastroenterology 2016, 151, 986–998.e4. [Google Scholar] [CrossRef]
- Svicher, V.; Salpini, R.; Piermatteo, L.; Carioti, L.; Battisti, A.; Colagrossi, L.; Scutari, R.; Surdo, M.; Cacciafesta, V.; Nuccitelli, A.; et al. Whole exome HBV DNA integration is independent of the intrahepatic HBV reservoir in HBeAg-negative chronic hepatitis B. Gut 2021, 70, 2337–2348. [Google Scholar] [CrossRef] [PubMed]
- Maini, M.; Boni, C.; Lee, C.K.; Larrubia, J.; Reignat, S.; Ogg, G.S.; King, A.S.; Herberg, J.; Gilson, R.; Alisa, A.; et al. The Role of Virus-Specific Cd8+ Cells in Liver Damage and Viral Control during Persistent Hepatitis B Virus Infection. J. Exp. Med. 2000, 191, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Heim, K.; Neumann-Haefelin, C.; Thimme, R.; Hofmann, M. Heterogeneity of HBV-Specific CD8+ T-Cell Failure: Implications for Immunotherapy. Front. Immunol. 2019, 10, 2240. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Peng, H.; Sun, C.; Li, F.; Zheng, M.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. Breakdown of adaptive immunotolerance induces hepatocellular carcinoma in HBsAg-tg mice. Nat. Commun. 2019, 10, 221. [Google Scholar] [CrossRef]
- Fu, J.; Zhang, Z.; Zhou, L.; Qi, Z.; Xing, S.; Lv, J.; Shi, J.; Fu, B.; Liu, Z.; Zhang, J.-Y.; et al. Impairment of CD4+cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatology 2012, 58, 139–149. [Google Scholar] [CrossRef]
- Moeini, A.; Torrecilla, S.; Tovar, V.; Montironi, C.; Andreu-Oller, C.; Peix, J.; Higuera, M.; Pfister, D.; Ramadori, P.; Pinyol, R.; et al. An Immune Gene Expression Signature Associated With Development of Human Hepatocellular Carcinoma Identifies Mice That Respond to Chemopreventive Agents. Gastroenterology 2019, 157, 1383–1397.e11. [Google Scholar] [CrossRef]
- Sia, D.; Jiao, Y.; Martinez-Quetglas, I.; Kuchuk, O.; Villacorta-Martin, C.; de Moura, M.C.; Putra, J.; Campreciós, G.; Bassaganyas, L.; Akers, N.; et al. Identification of an Immune-specific Class of Hepatocellular Carcinoma, Based on Molecular Features. Gastroenterology 2017, 153, 812–826. [Google Scholar] [CrossRef]
- Yang, P.; Li, Q.-J.; Feng, Y.; Zhang, Y.; Markowitz, G.J.; Ning, S.; Deng, Y.; Zhao, J.; Jiang, S.; Yuan, Y.; et al. TGF-β-miR-34a-CCL22 Signaling-Induced Treg Cell Recruitment Promotes Venous Metastases of HBV-Positive Hepatocellular Carcinoma. Cancer Cell 2012, 22, 291–303. [Google Scholar] [CrossRef]
- Trehanpati, N.; Vyas, A.K. Immune Regulation by T Regulatory Cells in Hepatitis B Virus-Related Inflammation and Cancer. Scand. J. Immunol. 2017, 85, 175–181. [Google Scholar] [CrossRef]
- Fu, J.; Xu, D.; Liu, Z.; Shi, M.; Zhao, P.; Fu, B.; Zhang, Z.; Yang, H.; Zhang, H.; Zhou, C.; et al. Increased Regulatory T Cells Correlate With CD8 T-Cell Impairment and Poor Survival in Hepatocellular Carcinoma Patients. Gastroenterology 2007, 132, 2328–2339. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Tangye, S.G.; Ma, C.; Brink, R.; Deenick, E.K. The good, the bad and the ugly—TFH cells in human health and disease. Nat. Rev. Immunol. 2013, 13, 412–426. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.-Q.; Tong, D.-N.; Guan, J.; Tan, H.-W.; Zhao, L.-D.; Zhu, Y.; Yao, J.; Yang, J.; Zhang, Z.-Y. Follicular helper T cell exhaustion induced by PD-L1 expression in hepatocellular carcinoma results in impaired cytokine expression and B cell help, and is associated with advanced tumor stages. Am. J. Transl. Res. 2016, 8, 2926–2936. [Google Scholar] [PubMed]
- Jia, Y.; Zeng, Z.; Li, Y.; Li, Z.; Jin, L.; Zhang, Z.; Wang, L.; Wang, F.-S. Impaired Function of CD4+ T Follicular Helper (Tfh) Cells Associated with Hepatocellular Carcinoma Progression. PLoS ONE 2015, 10, e0117458. [Google Scholar] [CrossRef]
