
Lynch-like Syndrome: Potential Mechanisms and Management

 , , , , and
, , , , and 
Simple Summary
Abstract
1. Introduction
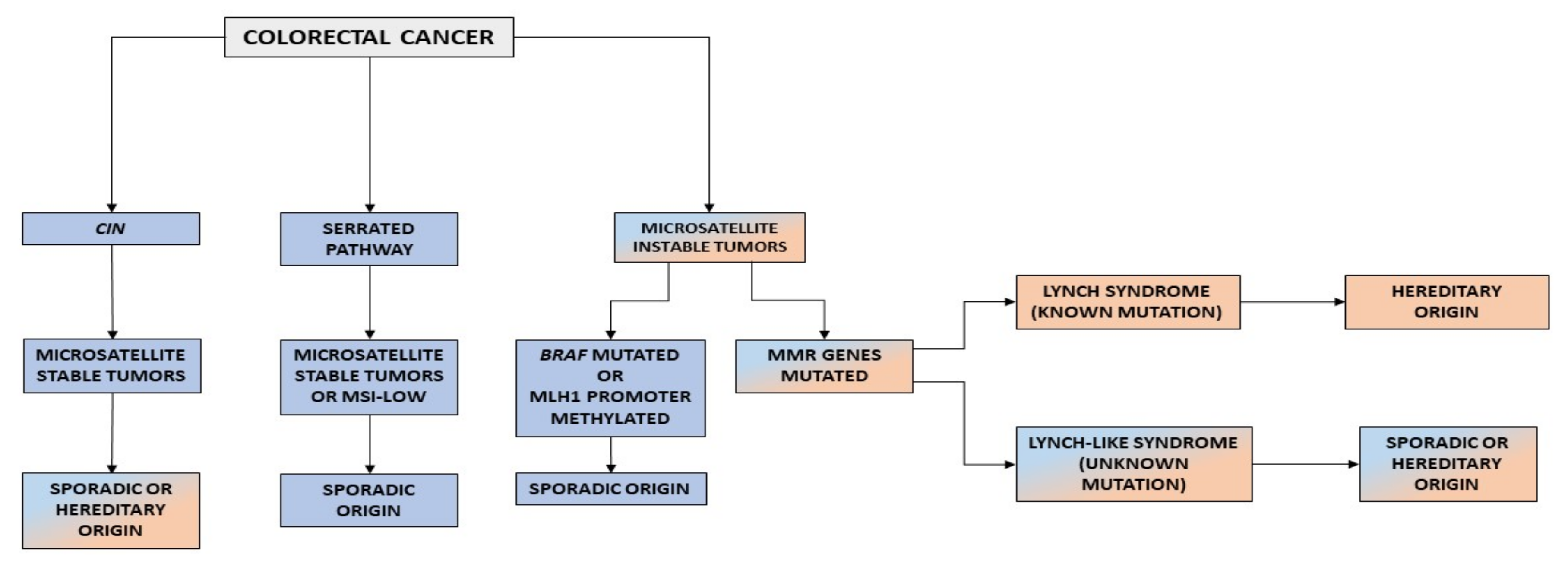
2. Carcinogenic Pathways in Colorectal Cancer
3. Lynch Syndrome
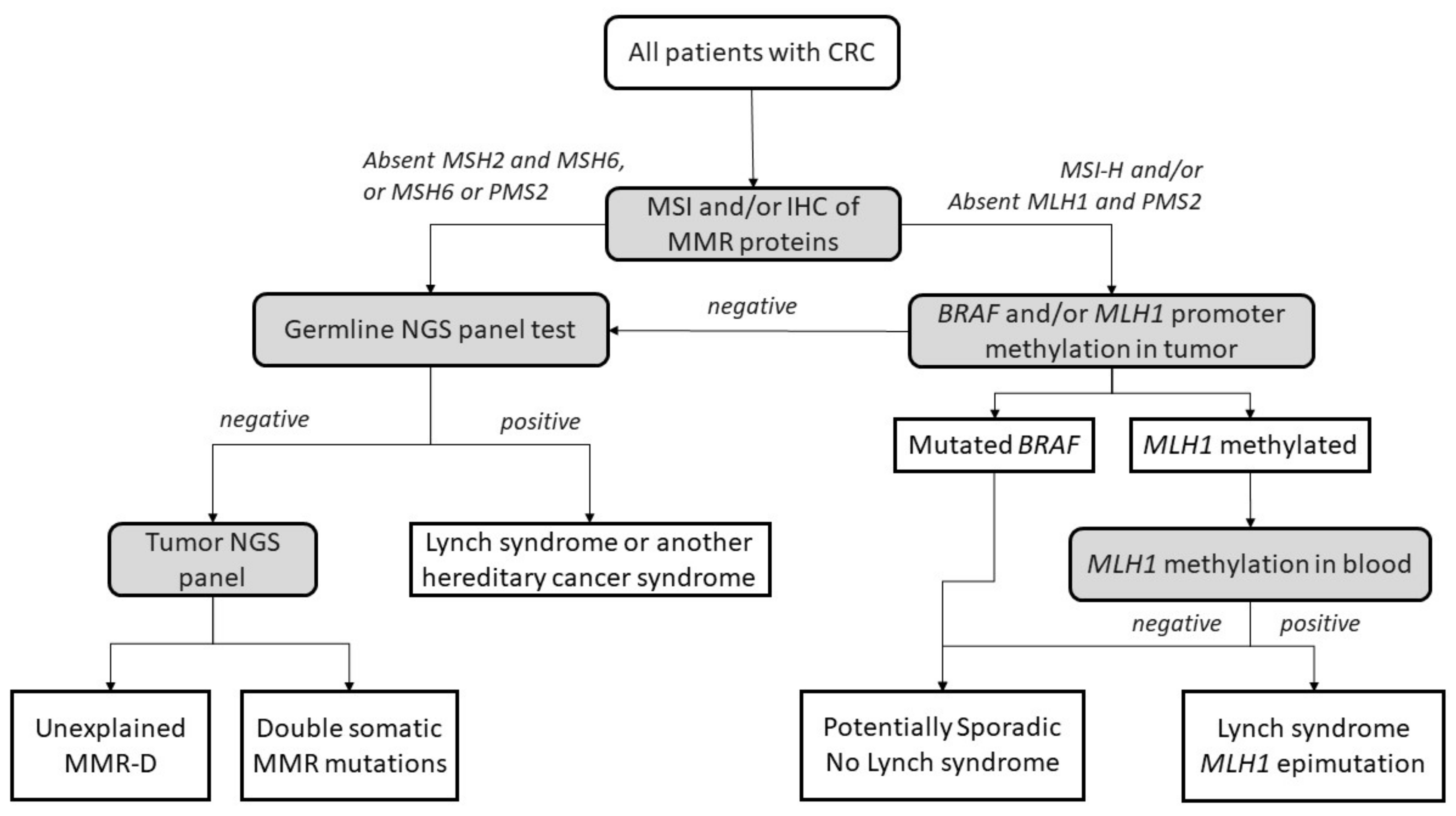
4. Lynch-like Syndrome
4.1. Demographics
4.2. Family History
4.3. Pathology and IHC
4.4. Cancer Risk
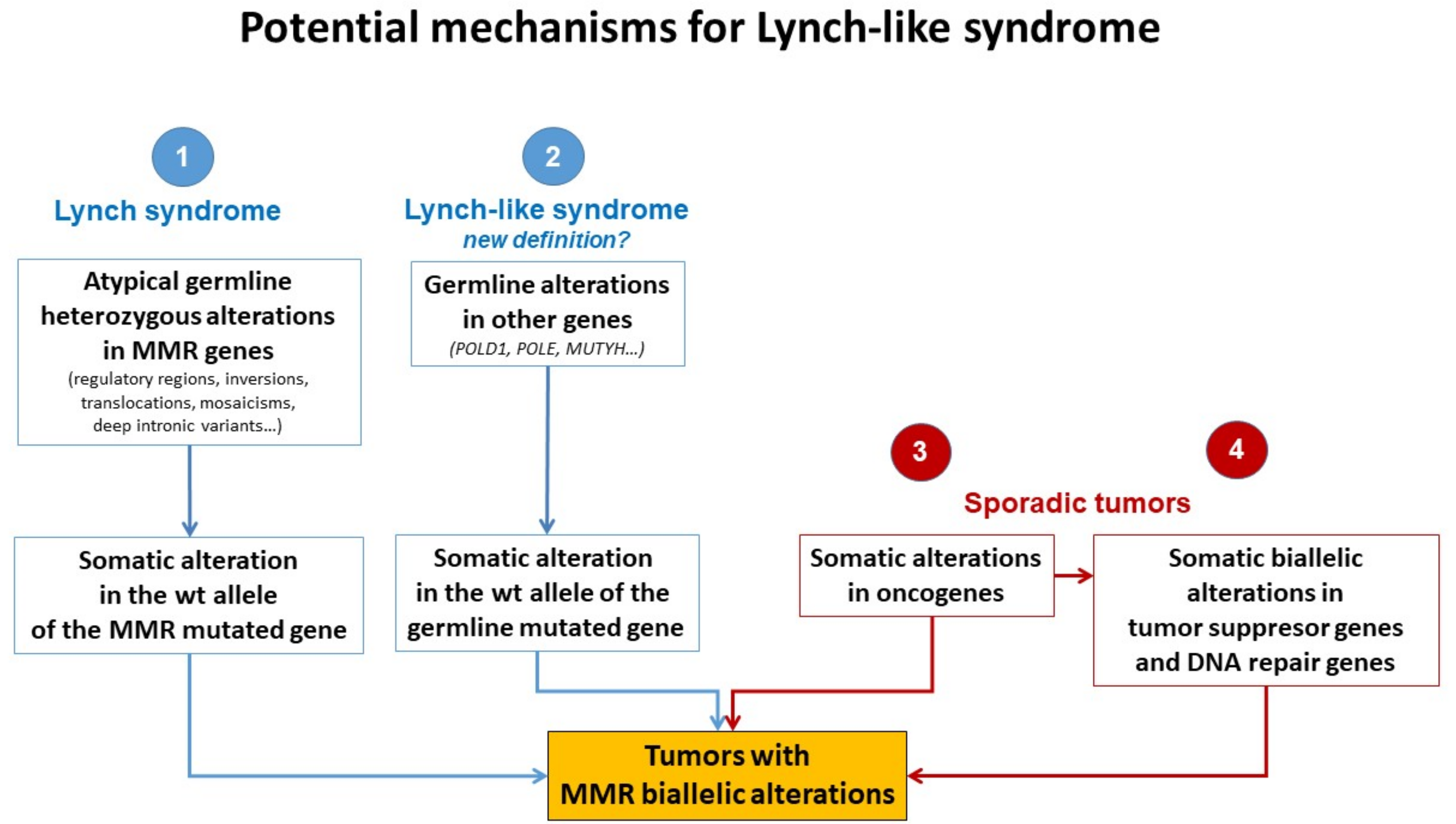
5. Potential Causes of Lynch-like Syndrome
5.1. Germline Mutations in Other Genes Affecting the MMR System
5.2. Hereditary Cases: Unknown Mutations in MMR Genes
5.3. Somatic Alteration in Other Cancer Genes or Epigenetic Structures
5.4. Somatic Biallelic Alteration in MMR
6. Future Research
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rodríguez–Soler, M.; Pérez–Carbonell, L.; Guarinos, C.; Zapater, P.; Castillejo, A.; Barberá, V.M.; Juárez, M.; Bessa, X.; Xicola, R.M.; Clofent, J.; et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology 2013, 144, 926–932. [Google Scholar] [CrossRef]
