
Epigenetics of Dendritic Cells in Tumor Immunology


{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Mechanisms Underlying Epigenetic Dysregulation
3. Dendritic Cell Subtypes and Epigenetic Regulation
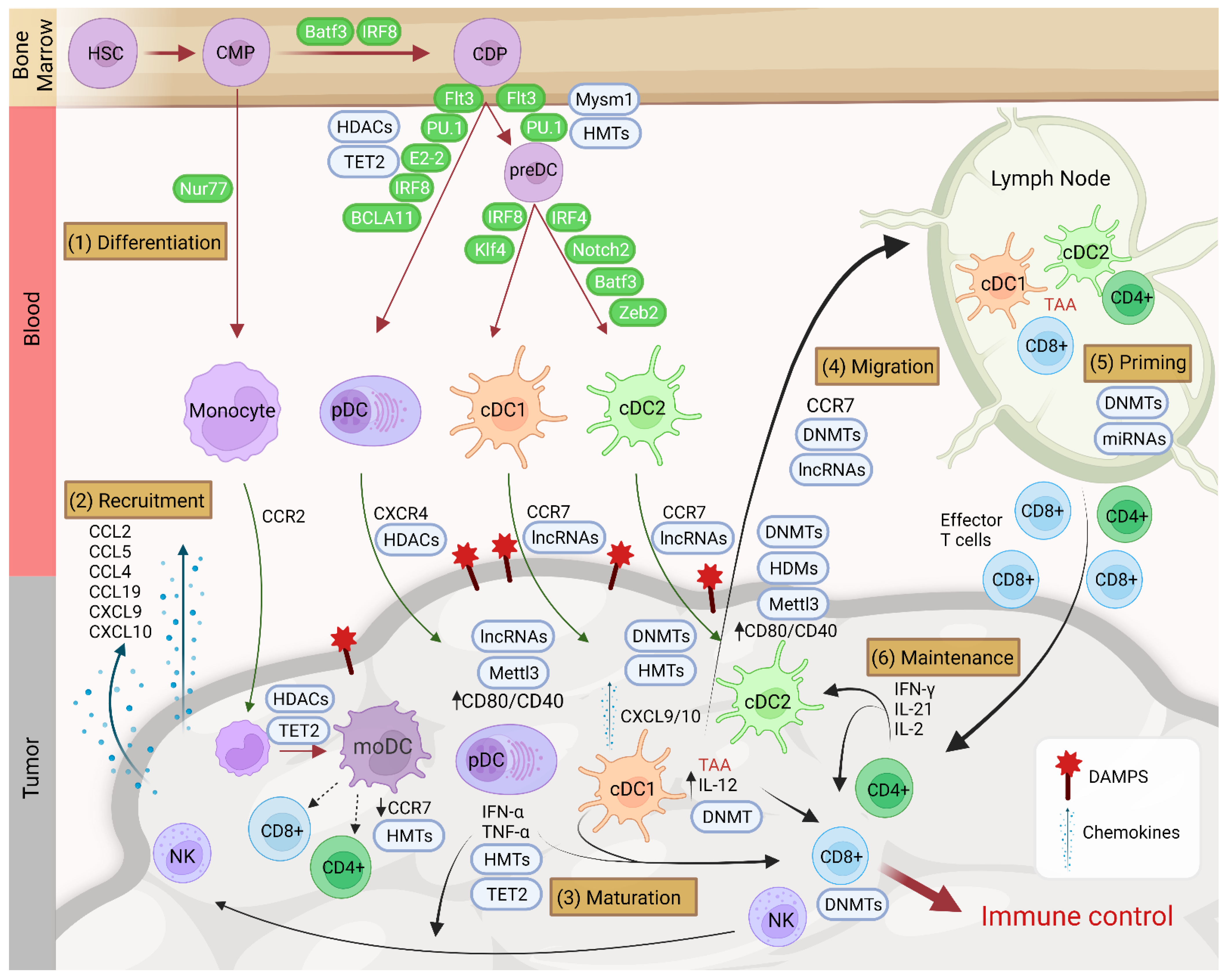
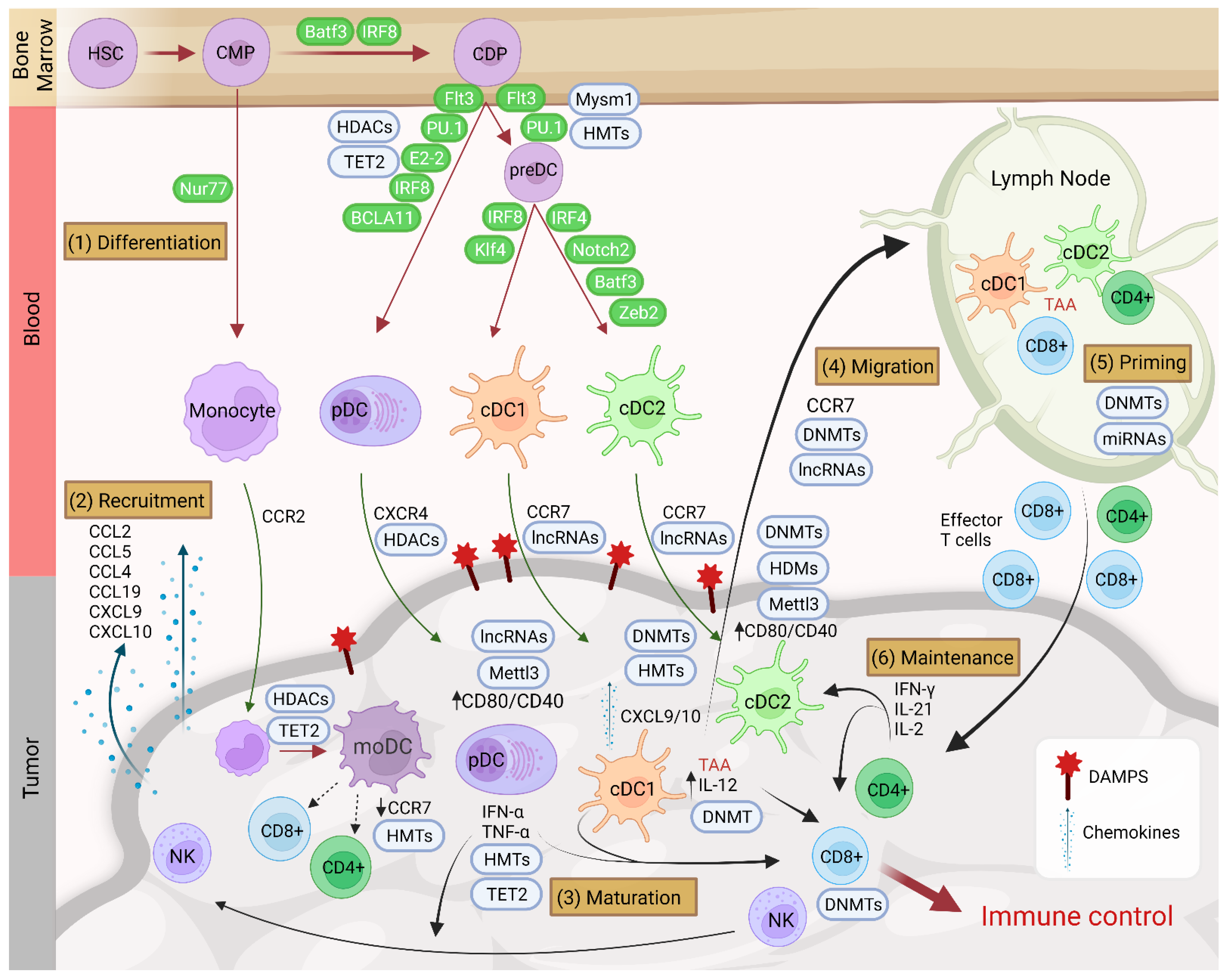
4. Recruitment of Dendritic Cells in the TME and Involvement of Epigenetics
5. Functions of Dendritic Cells in the TME
6. Epigenetic Impact of the TME on Dendritic Cells
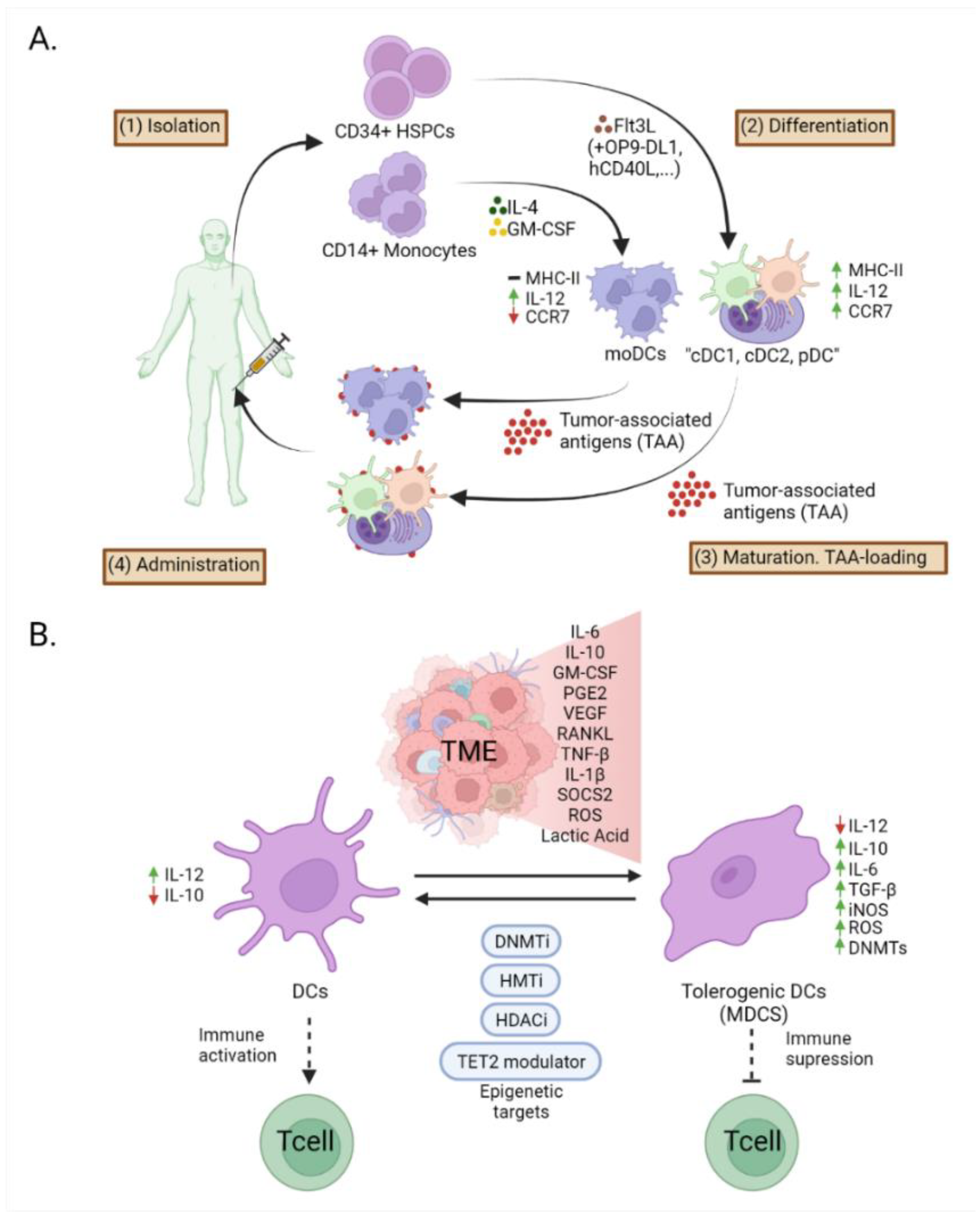
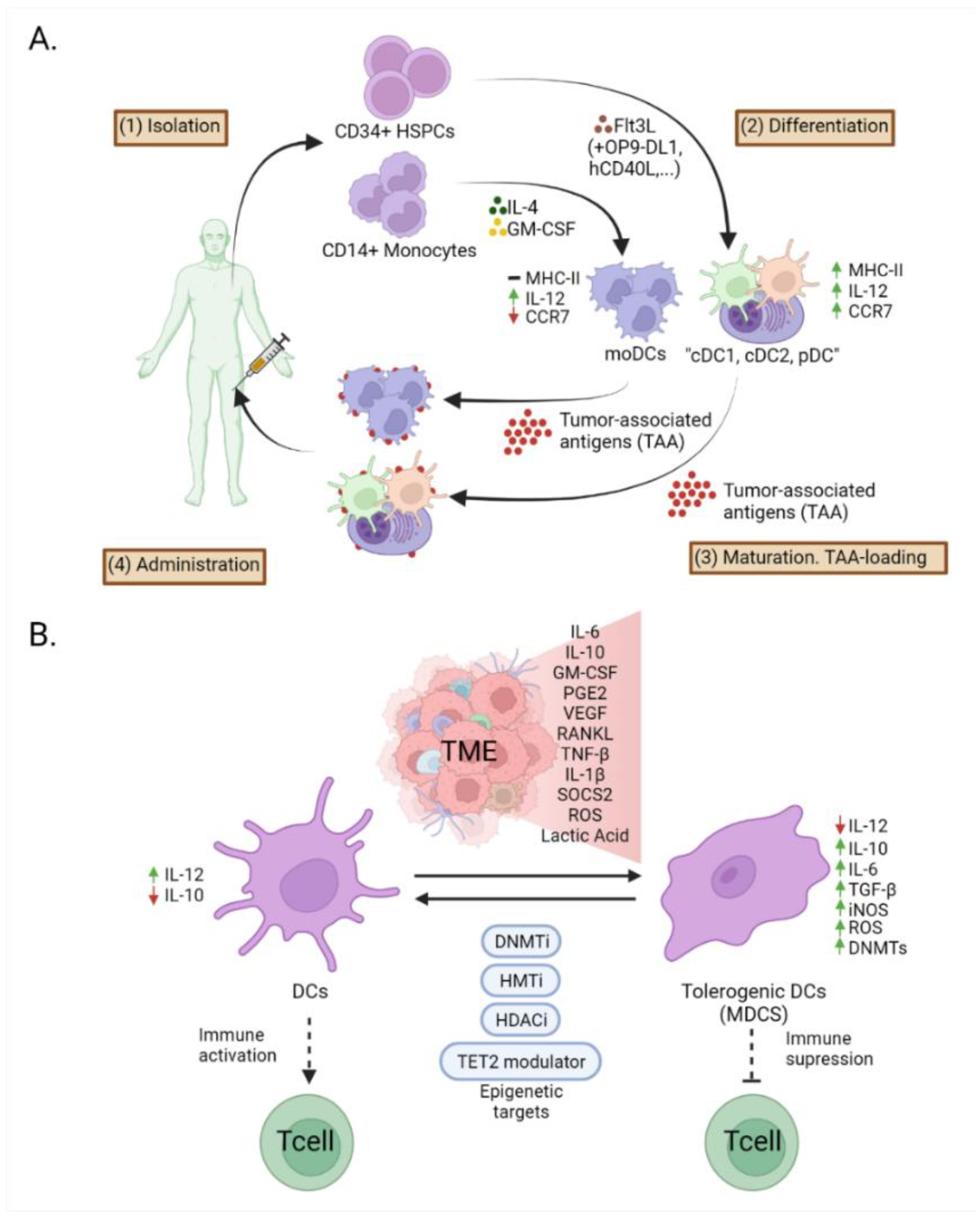
7. Epigenetic Modifications in Dendritic Cells in Cancer Immunotherapy
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Janco, J.M.T.; Lamichhane, P.; Karyampudi, L.; Knutson, K.L. Tumor-Infiltrating Dendritic Cells in Cancer Pathogenesis. J. Immunol. 2015, 194, 2985–2991. [Google Scholar] [CrossRef] [Green Version]
- Ugel, S.; De Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [Green Version]
- Alvarez_Errico, D.; Vento-Tormo, R.; Sieweke, M.; Ballestar, E. Epigenetic control of myeloid cell differentiation, identity and function. Nat. Rev. Immunol. 2015, 15, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Park, S.H. Epigenetic Regulation of Myeloid Cells. Microbiol. Spectr. 2016, 4, 571–590. [Google Scholar] [CrossRef] [Green Version]
- Roulois, D.; Loo Yau, H.; Singhania, R.; Wang, Y.; Danesh, A.; Shen, S.Y.; Han, H.; Liang, G.; Jones, P.A.; Pugh, T.J.; et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015, 162, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA Methyltransferases Dnmt3a and Dnmt3b Are Essential for De Novo Methylation and Mammalian Development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Wu, H.; Diep, D.; Yamaguchi, S.; D’Alessio, A.C.; Fung, H.-L.; Zhang, K.; Zhang, Y. Genome-wide Analysis Reveals TET- and TDG-Dependent 5-Methylcytosine Oxidation Dynamics. Cell 2013, 153, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Wajed, S.A.; Laird, P.W.; Demeester, T.R. DNA Methylation: An Alternative Pathway to Cancer. Ann. Surg. 2001, 234, 10–20. [Google Scholar] [CrossRef]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet. 2006, 15, R17–R29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Eichhorn, S.W.; Guo, H.; McGeary, S.E.; Rodriguez-Mias, R.A.; Shin, C.; Baek, D.; Hsu, S.-H.; Ghoshal, K.; Villén, J.; Bartel, D.P. mRNA Destabilization Is the Dominant Effect of Mammalian MicroRNAs by the Time Substantial Repression Ensues. Mol. Cell 2014, 56, 104–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birnberg, T.; Bar-On, L.; Sapoznikov, A.; Caton, M.L.; Cervantes-Barragán, L.; Makia, D.; Krauthgamer, R.; Brenner, O.; Ludewig, B.; Brockschnieder, D.; et al. Lack of Conventional Dendritic Cells Is Compatible with Normal Development and T Cell Homeostasis, but Causes Myeloid Proliferative Syndrome. Immunity 2008, 29, 986–997. [Google Scholar] [CrossRef] [Green Version]
- Hildner, K.; Edelson, B.T.; Purtha, W.E.; Diamond, M.; Matsushita, H.; Kohyama, M.; Calderon, B.; Schraml, B.U.; Unanue, E.R.; Diamond, M.S.; et al. Batf3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 2008, 322, 1097–1100. [Google Scholar] [CrossRef] [Green Version]
- Cella, M.; Salio, M.; Sakakibara, Y.; Langen, H.; Julkunen, I.; Lanzavecchia, A. Maturation, Activation, and Protection of Dendritic Cells Induced by Double-stranded RNA. J. Exp. Med. 1999, 189, 821–829. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Ulm, A.; Somineni, H.K.; Oh, S.; Weirauch, M.T.; Zhang, H.-X.; Chen, X.; Lehn, A.M.; Janssen, E.M.; Ji, H. DNA methylation dynamics during ex vivo differentiation and maturation of human dendritic cells. Epigenet. Chromatin 2014, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Yi, L.; Li, Z.; Hu, T.; Liu, J.; Li, N.; Cao, X.; Liu, S. Intracellular HSP70L1 inhibits human dendritic cell maturation by promoting suppressive H3K27me3 and H2AK119Ub1 histone modifications. Cell. Mol. Immunol. 2019, 17, 85–94. [Google Scholar] [CrossRef]
- Boukhaled, G.M.; Cordeiro, B.; Deblois, G.; Dimitrov, V.; Bailey, S.D.; Holowka, T.; Domi, A.; Guak, H.; Chiu, H.-H.; Everts, B.; et al. The Transcriptional Repressor Polycomb Group Factor 6, PCGF6, Negatively Regulates Dendritic Cell Activation and Promotes Quiescence. Cell Rep. 2016, 16, 1829–1837. [Google Scholar] [CrossRef] [Green Version]
- Reis e Sousa, C. Toll-like receptors and dendritic cells: For whom the bug tolls. Semin. Immunol. 2004, 16, 27–34. [Google Scholar] [CrossRef]
- Scharer, C.; Barwick, B.; Youngblood, B.; Ahmed, R.; Boss, J.M. Global DNA Methylation Remodeling Accompanies CD8 T Cell Effector Function. J. Immunol. 2013, 191, 3419–3429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladle, B.H.; Li, K.-P.; Phillips, M.J.; Pucsek, A.B.; Haile, A.; Powell, J.D.; Jaffee, E.M.; Hildeman, D.A.; Gamper, C.J. De novo DNA methylation by DNA methyltransferase 3a controls early effector CD8+ T-cell fate decisions following activation. Proc. Natl. Acad. Sci. USA 2016, 113, 10631–10636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russ, B.E.; Olshanksy, M.; Smallwood, H.S.; Li, J.; Denton, A.; Prier, J.; Stock, A.T.; Croom, H.A.; Cullen, J.G.; Nguyen, M.L.; et al. Distinct Epigenetic Signatures Delineate Transcriptional Programs during Virus-Specific CD8+ T Cell Differentiation. Immunity 2014, 41, 853–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, Y.; Wang, Z.; Zang, C.; Wood, W.H.; Schones, D.; Cui, K.; Roh, T.-Y.; Lhotsky, B.; Wersto, R.P.; Peng, W.; et al. Genome-wide Analysis of Histone Methylation Reveals Chromatin State-Based Regulation of Gene Transcription and Function of Memory CD8+ T Cells. Immunity 2009, 30, 912–925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussenzweig, M.C.; Steinman, R.M.; Gutchinov, B.; Cohn, A.Z. Dendritic cells are accessory cells for the development of anti-trinitrophenyl cytotoxic T lymphocytes. J. Exp. Med. 1980, 152, 1070–1084. [Google Scholar] [CrossRef] [Green Version]
