Development of Gene Expression-Based Random Forest Model for Predicting Neoadjuvant Chemotherapy Response in Triple-Negative Breast Cancer


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Datasets
2.2. Preprocessing
2.3. Collection of Prior Breast Cancer Markers
2.4. Differential Expression Analysis
2.5. Model Training
2.6. Selection of an Optimal Threshold
2.7. Model Validation
2.8. Comparative Analysis
2.9. Survival Analysis
2.10. Chemosensitivity Analysis
2.11. Function Enrichment Analysis
2.12. Visualize Error Matrix and Score Distributions
3. Results
3.1. Patient Characteristics
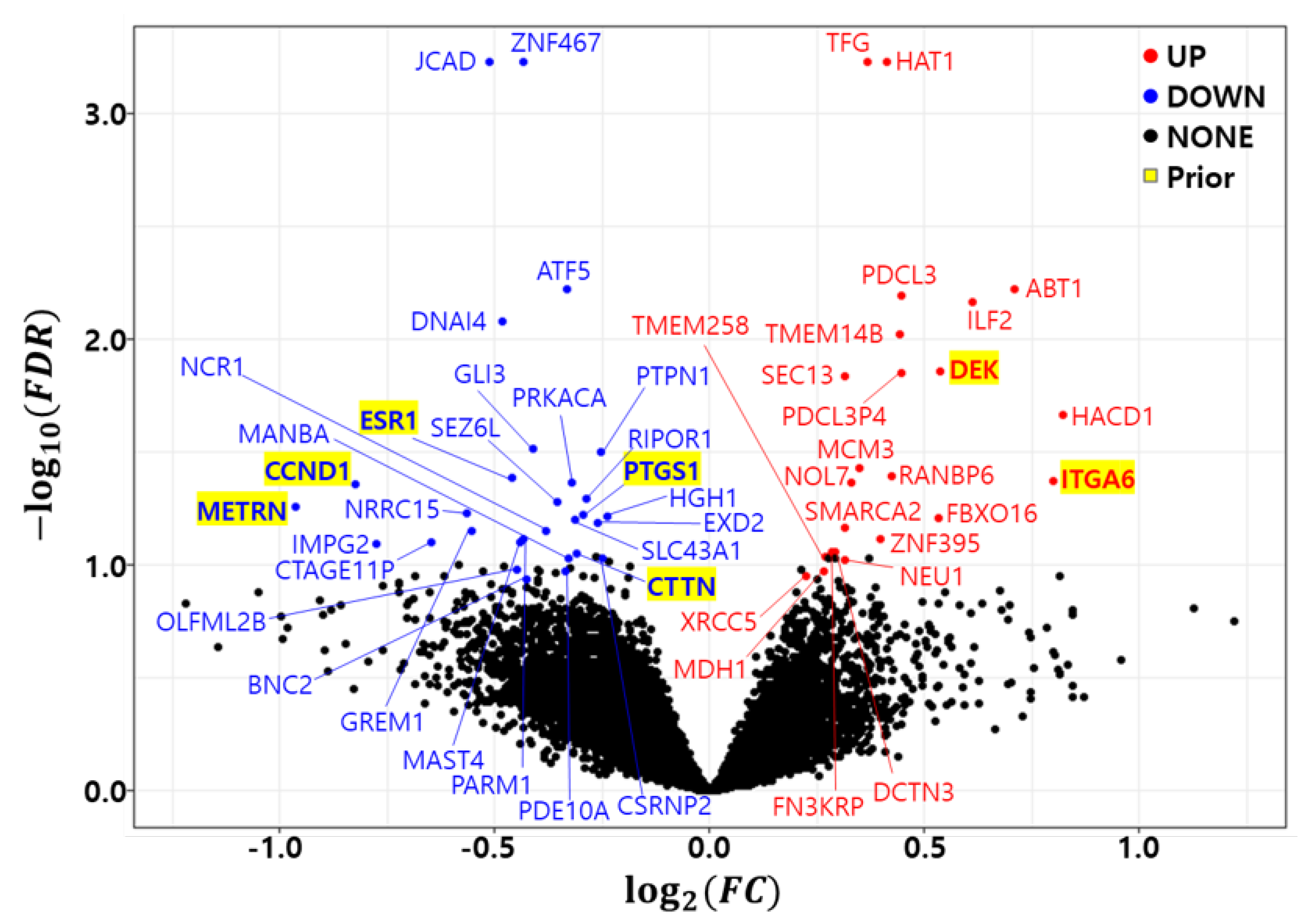
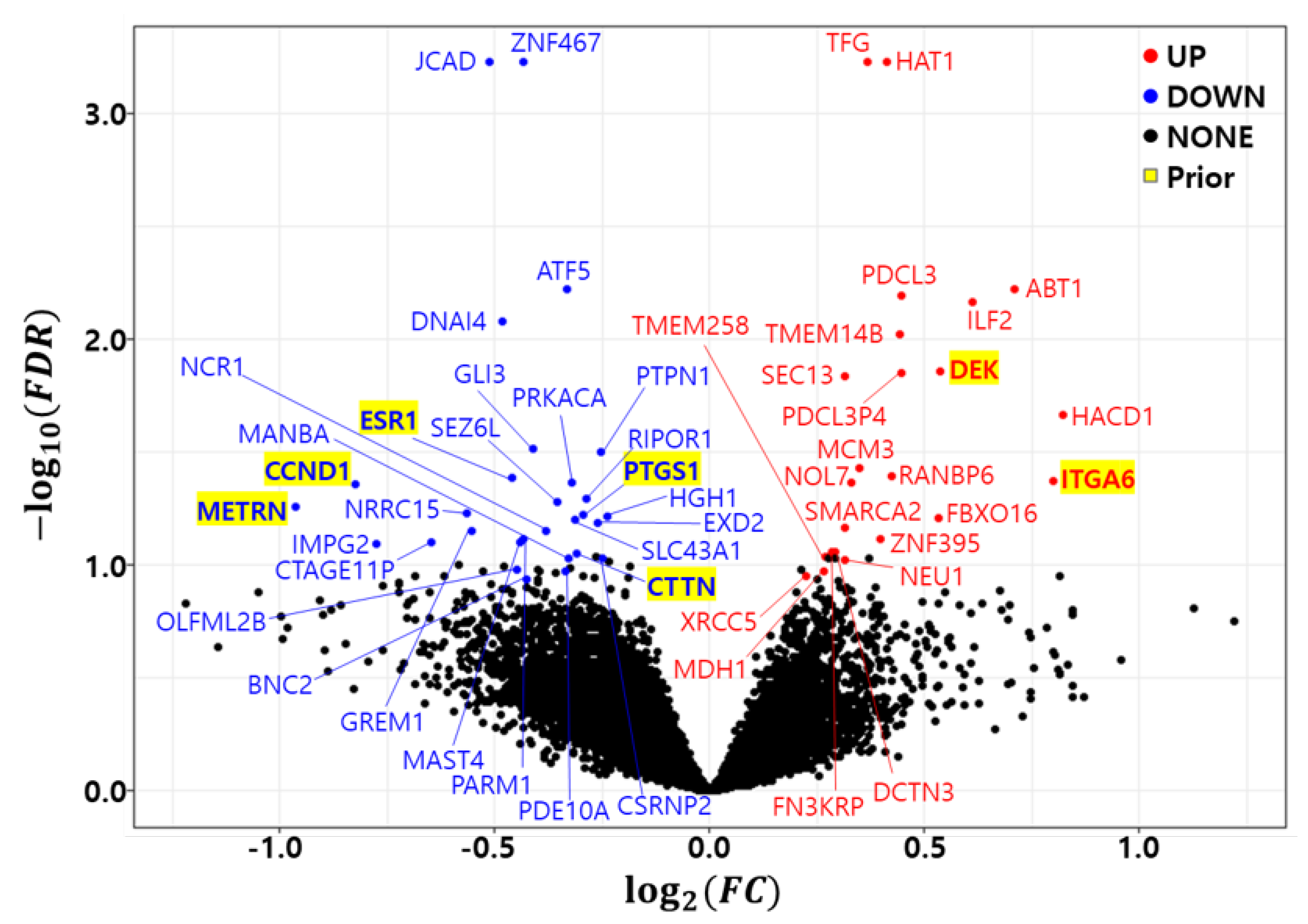
3.2. Differential Expression Analysis
3.3. Optimal Marker Set and Model Selection by RF-RFE
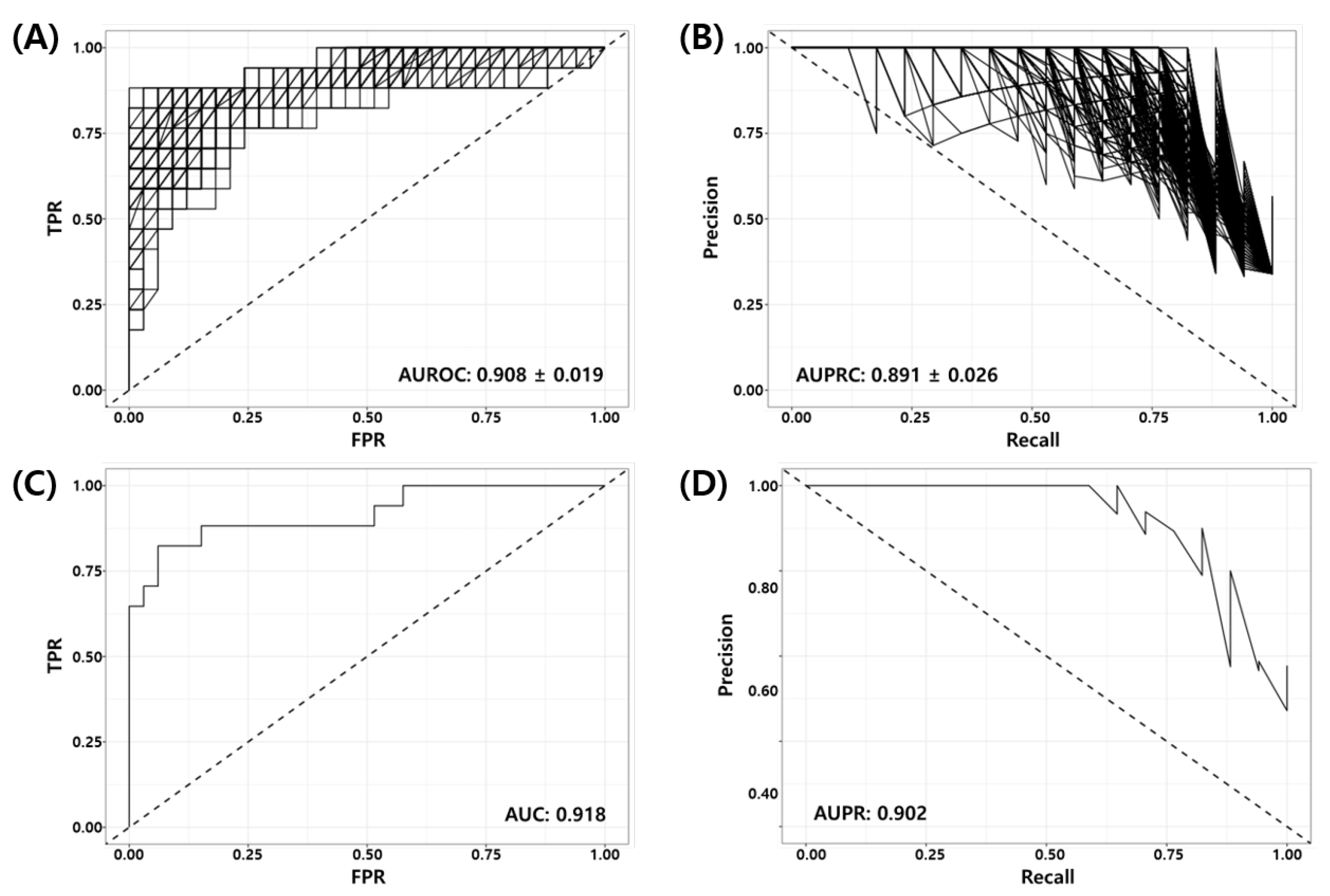
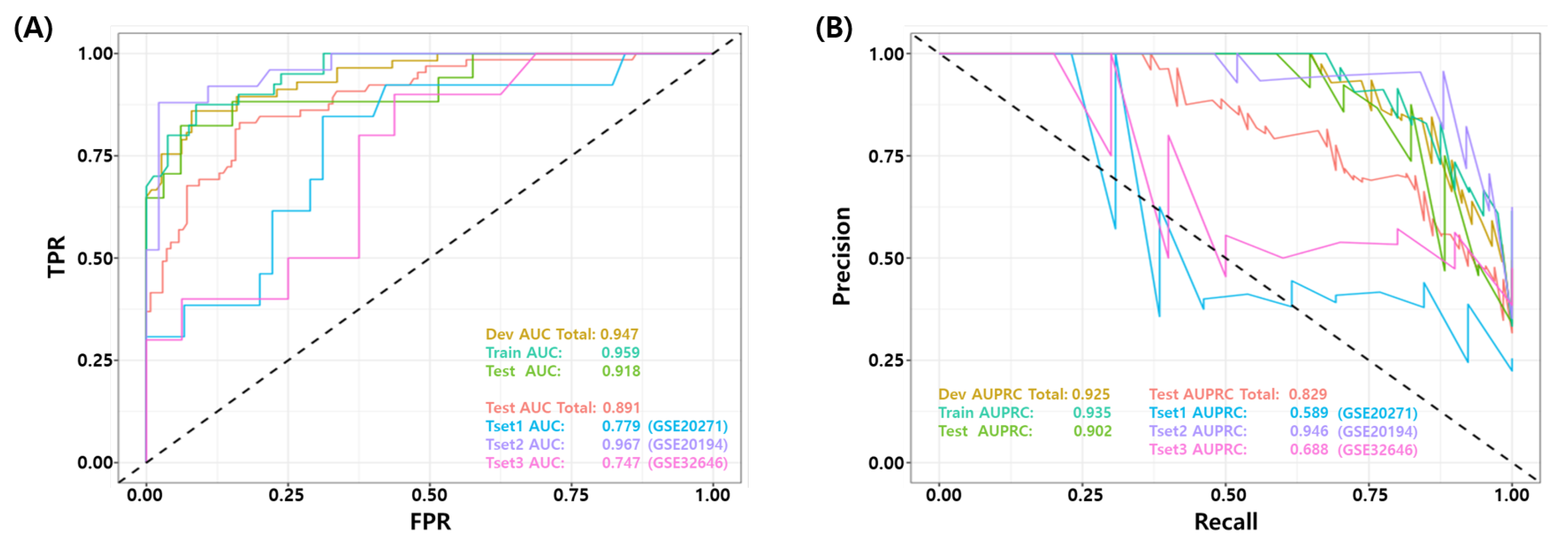
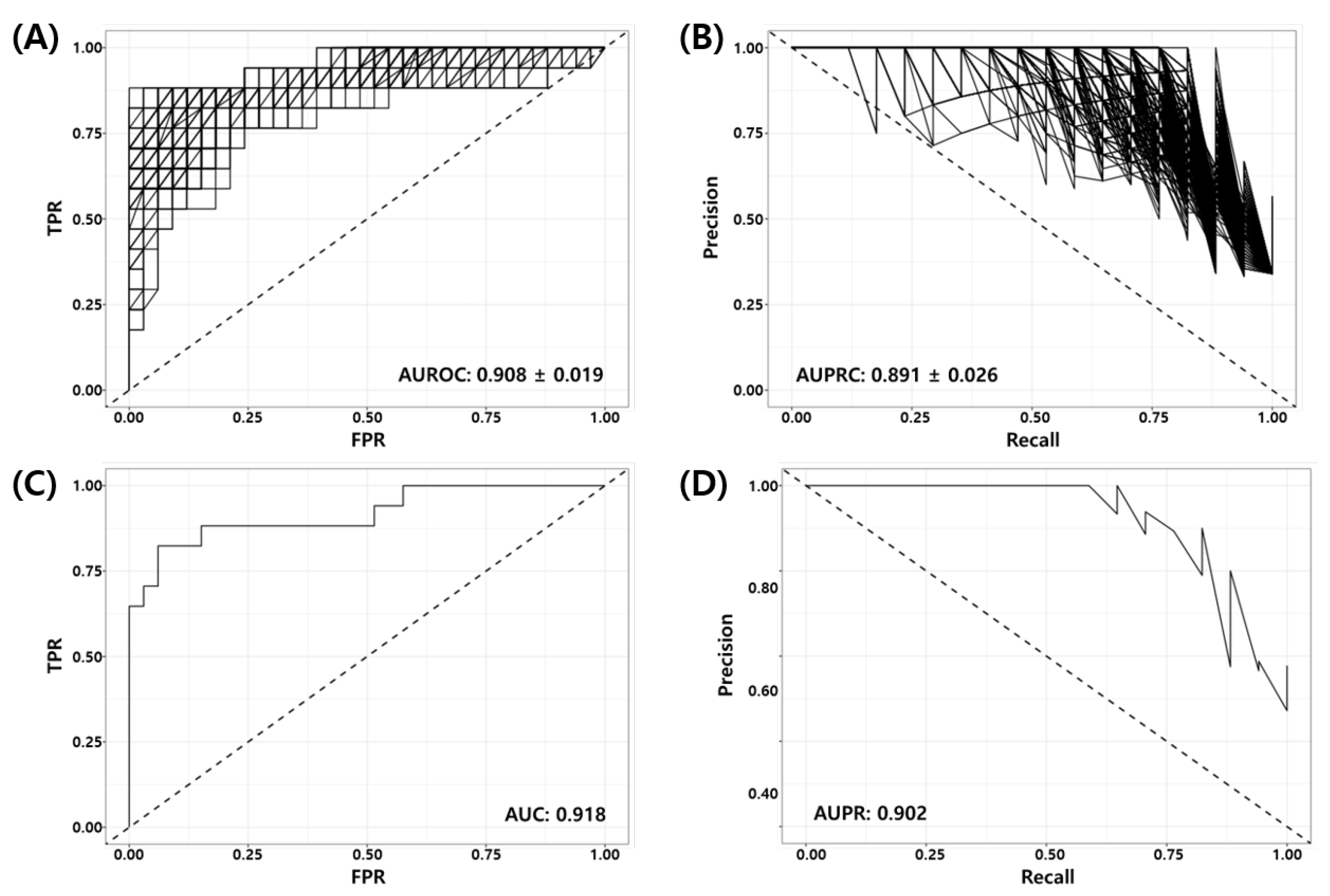
3.4. Validation of the Final Model Using Independent Test Datasets
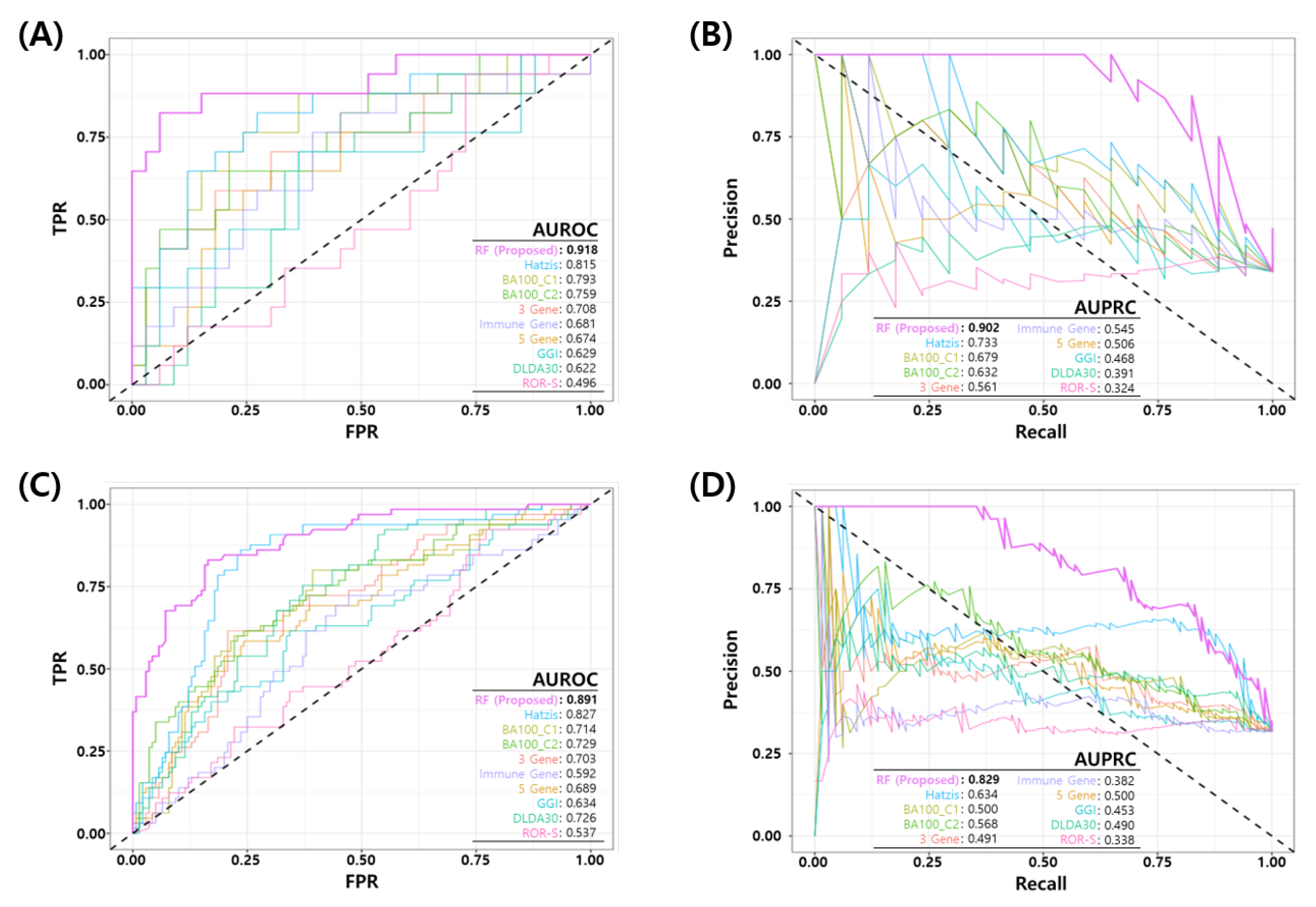
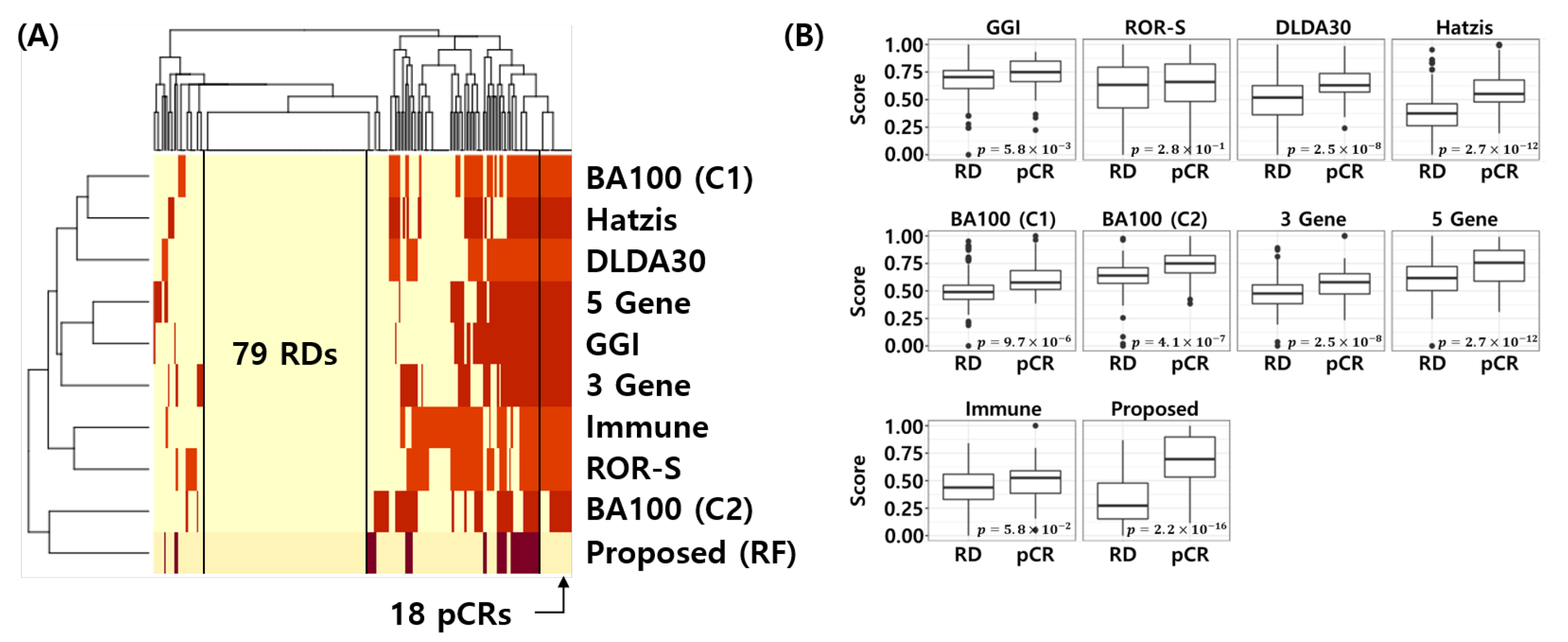
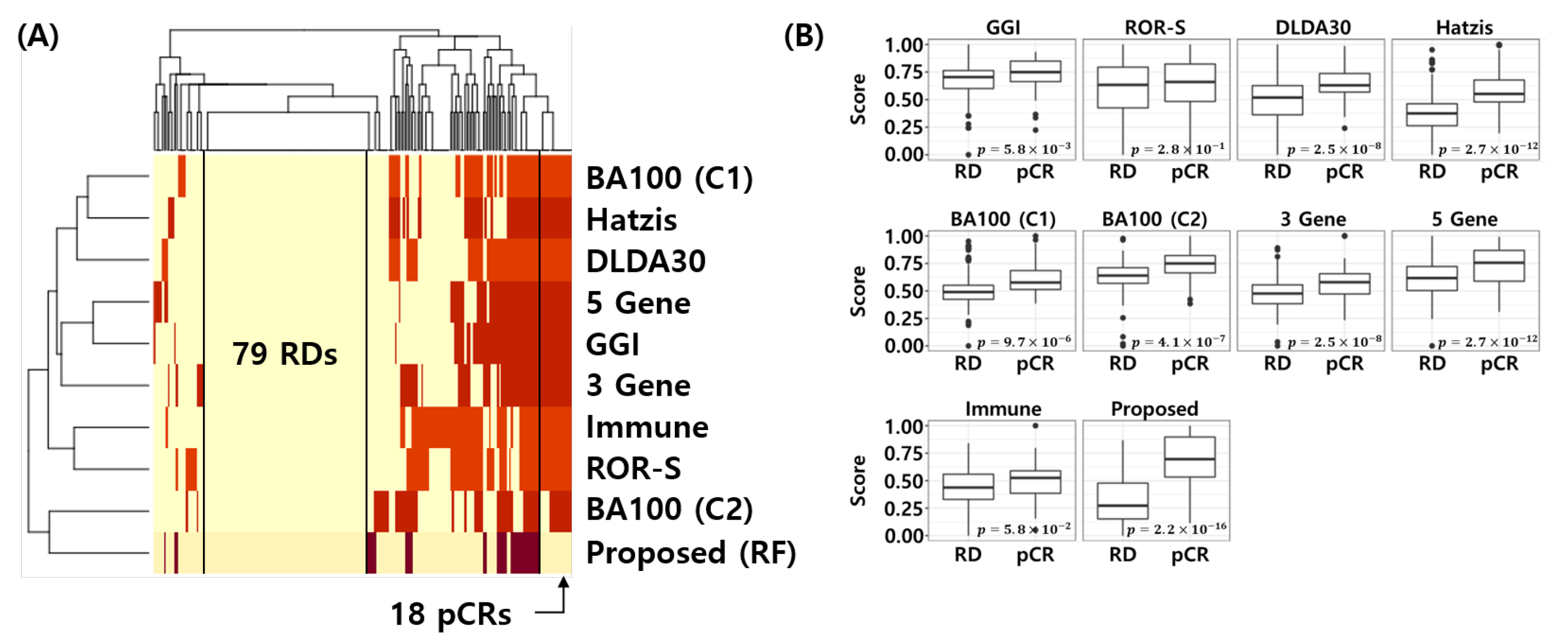
3.5. Comparative Analysis
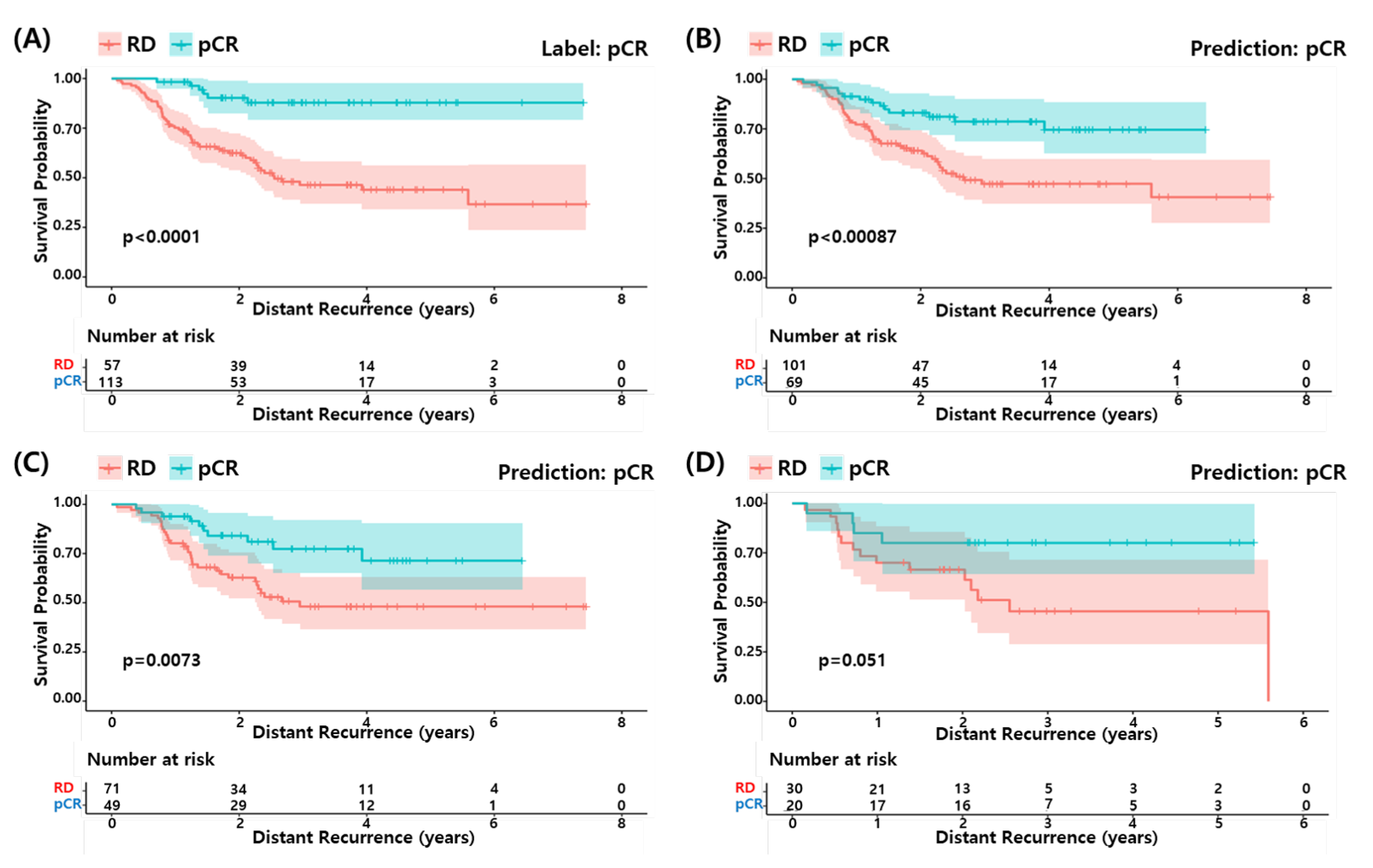
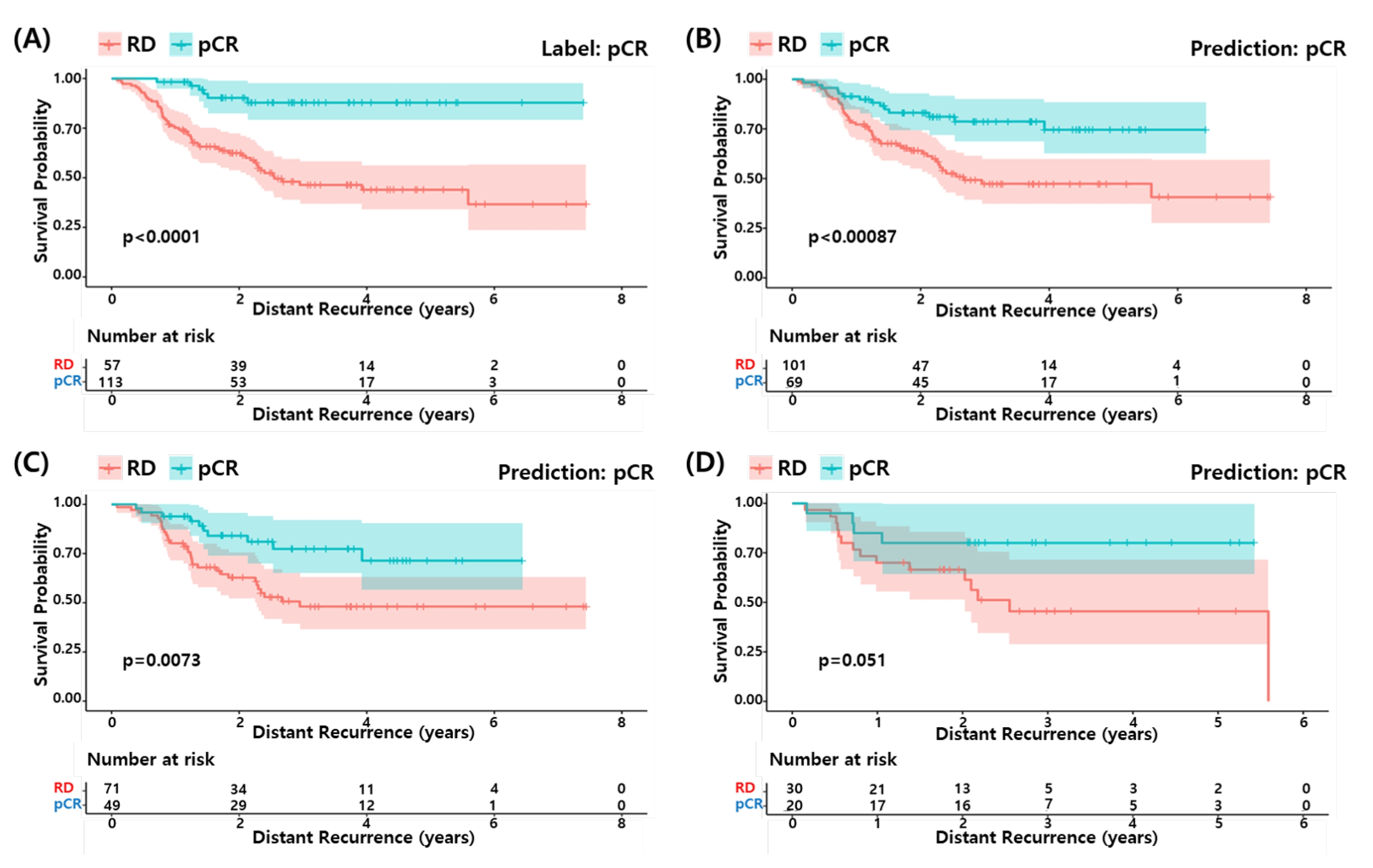
3.6. Survival Analysis
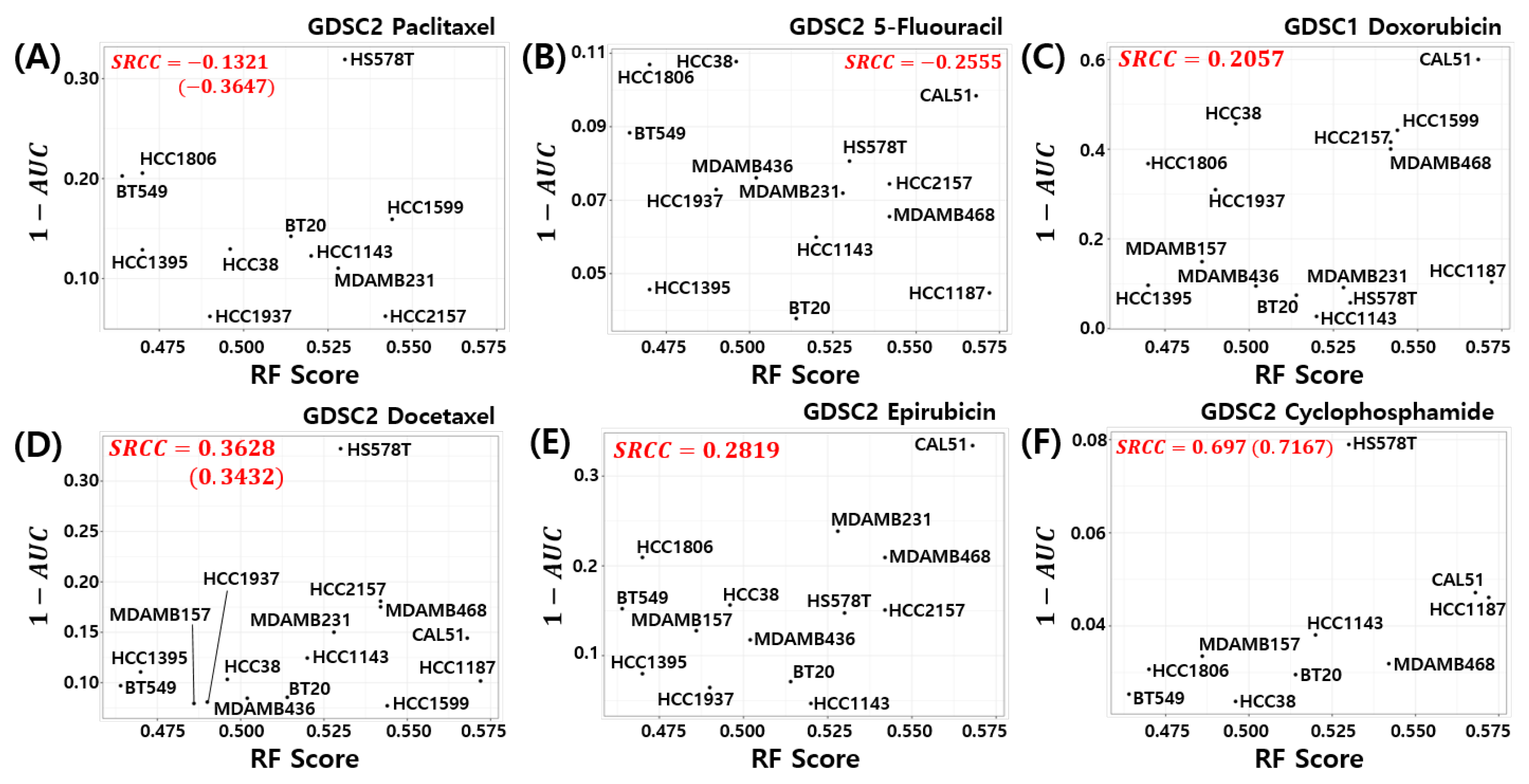
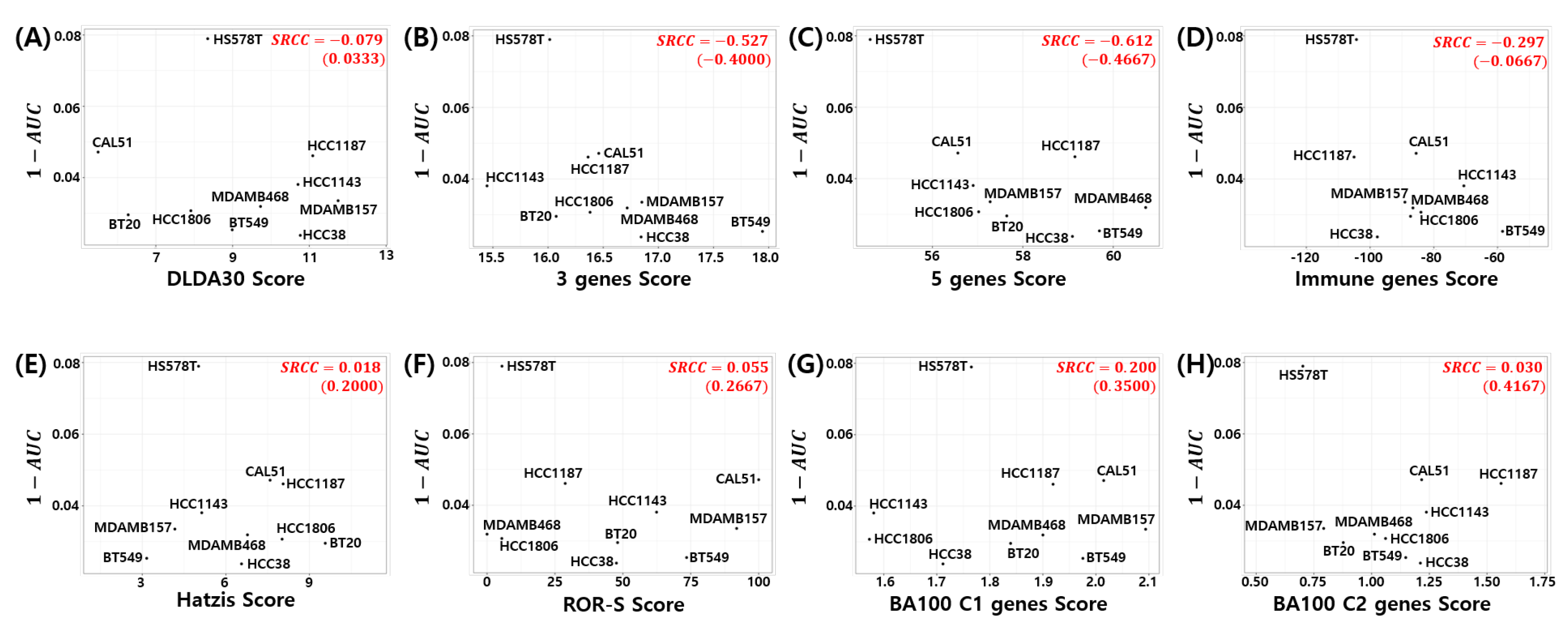
3.7. Relation to Chemosensitivity in TNBC Cell Models
3.8. Cellular Functions Associated with the 86 Genes of the Proposed Model
3.9. Visualization of the Error Matrix and Score Distributions
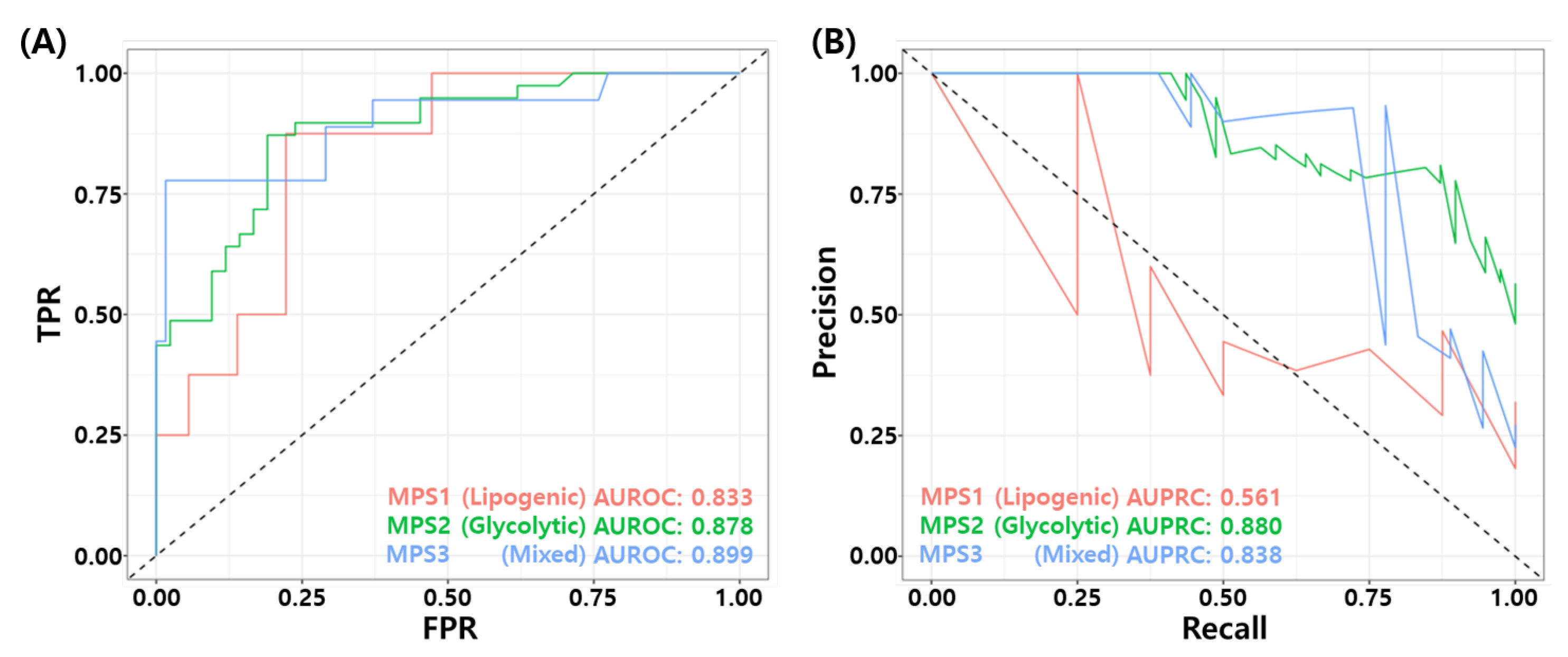
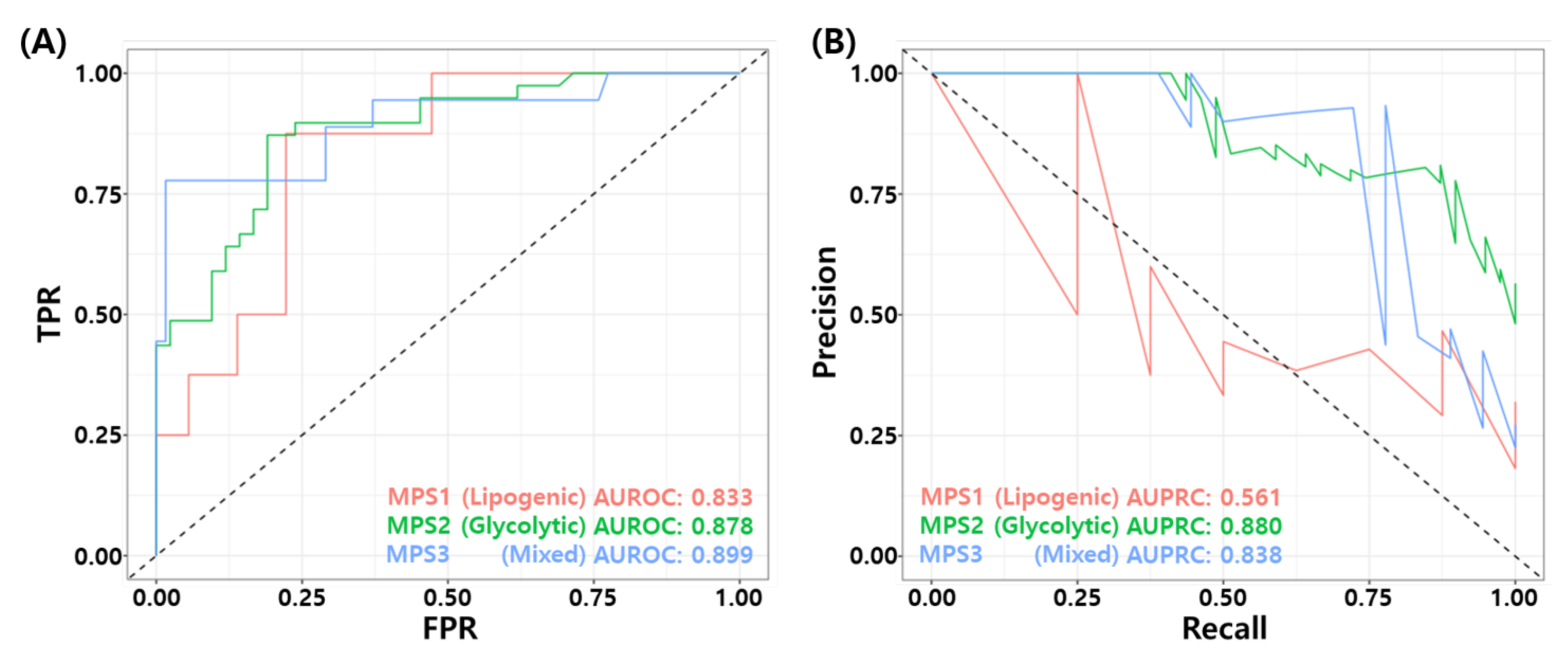
3.10. Association for Metabolic Pathway Based Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Bauer, K.R.; Brown, M.; Cress, R.D.; Parise, C.A.; Caggiano, V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California cancer Registry. Cancer 2007, 109, 1721–1728. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.G. Targeted therapies for triple-negative breast cancer. Curr. Treat. Options Oncol. 2019, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Korde, L.A.; Somerfield, M.R.; Carey, L.A.; Crews, J.R.; Denduluri, N.; Hwang, E.S.; Khan, S.A.; Loibl, S.; Morris, E.A.; Perez, A.; et al. Neoadjuvant chemotherapy, endocrine therapy, and targeted therapy for breast cancer: ASCO guideline. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2021, 39, 1485–1505. [Google Scholar] [CrossRef] [PubMed]
- Eikesdal, H.P.; Yndestad, S.; Elzawahry, A.; Llop-Guevara, A.; Gilje, B.; Blix, E.S.; Espelid, H.; Lundgren, S.; Geisler, J.; Vagstad, G.; et al. Olaparib monotherapy as primary treatment in unselected triple negative breast cancer. Ann. Oncol. 2021, 32, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Cortes, J.; Pusztai, L.; McArthur, H.; Kümmel, S.; Bergh, J.; Denkert, C.; Park, Y.H.; Hui, R.; Harbeck, N.; et al. Pembrolizumab for early triple-negative breast cancer. N. Engl. J. Med. 2020, 382, 810–821. [Google Scholar] [CrossRef]
- Bardia, A.; Hurvitz, S.A.; Tolaney, S.M.; Loirat, D.; Punie, K.; Oliveira, M.; Brufsky, A.; Sardesai, S.D.; Kalinsky, K.; Zelnak, A.B.; et al. Sacituzumab govitecan in metastatic triple-negative breast cancer. N. Engl. J. Med. 2021, 384, 1529–1541. [Google Scholar] [CrossRef]
- Vagia, E.; Mahalingam, D.; Cristofanilli, M. The landscape of targeted therapies in TNBC. Cancers 2020, 12, 916. [Google Scholar] [CrossRef] [Green Version]
- Isakoff, S.J. Triple negative breast cancer: Role of specific chemotherapy agents. Cancer J. 2010, 16, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapoport, B.; Barnard-Tidy, J.; Van Eeden, R.; Smit, T.; Nayler, S.; Benn, C. Pathological complete response in early breast cancer patients undergoing neoadjuvant chemotherapy: Focus on Ki-67 and molecular subtypes. Ann. Oncol. 2019, 30, iii37. [Google Scholar] [CrossRef]
- Esserman, L.J.; Berry, D.A.; DeMichele, A.; Carey, L.; Davis, S.E.; Buxton, M.; Hudis, C.; Gray, J.W.; Perou, C.; Yau, C.; et al. Pathologic complete response predicts recurrence-free survival more effectively by cancer subset: Results from the I-SPY 1 TRIAL—CALGB 150007/150012, ACRIN 6657. J. Clin. Oncol. 2012, 30, 3242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Minckwitz, G.; Untch, M.; Blohmer, J.U.; Costa, S.D.; Eidtmann, H.; Fasching, P.A.; Gerber, B.; Eiermann, W.; Hilfrich, J.; Huober, J.; et al. Definition and impact of pathologic complete response on prognosis after neoadjuvant chemotherapy in various intrinsic breast cancer subtypes. J. Clin. Oncol. 2012, 30, 1796–1804. [Google Scholar] [CrossRef] [Green Version]