- Mensenkamp, A.R.; Vogelaar, I.P.; van Zelst–Stams, W.A.; Goossens, M.; Ouchene, H.; Hendriks–Cornelissen, S.J.; Kwint, M.P.; Hoogerbrugge, N.; Nagtegaal, I.D.; Ligtenberg, M.J. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in lynch syndrome-like tumors. Gastroenterology 2014, 146, 643–646. [Google Scholar] [CrossRef]
- Geurts-Giele, W.R.R.; Leenen, C.H.M.; Dubbink, H.J.; Meijssen, I.C.; Post, E.; Sleddens, H.F.B.M.; Kuipers, E.J.; Goverde, A.; Ouweland, A.M.W.V.D.; Van Lier, M.G.F.; et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J. Pathol. 2014, 234, 548–559. [Google Scholar] [CrossRef] [PubMed]
- Picó, M.D.; Sánchez-Heras, A.B.; Castillejo, A.; Giner-Calabuig, M.; Alustiza, M.; Sánchez, A.; Moreira, L.; Pellise, M.; Castells, A.; Llort, G.; et al. Risk of Cancer in Family Members of Patients with Lynch-Like Syndrome. Cancers 2020, 12, 2225. [Google Scholar] [CrossRef]
- World Health Organisation. Globocan. Int. Agency Res. 2020, 419, 3–4. Available online: https://ascopost.com/news/december-2020/globocan-2020-database-provides-latest-global-data-on-cancer-burden-cancer-deaths (accessed on 23 January 2022).