- Mildner, A.; Jung, S. Development and Function of Dendritic Cell Subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Joffre, O.; Segura, E.; Savina, A.; Amigorena, S. Cross-presentation by dendritic cells. Nat. Rev. Immunol. 2012, 12, 557–569. [Google Scholar] [CrossRef]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The Dendritic Cell Lineage: Ontogeny and Function of Dendritic Cells and Their Subsets in the Steady State and the Inflamed Setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [Green Version]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef]
- Ferris, S.T.; Durai, V.; Wu, R.; Theisen, D.J.; Ward, J.; Bern, M.D.; Iv, J.T.D.; Bagadia, P.; Liu, T.; Briseño, C.G.; et al. cDC1 prime and are licensed by CD4+ T cells to induce anti-tumour immunity. Nature 2020, 584, 624–629. [Google Scholar] [CrossRef]
- Binnewies, M.; Mujal, A.M.; Pollack, J.L.; Combes, A.J.; Hardison, E.A.; Barry, K.C.; Tsui, J.; Ruhland, M.K.; Kersten, K.; Abushawish, M.A.; et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell 2019, 177, 556–571. [Google Scholar] [CrossRef] [PubMed]
- Grajales-Reyes, G.E.; Iwata, A.; Albring, J.C.; Wumesh, K.C.; Tussiwand, R.; Kc, W.; Kretzer, N.M.; Briseno, C.; Durai, V.; Bagadia, P.; et al. Batf3 maintains autoactivation of Irf8 for commitment of a CD8α+ conventional DC clonogenic progenitor. Nat. Immunol. 2015, 16, 708–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, T.L.; Grajales-Reyes, G.E.; Wu, X.; Tussiwand, R.; Briseño, C.G.; Iwata, A.; Kretzer, N.M.; Durai, V.; Murphy, K.M. Transcriptional Control of Dendritic Cell Development. Annu. Rev. Immunol. 2016, 34, 93–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlitzer, A.; McGovern, N.; Teo, P.; Zelante, T.; Atarashi, K.; Low, D.; Ho, A.W.S.; See, P.; Shin, A.; Wasan, P.S.; et al. IRF4 Transcription Factor-Dependent CD11b+ Dendritic Cells in Human and Mouse Control Mucosal IL-17 Cytokine Responses. Immunity 2013, 38, 970–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.W.; Tjota, M.Y.; Clay, B.S.; Vander Lugt, B.; Bandukwala, H.S.; Hrusch, C.L.; Decker, D.C.; Blaine, K.M.; Fixsen, B.R.; Singh, H.; et al. Transcription factor IRF4 drives dendritic cells to promote Th2 differentiation. Nat. Commun. 2013, 4, 2990. [Google Scholar] [CrossRef] [Green Version]
- Kurotaki, D.; Kawase, W.; Sasaki, H.; Nakabayashi, J.; Nishiyama, A.; Morse, H.C.; Ozato, K.; Suzuki, Y.; Tamura, T. Epigenetic control of early dendritic cell lineage specification by the transcription factor IRF8 in mice. Blood 2019, 133, 1803–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chopin, M.; Lun, A.T.; Zhan, Y.; Schreuder, J.; Coughlan, H.; D’Amico, A.; Mielke, L.; Almeida, F.F.; Kueh, A.J.; Dickins, R.; et al. Transcription Factor PU.1 Promotes Conventional Dendritic Cell Identity and Function via Induction of Transcriptional Regulator DC-SCRIPT. Immunity 2019, 50, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Won, H.; Nandakumar, V.; Yates, P.; Sanchez, S.; Jones, L.; Huang, X.F.; Chen, S.-Y. Epigenetic control of dendritic cell development and fate determination of common myeloid progenitor by Mysm1. Blood 2014, 124, 2647–2656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Q.; Chauvistré, H.; Costa, I.G.; Gusmao, E.G.; Mitzka, S.; Hänzelmann, S.; Baying, B.; Klisch, T.; Moriggl, R.; Hennuy, B.; et al. Epigenetic program and transcription factor circuitry of dendritic cell development. Nucleic Acids Res. 2015, 43, 9680–9693. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, P.; Alberti-Servera, L.; Eremin, A.; Grajales-Reyes, G.E.; Ivanek, R.; Tussiwand, R. Distinct progenitor lineages contribute to the heterogeneity of plasmacytoid dendritic cells. Nat. Immunol. 2018, 19, 711–722. [Google Scholar] [CrossRef]
- Dress, R.J.; Dutertre, C.-A.; Giladi, A.; Schlitzer, A.; Low, I.; Shadan, N.B.; Tay, A.; Lum, J.; Kairi, M.F.B.M.; Hwang, Y.Y.; et al. Plasmacytoid dendritic cells develop from Ly6D+ lymphoid progenitors distinct from the myeloid lineage. Nat. Immunol. 2019, 20, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Reizis, B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity 2019, 50, 37–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Y.; Gaugler, B.; Mohty, M.; Malard, F. Plasmacytoid dendritic cell biology and its role in immune-mediated diseases. Clin. Transl. Immunol. 2020, 9, 1139. [Google Scholar] [CrossRef]
- Musumeci, A.; Lutz, K.; Winheim, E.; Krug, A.B. What Makes a pDC: Recent Advances in Understanding Plasmacytoid DC Development and Heterogeneity. Front. Immunol. 2019, 10, 1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sichien, D.; Scott, C.; Martens, L.; Vanderkerken, M.; Van Gassen, S.; Plantinga, M.; Joeris, T.; De Prijck, S.; Vanhoutte, L.; Vanheerswynghels, M.; et al. IRF8 Transcription Factor Controls Survival and Function of Terminally Differentiated Conventional and Plasmacytoid Dendritic Cells, Respectively. Immunity 2016, 45, 626–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, H.S.; Cisse, B.; Bunin, A.; Lewis, K.L.; Reizis, B. Continuous Expression of the Transcription Factor E2-2 Maintains the Cell Fate of Mature Plasmacytoid Dendritic Cells. Immunity 2010, 33, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Schiavoni, G.; Mattei, F.; Sestili, P.; Borghi, P.; Venditti, M.; Morse, H.; Belardelli, F.; Gabriele, L. ICSBP Is Essential for the Development of Mouse Type I Interferon-producing Cells and for the Generation and Activation of CD8α+ Dendritic Cells. J. Exp. Med. 2002, 196, 1415–1425. [Google Scholar] [CrossRef] [Green Version]
- Chauvistré, H.; Küstermann, C.; Rehage, N.; Klisch, T.; Mitzka, S.; Felker, P.; Rose-John, S.; Zenke, M.; Seré, K.M. Dendritic cell development requires histone deacetylase activity. Eur. J. Immunol. 2014, 44, 2478–2488. [Google Scholar] [CrossRef] [Green Version]