- Asaoka, M.; Narui, K.; Suganuma, N.; Chishima, T.; Yamada, A.; Sugae, S.; Kawai, S.; Uenaka, N.; Teraoka, S.; Miyahara, K.; et al. Clinical and pathological predictors of recurrence in breast cancer patients achieving pathological complete response to neoadjuvant chemotherapy. Eur. J. Surg. Oncol. 2019, 45, 2289–2294. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Bagegni, N.A.; Tao, Y.; Ademuyiwa, F.O. Clinical outcomes with neoadjuvant versus adjuvant chemotherapy for triple negative breast cancer: A report from the National Cancer Database. PLoS ONE 2019, 14, e0222358. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Chen, X.; Deng, F.; Zhang, J.; Zhang, W.; Tang, J. Predictors of Neoadjuvant Chemotherapy Response in Breast Cancer: A Review. Oncotargets Ther. 2020, 13, 5887. [Google Scholar] [CrossRef]
- Kwa, M.; Makris, A.; Esteva, F.J. Clinical utility of gene-expression signatures in early stage breast cancer. Nat. Rev. Clin. Oncol. 2017, 14, 595–610. [Google Scholar] [CrossRef]
- Hatzis, C.; Pusztai, L.; Valero, V.; Booser, D.J.; Esserman, L.; Lluch, A.; Vidaurre, T.; Holmes, F.; Souchon, E.; Wang, H.; et al. A genomic predictor of response and survival following taxane-anthracycline chemotherapy for invasive breast cancer. JAMA 2011, 305, 1873–1881. [Google Scholar] [CrossRef] [Green Version]
- Oshi, M.; Angarita, F.A.; Tokumaru, Y.; Yan, L.; Matsuyama, R.; Endo, I.; Takabe, K. A Novel Three-Gene Score as a Predictive Biomarker for Pathologically Complete Response after Neoadjuvant Chemotherapy in Triple-Negative Breast Cancer. Cancers 2021, 13, 2401. [Google Scholar] [CrossRef]
- Oshi, M.; Gandhi, S.; Angarita, F.A.; Kim, T.H.; Tokumaru, Y.; Yan, L.; Matsuyama, R.; Endo, I.; Takabe, K. A novel five-gene score to predict complete pathological response to neoadjuvant chemotherapy in ER-positive/HER2-negative breast cancer. Am. J. Cancer Res. 2021, 11, 3611. [Google Scholar]
- Fu, C.; Liu, Y.; Han, X.; Pan, Y.; Wang, H.Q.; Wang, H.; Dai, H.; Yang, W. An Immune-Associated Genomic Signature Effectively Predicts Pathologic Complete Response to Neoadjuvant Paclitaxel and Anthracycline-Based Chemotherapy in Breast Cancer. Front. Immunol. 2021, 12, 3453. [Google Scholar] [CrossRef]
- Liedtke, C.; Hatzis, C.; Symmans, W.F.; Desmedt, C.; Haibe-Kains, B.; Valero, V.; Kuerer, H.; Hortobagyi, G.N.; Piccart-Gebhart, M.; Sotiriou, C.; et al. Genomic grade index is associated with response to chemotherapy in patients with breast cancer. J. Clin. Oncol. 2009, 27, 3185. [Google Scholar] [CrossRef] [Green Version]
- Hess, K.R.; Anderson, K.; Symmans, W.F.; Valero, V.; Ibrahim, N.; Mejia, J.A.; Booser, D.; Theriault, R.L.; Buzdar, A.U.; Dempsey, P.J.; et al. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J. Clin. Oncol. 2006, 24, 4236–4244. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.; Popescu, B.E. Gradient Directed Regularization for Linear Regression and Classification; Technical Report; Statistics Department, Stanford University: Stanford, CA, USA, 2003. [Google Scholar]
- Ma, S.; Huang, J. Regularized ROC method for disease classification and biomarker selection with microarray data. Bioinformatics 2005, 21, 4356–4362. [Google Scholar] [CrossRef]
- Fournier, M.V.; Goodwin, E.C.; Chen, J.; Obenauer, J.C.; Tannenbaum, S.H.; Brufsky, A.M. A predictor of pathological complete response to neoadjuvant chemotherapy stratifies triple negative breast cancer patients with high risk of recurrence. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160. [Google Scholar] [CrossRef]
- Li, Y.; Bellotti, T.; Adams, N. Issues using logistic regression with class imbalance, with a case study from credit risk modelling. Found. Data Sci. 2019, 1, 389. [Google Scholar] [CrossRef] [Green Version]
- Sota, Y.; Naoi, Y.; Tsunashima, R.; Kagara, N.; Shimazu, K.; Maruyama, N.; Shimomura, A.; Shimoda, M.; Kishi, K.; Baba, Y.; et al. Construction of novel immune-related signature for prediction of pathological complete response to neoadjuvant chemotherapy in human breast cancer. Ann. Oncol. 2014, 25, 100–106. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef] [Green Version]
- Tabchy, A.; Valero, V.; Vidaurre, T.; Lluch, A.; Gomez, H.; Martin, M.; Qi, Y.; Barajas-Figueroa, L.J.; Souchon, E.; Coutant, C.; et al. Evaluation of a 30-gene paclitaxel, fluorouracil, doxorubicin, and cyclophosphamide chemotherapy response predictor in a multicenter randomized trial in breast cancer. Clin. Cancer Res. 2010, 16, 5351–5361. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Campbell, G.; Jones, W.D.; Campagne, F.; Wen, Z.; Walker, S.J.; Su, Z.; Chu, T.M.; Goodsaid, F.M.; Pusztai, L.; et al. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models. Nat. Biotechnol. 2010, 28, 827–838. [Google Scholar]