- Valle, L. Genetic predisposition to colorectal cancer: Where we stand and future perspectives. World J. Gastroenterol. 2014, 20, 9828–9849. [Google Scholar] [CrossRef]
- Sieber, O.M.; Heinimann, K.; Tomlinson, I.P.M. Genomic Instability—The Engine of Tumorigenesis? Nat. Rev. Cancer 2003, 3, 701–708. [Google Scholar] [CrossRef]
- Grady, W.M.; Carethers, J.M. Genomic and Epigenetic Instability in Colorectal Cancer Pathogenesis. Gastroenterology 2008, 135, 1079–1099. [Google Scholar] [CrossRef]
- Yuen, K.W.; Desai, A. The wages of CIN. J. Cell Biol. 2008, 180, 661–663. [Google Scholar] [CrossRef]
- Popat, S.; Hubner, R.; Houlston, R.S. Systematic Review of Microsatellite Instability and Colorectal Cancer Prognosis. J. Clin. Oncol. 2005, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of Chromosomal Instability. Curr. Biol. 2010, 20, R285–R295. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Gil, J.; Schwartz, S.; Perucho, M. Frameshift mutations in Fas, Apaf-1, and Bcl-10 in gastro-intestinal cancer of the microsatellite mutator phenotype. Cell Death Differ. 2000, 7, 238–239. [Google Scholar] [CrossRef]
- Bettington, M.; Walker, N.; Clouston, A.; Brown, I.; Leggett, B.; Whitehall, V. The serrated pathway to colorectal carcinoma: Current concepts and challenges. Histopathology 2013, 62, 367–386. [Google Scholar] [CrossRef]
- Togo, G.; Shiratori, Y.; Okamoto, M.; Yamaji, Y.; Matsumura, M.; Sano, T.; Motojima, T.; Omata, M. Relationship between grade of microsatellite instability and target genes of mismatch repair pathways in sporadic colorectal carcinoma. Am. J. Dig. Dis. 2001, 46, 1615–1622. [Google Scholar]
- Boland, C.R.; Goel, A. Microsatellite Instability in Colorectal Cancer. Gastroenterology 2010, 138, 2073–2087. [Google Scholar] [CrossRef]
- Vilar, E.; Gruber, S.B. Microsatellite instability in colorectal cancer—The stable evidence. Nat. Rev. Clin. Oncol. 2010, 7, 153–162. [Google Scholar] [CrossRef]
- Imai, K.; Yamamoto, H. Carcinogenesis and microsatellite instability: The interrelationship between genetics and epigenetics. Carcinogenesis 2008, 29, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-S.; Lee, C.W.; Shun, C.-T.; Wang, H.P.; Lee, W.J.; Sheu, J.-C.; Lin, J.T. Clinicopathological significance of altered loci of replication error and microsatellite instability-associated mutations in gastric cancer. Cancer Res. 1998, 58, 1494–1497. [Google Scholar]
- Xicola, R.M.; Llor, X.; Pons, E.; Castells, A.; Alenda, C.; Piñol, V.; Andreu, M.; Castellvi-Bel, S.; Payá, A.; Jover, R.; et al. Performance of Different Microsatellite Marker Panels for Detection of Mismatch Repair–Deficient Colorectal Tumors. JNCI J. Natl. Cancer Inst. 2007, 99, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Young, J.; Simms, L.A.; Biden, K.G.; Wynter, C.; Whitehall, V.; Karamatic, R.; George, J.; Goldblatt, J.; Walpole, I.; Robin, S.-A.; et al. Features of Colorectal Cancers with High-Level Microsatellite Instability Occurring in Familial and Sporadic Settings: Parallel Pathways of Tumorigenesis. Am. J. Pathol. 2001, 159, 2107–2116. [Google Scholar] [CrossRef]
- Inamura, K. Colorectal Cancers: An Update on Their Molecular Pathology. Cancers 2018, 10, 26. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; De La Chapelle, A. Genetic susceptibility to non-polyposis colorectal cancer. J. Med. Genet. 1999, 36, 801–818. [Google Scholar] [PubMed]
- De La Chapelle, A. Genetic predisposition to colorectal cancer. Nat. Rev. Cancer 2004, 4, 769–780. [Google Scholar] [CrossRef]
- Gazzoli, I.; Loda, M.; Garber, J.; Syngal, S.; Kolodner, R.D. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002, 62, 3925–3928. [Google Scholar]
- Hitchins, M.; Williams, R.; Cheong, K.; Halani, N.; Lin, V.A.; Packham, D.; Ku, S.; Buckle, A.; Hawkins, N.; Burn, J.; et al. MLH1 Germline Epimutations as a Factor in Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 2005, 129, 1392–1399. [Google Scholar] [CrossRef]
- Hitchins, M.P.; Wong, J.J.; Suthers, G.; Suter, C.M.; Martin, D.I.; Hawkins, N.J.; Ward, R.L. Inheritance of a Cancer-Associated MLH1 Germ-Line Epimutation. N. Engl. J. Med. 2007, 356, 697–705. [Google Scholar] [CrossRef]
- Morak, M.; Schackert, H.K.; Rahner, N.; Betz, B.; Ebert, M.; Walldorf, C.; Royer-Pokora, B.; Schulmann, K.; von Knebel-Doeberitz, M.; Dietmaier, W.; et al. Further evidence for heritability of an epimutation in one of 12 cases with MLH1 promoter methylation in blood cells clinically displaying HNPCC. Eur. J. Hum. Genet. 2008, 16, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Hitchins, M.P.; Ward, R. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J. Med. Genet. 2009, 46, 793–802. [Google Scholar] [CrossRef]