- Liao, F.-T.; Lee, Y.-J.; Ko, J.-L.; Tsai, C.-C.; Tseng, C.-J.; Sheu, G.-T. Hepatitis delta virus epigenetically enhances clusterin expression via histone acetylation in human hepatocellular carcinoma cells. J. Gen. Virol. 2009, 90, 1124–1134. [Google Scholar] [CrossRef]
- Choi, S.; Jeong, S.; Hwang, S.B. Large Hepatitis Delta Antigen Modulates Transforming Growth Factor-β Signaling Cascades: Implication of Hepatitis Delta Virus–Induced Liver Fibrosis. Gastroenterology 2007, 132, 343–357. [Google Scholar] [CrossRef]
- Rehermann, B. Pathogenesis of chronic viral hepatitis: Differential roles of T cells and NK cells. Nat. Med. 2013, 19, 859–868. [Google Scholar] [CrossRef]
- Pikarsky, E.; Porat, R.M.; Stein, I.; Abramovitch, R.; Amit, S.; Kasem, S.; Gutkovich-Pyest, E.; Urieli-Shoval, S.; Galun, E.; Ben-Neriah, Y. NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 2004, 431, 461–466. [Google Scholar] [CrossRef]
- Zaki, M.Y.W.; Mahdi, A.K.; Patman, G.L.; Whitehead, A.; Maurício, J.P.; McCain, M.V.; Televantou, D.; Abou-Beih, S.; Ramon-Gil, E.; Watson, R.; et al. Key features of the environment promoting liver cancer in the absence of cirrhosis. Sci. Rep. 2021, 11, 1–17. [Google Scholar] [CrossRef]
- Nakagawa, H.; Maeda, S.; Yoshida, H.; Tateishi, R.; Masuzaki, R.; Ohki, T.; Hayakawa, Y.; Kinoshita, H.; Yamakado, M.; Kato, N.; et al. Serum IL-6 levels and the risk for hepatocarcinogenesis in chronic hepatitis C patients: An analysis based on gender differences. Int. J. Cancer 2009, 125, 2264–2269. [Google Scholar] [CrossRef]
- Tacke, R.S.; Tosello-Trampont, A.; Nguyen, V.; Mullins, D.W.; Hahn, Y.S. Extracellular Hepatitis C Virus Core Protein Activates STAT3 in Human Monocytes/Macrophages/Dendritic Cells via an IL-6 Autocrine Pathway. J. Biol. Chem. 2011, 286, 10847–10855. [Google Scholar] [CrossRef] [PubMed]
- Hoshida, Y.; Villanueva, A.; Kobayashi, M.; Peix, J.; Chiang, D.Y.; Camargo, A.; Gupta, S.; Moore, J.; Wrobel, M.J.; Lerner, J.; et al. Gene Expression in Fixed Tissues and Outcome in Hepatocellular Carcinoma. New Engl. J. Med. 2008, 359, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Park, K.-J.; Choi, S.-H.; Choi, D.-H.; Park, J.-M.; Yie, S.W.; Lee, S.Y.; Hwang, S.B. Hepatitis C Virus NS5A Protein Modulates c-Jun N-terminal Kinase through Interaction with Tumor Necrosis Factor Receptor-associated Factor 2. J. Biol. Chem. 2003, 278, 30711–30718. [Google Scholar] [CrossRef] [PubMed]
- Das, M.; Garlick, D.S.; Greiner, D.L.; Davis, R.J. The role of JNK in the development of hepatocellular carcinoma. Genes Dev. 2011, 25, 634–645. [Google Scholar] [CrossRef]
- Fuertes, M.B.; Woo, S.-R.; Burnett, B.; Fu, Y.-X.; Gajewski, T.F. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2012, 34, 67–73. [Google Scholar] [CrossRef]
- Aroucha, D.; Carmo, R.D.; Moura, P.; Silva, J.; Vasconcelos, L.; Cavalcanti, M.; Muniz, M.T.C.; Aroucha, M.; Siqueira, E.; Cahú, G.; et al. High tumor necrosis factor-α/interleukin-10 ratio is associated with hepatocellular carcinoma in patients with chronic hepatitis C. Cytokine 2013, 62, 421–425. [Google Scholar] [CrossRef]
- Haybaeck, J.; Zeller, N.; Wolf, M.J.; Weber, A.; Wagner, U.; Kurrer, M.O.; Bremer, J.; Iezzi, G.; Graf, R.; Clavien, P.-A.; et al. A Lymphotoxin-Driven Pathway to Hepatocellular Carcinoma. Cancer Cell 2009, 16, 295–308. [Google Scholar] [CrossRef]
- Ramzan, M.; Sturm, N.; Decaens, T.; Bioulac-Sage, P.; Bancel, B.; Merle, P.; Tran Van Nhieu, J.; Slama, R.; Letoublon, C.; Zarski, J.P. Liver-infiltrating CD 8+ lymphocytes as prognostic factor for tumour recurrence in hepatitis C virus-related hepatocellular carcinoma. Liver Int. 2016, 36, 434–444. [Google Scholar] [CrossRef]