- Schlitzer, A.; McGovern, N.; Ginhoux, F. Dendritic cells and monocyte-derived cells: Two complementary and integrated functional systems. Semin. Cell Dev. Biol. 2015, 41, 9–22. [Google Scholar] [CrossRef]
- Mildner, A.; Yona, S.; Jung, S. A Close Encounter of the Third Kind. Adv. Immunol. 2013, 120, 69–103. [Google Scholar] [CrossRef]
- Bosteels, C.; Neyt, K.; Vanheerswynghels, M.; van Helden, M.J.; Sichien, D.; Debeuf, N.; De Prijck, S.; Bosteels, V.; Vandamme, N.; Martens, L.; et al. Inflammatory Type 2 cDCs Acquire Features of cDC1s and Macrophages to Orchestrate Immunity to Respiratory Virus Infection. Immunity 2020, 52, 1039–1056. [Google Scholar] [CrossRef]
- Naik, S.H.; Sathe, P.; Park, H.-Y.; Metcalf, D.; Proietto, I.A.; Dakic, A.; Carotta, S.; O’Keeffe, M.; Bahlo, M.; Papenfuss, A.; et al. Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat. Immunol. 2007, 8, 1217–1226. [Google Scholar] [CrossRef] [PubMed]
- Meredith, M.M.; Liu, K.; Darrasse-Jeze, G.; Kamphorst, A.O.; Schreiber, H.A.; Guermonprez, P.; Idoyaga, J.; Cheong, C.; Yao, K.-H.; Niec, R.; et al. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J. Exp. Med. 2012, 209, 1153–1165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallusto, F.; Lanzavecchia, A. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 1994, 179, 1109–1118. [Google Scholar] [CrossRef] [Green Version]
- Vento-Tormo, R.; Company, C.; Rodríguez-Ubreva, J.; De La Rica, L.; Urquiza, J.M.; Javierre, B.M.; Sabarinathan, R.; Luque, A.; Esteller, M.; Aran, J.M.; et al. IL-4 orchestrates STAT6-mediated DNA demethylation leading to dendritic cell differentiation. Genome Biol. 2016, 17, 4. [Google Scholar] [CrossRef] [Green Version]
- Nencioni, A.; Beck, J.; Werth, D.; Grünebach, F.; Patrone, F.; Ballestrero, A.; Brossart, P. Histone Deacetylase Inhibitors Affect Dendritic Cell Differentiation and Immunogenicity. Clin. Cancer Res. 2007, 13, 3933–3941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Jerby-Arnon, L.; Shah, P.; Cuoco, M.; Rodman, C.; Su, M.-J.; Melms, J.; Leeson, R.; Kanodia, A.; Mei, S.; Lin, J.-R.; et al. A Cancer Cell Program Promotes T Cell Exclusion and Resistance to Checkpoint Blockade. Cell 2018, 175, 984–997. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Ramnarayanan, K.; Sundar, R.; Padmanbahan, N.; Srivastava, S.; Koiwa, M.; Yasuda, T.; Koh, V.; Huang, K.K.; Tay, S.T.; et al. Single-cell atlas of lineage states, tumor microenvironment and subtype-specific expression programs in gastric cancer. Cancer Discov. 2021. [Google Scholar] [CrossRef]
- Massalha, H.; Halpern, K.B.; Abu-Gazala, S.; Jana, T.; Massasa, E.E.; Moor, E.A.; Buchauer, L.; Rozenberg, M.; Pikarsky, E.; Amit, I.; et al. A single cell atlas of the human liver tumor microenvironment. Mol. Syst. Biol. 2020, 16, e9682. [Google Scholar] [CrossRef]
- Engblom, C.; Pfirschke, C.; Pittet, M.J. The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 2016, 16, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Garris, C.S.; Luke, J.J. Dendritic Cells, the T-cell-inflamed Tumor Microenvironment, and Immunotherapy Treatment Response. Clin. Cancer Res. 2020, 26, 3901–3907. [Google Scholar] [CrossRef] [Green Version]
- Perrot, I.; Blanchard, D.; Freymond, N.; Isaac, S.; Guibert, B.; Pachéco, Y.; Lebecque, S. Dendritic Cells Infiltrating Human Non-Small Cell Lung Cancer Are Blocked at Immature Stage. J. Immunol. 2007, 178, 2763–2769. [Google Scholar] [CrossRef] [Green Version]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103+/CD141+ Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103+ Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villablanca, E.; Raccosta, L.; Zhou, D.; Fontana, R.; Maggioni, D.; Negro, A.; Sanvito, F.; Ponzoni, M.; Valentinis, B.; Bregni, M.; et al. Tumor-mediated liver X receptor-α activation inhibits CC chemokine receptor-7 expression on dendritic cells and dampens antitumor responses. Nat. Med. 2010, 16, 98–105. [Google Scholar] [CrossRef]
- Sluijter, B.J.; Hout, M.F.V.D.; Koster, B.; Van Leeuwen, P.A.; Schneiders, F.L.; Van De Ven, R.; Molenkamp, B.G.; Vosslamber, S.; Verweij, C.L.; Tol, M.P.V.D.; et al. Arming the Melanoma Sentinel Lymph Node through Local Administration of CpG-B and GM-CSF: Recruitment and Activation of BDCA3/CD141+ Dendritic Cells and Enhanced Cross-Presentation. Cancer Immunol. Res. 2015, 3, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Xue, Y.; Han, Y.; Lin, L.; Wu, C.; Xu, S.; Jiang, Z.; Xu, J.; Liu, Q.; Cao, X. The STAT3-Binding Long Noncoding RNA lnc-DC Controls Human Dendritic Cell Differentiation. Science 2014, 344, 310–313. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, X.; Chen, K.; Cheng, Y.; Liu, S.; Xia, M.; Chen, Y.; Zhu, H.; Li, Z.; Cao, X. CCR7 Chemokine Receptor-Inducible lnc-Dpf3 Restrains Dendritic Cell Migration by Inhibiting HIF-1α-Mediated Glycolysis. Immunity 2019, 50, 600–615. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, G.; Benzi, A.; Sturla, L.; Marubbi, D.; Frumento, D.; Spinelli, S.; Abbotto, E.; Ivaldi, F.; Von Holtey, M.; Murone, M.; et al. Sirt6 inhibition delays the onset of experimental autoimmune encephalomyelitis by reducing dendritic cell migration. J. Neuroinflamm. 2020, 17, 228. [Google Scholar] [CrossRef]
- Tsou, C.-L.; Peters, W.; Si, Y.; Slaymaker, S.; Aslanian, A.M.; Weisberg, S.; Mack, M.; Charo, I.F. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Investig. 2007, 117, 902–909. [Google Scholar] [CrossRef] [Green Version]
- Morse, M.A.; Coleman, R.E.; Akabani, G.; Niehaus, N.; Coleman, D.; Lyerly, H.K. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 1999, 59, 56–58. [Google Scholar]
- Shinde, P.; Fernandes, S.; Melinkeri, S.; Kale, V.; Limaye, L. Compromised functionality of monocyte-derived dendritic cells in multiple myeloma patients may limit their use in cancer immunotherapy. Sci. Rep. 2018, 8, 5705. [Google Scholar] [CrossRef] [PubMed]