- Miyake, T.; Nakayama, T.; Naoi, Y.; Yamamoto, N.; Otani, Y.; Kim, S.J.; Shimazu, K.; Shimomura, A.; Maruyama, N.; Tamaki, Y.; et al. GSTP 1 expression predicts poor pathological complete response to neoadjuvant chemotherapy in ER-negative breast cancer. Cancer Sci. 2012, 103, 913–920. [Google Scholar] [CrossRef]
- Gautier, L.; Cope, L.; Bolstad, B.M.; Irizarry, R.A. Affy—analysis of Affymetrix GeneChip data at the probe level. Bioinformatics 2004, 20, 307–315. [Google Scholar] [CrossRef]
- Perscheid, C. Integrative biomarker detection on high-dimensional gene expression data sets: A survey on prior knowledge approaches. Briefings Bioinform. 2021, 22, bbaa151. [Google Scholar] [CrossRef]
- Harbeck, N.; Gnant, M. Breast cancer. Lancet 2016, 389, 1134–1150. [Google Scholar] [CrossRef]
- Bertucci, F.; Ng, C.K.; Patsouris, A.; Droin, N.; Piscuoglio, S.; Carbuccia, N.; Soria, J.C.; Dien, A.T.; Adnani, Y.; Kamal, M.; et al. Genomic characterization of metastatic breast cancers. Nature 2019, 569, 560–564. [Google Scholar] [CrossRef]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic signaling pathways in the cancer genome atlas. Cell 2018, 173, 321–337. [Google Scholar] [CrossRef] [Green Version]
- Leo, C.P.; Leo, C.; Szucs, T.D. Breast cancer drug approvals by the US FDA from 1949 to 2018. Nat. Rev. Drug Discov. 2020, 19, 11–12. [Google Scholar] [CrossRef]
- Dai, X.; Xiang, L.; Li, T.; Bai, Z. Cancer hallmarks, biomarkers and breast cancer molecular subtypes. J. Cancer 2016, 7, 1281. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Koboldt, D.; Fulton, R.; McLellan, M.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.; Fulton, L.; Dooling, D.; Ding, L.; Mardis, E.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Ursu, O.; Holmes, J.; Knockel, J.; Bologa, C.G.; Yang, J.J.; Mathias, S.L.; Nelson, S.J.; Oprea, T.I. DrugCentral: Online drug compendium. Nucleic Acids Res. 2016, 45, D932–D939. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Freshour, S.L.; Kiwala, S.; Cotto, K.C.; Coffman, A.C.; McMichael, J.F.; Song, J.J.; Griffith, M.; Griffith, O.L.; Wagner, A.H. Integration of the Drug–Gene Interaction Database (DGIdb 4.0) with open crowdsource efforts. Nucleic Acids Res. 2021, 49, D1144–D1151. [Google Scholar] [CrossRef]
- Paik, S.; Shak, S.; Tang, G.; Kim, C.; Baker, J.; Cronin, M.; Baehner, F.L.; Walker, M.G.; Watson, D.; Park, T.; et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N. Engl. J. Med. 2004, 351, 2817–2826. [Google Scholar] [CrossRef] [Green Version]
- Van’t Veer, L.J.; Dai, H.; Van De Vijver, M.J.; He, Y.D.; Hart, A.A.; Mao, M.; Peterse, H.L.; Van Der Kooy, K.; Marton, M.J.; Witteveen, A.T.; et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002, 415, 530–536. [Google Scholar] [CrossRef] [Green Version]
- Filipits, M.; Rudas, M.; Jakesz, R.; Dubsky, P.; Fitzal, F.; Singer, C.F.; Dietze, O.; Greil, R.; Jelen, A.; Sevelda, P.; et al. A new molecular predictor of distant recurrence in ER-positive, HER2-negative breast cancer adds independent information to conventional clinical risk factors. Clin. Cancer Res. 2011, 17, 6012–6020. [Google Scholar] [CrossRef] [Green Version]
- Jerevall, P.L.; Ma, X.J.; Li, H.; Salunga, R.; Kesty, N.C.; Erlander, M.G.; Sgroi, D.; Holmlund, B.; Skoog, L.; Fornander, T.; et al. Prognostic utility of HOXB13: IL17BR and molecular grade index in early-stage breast cancer patients from the Stockholm trial. Br. J. Cancer 2011, 104, 1762–1769. [Google Scholar] [CrossRef]
- Naoi, Y.; Kishi, K.; Tanei, T.; Tsunashima, R.; Tominaga, N.; Baba, Y.; Kim, S.J.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Development of 95-gene classifier as a powerful predictor of recurrences in node-negative and ER-positive breast cancer patients. Breast Cancer Res. Treat. 2011, 128, 633–641. [Google Scholar] [CrossRef]
- Oh, E.; Choi, Y.L.; Park, T.; Lee, S.; Nam, S.J.; Shin, Y.K. A prognostic model for lymph node-negative breast cancer patients based on the integration of proliferation and immunity. Breast Cancer Res. Treat. 2012, 132, 499–509. [Google Scholar] [CrossRef]
- Sotiriou, C.; Wirapati, P.; Loi, S.; Harris, A.; Fox, S.; Smeds, J.; Nordgren, H.; Farmer, P.; Praz, V.; Haibe-Kains, B.; et al. Gene expression profiling in breast cancer: Understanding the molecular basis of histologic grade to improve prognosis. J. Natl. Cancer Inst. 2006, 98, 262–272. [Google Scholar] [CrossRef]