- Chan, T.L.; Yuen, S.T.; Kong, C.K.; Chan, Y.W.; Chan, A.S.; Ng, W.F.; Tsui, W.Y.; Lo, M.W.; Tam, W.Y.; Li, V.S.; et al. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat. Genet. 2006, 38, 1178–1183. [Google Scholar] [CrossRef]
- Valle, L.; Carbonell, P.; Fernández, V.; Dotor, A.; Sanz, M.; Benítez, J.; Urioste, M. MLH1 germline epimutations in selected patients with early-onset non-polyposis colorectal cancer. Clin. Genet. 2007, 71, 232–237. [Google Scholar] [CrossRef]
- Castillejo, A.; Vargas-Parra, G.; Castillejo, M.I.; Navarro, M.; Barbera, V.-M.; González, S.; Hernández-Illán, E.; Brunet, J.; Cajal, T.R.Y.; Balmaña, J.; et al. Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur. J. Cancer 2014, 50, 2241–2250. [Google Scholar] [CrossRef] [PubMed]
- Cini, G.; Carnevali, I.; Quaia, M.; Chiaravalli, A.M.; Sala, P.; Giacomini, E.; Maestro, R.; Tibiletti, M.G.; Viel, A. Concomitant mutation and epimutation of the MLH1 gene in a Lynch syndrome family. Carcinogenesis 2015, 36, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Kidambi, T.D.; Blanco, A.; Van Ziffle, J.; Terdiman, J.P. Constitutional MLH1 methylation presenting with colonic polyposis syndrome and not Lynch syndrome. Fam. Cancer 2016, 15, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Hitchins, M.P. Finding the needle in a haystack: Identification of cases of Lynch syndrome with MLH1 epimutation. Fam. Cancer 2016, 15, 413–422. [Google Scholar] [CrossRef]
- Crépin, M.; Dieu, M.-C.; Lejeune, S.; Escande, F.; Boidin, D.; Porchet, N.; Morin, G.; Manouvrier, S.; Mathieu, M.; Buisine, M.-P. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum. Mutat. 2011, 33, 180–188. [Google Scholar] [CrossRef]
- Castillejo, A.; Hernández-Illán, E.; Rodriguez-Soler, M.; Pérez-Carbonell, L.; Egoavil, C.; Barberá, V.M.; Castillejo, M.-I.; Guarinos, C.; Martínez-De-Dueñas, E.; Juan, M.-J.; et al. Prevalence ofMLH1constitutional epimutations as a cause of Lynch syndrome in unselected versus selected consecutive series of patients with colorectal cancer. J. Med. Genet. 2015, 52, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Vasen, A.H.F.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut 2013, 62, 812–823. [Google Scholar] [CrossRef] [PubMed]
- Lynch, H.T.; Boland, C.R.; Rodriguez-Bigas, M.A.; Amos, C.; Lynch, J.P.; Lynch, P.M. Who should be sent for genetic testing in hereditary colorectal cancer syndromes? J. Clin. Oncol. 2007, 25, 3534–3542. [Google Scholar] [CrossRef]
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Nakagawa, H.; Sotamaa, K.; Prior, T.W.; Westman, J.; et al. Screening for the Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer). N. Engl. J. Med. 2005, 352, 1851–1860. [Google Scholar] [CrossRef]
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Cancer 2015, 15, 181–194. [Google Scholar] [CrossRef]
- Risio, M.; Reato, G.; Di Celle, P.F.; Fizzotti, M.; Rossini, F.P.; Foà, R. Microsatellite instability is associated with the histological features of the tumor in nonfamilial colorectal cancer. Cancer Res. 1996, 56, 5470–5474. [Google Scholar]
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Schwitalle, Y.; Kloor, M.; Eiermann, S.; Linnebacher, M.; Kienle, P.; Knaebel, H.P.; Tariverdian, M.; Benner, A.; von Knebel Doeberitz, M. Immune Response Against Frameshift-Induced Neopeptides in HNPCC Patients and Healthy HNPCC Mutation Carriers. Gastroenterology 2008, 134, 988–997. [Google Scholar] [CrossRef]
- Mas-Moya, J.; Dudley, B.; Brand, R.E.; Thull, D.; Bahary, N.; Nikiforova, M.N.; Pai, R.K. Clinicopathological comparison of colorectal and endometrial carcinomas in patients with Lynch-like syndrome versus patients with Lynch syndrome. Hum. Pathol. 2015, 46, 1616–1625. [Google Scholar] [CrossRef] [PubMed]
- Boland, C.R.; Shike, M. Report from the Jerusalem Workshop on Lynch Syndrome-Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 2010, 138, 2197.e1. [Google Scholar] [CrossRef] [PubMed]
- Aran, V.; Victorino, A.P.; Thuler, L.C.; Gil Ferreira, C. Colorectal Cancer: Epidemiology, Disease Mechanisms and Interventions to Reduce Onset and Mortality. Clin. Color. Cancer 2016, 15, 195–203. [Google Scholar] [CrossRef]
- Sinicrope, F.A. Lynch Syndrome–Associated Colorectal Cancer. N. Engl. J. Med. 2018, 379, 764–773. [Google Scholar] [CrossRef]
- Hitchins, M.P. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat. Rev. Cancer 2015, 15, 625–634. [Google Scholar] [CrossRef]
- Valle, L.; Vilar, E.; Tavtigian, S.V.; Stoffel, E.M. Genetic predisposition to colorectal cancer: Syndromes, genes, classification of genetic variants and implications for precision medicine. J. Pathol. 2018, 247, 574–588. [Google Scholar] [CrossRef]
- Win, A.; Buchanan, D.; Rosty, C.; MacInnis, R.; Dowty, J.; Dite, G.; Giles, G.; Southey, M.C.; Young, J.; Clendenning, M.; et al. Role of tumour molecular and pathology features to estimate colorectal cancer risk for first-degree relatives. Gut 2014, 64, 101–110. [Google Scholar] [CrossRef]