- Moran, T.P.; Nakano, H.; Kondilis-Mangum, H.D.; Wade, P.A.; Cook, N.N. Epigenetic control of Ccr7 expression in distinct lineages of lung dendritic cells. J. Immunol. 2014, 193, 4904–4913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williford, J.-M.; Ishihara, J.; Ishihara, A.; Mansurov, A.; Hosseinchi, P.; Marchell, T.M.; Potin, L.; Swartz, M.A.; Hubbell, J.A. Recruitment of CD103+ dendritic cells via tumor-targeted chemokine delivery enhances efficacy of checkpoint inhibitor immunotherapy. Sci. Adv. 2019, 5, eaay1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böttcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis e Sousa, C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Shen, L.; Gan, J.; He, L.; Lin, J.; Guo, S.; Xiong, Z.; Lin, J.; Zhang, S. Integrative analysis identifies DNMTs against immune-infiltrating neutrophils and dendritic cells in colorectal cancer. Epigenetics 2019, 14, 392–404. [Google Scholar] [CrossRef]
- Lodewijk, I.; Nunes, S.P.; Henrique, R.; Jerónimo, C.; Dueñas, M.; Paramio, J.M. Tackling tumor microenvironment through epigenetic tools to improve cancer immunotherapy. Clin. Epigenet. 2021, 13, 1–24. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.D.; Rodriguez, P.C.; Koehn, B.H.; Baban, B.; Cui, Y.; Guo, G.; Shimoda, M.; Pacholczyk, R.; Shi, H.; Lee, E.-J.; et al. Activation of p53 in Immature Myeloid Precursor Cells Controls Differentiation into Ly6c+CD103+ Monocytic Antigen-Presenting Cells in Tumors. Immunity 2018, 48, 91–106. [Google Scholar] [CrossRef] [Green Version]
- Noubade, R.; Majri-Morrison, S.; Tarbell, K.V. Beyond cDC1: Emerging Roles of DC Crosstalk in Cancer Immunity. Front. Immunol. 2019, 10, 1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, P.; Shin, E.-C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC Class I Antigen Processing and Presenting Machinery: Organization, Function, and Defects in Tumor Cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, N.; Nixon, M.J.; Gonzalez-Ericsson, P.I.; Sanchez, V.; Opalenik, S.R.; Li, H.; Zahnow, C.A.; Nickels, M.L.; Liu, F.; Tantawy, M.N.; et al. DNA methyltransferase inhibition upregulates MHC-I to potentiate cytotoxic T lymphocyte responses in breast cancer. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Magner, W.J.; Kazim, A.L.; Stewart, C.; Romano, M.A.; Catalano, G.; Grande, C.; Keiser, N.; Santaniello, F.; Tomasi, T.B. Activation of MHC Class I, II, and CD40 Gene Expression by Histone Deacetylase Inhibitors. J. Immunol. 2000, 165, 7017–7024. [Google Scholar] [CrossRef] [PubMed]
- Kortenhorst, M.S.; Wissing, M.D.; Rodriguez, R.; Kachhap, S.K.; Jans, J.; Van Der Groep, P.; Verheul, H.; Gupta, A.; Aiyetan, O.P.; Van Der Wall, E.; et al. Analysis of the genomic response of human prostate cancer cells to histone deacetylase inhibitors. Epigenetics 2013, 8, 907–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardner, A.; Ruffell, B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016, 37, 855–865. [Google Scholar] [CrossRef] [Green Version]
- Antony, P.A.; Piccirillo, C.A.; Akpinarli, A.; Finkelstein, S.E.; Speiss, P.J.; Surman, D.R.; Palmer, D.; Chan, C.-C.; Klebanoff, C.; Overwijk, W.W.; et al. CD8+T Cell Immunity against a Tumor/Self-Antigen Is Augmented by CD4+T Helper Cells and Hindered by Naturally Occurring T Regulatory Cells. J. Immunol. 2005, 174, 2591–2601. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.C.; Bevan, M.J. Defective CD8 T Cell Memory Following Acute Infection Without CD4 T Cell Help. Science 2003, 300, 339–342. [Google Scholar] [CrossRef] [Green Version]
- Ferlazzo, G.; Morandi, B. Cross-Talks between Natural Killer Cells and Distinct Subsets of Dendritic Cells. Front. Immunol. 2014, 5, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garris, C.S.; Arlauckas, S.P.; Kohler, R.H.; Trefny, M.P.; Garren, S.; Piot, C.; Engblom, C.; Pfirschke, C.; Siwicki, M.; Gungabeesoon, J.; et al. Successful Anti-PD-1 Cancer Immunotherapy Requires T Cell-Dendritic Cell Crosstalk Involving the Cytokines IFN-γ and IL-12. Immunity 2018, 49, 1148–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.; Liu, J.; Liu, S.; Xia, M.; Zhang, X.; Han, D.; Jiang, Y.; Wang, C.; Cao, X. Methyltransferase SETD2-Mediated Methylation of STAT1 Is Critical for Interferon Antiviral Activity. Cell 2017, 170, 492–506. [Google Scholar] [CrossRef] [Green Version]
- Lotze, M.T. Getting to the Source: Dendritic Cells as Therapeutic Reagents for the Treatment of Patients with Cancer. Ann. Surg. 1997, 226, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Mattiuz, R.; Brousse, C.; Ambrosini, M.; Cancel, J.; Bessou, G.; Mussard, J.; Sanlaville, A.; Caux, C.; Bendriss-Vermare, N.; Valladeau-Guilemond, J.; et al. Type 1 conventional dendritic cells and interferons are required for spontaneous CD4+ and CD8+ T-cell protective responses to breast cancer. Clin. Transl. Immunol. 2021, 10, 1305. [Google Scholar] [CrossRef]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-redundant requirement for CXCR3 signalling during tumoricidal T-cell trafficking across tumour vascular checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef]
- Peng, D.; Kryczek, I.; Nagarsheth, N.; Zhao, L.; Wei, S.; Wang, W.; Sun, Y.; Zhao, E.; Vatan, L.; Szeliga, W.; et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature 2015, 527, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Bischoff, P.; Trinks, A.; Obermayer, B.; Pett, J.P.; Wiederspahn, J.; Uhlitz, F.; Liang, X.; Lehmann, A.; Jurmeister, P.; Elsner, A.; et al. Single-cell RNA sequencing reveals distinct tumor microenvironmental patterns in lung adenocarcinoma. Oncogene 2021, 40, 6748–6758. [Google Scholar] [CrossRef]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Cachot, A.; Bilous, M.; Liu, Y.-C.; Li, X.; Saillard, M.; Cenerenti, M.; Rockinger, G.A.; Wyss, T.; Guillaume, P.; Schmidt, J.; et al. Tumor-specific cytolytic CD4 T cells mediate immunity against human cancer. Sci. Adv. 2021, 7, eabe3348. [Google Scholar] [CrossRef]