- Kohl, M. MKinfer: Inferential Statistics, R Package Version 0.6; 2020. Available online: https://cran.r-project.org/web/packages/MKinfer/index.html (accessed on 15 December 2021).
- Janssen, A. Studentized permutation tests for non-iid hypotheses and the generalized Behrens-Fisher problem. Stat. Probab. Lett. 1997, 36, 9–21. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Chawla, N.V.; Bowyer, K.W.; Hall, L.O.; Kegelmeyer, W.P. SMOTE: Synthetic minority over-sampling technique. J. Artif. Intell. Res. 2002, 16, 321–357. [Google Scholar] [CrossRef]
- Torgo, L. Data Mining with R: Learning with Case Studies; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Karapanagiotis, S.; Benedetto, U.; Mukherjee, S.; Kirk, P.D.; Newcombe, P.J. Tailored Bayes: A risk modelling framework under unequal misclassification costs. arXiv 2021, arXiv:2104.01822. [Google Scholar] [CrossRef]
- Tong, X.; Feng, Y.; Li, J.J. Neyman-Pearson classification algorithms and NP receiver operating characteristics. Sci. Adv. 2018, 4, eaao1659. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Li, J.; Zhu, Q.; Chang, C. Prediction of pathologic complete response by ultrasonography and magnetic resonance imaging after neoadjuvant chemotherapy in patients with breast cancer. Cancer Manag. Res. 2020, 12, 2603. [Google Scholar] [CrossRef] [Green Version]
- Gendoo, D.M.; Ratanasirigulchai, N.; Schröder, M.S.; Paré, L.; Parker, J.S.; Prat, A.; Haibe-Kains, B. Genefu: An R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 2016, 32, 1097–1099. [Google Scholar] [CrossRef] [Green Version]
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Survminer: Drawing Survival Curves using ‘ggplot2’. R Package Version 0.4.9. 2021. Available online: https://cran.r-project.org/web/packages/survminer/index.html (accessed on 15 December 2021).
- Yang, W.; Soares, J.; Greninger, P.; Edelman, E.J.; Lightfoot, H.; Forbes, S.; Bindal, N.; Beare, D.; Smith, J.A.; Thompson, I.R.; et al. Genomics of Drug Sensitivity in Cancer (GDSC): A resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2012, 41, D955–D961. [Google Scholar] [CrossRef] [Green Version]
- Bouhaddou, M.; DiStefano, M.S.; Riesel, E.A.; Carrasco, E.; Holzapfel, H.Y.; Jones, D.C.; Smith, G.R.; Stern, A.D.; Somani, S.S.; Thompson, T.V.; et al. Drug response consistency in CCLE and CGP. Nature 2016, 540, E9–E10. [Google Scholar] [CrossRef]
- Rees, M.G.; Seashore-Ludlow, B.; Cheah, J.H.; Adams, D.J.; Price, E.V.; Gill, S.; Javaid, S.; Coletti, M.E.; Jones, V.L.; Bodycombe, N.E.; et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat. Chem. Biol. 2016, 12, 109–116. [Google Scholar] [CrossRef]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [Green Version]
- Chavez, K.J.; Garimella, S.V.; Lipkowitz, S. Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 2010, 32, 35. [Google Scholar] [CrossRef] [Green Version]
- Khatri, P.; Drăghici, S. Ontological analysis of gene expression data: Current tools, limitations, and open problems. Bioinformatics 2005, 21, 3587–3595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, G.O. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [Green Version]
- Anand, K.; Patel, T.; Niravath, P.; Rodriguez, A.; Darcourt, J.; Belcheva, A.; Boone, T.; Ensor, J.; Chang, J. Targeting mTOR and DNA repair pathways in residual triple negative breast cancer post neoadjuvant chemotherapy. Sci. Rep. 2021, 11, 1–8. [Google Scholar]
- Radosevic-Robin, N.; Selenica, P.; Zhu, Y.; Won, H.H.; Berger, M.F.; Ferrando, L.; Cocco, E.; Privat, M.; Ponelle-Chachuat, F.; Abrial, C.; et al. Recurrence biomarkers of triple negative breast cancer treated with neoadjuvant chemotherapy and anti-EGFR antibodies. NPJ Breast Cancer 2021, 7, 1–10. [Google Scholar] [CrossRef]
- Gong, Y.; Ji, P.; Yang, Y.S.; Xie, S.; Yu, T.J.; Xiao, Y.; Jin, M.L.; Ma, D.; Guo, L.W.; Pei, Y.C.; et al. Metabolic-pathway-based subtyping of triple-negative breast cancer reveals potential therapeutic targets. Cell Metab. 2021, 33, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Ismaila, N.; Henry, N.L.; Somerfield, M.R.; Bast, R.C.; Barlow, W.; Collyar, D.E.; Hammond, M.E.; Kuderer, N.M.; Liu, M.C.; et al. Use of biomarkers to guide decisions on adjuvant systemic therapy for women with early-stage invasive breast cancer: ASCO clinical practice guideline update—integration of results from TAILORx. J. Clin. Oncol. 2019, 37, 1956–1964. [Google Scholar] [CrossRef] [Green Version]