- Overbeek, L.I.H.; Kets, C.M.; Hebeda, K.M.; Bodmer, D.; Van Der Looij, E.; Willems, R.; Goossens, M.; Arts, N.; Brunner, H.G.; Van Krieken, J.H.J.M.; et al. Patients with an unexplained microsatellite instable tumour have a low risk of familial cancer. Br. J. Cancer 2007, 96, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M. Differentiating Lynch-Like From Lynch Syndrome. Gastroenterology 2014, 146, 602–604. [Google Scholar] [CrossRef]
- Pérez-Carbonell, L.; Ruiz-Ponte, C.; Guarinos, C.; Alenda, C.; Payá, A.; Brea-Fernández, A.; Egoavil, C.; Castillejo, A.; Barbera, V.-M.; Bessa, X.; et al. Comparison between universal molecular screening for Lynch syndrome and revised Bethesda guidelines in a large population-based cohort of patients with colorectal cancer. Gut 2011, 61, 865–872. [Google Scholar] [CrossRef]
- PPicó, M.D.; Castillejo, A.; Murcia, O.; Giner-Calabuig, M.; Alustiza, M.; Sánchez, A.; Moreira, L.; Pellise, M.; Castells, A.; Carrillo-Palau, M.; et al. Clinical and Pathological Characterization of Lynch-Like Syndrome. Clin. Gastroenterol. Hepatol. 2019, 18, 368–374.e1. [Google Scholar] [CrossRef]
- Xavier, A.; Olsen, M.F.; Lavik, L.A.; Johansen, J.; Singh, A.K.; Sjursen, W.; Scott, R.J.; Talseth-Palmer, B.A. Comprehensive mismatch repair gene panel identifies variants in patients with Lynch-like syndrome. Mol. Genet. Genom. Med. 2019, 7, e850. [Google Scholar] [CrossRef] [PubMed]
- Porkka, N.; Lahtinen, L.; Ahtiainen, M.; Böhm, J.P.; Kuopio, T.; Eldfors, S.; Mecklin, J.P.; Seppälä, T.T.; Peltomäki, P. Epidemiological, clinical and molecular characterization of Lynch-like syndrome: A population-based study. Int. J. Cancer 2019, 145, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Ladabaum, U. What Is Lynch-like Syndrome and How Should We Manage It? Clin. Gastroenterol. Hepatol. 2020, 18, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Sourrouille, I.; Coulet, F.; Lefevre, J.H.; Colas, C.; Eyries, M.; Svrcek, M.; Bardier-Dupas, A.; Parc, Y.; Soubrier, F. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam. Cancer 2012, 12, 27–33. [Google Scholar] [CrossRef]
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; de la Chapelle, A.; Pritchard, C.C. Colon and Endometrial Cancers With Mismatch Repair Deficiency Can Arise From Somatic, Rather Than Germline, Mutations. Gastroenterology 2014, 147, 1308–1316.e1. [Google Scholar] [CrossRef]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–143. [Google Scholar] [CrossRef]
- Elsayed, F.A.; Kets, C.M.; Ruano, D.; Akker, B.V.D.; Mensenkamp, A.; Schrumpf, M.; Nielsen, M.; Wijnen, J.T.; Tops, C.M.; Ligtenberg, M.J.; et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 2014, 23, 1080–1084. [Google Scholar] [CrossRef]
- Morak, M.; Heidenreich, B.; Keller, G.; Hampel, H.; Laner, A.; De La Chapelle, A.; Holinski-Feder, E. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur. J. Hum. Genet. 2014, 22, 1334–1337. [Google Scholar] [CrossRef] [PubMed]
- Bellido, F.; Pineda, M.; Aiza, G.; Valdés-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Spier, I.; Holzapfel, S.; Altmüller, J.; Zhao, B.; Horpaopan, S.; Vogt, S.; Chen, S.; Morak, M.; Raeder, S.; Kayser, K.; et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int. J. Cancer 2015, 137, 320–331. [Google Scholar] [CrossRef]
- Giner-Calabuig, M. Novel Germline and Somatic Processes in Mismatch Repair Deficient Tumors. Ph.D. Thesis, Universidad de Alicante, Alicante, Spain, 30 November 2020. [Google Scholar]
- Keijzers, G.; Liu, D.; Rasmussen, L.J. Exonuclease 1 and its versatile roles in DNA repair. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 440–451. [Google Scholar] [CrossRef]
- Nicolas, E.; Golemis, E.; Arora, S. POLD1: Central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene 2016, 590, 128–141. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Putnam, C.; Kane, M.F.; Zhang, W.; Edelmann, L.; Russell, R.; Carrión, D.V.; Chin, L.; Kucherlapati, R.; Kolodner, R.D.; et al. Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat. Genet. 2005, 37, 750–755. [Google Scholar] [CrossRef]
- Huang, X.; Gao, Y.; He, J.; Cai, J.; Ta, N.; Jiang, H.; Zhu, J.; Zheng, J. The association between RFC1 G80A polymorphism and cancer susceptibility: Evidence from 33 studies. J. Cancer 2016, 7, 144–152. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000, 14, 927–939. [Google Scholar] [CrossRef]
- Golubicki, M.; Díaz-Gay, M.; Bonjoch, L.; Franch-Expósito, S.; Muñoz, J.; Cuatrecasas, M.; Ocaña, T.; Iseas, S.; Mendez, G.; Carballido, M.; et al. Comprehensive Genomic Characterization of Fifteen Early-Onset Lynch-Like Syndrome Colorectal Cancers. Cancers 2021, 13, 1259. [Google Scholar] [CrossRef]