- Sommermeyer, D.; Hudecek, M.; Kosasih, P.L.; Gogishvili, T.; Maloney, D.G.; Turtle, C.J.; Riddell, S.R. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia 2016, 30, 492–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerner, M.Y.; Casey, K.A.; Mescher, M.F. Defective MHC Class II Presentation by Dendritic Cells Limits CD4 T Cell Help for Antitumor CD8 T Cell Responses. J. Immunol. 2008, 181, 155–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, R.; Sherman, L.A. CD4+ T-Cell Help in the Tumor Milieu Is Required for Recruitment and Cytolytic Function of CD8+ T Lymphocytes. Cancer Res. 2010, 70, 8368–8377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Akpinarli, A.; Maris, C.; Hipkiss, E.L.; Lane, M.; Kwon, E.-K.M.; Muranski, P.; Restifo, N.P.; Antony, P.A. Naive tumor-specific CD4+ T cells differentiated in vivo eradicate established melanoma. J. Exp. Med. 2010, 207, 651–667. [Google Scholar] [CrossRef] [Green Version]
- Duong, E.; Fessenden, T.B.; Lutz, E.; Dinter, T.; Yim, L.; Blatt, S.; Bhutkar, A.; Wittrup, K.D.; Spranger, S. Type I interferon activates MHC class I-dressed CD11b+ conventional dendritic cells to promote protective anti-tumor CD8+ T cell immunity. Immunity 2021, 55, 308–323. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-J. IPC: Professional Type 1 Interferon-Producing Cells and Plasmacytoid Dendritic Cell Precursors. Annu. Rev. Immunol. 2005, 23, 275–306. [Google Scholar] [CrossRef] [PubMed]
- Jego, G.; Palucka, A.; Blanck, J.-P.; Chalouni, C.; Pascual, V.; Banchereau, J. Plasmacytoid Dendritic Cells Induce Plasma Cell Differentiation through Type I Interferon and Interleukin 6. Immunity 2003, 19, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Fang, T.C.; Schaefer, U.; Mecklenbrauker, I.; Stienen, A.; Dewell, S.; Chen, M.S.; Rioja, I.; Parravicini, V.; Prinjha, R.K.; Chandwani, R.; et al. Histone H3 lysine 9 di-methylation as an epigenetic signature of the interferon response. J. Exp. Med. 2012, 209, 661–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Wan, X.; Deng, Z.; Shi, L.; Hao, C.; Zhou, Z.; Zhou, C.; Fang, Y.; Liu, J.; Yang, J.; et al. Epigenetic regulator CXXC5 recruits DNA demethylase Tet2 to regulate TLR7/9-elicited IFN response in pDCs. J. Exp. Med. 2017, 214, 1471–1491. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, S.; Yang, Y.; Zhu, S.; Zhang, M.; Qiao, Y.; Liu, Y.-J.; Chen, J. TLR-activated plasmacytoid dendritic cells inhibit breast cancer cell growth in vitro and in vivo. Oncotarget 2016, 8, 11708–11718. [Google Scholar] [CrossRef] [Green Version]
- Conrad, C.; Gregorio, J.; Wang, Y.-H.; Ito, T.; Meller, S.; Hanabuchi, S.; Anderson, S.; Atkinson, N.; Ramirez, P.T.; Liu, Y.-J.; et al. Plasmacytoid Dendritic Cells Promote Immunosuppression in Ovarian Cancer via ICOS Costimulation of Foxp3+ T-Regulatory Cells. Cancer Res. 2012, 72, 5240–5249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poropatich, K.; Dominguez, D.; Chan, W.-C.; Andrade, J.; Zha, Y.; Wray, B.; Miska, J.; Qin, L.; Cole, L.; Coates, S.; et al. OX40+ plasmacytoid dendritic cells in the tumor microenvironment promote antitumor immunity. J. Clin. Investig. 2020, 130, 3528–3542. [Google Scholar] [CrossRef] [PubMed]
- Sisirak, V.; Vey, N.; Goutagny, N.; Renaudineau, S.; Malfroy, M.; Thys, S.; Treilleux, I.; Labidi-Galy, S.I.; Bachelot, T.; Dezutter-Dambuyant, C.; et al. Breast cancer-derived transforming growth factor-β and tumor necrosis factor-α compromise interferon-α production by tumor-associated plasmacytoid dendritic cells. Int. J. Cancer 2013, 133, 771–778. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, S.; Yang, J.; Ronchese, F. Monocyte-Derived Dendritic Cells Are Essential for CD8+ T Cell Activation and Antitumor Responses After Local Immunotherapy. Front. Immunol. 2015, 6, 584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davison, G.M.; Novitzky, N.; Abdulla, R. Monocyte derived dendritic cells have reduced expression of co-stimulatory molecules but are able to stimulate autologous T-cells in patients with MDS. Hematol. Stem Cell Ther. 2013, 6, 49–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Kim, G.; Kim, N.; Chu, H.; Park, B.-C.; Yang, J.S.; Han, S.H.; Yun, C.-H. Human CD141+ dendritic cells generated from adult peripheral blood monocytes. Cytotherapy 2019, 21, 1049–1063. [Google Scholar] [CrossRef]
- Wakim, L.M.; Waithman, J.; van Rooijen, N.; Heath, W.R.; Carbone, F.R. Dendritic Cell-Induced Memory T Cell Activation in Nonlymphoid Tissues. Science 2008, 319, 198–202. [Google Scholar] [CrossRef] [Green Version]
- De Vries, I.J.M.; Krooshoop, D.J.E.B.; Scharenborg, N.M.; Lesterhuis, W.J.; Diepstra, J.H.S.; Van Muijen, G.N.P.; Strijk, S.P.; Ruers, T.J.; Boerman, O.C.; Oyen, W.J.G.; et al. Effective migration of antigen-pulsed dendritic cells to lymph nodes in melanoma patients is determined by their maturation state. Cancer Res. 2003, 63, 12–17. [Google Scholar]
- Zhang, Q.; Zhao, K.; Shen, Q.; Han, Y.; Gu, Y.; Li, X.; Zhao, D.; Liu, Y.; Wang, C.; Zhang, X.; et al. Tet2 is required to resolve inflammation by recruiting Hdac2 to specifically repress IL-6. Nature 2015, 525, 389–393. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Liu, Y.; Mei, S.; Zhang, M.; Xin, J.; Zhang, Y.; Yang, R. MicroRNA-22 Impairs Anti-Tumor Ability of Dendritic Cells by Targeting p38. PLoS ONE 2015, 10, e0121510. [Google Scholar] [CrossRef] [Green Version]
- Ruffell, B.; Chang-Strachan, D.; Chan, V.; Rosenbusch, A.; Ho, C.M.; Pryer, N.; Daniel, D.; Hwang, E.S.; Rugo, H.S.; Coussens, L.M. Macrophage IL-10 Blocks CD8+ T Cell-Dependent Responses to Chemotherapy by Suppressing IL-12 Expression in Intratumoral Dendritic Cells. Cancer Cell 2014, 26, 623–637. [Google Scholar] [CrossRef] [Green Version]