- Meti, N.; Saednia, K.; Lagree, A.; Tabbarah, S.; Mohebpour, M.; Kiss, A.; Lu, F.I.; Slodkowska, E.; Gandhi, S.; Jerzak, K.J.; et al. Machine learning frameworks to predict neoadjuvant chemotherapy response in breast cancer using clinical and pathological features. JCO Clin. Cancer Inf. 2021, 5, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chu, Y.; Wang, B.; Jiang, T. A Narrative Review of Ultrasound Technologies for the Prediction of Neoadjuvant Chemotherapy Response in Breast Cancer. Cancer Manag. Res. 2021, 13, 7885. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner, A.; Tausch, C.; Hosch, S.; Papassotiropoulos, B.; Varga, Z.; Rageth, C.; Baege, A. Ultrasound-based prediction of pathologic response to neoadjuvant chemotherapy in breast cancer patients. Breast 2018, 39, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Comes, M.C.; Fanizzi, A.; Bove, S.; Didonna, V.; Diotaiuti, S.; La Forgia, D.; Latorre, A.; Martinelli, E.; Mencattini, A.; Nardone, A.; et al. Early prediction of neoadjuvant chemotherapy response by exploiting a transfer learning approach on breast DCE-MRIs. Sci. Rep. 2021, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, H.; Islam, S.; Alam, N.; Roy, A.; Roychoudhury, S.; Panda, C.K. Hypomethylation of mismatch repair genes MLH1 and MSH2 is associated with chemotolerance of breast carcinoma: Clinical significance. J. Surg. Oncol. 2019, 119, 88–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Reilly, E.A.; Gubbins, L.; Sharma, S.; Tully, R.; Guang, M.H.Z.; Weiner-Gorzel, K.; McCaffrey, J.; Harrison, M.; Furlong, F.; Kell, M.; et al. The fate of chemoresistance in triple negative breast cancer (TNBC). BBA Clin. 2015, 3, 257–275. [Google Scholar] [CrossRef] [Green Version]
- Ren, X.y.; Song, Y.; Wang, J.; Chen, L.y.; Pang, J.y.; Zhou, L.R.; Shen, S.j.; Cao, X.; Wang, Y.x.; Shao, M.m.; et al. Mismatch repair deficiency and microsatellite instability in triple-negative breast cancer: A retrospective study of 440 patients. Front. Oncol. 2021, 11, 570623. [Google Scholar] [CrossRef]
- Schafer, J.M.; Lehmann, B.D.; Gonzalez-Ericsson, P.I.; Marshall, C.B.; Beeler, J.S.; Redman, L.N.; Jin, H.; Sanchez, V.; Stubbs, M.C.; Scherle, P.; et al. Targeting MYCN-expressing triple-negative breast cancer with BET and MEK inhibitors. Sci. Transl. Med. 2020, 12, 534. [Google Scholar] [CrossRef] [PubMed]
- Vishnubalaji, R.; Alajez, N.M. Transcriptional landscape associated with TNBC resistance to neoadjuvant chemotherapy revealed by single-cell RNA-seq. Mol.-Ther.-Oncolytics 2021, 23, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Llop-Guevara, A.; Loibl, S.; Villacampa, G.; Vladimirova, V.; Schneeweiss, A.; Karn, T.; Zahm, D.M.; Herencia-Ropero, A.; Jank, P.; van Mackelenbergh, M.; et al. Association of RAD51 with homologous recombination deficiency (HRD) and clinical outcomes in untreated triple-negative breast cancer (TNBC): Analysis of the GeparSixto randomized clinical trial. Ann. Oncol. 2021, 32, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Opyrchal, M.; Salisbury, J.L.; Iankov, I.; Goetz, M.P.; McCubrey, J.; Gambino, M.W.; Malatino, L.; Puccia, G.; Ingle, J.N.; Galanis, E.; et al. Inhibition of Cdk2 kinase activity selectively targets the CD44+/CD24-/Low stem-like subpopulation and restores chemosensitivity of SUM149PT triple-negative breast cancer cells. Int. J. Oncol. 2014, 45, 1193–1199. [Google Scholar] [CrossRef] [Green Version]
- Nie, L.; Wei, Y.; Zhang, F.; Hsu, Y.H.; Chan, L.C.; Xia, W.; Ke, B.; Zhu, C.; Deng, R.; Tang, J.; et al. CDK2-mediated site-specific phosphorylation of EZH2 drives and maintains triple-negative breast cancer. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Tarasewicz, E.; Rivas, L.; Hamdan, R.; Dokic, D.; Parimi, V.; Bernabe, B.P.; Thomas, A.; Shea, L.D.; Jeruss, J.S. Inhibition of CDK-mediated phosphorylation of Smad3 results in decreased oncogenesis in triple negative breast cancer cells. Cell Cycle 2014, 13, 3191–3201. [Google Scholar] [CrossRef] [Green Version]
- Tormo, E.; Ballester, S.; Adam-Artigues, A.; Burgués, O.; Alonso, E.; Bermejo, B.; Menéndez, S.; Zazo, S.; Madoz-Gúrpide, J.; Rovira, A.; et al. The miRNA-449 family mediates doxorubicin resistance in triple-negative breast cancer by regulating cell cycle factors. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Jusino, S.; Rivera-Rivera, Y.; Chardón-Colón, C.; Ruiz-Justiz, A.J.; Vélez-Velázquez, J.; Isidro, A.; Cruz-Robles, M.E.; Bonilla-Claudio, M.; Armaiz-Pena, G.N.; Saavedra, H.I. E2F3 drives the epithelial-to-mesenchymal transition, cell invasion, and metastasis in breast cancer. Exp. Biol. Med. 2021, 246, 2057–2071. [Google Scholar] [CrossRef]