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. J. Clin. Oncol. 2017, 35, 1086–1095. [Google Scholar] [CrossRef]
- Arora, S.; Yan, H.; Cho, I.; Fan, H.-Y.; Luo, B.; Gai, X.; Bodian, D.L.; Vockley, J.G.; Zhou, Y.; Handorf, E.A.; et al. Genetic Variants That Predispose to DNA Double-Strand Breaks in Lymphocytes From a Subset of Patients With Familial Colorectal Carcinomas. Gastroenterology 2015, 149, 1872–1883.e9. [Google Scholar] [CrossRef] [PubMed]
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J.; Hartman, A.-R.; Syngal, S. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology 2015, 149, 604–613.e20. [Google Scholar] [CrossRef] [PubMed]
- Weren, R.D.; Ligtenberg, M.J.; Kets, C.M.; De Voer, R.M.; Verwiel, E.T.; Spruijt, L.; van Zelst-Stams, W.A.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015, 47, 668–671. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, Y.; Halpern, N.; Hubert, A.; Adler, S.N.; Cohen, S.; Plesser-Duvdevani, M.; Pappo, O.; Shaag, A.; Meiner, V. Mutated MCM9 is associated with predisposition to hereditary mixed polyposis and colorectal cancer in addition to primary ovarian failure. Cancer Genet. 2015, 208, 621–624. [Google Scholar] [CrossRef]
- Seguí, N.; Mina, L.B.; Lázaro, C.; Sanz-Pamplona, R.; Pons, T.; Navarro, M.; Bellido, F.; López-Doriga, A.; Valdés-Mas, R.; Pineda, M.; et al. Germline Mutations in FAN1 Cause Hereditary Colorectal Cancer by Impairing DNA Repair. Gastroenterology 2015, 149, 563–566. [Google Scholar] [CrossRef]
- De Voer, R.M.; van Kessel, A.G.; Weren, R.D.; Ligtenberg, M.J.; Smeets, D.; Fu, L.; Vreede, L.; Kamping, E.J.; Verwiel, E.T.; Hahn, M.M.; et al. Germline mutations in the spindle assembly checkpoint genes BUB1 and BUB3 are risk factors for colorectal cancer. Gastroenterology 2013, 145, 544–547. [Google Scholar] [CrossRef]
- Vargas-Parra, G.M.; González-Acosta, M.; Thompson, B.A.; Gómez, C.; Fernández, A.; Dámaso, E.; Pons, T.; Morak, M.; Del Valle, J.; Iglesias, S.; et al. Elucidating the molecular basis of MSH2-deficient tumors by combined germline and somatic analysis. Int. J. Cancer 2017, 141, 1365–1380. [Google Scholar] [CrossRef]
- Xicola, R.M.; Clark, J.R.; Carroll, T.; Alvikas, J.; Marwaha, P.; Regan, M.R.; Lopez-Giraldez, F.; Choi, J.; Emmadi, R.; Alagiozian-Angelova, V.; et al. Implication of DNA repair genes in Lynch-like syndrome. Fam. Cancer 2019, 18, 331–342. [Google Scholar] [CrossRef]
- Clendenning, M.; Buchanan, D.; Walsh, M.D.; Nagler, B.; Rosty, C.; Thompson, B.; Spurdle, A.; Hopper, J.L.; Jenkins, M.; Young, J. Mutation deep within an intron of MSH2 causes Lynch syndrome. Fam. Cancer 2011, 10, 297–301. [Google Scholar] [CrossRef][Green Version]
- Mork, M.E.; Rodriguez, A.; Taggart, M.W.; Rodriguez-Bigas, M.A.; Lynch, P.M.; Bannon, S.A.; You, Y.N.; Vilar, E. Identification of MSH2 inversion of exons 1–7 in clinical evaluation of families with suspected Lynch syndrome. Fam. Cancer 2016, 16, 357–361. [Google Scholar] [CrossRef]
- Wagner, A.; van der Klift, H.; Franken, P.; Wijnen, J.; Breukel, C.; Bezrookove, V.; Smits, R.; Kinarsky, Y.; Barrows, A.; Franklin, B.; et al. A 10-Mb paracentric inversion of chromosome arm 2p inactivatesMSH2 and is responsible for hereditary nonpolyposis colorectal cancer in a North-American kindred. Genes Chromosom. Cancer 2002, 35, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Rhees, J.; Arnold, M.; Boland, C.R. Inversion of exons 1–7 of the MSH2 gene is a frequent cause of unexplained Lynch syndrome in one local population. Fam. Cancer 2013, 13, 219–225. [Google Scholar] [CrossRef]
- Liu, Q.; Hesson, L.B.; Nunez, A.C.; Packham, D.; Williams, R.; Ward, R.L.; Sloane, M.A. A cryptic paracentric inversion ofMSH2exons 2–6 causes Lynch syndrome. Carcinogenesis 2015, 37, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Morak, M.; Koehler, U.; Schackert, H.K.; Steinke, V.; Royer-Pokora, B.; Schulmann, K.; Kloor, M.; Höchter, W.; Weingart, J.; Keiling, C.; et al. Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J. Med. Genet. 2011, 48, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Brieger, A.; Plotz, G.; Weber, N.; Passmann, S.; Dingermann, T.; Zeuzem, S.; Trojan, J.; Marschalek, R. An Interstitial Deletion at 3p21.3 Results in the Genetic Fusion of MLH1 and ITGA9 in a Lynch Syndrome Family. Clin. Cancer Res. 2009, 15, 762–769. [Google Scholar] [CrossRef]
- Van der Klift, H.M.; Tops, C.M.; Hes, F.J.; Devilee, P.; Wijnen, J.T. Insertion of an SVA element, a nonautonomous retrotransposon, inPMS2intron 7 as a novel cause of lynch syndrome. Hum. Mutat. 2012, 33, 1051–1055. [Google Scholar] [CrossRef]
- Liu, Q.; Thompson, B.; Ward, R.; Hesson, L.B.; Sloane, M.A. Understanding the Pathogenicity of Noncoding Mismatch Repair Gene Promoter Variants in Lynch Syndrome. Hum. Mutat. 2016, 37, 417–426. [Google Scholar] [CrossRef]