- De Smedt, T.; van Mechelen, M.; De Becker, G.; Urbain, J.; Leo, O.; Moser, M. Effect of interleukin-10 on dendritic cell maturation and function. Eur. J. Immunol. 1997, 27, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.; O’Brien, T.F.; Wright, G.; Yang, J.; Shin, J.; Wright, K.L.; Zhong, X.-P. Critical Role of the Tumor Suppressor Tuberous Sclerosis Complex 1 in Dendritic Cell Activation of CD4 T Cells by Promoting MHC Class II Expression via IRF4 and CIITA. J. Immunol. 2013, 191, 699–707. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.-E.; Yu, H.-N.; Yoon, C.-H.; Bae, Y.-S. Tumor-mediated down-regulation of MHC class II in DC development is attributable to the epigenetic control of the CIITA type I promoter. Eur. J. Immunol. 2009, 39, 858–868. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, B.A.; Purves, T.; Palmer, S.; Sullenger, B.; Welty-Wolf, K.; Haines, K.; Agarwal, S.; Kasotakis, G. IL-10 and class 1 histone deacetylases act synergistically and independently on the secretion of proinflammatory mediators in alveolar macrophages. PLoS ONE 2021, 16, e0245169. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Matsuoka, K.; Sheikh, S.Z.; Russo, S.M.; Mishima, Y.; Collins, C.; Dezoeten, E.F.; Karp, C.; Ting, J.P.Y.; Sartor, R.B.; et al. IL-10 RegulatesIl12bExpression via Histone Deacetylation: Implications for Intestinal Macrophage Homeostasis. J. Immunol. 2012, 189, 1792–1799. [Google Scholar] [CrossRef] [Green Version]
- Villagra, A.; Cheng, F.; Wang, H.-W.; Suarez, I.; Glozak, M.; Maurin, M.; Nguyen, D.; Wright, K.L.; Atadja, P.W.; Bhalla, K.; et al. The histone deacetylase HDAC11 regulates the expression of interleukin 10 and immune tolerance. Nat. Immunol. 2008, 10, 92–100. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, H.; Ohno, Y.; Toyoshima, Y.; Ohtake, J.; Homma, S.; Kawamura, H.; Takahashi, N.; Taketomi, A. Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 2017, 108, 1947–1952. [Google Scholar] [CrossRef]
- Català-Moll, F.; Ferreté-Bonastre, A.G.; Godoy-Tena, G.; Morante-Palacios, O.; Ciudad, L.; Barberà, L.; Fondelli, F.; Martínez-Cáceres, E.M.; Rodríguez-Ubreva, J.; Li, T.; et al. Vitamin D receptor, STAT3, and TET2 cooperate to establish tolerogenesis. Cell Rep. 2022, 38, 110244. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.; Keskinov, A.A.; Shurin, G.V.; Shurin, M. Tumor-derived factors modulating dendritic cell function. Cancer Immunol. Immunother. 2016, 65, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Pugh, S.; Thomas, A.G. Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2. Gut 1994, 35, 675–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sombroek, C.C.; Stam, A.G.M.; Masterson, A.J.; Lougheed, S.M.; Schakel, M.J.A.G.; Meijer, C.J.L.M.; Pinedo, H.M.; Eertwegh, A.J.M.V.D.; Scheper, R.J.; De Gruijl, T.D. Prostanoids Play a Major Role in the Primary Tumor-Induced Inhibition of Dendritic Cell Differentiation. J. Immunol. 2002, 168, 4333–4343. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Wang, D.; Kim, S.-H.; Katoh, H.; Dubois, R.N. Prostaglandin E2 promotes intestinal tumor growth via DNA methylation. Nat. Med. 2012, 18, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998, 92, 4150–4166. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, A.S.; Somja, J.; Duray, A.; Guénin, S.; Roncarati, P.; Delvenne, O.P.; Herfs, M.F.; Hubert, P.M. Cervical (pre)neoplastic microenvironment promotes the emergence of tolerogenic dendritic cells via RANKL secretion. OncoImmunology 2015, 4, e1008334. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Gerber, H.-P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K.; Arihiro, K. Tumor-Driven Evolution of Immunosuppressive Networks during Malignant Progression. Cancer Res. 2006, 66, 5527–5536. [Google Scholar] [CrossRef] [Green Version]
- Roland, C.L.; Lynn, K.D.; Toombs, J.E.; Dineen, S.; Udugamasooriya, D.G.; Brekken, R.A. Cytokine Levels Correlate with Immune Cell Infiltration after Anti-VEGF Therapy in Preclinical Mouse Models of Breast Cancer. PLoS ONE 2009, 4, e7669. [Google Scholar] [CrossRef]
- Kel, J.M.; Girard-Madoux, M.J.H.; Reizis, B.; Clausen, B.E.; Van De Laar, L.; Buitenhuis, M.; Wensveen, F.M.; Janssen, H.L.; Coffer, P.J.; Woltman, A.M. TGF-β Is Required to Maintain the Pool of Immature Langerhans Cells in the Epidermis. J. Immunol. 2010, 185, 3248–3255. [Google Scholar] [CrossRef] [Green Version]
- Lievens, D.; Habets, K.L.; Robertson, A.-K.; Laouar, Y.; Winkels, H.; Rademakers, T.; Beckers, L.; Wijnands, E.; Boon, L.; Mosaheb, M.; et al. Abrogated transforming growth factor beta receptor II (TGFβRII) signalling in dendritic cells promotes immune reactivity of T cells resulting in enhanced atherosclerosis. Eur. Heart J. 2013, 34, 3717–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Min, S.; Lui, Y.; Sun, J.; Su, X.; Liu, Y.; Zhang, Y.; Han, D.; Che, Y.; Zhao, C.; et al. Global mapping of H3K4me3 and H3K27me3 reveals chromatin state-based regulation of human monocyte-derived dendritic cells in different environments. Genes Immun. 2012, 13, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Hmadcha, A.; Bedoya, F.; Sobrino, F.; Pintado, E. Methylation-Dependent Gene Silencing Induced by Interleukin 1β via Nitric Oxide Production. J. Exp. Med. 1999, 190, 1595–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; Shields, C.E.D.; Niknafs, N.; Yen, R.-W.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300. [Google Scholar] [CrossRef] [Green Version]
- Nirschl, C.J.; Suárez-Fariñas, M.; Izar, B.; Prakadan, S.; Dannenfelser, R.; Tirosh, I.; Liu, Y.; Zhu, Q.; Devi, K.S.P.; Carroll, S.L.; et al. IFNγ-Dependent Tissue-Immune Homeostasis Is Co-opted in the Tumor Microenvironment. Cell 2017, 170, 127–141. [Google Scholar] [CrossRef] [Green Version]