- Issac, M.S.M.; Yousef, E.; Tahir, M.R.; Gaboury, L.A. MCM2, MCM4, and MCM6 in breast cancer: Clinical utility in diagnosis and prognosis. Neoplasia 2019, 21, 1015–1035. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study | Abb. | Target Population | Preprocessing | Batch Correction | Performance (AUC) | |
|---|---|---|---|---|---|---|
| Hess et al., 2006, JCO [23] | DLDA30 | 30 | All | dChip | Not Reported | 0.877 |
| Parker et al., 2009, JCO [27] | ROR-S | 52 | All | Not Reported | Not Reported | 0.781 |
| Liedtke et al., 2009, JCO [22] | GGI | 97 | All | Not Reported | Not Reported | 0.735 |
| Hatzis et al., 2011, JAMA [18] | Hatzis | 206 | HER2- | MAS5 | Not Reported | Not Reported |
| Fournier et al., 2019, Sci. Rep. [26] | BA100 | 32 | TNBC | MAS5 | ComBat, QtNorm | Not Reported |
| Masanori et al., 2021, Cancers [19] | 3 gene | 3 | TNBC | Not Reported | Not Reported | 0.735 |
| Masanori et al., 2021, Am. J. Cancer Res. [20] | 5 gene | 5 | ER+/HER2- | Not Reported | Not Reported | 0.813 |
| Changfang Fu et al., 2021, Front. Immunol. [21] | Immune gene | 25 | All | RMA | ComBat | 0.956 |
| Study | GSE | GPL | Platform | NAC Regimen | pCR | RD | Total |
|---|---|---|---|---|---|---|---|
| Hatzis et al., 2011, JAMA [18] | GSE25066 | GPL96 | Affymetrix HG U133A | T/FAC (75%), T/FEC (18%), other (7%) | 57 | 113 | 170 |
| Tabchy A et al., 2010, Clin. Cancer. Res. [31] | GSE20271 | GPL96 | Affymetrix HG U133A | T/FAC (36.2%), T/FEC (27.6%), FAC (10.3%), FEC (17.2%), other (1.70%) | 13 | 45 | 58 |
| Shi L et al., 2010, Nat. Biotech. [32] | GSE20194 | GPL96 | Affymetrix HG U133A | T/FAC (67.6%), T/FEC (16.9%), FAC (4.23%), FEC (1.41%), other (9.87%) | 25 | 46 | 71 |
| Miyake T et al., 2012, Cancer Sci. [33] | GSE32646 | GPL570 | Affymetrix HG U133 Plus 2.0 | T/FEC (100%) | 10 | 16 | 26 |
| Year | Authors | Title | Journal | References |
|---|---|---|---|---|
| 2019 | Nadia Harbeck et al. | Breast Cancer | Nat. Rev. Dis. Primers | PMID: 31548545 [36] |
| 2019 | Francois Bertucci et al. | Genomic Characterization of Metastatic Breast Cancers | Nature | PMID: 31118521 [37] |
| 2018 | Francisco Sanchez-Vega et al. | Oncogenic Signaling Pathways in The Cancer Genome Atlas | Cell | PMID: 29625050 [38] |
| 2018 | Chandra P. Leo et al. | Breast Cancer Drug Approvals by the US FDA From 1949 to 2018 | NRDD | PMID: 31907423 [39] |
| 2016 | Xiaofeng Dai et al. | Cancer Hallmarks, Biomarkers and Breast Cancer Molecular Subtypes | J. Cancer | PMID: 27390604 [40] |
| 2016 | Serena Nik-Zainal et al. | Landscape of Somatic Mutations in 560 Breast Cancer Whole-Genome Sequences | Nature | PMID: 27135926 [41] |
| 2012 | Cancer Genome Atlas Network | Comprehensive Molecular Portraits of Human Breast Tumours | Nature | PMID: 23000897 [42] |
| Characteristic | GSE25066 | GSE20271 | GSE20194 | GSE32646 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pCR | RD | p-Value | pCR | RD | p-Value | pCR | RD | p-Value | pCR | RD | p-Value | |
| N | 57 | 113 | 13 | 45 | 25 | 46 | 10 | 16 | ||||
| Age (Med, IQR) | 48 (41–53) | 50 (40–60) | 0.1 | 53 (49–58) | 51 (40–58) | 0.47 | 48 (44–53) | 51 (42–61) | 0.17 | 60 (54–67) | 56 (43–63) | 0.46 |
| T Stage | 0.28 | 0.42 | 0.95 | 0.73 | ||||||||
| T1 | 3 (5%) | 5 (4%) | 0 (0%) | 1 (2%) | 2 (8%) | 4 (9%) | 1 (10%) | 0 (0%) | ||||
| T2 | 32 (56%) | 47 (42%) | 8 (62%) | 15 (33%) | 13 (52%) | 20 (43%) | 8 (80%) | 12 (75%) | ||||
| T3 | 15 (26%) | 38 (34%) | 2 (15%) | 13 (29%) | 5 (20%) | 9 (20%) | 1 (10%) | 3 (19%) | ||||
| T4 | 7 (12%) | 23 (20%) | 3 (23%) | 16 (36%) | 5 (20%) | 12 (26%) | 0 (0%) | 1 (6%) | ||||
| Missing | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (2%) | ||||||
| N Stage | 0.86 | 0.61 | 0.32 | NA | ||||||||
| N0 | 15 (26%) | 26 (23%) | 4 (31%) | 14 (31%) | 2 (8%) | 9 (20%) | ||||||
| N1 | 26 (46%) | 52 (46%) | 7 (54%) | 16 (36%) | 16 (64%) | 18 (39%) | ||||||
| N2 | 8 (14%) | 21 (19%) | 2 (15%) | 12 (27%) | 3 (12%) | 8 (17%) | ||||||
| N3 | 8 (14%) | 14 (12%) | 0 (0%) | 3 (7%) | 3 (12%) | 7 (15%) | ||||||
| Missing | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (4%) | 4 (9%) | ||||||
| Grade | 0.34 | 1 | 0.19 | 0.32 | ||||||||
| G1 | 0 (0%) | 1 (1%) | 0 (0%) | 1 (2%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (6%) | ||||
| G2 | 4 (7%) | 16 (14%) | 2 (15%) | 6 (13%) | 2 (8%) | 9 (20%) | 4 (40%) | 10 (62%) | ||||
| G3 | 48 (84%) | 86 (76%) | 9 (69%) | 26 (58%) | 22 (88%) | 31 (67%) | 6 (60%) | 5 (31%) | ||||
| Missing | 5 (9%) | 10 (9%) | 2 (15%) | 12 (27%) | 1 (4%) | 6 (13%) | 0 (0%) | 0 (0%) | ||||
| Tumor Stage | 0.79 | NA | NA | 0.54 | ||||||||
| I | 1 (2%) | 2 (2%) | 0 (0%) | 0 (0%) | ||||||||
| IIA | 10 (18%) | 15 (13%) | 1 (10%) | 4 (25%) | ||||||||