- Piconese, S.; Timperi, E.; Pacella, I.; Schinzari, V.; Tripodo, C.; Rossi, M.; Guglielmo, N.; Mennini, G.; Grazi, G.L.; Di Filippo, S.; et al. Human OX40 tunes the function of regulatory T cells in tumor and nontumor areas of hepatitis C virus-infected liver tissue. Hepatology 2014, 60, 1494–1507. [Google Scholar] [CrossRef]
- Lu, L.; Jiang, J.; Zhan, M.; Zhang, H.; Wang, Q.; Sun, S.; Guo, X.; Yin, H.; Wei, Y.; Li, S.; et al. Targeting Tumor-Associated Antigens in Hepatocellular Carcinoma for Immunotherapy: Past Pitfalls and Future Strategies. Hepatology 2020, 73, 821–832. [Google Scholar] [CrossRef]
- Flecken, T.; Schmidt, N.; Hild, S.; Gostick, E.; Drognitz, O.; Zeiser, R.; Schemmer, P.; Bruns, H.; Eiermann, T.; Price, D.A.; et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8 + T-cell responses in hepatocellular carcinoma. Hepatology 2013, 59, 1415–1426. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Haining, W.N.; Held, W.; Hogan, P.G.; Kallies, A.; Lugli, E.; Lynn, R.C.; Philip, M.; Rao, A.; Restifo, N.P.; et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 2019, 19, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Lok, A.S.-F. Hepatitis B Treatment: What We Know Now and What Remains to Be Researched. Hepatol. Commun. 2018, 3, 8–19. [Google Scholar] [CrossRef]
- Feld, J.J.; Jacobson, I.M.; Hézode, C.; Asselah, T.; Ruane, P.J.; Gruener, N.; Abergel, A.; Mangia, A.; Lai, C.-L.; Chan, H.L.; et al. Sofosbuvir and Velpatasvir for HCV Genotype 1, 2, 4, 5, and 6 Infection. New Engl. J. Med. 2015, 373, 2599–2607. [Google Scholar] [CrossRef]
- Bruix, J.; Cheng, A.-L.; Meinhardt, G.; Nakajima, K.; De Sanctis, Y.; Llovet, J. Prognostic factors and predictors of sorafenib benefit in patients with hepatocellular carcinoma: Analysis of two phase III studies. J. Hepatol. 2017, 67, 999–1008. [Google Scholar] [CrossRef]
- Eldafashi, N.; Darlay, R.; Shukla, R.; McCain, M.; Watson, R.; Liu, Y.; McStraw, N.; Fathy, M.; Fawzy, M.; Zaki, M.; et al. A PDCD1 Role in the Genetic Predisposition to NAFLD-HCC? Cancers 2021, 13, 1412. [Google Scholar] [CrossRef]
- Prenner, S.B.; VanWagner, L.B.; Flamm, S.L.; Salem, R.; Lewandowski, R.J.; Kulik, L. Hepatocellular carcinoma decreases the chance of successful hepatitis C virus therapy with direct-acting antivirals. J. Hepatol. 2017, 66, 1173–1181. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaki, M.Y.W.; Fathi, A.M.; Samir, S.; Eldafashi, N.; William, K.Y.; Nazmy, M.H.; Fathy, M.; Gill, U.S.; Shetty, S. Innate and Adaptive Immunopathogeneses in Viral Hepatitis; Crucial Determinants of Hepatocellular Carcinoma. Cancers 2022, 14, 1255. https://doi.org/10.3390/cancers14051255
Zaki MYW, Fathi AM, Samir S, Eldafashi N, William KY, Nazmy MH, Fathy M, Gill US, Shetty S. Innate and Adaptive Immunopathogeneses in Viral Hepatitis; Crucial Determinants of Hepatocellular Carcinoma. Cancers. 2022; 14(5):1255. https://doi.org/10.3390/cancers14051255
Chicago/Turabian StyleZaki, Marco Y. W., Ahmed M. Fathi, Samara Samir, Nardeen Eldafashi, Kerolis Y. William, Maiiada Hassan Nazmy, Moustafa Fathy, Upkar S. Gill, and Shishir Shetty. 2022. "Innate and Adaptive Immunopathogeneses in Viral Hepatitis; Crucial Determinants of Hepatocellular Carcinoma" Cancers 14, no. 5: 1255. https://doi.org/10.3390/cancers14051255
APA StyleZaki, M. Y. W., Fathi, A. M., Samir, S., Eldafashi, N., William, K. Y., Nazmy, M. H., Fathy, M., Gill, U. S., & Shetty, S. (2022). Innate and Adaptive Immunopathogeneses in Viral Hepatitis; Crucial Determinants of Hepatocellular Carcinoma. Cancers, 14(5), 1255. https://doi.org/10.3390/cancers14051255