- Wilding, J.L.; McGowan, S.; Liu, Y.; Bodmer, W.F. Replication error deficient and proficient colorectal cancer gene expression differences caused by 3’UTR polyT sequence deletions. Proc. Natl. Acad. Sci. USA 2010, 107, 21058–21063. [Google Scholar] [CrossRef]
- Valeri, N.; Gasparini, P.; Braconi, C.; Paone, A.; Lovat, F.; Fabbri, M.; Sumani, K.M.; Alder, H.; Amadori, D.; Patel, T.; et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc. Natl. Acad. Sci. USA 2010, 107, 21098–21103. [Google Scholar] [CrossRef] [PubMed]
- Pastrello, C.; Fornasarig, M.; Pin, E.; Berto, E.; Pivetta, B.; Viel, A. Somatic mosaicism in a patient with Lynch syndrome. Am. J. Med. Genet. Part A 2009, 149A, 212–215. [Google Scholar] [CrossRef] [PubMed]
- Geurts-Giele, W.R.; Rosenberg, E.H.; Van Rens, A.; Van Leerdam, M.E.; Dinjens, W.N.; Bleeker, F.E. Somatic mosaicism by a de novo MLH1 mutation as a cause of Lynch syndrome. Mol. Genet. Genom. Med. 2019, 7, e00699. [Google Scholar] [CrossRef]
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.-P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capellá, G.; den Dunnen, J.T.; et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014, 46, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, J.; Vermaut, C.; Buisine, M.-P. Diagnosis of Lynch Syndrome and Strategies to Distinguish Lynch-Related Tumors from Sporadic MSI/dMMR Tumors. Cancers 2021, 13, 467. [Google Scholar] [CrossRef]
- Edmunds, J.W.; Mahadevan, L.C.; Clayton, A.L. Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 2007, 27, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Mao, G.; Tong, D.; Huang, J.; Gu, L.; Yang, W.; Li, G.-M. The Histone Mark H3K36me3 Regulates Human DNA Mismatch Repair through Its Interaction with MutSα. Cell 2013, 153, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.; Li, J.; Lee, S.; Tong, D.; Gu, L.; Li, G.-M. Phosphorylation of PCNA by EGFR inhibits mismatch repair and promotes misincorporation during DNA synthesis. Proc. Natl. Acad. Sci. USA 2015, 112, 5667–5672. [Google Scholar] [CrossRef]
- Wu, J.N.; Roberts, C.W.M. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43. [Google Scholar] [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562. [Google Scholar] [CrossRef]
- Billingsley, C.C.; Cohn, D.E.; Mutch, D.G.; Stephens, J.A.; Suarez, A.A.; Goodfellow, P.J. Polymerase ε (POLE) mutations in endometrial cancer: Clinical outcomes and implications for Lynch syndrome testing. Cancer 2015, 121, 386–394. [Google Scholar] [CrossRef] [PubMed]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672. [Google Scholar] [CrossRef]
- Li, S.K.; Martin, A. Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored. Trends Mol. Med. 2016, 22, 274–289. [Google Scholar] [CrossRef] [PubMed]
- Tseng-Rogenski, S.S.; Hamaya, Y.; Choi, D.Y.; Carethers, J.M. Interleukin 6 Alters Localization of hMSH3, Leading to DNA Mismatch Repair Defects in Colorectal Cancer Cells. Gastroenterology 2015, 148, 579–589. [Google Scholar] [CrossRef]
- Chang, C.L.; Marra, G.; Chauhan, D.P.; Ha, H.T.; Chang, D.K.; Ricciardiello, L.; Randolph, A.; Carethers, J.M.; Boland, C.R. Oxidative stress inactivates the human DNA mismatch repair system. Am. J. Physiol. Cell Physiol. 2002, 283, C148–C154. [Google Scholar] [CrossRef]
- Greger, V.; Passarge, E.; Messmer, E.; Horsthemke, B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Qual. Life Res. 1989, 83, 155–158. [Google Scholar] [CrossRef]
- Sakai, T.; Toguchida, J.; Ohtani, N.; Yandell, D.W.; Rapaport, J.M.; Dryja, T.P. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am. J. Hum. Genet. 1991, 48, 880–888. [Google Scholar]
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.; Linehan, W.M. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704. [Google Scholar] [CrossRef]
- Dobrovic, A.; Simpfendorfer, D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 1997, 57, 3347–3350. [Google Scholar] [PubMed]
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997, 57, 808–811. [Google Scholar]
- Buckley, A.R.; Ideker, T.; Carter, H.; Harismendy, O.; Schork, N.J. Exome-wide analysis of bi-allelic alterations identifies a Lynch phenotype in The Cancer Genome Atlas. Genome Med. 2018, 10, 69. [Google Scholar] [CrossRef]
- Jansen, A.M.; Van Wezel, T.; Akker, B.E.V.D.; García, M.V.; Ruano, D.; Tops, C.M.; Wagner, A.; Letteboer, T.G.; Gomez-Garcia, E.; Devilee, P.; et al. Combined mismatch repair and POLE/POLD1 defects explain unresolved suspected Lynch syndrome cancers. Eur. J. Hum. Genet. 2015, 24, 1089–1092. [Google Scholar] [CrossRef] [PubMed]
- Lefol, C.; Sohier, E.; Baudet, C.; Naïbo, P.; Ruano, E.; Grand-Masson, C.; Viari, A.; Wang, Q. Acquired somatic MMR deficiency is a major cause of MSI tumor in patients suspected for “Lynch-like syndrome” including young patients. Eur. J. Hum. Genet. 2020, 29, 482–488. [Google Scholar] [CrossRef]