| IIB | 20 (35%) | 34 (30%) | 8 (80%) | 8 (50%) | ||||||||
| IIIA | 11 (19%) | 32 (28%) | 1 (10%) | 3 (19%) | ||||||||
| IIIB | 10 (18%) | 21 (19%) | 0 (0%) | 1 (6%) | ||||||||
| IIIC | 5 (9%) | 7 (6%) | 0 (0%) | 0 (0%) | ||||||||
| Inflammatory | 0 (0%) | 2 (2%) | 0 (0%) | 0 (0%) | ||||||||
| Symbol | NCBI ID | Count | FC | FDR | Direction | Symbol | NCBI ID | Count | FC | FDR | Direction |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HAT1 | 8520 | 10 | 1.330 | 0.00 | Up | LRRC15 | 131578 | 3 | 0.676 | 5.89 × 10 | Down |
| TFG | 10342 | 10 | 1.288 | 0.00 | Up | PTGS1 | 5742 | 3 | 0.816 | 6.03 × 10 | Down |
| JCAD | 57608 | 10 | 0.701 | 0.00 | Down | HGH1 | 51236 | 3 | 0.847 | 6.05 × 10 | Down |
| ZNF467 | 168544 | 10 | 0.740 | 0.00 | Down | FBXO16 | 157574 | 3 | 1.447 | 6.16 × 10 | Up |
| ATF5 | 22809 | 9 | 0.795 | 5.97 × 10 | Down | SLC43A1 | 8501 | 3 | 0.806 | 6.25 × 10 | Down |
| ABT1 | 29777 | 9 | 1.636 | 5.97 × 10 | Up | EXD2 | 55218 | 2 | 0.834 | 6.47 × 10 | Down |
| PDCL3 | 79031 | 9 | 1.361 | 6.39 × 10 | Up | SMARCA2 | 6595 | 2 | 1.243 | 6.80 × 10 | Up |
| ILF2 | 3608 | 9 | 1.528 | 6.87 × 10 | Up | GREM1 | 26585 | 2 | 0.681 | 7.02 × 10 | Down |
| DNAI4 | 79819 | 9 | 0.716 | 8.33 × 10 | Down | NCR1 | 9437 | 2 | 0.768 | 7.05 × 10 | Down |
| TMEM14B | 81853 | 9 | 1.360 | 9.48 × 10 | Up | PARM1 | 25849 | 2 | 0.741 | 7.67 × 10 | Down |
| DEK | 7913 | 8 | 1.450 | 1.38 × 10 | Up | ZNF395 | 55893 | 2 | 1.317 | 7.72 × 10 | Up |
| PDCL3P4 | 285359 | 8 | 1.361 | 1.41 × 10 | Up | MAST4 | 375449 | 1 | 0.737 | 7.90 × 10 | Down |
| SEC13 | 6396 | 8 | 1.244 | 1.45 × 10 | Up | CTAGE11P | 647288 | 1 | 0.638 | 7.94 × 10 | Down |
| HACD1 | 9200 | 7 | 1.768 | 2.15 × 10 | Up | IMPG2 | 50939 | 1 | 0.585 | 8.02 × 10 | Down |
| GLI3 | 2737 | 6 | 0.752 | 3.03 × 10 | Down | FN3KRP | 79672 | 1 | 1.218 | 8.72 × 10 | Up |
| PTPN1 | 5770 | 6 | 0.838 | 3.15 × 10 | Down | DCTN3 | 11258 | 1 | 1.225 | 8.80 × 10 | Up |
| MCM3 | 4172 | 6 | 1.274 | 3.71 × 10 | Up | CTTN | 2017 | 1 | 0.808 | 8.93 × 10 | Down |
| RANBP6 | 26953 | 5 | 1.341 | 4.04 × 10 | Up | TMEM258 | 746 | 1 | 1.205 | 9.16 × 10 | Up |
| ESR1 | 2099 | 5 | 0.728 | 4.13 × 10 | Down | MANBA | 4126 | 1 | 0.798 | 9.35 × 10 | Down |
| ITGA6 | 3655 | 5 | 1.740 | 4.23 × 10 | Up | CSRNP2 | 81566 | 1 | 0.842 | 9.40 × 10 | Down |
| NOL7 | 51406 | 4 | 1.256 | 4.32 × 10 | Up | NEU1 | 4758 | 1 | 1.245 | 9.44 × 10 | Up |
| PRKACA | 5566 | 4 | 0.801 | 4.34 × 10 | Down | OLFML2B | 25903 | 1 | 0.733 | 1.04 × 10 | Down |
| CCND1 | 595 | 4 | 0.564 | 4.41 × 10 | Down | MDH1 | 4190 | 1 | 1.203 | 1.06 × 10 | Up |
| RIPOR1 | 79567 | 4 | 0.820 | 5.07 × 10 | Down | PDE10A | 10846 | 1 | 0.793 | 1.06 × 10 | Down |
| SEZ6L | 23544 | 4 | 0.782 | 5.23 × 10 | Down | XRCC5 | 7520 | 1 | 1.167 | 1.12 × 10 | Up |
| METRN | 79006 | 3 | 0.513 | 5.56 × 10 | Down | BNC2 | 54796 | 1 | 0.744 | 1.15 × 10 | Down |
| Dataset | pCR (Positive) | RD (Negative) | TP | FP | FN | TN | ACC | BACC | TPR | TNR | PPV | NPV | FNR | F1 | MCC | Yoden’s Index | AUROC | AUPRC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GSE25066 | 17 | 33 | 14 | 3 | 3 | 30 | 0.880 | 0.866 | 0.824 | 0.909 | 0.909 | 0.909 | 0.177 | 0.824 | 0.733 | 0.733 | 0.918 | 0.902 |
| GSE20271 | 13 | 45 | 5 | 4 | 8 | 41 | 0.793 | 0.648 | 0.385 | 0.911 | 0.911 | 0.837 | 0.615 | 0.455 | 0.341 | 0.296 | 0.779 | 0.589 |
| GSE20194 | 25 | 46 | 22 | 4 | 3 | 42 | 0.901 | 0.897 | 0.880 | 0.913 | 0.913 | 0.933 | 0.120 | 0.863 | 0.786 | 0.793 | 0.967 | 0.946 |
| GSE32646 | 10 | 16 | 4 | 1 | 6 | 15 | 0.731 | 0.669 | 0.400 | 0.938 | 0.938 | 0.714 | 0.600 | 0.533 | 0.417 | 0.338 | 0.747 | 0.688 |
| Total | 65 | 140 | 45 | 13 | 20 | 127 | 0.839 | 0.800 | 0.692 | 0.907 | 0.907 | 0.864 | 0.308 | 0.732 | 0.619 | 0.599 | 0.891 | 0.829 |
| Dataset | pCR | RD | TP | FP | FN | TN | ACC | BACC | TPR | TNR | PPV | NPV | FNR | F1 | MCC | Yoden’s Index | AUROC | AUPRC | Method |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Test Set (GSE25066) | 17 | 33 | 5 | 2 | 12 | 31 | 0.720 | 0.617 | 0.294 | 0.939 | 0.939 | 0.721 | 0.706 | 0.417 | 0.319 | 0.234 | 0.629 | 0.468 | GGI |
| 2 | 3 | 15 | 30 | 0.640 | 0.513 | 0.118 | 0.909 | 0.909 | 0.667 | 0.882 | 0.182 | 0.042 | 0.027 | 0.496 | 0.324 | ROR-S | |||
| 7 | 2 | 10 | 31 | 0.760 | 0.676 | 0.412 | 0.939 | 0.939 | 0.756 | 0.588 | 0.539 | 0.433 | 0.351 | 0.815 | 0.733 | Hatzis | |||
| 7 | 2 | 10 | 31 | 0.760 | 0.676 | 0.412 | 0.939 | 0.939 | 0.756 | 0.588 | 0.539 | 0.433 | 0.351 | 0.708 | 0.561 | 3 Gene | |||
| 2 | 1 | 15 | 32 | 0.680 | 0.544 | 0.118 | 0.970 | 0.970 | 0.681 | 0.882 | 0.200 | 0.174 | 0.087 | 0.674 | 0.506 | 5 Gene | |||