- Gottfried, E.; Kunz-Schughart, L.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef]
- Kang, K.A.; Zhang, R.; Kim, G.Y.; Bae, S.C.; Hyun, J.W. Epigenetic changes induced by oxidative stress in colorectal cancer cells: Methylation of tumor suppressor RUNX3. Tumor Biol. 2012, 33, 403–412. [Google Scholar] [CrossRef]
- Zhang, R.; Kang, K.A.; Kim, K.C.; Na, S.-Y.; Chang, W.Y.; Kim, G.Y.; Kim, H.S.; Hyun, J.W. Oxidative stress causes epigenetic alteration of CDX1 expression in colorectal cancer cells. Gene 2013, 524, 214–219. [Google Scholar] [CrossRef]
- O’Hagan, H.; Wang, W.; Sen, S.; Shields, C.D.; Lee, S.; Zhang, Y.W.; Clements, E.G.; Cai, Y.; Van Neste, L.; Easwaran, H.; et al. Oxidative Damage Targets Complexes Containing DNA Methyltransferases, SIRT1, and Polycomb Members to Promoter CpG Islands. Cancer Cell 2011, 20, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Obermajer, N.; Muthuswamy, R.; Lesnock, J.; Edwards, R.P.; Kalinski, P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011, 118, 5498–5505. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Bronte, V.; Chen, S.-H.; Colombo, M.P.; Ochoa, A.; Ostrand-Rosenberg, S.; Schreiber, H. The Terminology Issue for Myeloid-Derived Suppressor Cells. Cancer Res. 2007, 67, 425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Li, C.; Liu, T.; Dai, X.; Bazhin, A.V. Myeloid-Derived Suppressor Cells in Tumors: From Mechanisms to Antigen Specificity and Microenvironmental Regulation. Front. Immunol. 2020, 11, 1371. [Google Scholar] [CrossRef] [PubMed]
- Di Blasio, S.; Van Wigcheren, G.F.; Becker, A.; Van Duffelen, A.; Gorris, M.; Verrijp, K.; Stefanini, I.; Bakker, G.J.; Bloemendal, M.; Halilovic, A.; et al. The tumour microenvironment shapes dendritic cell plasticity in a human organotypic melanoma culture. Nat. Commun. 2020, 11, 2749. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K. Reactive Oxygen Species as Regulators of MDSC-Mediated Immune Suppression. Front. Immunol. 2018, 9, 2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sacchi, A.; Tumino, N.; Sabatini, A.; Cimini, E.; Casetti, R.; Bordoni, V.; Grassi, G.; Agrati, C. Myeloid-Derived Suppressor Cells Specifically Suppress IFN-γ Production and Antitumor Cytotoxic Activity of Vδ2 T Cells. Front. Immunol. 2018, 9, 1271. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Xu, B.; Luan, B.; Zhang, Y.; Li, Y.; Xiong, X.; Shi, H. Myeloid-derived suppressor cells (MDSCs) and mechanistic target of rapamycin (mTOR) signaling pathway interact through inducible nitric oxide synthase (iNOS) and nitric oxide (NO) in asthma. Am. J. Transl. Res. 2019, 11, 6170–6184. [Google Scholar]
- Redd, P.S.; Ibrahim, M.L.; Klement, J.D.; Sharman, S.K.; Paschall, A.V.; Yang, D.; Nayak-Kapoor, A.; Liu, K. SETD1B Activates iNOS Expression in Myeloid-Derived Suppressor Cells. Cancer Res. 2017, 77, 2834–2843. [Google Scholar] [CrossRef] [Green Version]
- Nair, V.S.; Saleh, R.; Toor, S.M.; Taha, R.Z.; Ahmed, A.A.; Kurer, M.A.; Murshed, K.; Alajez, N.M.; Abu Nada, M.; Elkord, E. Transcriptomic profiling disclosed the role of DNA methylation and histone modifications in tumor-infiltrating myeloid-derived suppressor cell subsets in colorectal cancer. Clin. Epigenet. 2020, 12, 13. [Google Scholar] [CrossRef] [Green Version]
- Fleming, V.; Hu, X.; Weber, R.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Targeting Myeloid-Derived Suppressor Cells to Bypass Tumor-Induced Immunosuppression. Front. Immunol. 2018, 9, 398. [Google Scholar] [CrossRef]
- Rosenzweig, J.M.; Glenn, J.D.; Calabresi, P.A.; Whartenby, K.A. KLF4 Modulates Expression of IL-6 in Dendritic Cells via Both Promoter Activation and Epigenetic Modification. J. Biol. Chem. 2013, 288, 23868–23874. [Google Scholar] [CrossRef] [Green Version]
- Sido, J.M.; Yang, X.; Nagarkatti, P.S.; Nagarkatti, M. Δ9-Tetrahydrocannabinol-mediated epigenetic modifications elicit myeloid-derived suppressor cell activation via STAT3/S100A8. J. Leukoc. Biol. 2015, 97, 677–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Liu, Q.; Mi, S.; Liang, X.; Zhang, Z.; Su, X.; Liu, J.; Chen, Y.; Wang, M.; Zhang, Y.; et al. Both miR-17-5p and miR-20a Alleviate Suppressive Potential of Myeloid-Derived Suppressor Cells by Modulating STAT3 Expression. J. Immunol. 2011, 186, 4716–4724. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Ubreva, J.; Català-Moll, F.; Obermajer, N.; Álvarez-Errico, D.; Ramirez, R.N.; Company, C.; Vento-Tormo, R.; Moreno-Bueno, G.; Edwards, R.P.; Mortazavi, A.; et al. Prostaglandin E2 Leads to the Acquisition of DNMT3A-Dependent Tolerogenic Functions in Human Myeloid-Derived Suppressor Cells. Cell Rep. 2017, 21, 154–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahakian, E.; Powers, J.; Chen, J.; Deng, S.L.; Cheng, F.; Distler, A.; Woods, D.M.; Rock-Klotz, J.; Sodre, A.L.; Youn, J.-I.; et al. Histone deacetylase 11: A novel epigenetic regulator of myeloid derived suppressor cell expansion and function. Mol. Immunol. 2015, 63, 579–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.N.; Chamberlain, E.D. Sipuleucel-T in the treatment of prostate cancer: An evidence-based review of its place in therapy. Core Évid. 2014, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mougel, A.; Terme, M.; Tanchot, C. Therapeutic Cancer Vaccine and Combinations with Antiangiogenic Therapies and Immune Checkpoint Blockade. Front. Immunol. 2019, 10, 467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau-Kilby, A.; Kretz, C.C.; Pechhold, S.; Price, J.D.; Dorta, S.; Ramos, H.; Trinchieri, G.; Tarbell, K.V. Interleukin-2 inhibits FMS-like tyrosine kinase 3 receptor ligand (flt3L)-dependent development and function of conventional and plasmacytoid dendritic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 2408–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inaba, K.; Yashiro, T.; Hiroki, I.; Watanabe, R.; Kasakura, K.; Nishiyama, C. Dual Roles of PU.1 in the Expression of PD-L2: Direct Transactivation with IRF4 and Indirect Epigenetic Regulation. J. Immunol. 2020, 205, ji1901008. [Google Scholar] [CrossRef]