- Elze, L.; Mensenkamp, A.R.; Nagtegaal, I.D.; van Zelst-Stams, W.A.; de Voer, R.M.; Ligtenberg, M.J.; Dommering, C.J.; Hoogerbrugge, N.; de Jong, M.M.; Bleeker, F.E.; et al. Somatic Nonepigenetic Mismatch Repair Gene Aberrations Underly Most Mismatch Repair–Deficient Lynch-Like Tumors. Gastroenterology 2021, 160, 1414–1416.e3. [Google Scholar] [CrossRef]
- Hemminger, J.A.; Pearlman, R.; Haraldsdottir, S.; Knight, D.; Jonasson, J.G.; Pritchard, C.C.; Hampel, H.; Frankel, W.L. Histology of colorectal adenocarcinoma with double somatic mismatch-repair mutations is indistinguishable from those caused by Lynch syndrome. Hum. Pathol. 2018, 78, 125–130. [Google Scholar] [CrossRef]
- Nagasaka, T.; Rhees, J.; Kloor, M.; Gebert, J.; Naomoto, Y.; Boland, C.R.; Goel, A. Somatic Hypermethylation of MSH2 Is a Frequent Event in Lynch Syndrome Colorectal Cancers. Cancer Res. 2010, 70, 3098–3108. [Google Scholar] [CrossRef] [PubMed]
- Rumilla, K.; Schowalter, K.V.; Lindor, N.M.; Thomas, B.C.; Mensink, K.A.; Gallinger, S.; Holter, S.; Newcomb, P.A.; Potter, J.; Jenkins, M.; et al. Frequency of Deletions of EPCAM (TACSTD1) in MSH2-Associated Lynch Syndrome Cases. J. Mol. Diagn. 2011, 13, 93–99. [Google Scholar] [CrossRef]
- Vymetalkova, V.P.; Slyskova, J.; Korenkova, V.; Bielik, L.; Langerova, L.; Prochazka, P.; Rejhova, A.; Schwarzova, L.; Pardini, B.; Naccarati, A.; et al. Molecular characteristics of mismatch repair genes in sporadic colorectal tumors in Czech patients. BMC Med. Genet. 2014, 15, 17. [Google Scholar] [CrossRef]
- Moura Lima, E.; Ferreira Leal, M.; de Arruda Cardoso Smith, M.; Rodríguez Burbano, R.; Pimentel de Assumpção, P.; Bello, M.J.; Rey, J.A.; Ferreira de Lima, F.; Casartelli, C. DNA mismatch repair gene methylation in gastric cancer in individuals from northern Brazil. Biocell 2008, 32, 237–243. [Google Scholar] [CrossRef]
- Truninger, K.; Menigatti, M.; Luz, J.; Russell, A.; Haider, R.; Gebbers, J.O.; Bannwart, F.; Yurtsever, H.; Neuweiler, J.; Riehle, H.M.; et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology 2005, 128, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Kamps, R.; Brandão, R.D.; van den Bosch, B.J.; Paulussen, A.D.; Xanthoulea, S.; Blok, M.J.; Romano, A. Next-Generation Sequencing in Oncology: Genetic Diagnosis, Risk Prediction and Cancer Classification. Int. J. Mol. Sci. 2017, 18, 308. [Google Scholar] [CrossRef] [PubMed]
- Golubicki, M.; Bonjoch, L.; Acuña-Ochoa, J.G.; Díaz-Gay, M.; Muñoz, J.; Cuatrecasas, M.; Ocaña, T.; Iseas, S.; Mendez, G.; Cisterna, D.; et al. Germline biallelic Mcm8 variants are associated with early-onset Lynch-like syndrome. JCI Insight 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Pearlman, R.; Beightol, M.; Zhao, W.; Jones, D.; Frankel, W.L.; Goodfellow, P.J.; Yilmaz, A.; Miller, K.; Bacher, J.; et al. Assessment of Tumor Sequencing as a Replacement for Lynch Syndrome Screening and Current Molecular Tests for Patients With Colorectal Cancer. JAMA Oncol. 2018, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Mutations in other Genes Affecting MMR System (Germline) | Unknown Mutations in MMR Genes (Germline) | Somatic Mutations in Cancer Genes (Somatic) | Biallelic Alteration in MMR (Somatic) |
|---|---|---|---|
| MUTYH | Mutation EXON 2 MSH2 | H3K36me3 | Double somatic hit |
| POLE/POLD1 | Inversion EXON 1-7 MSH2 | SETD2 | Somatic mutations in MMR genes |
| EXO1/RFC1/RPA1 | Inversion EXON 2-6 MSH2 | PCNA | Methylation in MMR genes |
| ERCC6/RAD54L/PALB2 | MLH1-LRRFIP2 fusion | ARID1A | |
| PIK3CA | MLH1 3′ UTR mutation | POLE | |
| FAN1/MCM9 | Deep intronic variant in MSH2 | IL-6 and oxidative stress | |
| NTHL1 | miRNA 21 AND miRNA 155 | Methylation in other genes | |
| BUB1/BUB3/WRN/MCPH1/REV3L | Mosaicism | ||
| VUS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Roca, A.; Giner-Calabuig, M.; Murcia, O.; Castillejo, A.; Soto, J.L.; García-Heredia, A.; Jover, R. Lynch-like Syndrome: Potential Mechanisms and Management. Cancers 2022, 14, 1115. https://doi.org/10.3390/cancers14051115
Martínez-Roca A, Giner-Calabuig M, Murcia O, Castillejo A, Soto JL, García-Heredia A, Jover R. Lynch-like Syndrome: Potential Mechanisms and Management. Cancers. 2022; 14(5):1115. https://doi.org/10.3390/cancers14051115
Chicago/Turabian StyleMartínez-Roca, Alejandro, Mar Giner-Calabuig, Oscar Murcia, Adela Castillejo, José Luis Soto, Anabel García-Heredia, and Rodrigo Jover. 2022. "Lynch-like Syndrome: Potential Mechanisms and Management" Cancers 14, no. 5: 1115. https://doi.org/10.3390/cancers14051115
APA StyleMartínez-Roca, A., Giner-Calabuig, M., Murcia, O., Castillejo, A., Soto, J. L., García-Heredia, A., & Jover, R. (2022). Lynch-like Syndrome: Potential Mechanisms and Management. Cancers, 14(5), 1115. https://doi.org/10.3390/cancers14051115