| 4 | 3 | 13 | 30 | 0.680 | 0.572 | 0.235 | 0.909 | 0.909 | 0.698 | 0.765 | 0.333 | 0.197 | 0.144 | 0.681 | 0.545 | Immune | |||
| 7 | 2 | 10 | 31 | 0.760 | 0.676 | 0.412 | 0.939 | 0.939 | 0.756 | 0.588 | 0.539 | 0.433 | 0.351 | 0.793 | 0.679 | BA100 C1 | |||
| 8 | 2 | 9 | 31 | 0.780 | 0.705 | 0.471 | 0.939 | 0.939 | 0.775 | 0.529 | 0.593 | 0.486 | 0.410 | 0.759 | 0.632 | BA100 C2 | |||
| 1 | 3 | 16 | 30 | 0.620 | 0.484 | 0.059 | 0.909 | 0.909 | 0.652 | 0.941 | 0.095 | -0.056 | -0.032 | 0.622 | 0.391 | DLDA30 | |||
| 14 | 2 | 3 | 31 | 0.900 | 0.882 | 0.824 | 0.939 | 0.939 | 0.912 | 0.177 | 0.849 | 0.775 | 0.763 | 0.918 | 0.902 | Proposed | |||
| All Testsets | 65 | 141 | 12 | 14 | 53 | 127 | 0.675 | 0.543 | 0.185 | 0.901 | 0.901 | 0.706 | 0.815 | 0.264 | 0.119 | 0.085 | 0.606 | 0.406 | GGI |
| 9 | 14 | 56 | 127 | 0.660 | 0.520 | 0.139 | 0.901 | 0.901 | 0.694 | 0.862 | 0.205 | 0.058 | 0.039 | 0.545 | 0.344 | ROR-S | |||
| 24 | 14 | 41 | 127 | 0.733 | 0.635 | 0.369 | 0.901 | 0.901 | 0.756 | 0.631 | 0.466 | 0.323 | 0.270 | 0.827 | 0.634 | Hatzis | |||
| 16 | 12 | 49 | 129 | 0.704 | 0.581 | 0.246 | 0.915 | 0.915 | 0.725 | 0.754 | 0.344 | 0.218 | 0.161 | 0.705 | 0.490 | 3 Gene | |||
| 18 | 13 | 47 | 128 | 0.709 | 0.592 | 0.277 | 0.908 | 0.908 | 0.731 | 0.723 | 0.375 | 0.240 | 0.185 | 0.691 | 0.499 | 5 Gene | |||
| 9 | 14 | 56 | 127 | 0.660 | 0.520 | 0.139 | 0.901 | 0.901 | 0.694 | 0.862 | 0.205 | 0.058 | 0.039 | 0.593 | 0.381 | Immune | |||
| 19 | 14 | 46 | 127 | 0.709 | 0.597 | 0.292 | 0.901 | 0.901 | 0.734 | 0.708 | 0.388 | 0.245 | 0.193 | 0.716 | 0.500 | BA100 C1 | |||
| 26 | 14 | 39 | 127 | 0.743 | 0.650 | 0.400 | 0.901 | 0.901 | 0.765 | 0.600 | 0.495 | 0.353 | 0.301 | 0.729 | 0.567 | BA100 C2 | |||
| 15 | 13 | 50 | 128 | 0.694 | 0.569 | 0.231 | 0.908 | 0.908 | 0.719 | 0.769 | 0.323 | 0.188 | 0.139 | 0.728 | 0.490 | DLDA30 | |||
| 45 | 13 | 20 | 128 | 0.840 | 0.800 | 0.692 | 0.908 | 0.908 | 0.865 | 0.308 | 0.732 | 0.620 | 0.600 | 0.892 | 0.829 | Proposed |
| SRC DB | Geneset Name | Category | p-Value | Gene Mapped | |||
|---|---|---|---|---|---|---|---|
| Gene Ontology | GO:0043570: maintenance of DNA repeat elements | Cell_Function | 5 | 2 | 0.400 | 8.78 × 10 | MSH2, MSH6 |
| Gene Ontology | GO:0032135: DNA insertion or deletion binding | Cell_Function | 6 | 2 | 0.333 | 1.31 × 10 | MSH2, MSH6 |
| Jensen_COMPARTMENTS | Mismatch_repair_complex | Cell_Localization | 6 | 2 | 0.333 | 1.31 × 10 | MSH2, MSH6 |
| TF_Perturbations_Followed_by_Expression | MYCN_SHRNA_IMR575_HUMAN_GSE80397_6HR_RNASEQ_UP | Transcription Factor | 6 | 2 | 0.333 | 1.31 × 10 | ENO1, MCM3 |
| CORUM | MSH2/6-BLM-p53-RAD51 complex (human) | Protein_Complex | 5 | 3 | 0.600 | 2.57 × 10 | RAD51, MSH2, MSH6 |
| CORUM | PCNA-MutS-alpha-MutL-alpha-DNA complex (human) | Protein_Complex | 5 | 2 | 0.400 | 8.78 × 10 | MSH2, MSH6 |
| CORUM | MCM complex (human) | Protein_Complex | 6 | 2 | 0.333 | 1.31 × 10 | MCM2, MCM3 |
| Reactome | R-HSA-68911: G2 Phase | Pathway | 5 | 2 | 0.400 | 8.78 × 10 | CDK2, E2F3 |
| MPS Subtypes | pCR | RD | TP | FP | FN | TN | ACC | BACC | TPR | TNR | PPV | NPV | FNR | F1 | MCC | Yoden’s Index | AUROC | AUPRC |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MPS1 (Lipogenic) | 8 | 36 | 3 | 3 | 5 | 33 | 0.818 | 0.646 | 0.375 | 0.917 | 0.917 | 0.868 | 0.625 | 0.429 | 0.328 | 0.292 | 0.833 | 0.561 |
| MPS2 (Glycolytic) | 39 | 42 | 23 | 4 | 16 | 38 | 0.753 | 0.747 | 0.590 | 0.905 | 0.905 | 0.704 | 0.410 | 0.697 | 0.524 | 0.495 | 0.878 | 0.880 |
| MPS3 (Mixed) | 18 | 62 | 14 | 6 | 4 | 56 | 0.875 | 0.841 | 0.778 | 0.903 | 0.903 | 0.933 | 0.222 | 0.737 | 0.657 | 0.681 | 0.899 | 0.838 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, S.; Yi, G. Development of Gene Expression-Based Random Forest Model for Predicting Neoadjuvant Chemotherapy Response in Triple-Negative Breast Cancer. Cancers 2022, 14, 881. https://doi.org/10.3390/cancers14040881
Park S, Yi G. Development of Gene Expression-Based Random Forest Model for Predicting Neoadjuvant Chemotherapy Response in Triple-Negative Breast Cancer. Cancers. 2022; 14(4):881. https://doi.org/10.3390/cancers14040881
Chicago/Turabian StylePark, Seongyong, and Gwansu Yi. 2022. "Development of Gene Expression-Based Random Forest Model for Predicting Neoadjuvant Chemotherapy Response in Triple-Negative Breast Cancer" Cancers 14, no. 4: 881. https://doi.org/10.3390/cancers14040881
APA StylePark, S., & Yi, G. (2022). Development of Gene Expression-Based Random Forest Model for Predicting Neoadjuvant Chemotherapy Response in Triple-Negative Breast Cancer. Cancers, 14(4), 881. https://doi.org/10.3390/cancers14040881