- Kirkling, M.E.; Cytlak, U.; Lau, C.M.; Lewis, K.L.; Resteu, A.; Khodadadi-Jamayran, A.; Siebel, C.W.; Salmon, H.; Merad, M.; Tsirigos, A.; et al. Notch Signaling Facilitates In Vitro Generation of Cross-Presenting Classical Dendritic Cells. Cell Rep. 2018, 23, 3658–3672. [Google Scholar] [CrossRef]
- Riol-Blanco, L.; Sánchez-Sánchez, N.; Torres, A.; Tejedor, A.; Narumiya, S.; Corbí, A.L.; Sánchez-Mateos, P.; Fernandez, J.L.R. The Chemokine Receptor CCR7 Activates in Dendritic Cells Two Signaling Modules That Independently Regulate Chemotaxis and Migratory Speed. J. Immunol. 2005, 174, 4070–4080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doñas, C.; Carrasco, M.; Fritz, M.; Prado, C.; Tejón, G.; Osorio-Barrios, F.; Manríquez, V.; Reyes, P.; Pacheco, R.; Bono, M.R.; et al. The histone demethylase inhibitor GSK-J4 limits inflammation through the induction of a tolerogenic phenotype on DCs. J. Autoimmun. 2016, 75, 105–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Xie, X.; Xiao, Y.; Hu, H.; Zou, Q.; Cheng, X.; Sun, S.-C. Epigenetic regulation of the expression of Il12 and Il23 and autoimmune inflammation by the deubiquitinase Trabid. Nat. Immunol. 2016, 17, 259–268. [Google Scholar] [CrossRef]
- Zhou, Z.; Chen, H.; Xie, R.; Wang, H.; Li, S.; Xu, Q.; Xu, N.; Cheng, Q.; Qian, Y.; Huang, R.; et al. Epigenetically modulated FOXM 1 suppresses dendritic cell maturation in pancreatic cancer and colon cancer. Mol. Oncol. 2019, 13, 873–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragale, A.; Romagnoli, G.; Licursi, V.; Buoncervello, M.; Del Vecchio, G.; Giuliani, C.; Parlato, S.; Leone, C.; De Angelis, M.; Canini, I.; et al. Antitumor Effects of Epidrug/IFNα Combination Driven by Modulated Gene Signatures in Both Colorectal Cancer and Dendritic Cells. Cancer Immunol. Res. 2017, 5, 604–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okada, N.; Mori, N.; Koretomo, R.; Okada, Y.; Nakayama, T.; Yoshie, O.; Mizuguchi, H.; Hayakawa, T.; Nakagawa, S.; Mayumi, T.; et al. Augmentation of the migratory ability of DC-based vaccine into regional lymph nodes by efficient CCR7 gene transduction. Gene Ther. 2004, 12, 129–139. [Google Scholar] [CrossRef]
- Guenther, C.; Faisal, I.; Fusciello, M.; Sokolova, M.; Harjunpää, H.; Ilander, M.; Tallberg, R.; Vartiainen, M.K.; Alon, R.; Gonzalez-Granado, J.-M.; et al. β2-Integrin Adhesion Regulates Dendritic Cell Epigenetic and Transcriptional Landscapes to Restrict Dendritic Cell Maturation and Tumor Rejection. Cancer Immunol. Res. 2021, 9, 1354–1369. [Google Scholar] [CrossRef]
- Wang, J.; Iwanowycz, S.; Yu, F.; Jia, X.; Leng, S.; Wang, Y.; Li, W.; Huang, S.; Ai, W.; Fan, D. microRNA-155 deficiency impairs dendritic cell function in breast cancer. OncoImmunology 2016, 5, e1232223. [Google Scholar] [CrossRef] [Green Version]
- Alcaraz-Serna, A.; Bustos-Morán, E.; Fernández-Delgado, I.; Calzada-Fraile, D.; Torralba, D.; Marina-Zárate, E.; Lorenzo-Vivas, E.; Vázquez, E.; de Alburquerque, J.B.; Ruef, N.; et al. Immune synapse instructs epigenomic and transcriptomic functional reprogramming in dendritic cells. Sci. Adv. 2021, 7, eabb9965. [Google Scholar] [CrossRef]
- Blaschke, K.; Ebata, K.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
- Magrì, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chilà, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707. [Google Scholar] [CrossRef]
- Lowenfeld, L.; Mick, R.; Datta, J.; Xu, S.; Fitzpatrick, E.; Fisher, C.S.; Fox, K.R.; DeMichele, A.; Zhang, P.J.; Weinstein, S.P.; et al. Dendritic Cell Vaccination Enhances Immune Responses and Induces Regression of HER2pos DCIS Independent of Route: Results of Randomized Selection Design Trial. Clin. Cancer Res. 2017, 23, 2961–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Almeida Nagata, D.E.; Chiang, E.Y.; Jhunjhunwala, S.; Caplazi, P.; Arumugam, V.; Modrusan, Z.; Chan, E.; Merchant, M.; Jin, L.; Arnott, D.; et al. Regulation of Tumor-Associated Myeloid Cell Activity by CBP/EP300 Bromodomain Modulation of H3K27 Acetylation. Cell Rep. 2019, 27, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orillion, A.; Hashimoto, A.; Damayanti, N.; Shen, L.; Adelaiye-Ogala, R.; Arisa, S.; Chintala, S.; Ordentlich, P.; Kao, C.; Elzey, B.; et al. Entinostat Neutralizes Myeloid-Derived Suppressor Cells and Enhances the Antitumor Effect of PD-1 Inhibition in Murine Models of Lung and Renal Cell Carcinoma. Clin. Cancer Res. 2017, 23, 5187–5201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christmas, B.J.; Rafie, C.I.; Hopkins, A.C.; Scott, B.A.; Ma, H.S.; Cruz, K.A.; Woolman, S.; Armstrong, T.D.; Connolly, R.M.; Azad, N.A.; et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yi, H.; Wang, C.; He, H.; Li, P.; Pan, H.; Sheng, N.; Ji, M.; Cai, L.; Ma, Y. Integrated Nanovaccine with MicroRNA-148a Inhibition Reprograms Tumor-Associated Dendritic Cells by Modulating miR-148a/DNMT1/SOCS1 Axis. J. Immunol. 2016, 197, 1231–1241. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godoy-Tena, G.; Ballestar, E. Epigenetics of Dendritic Cells in Tumor Immunology. Cancers 2022, 14, 1179. https://doi.org/10.3390/cancers14051179
Godoy-Tena G, Ballestar E. Epigenetics of Dendritic Cells in Tumor Immunology. Cancers. 2022; 14(5):1179. https://doi.org/10.3390/cancers14051179
Chicago/Turabian StyleGodoy-Tena, Gerard, and Esteban Ballestar. 2022. "Epigenetics of Dendritic Cells in Tumor Immunology" Cancers 14, no. 5: 1179. https://doi.org/10.3390/cancers14051179
APA StyleGodoy-Tena, G., & Ballestar, E. (2022). Epigenetics of Dendritic Cells in Tumor Immunology. Cancers, 14(5), 1179. https://doi.org/10.3390/cancers14051179