
Immunotherapy for Colorectal Cancer: Mechanisms and Predictive Biomarkers




Abstract
:Simple Summary
Abstract
1. Introduction
2. Colorectal Cancer Response to Immunotherapy
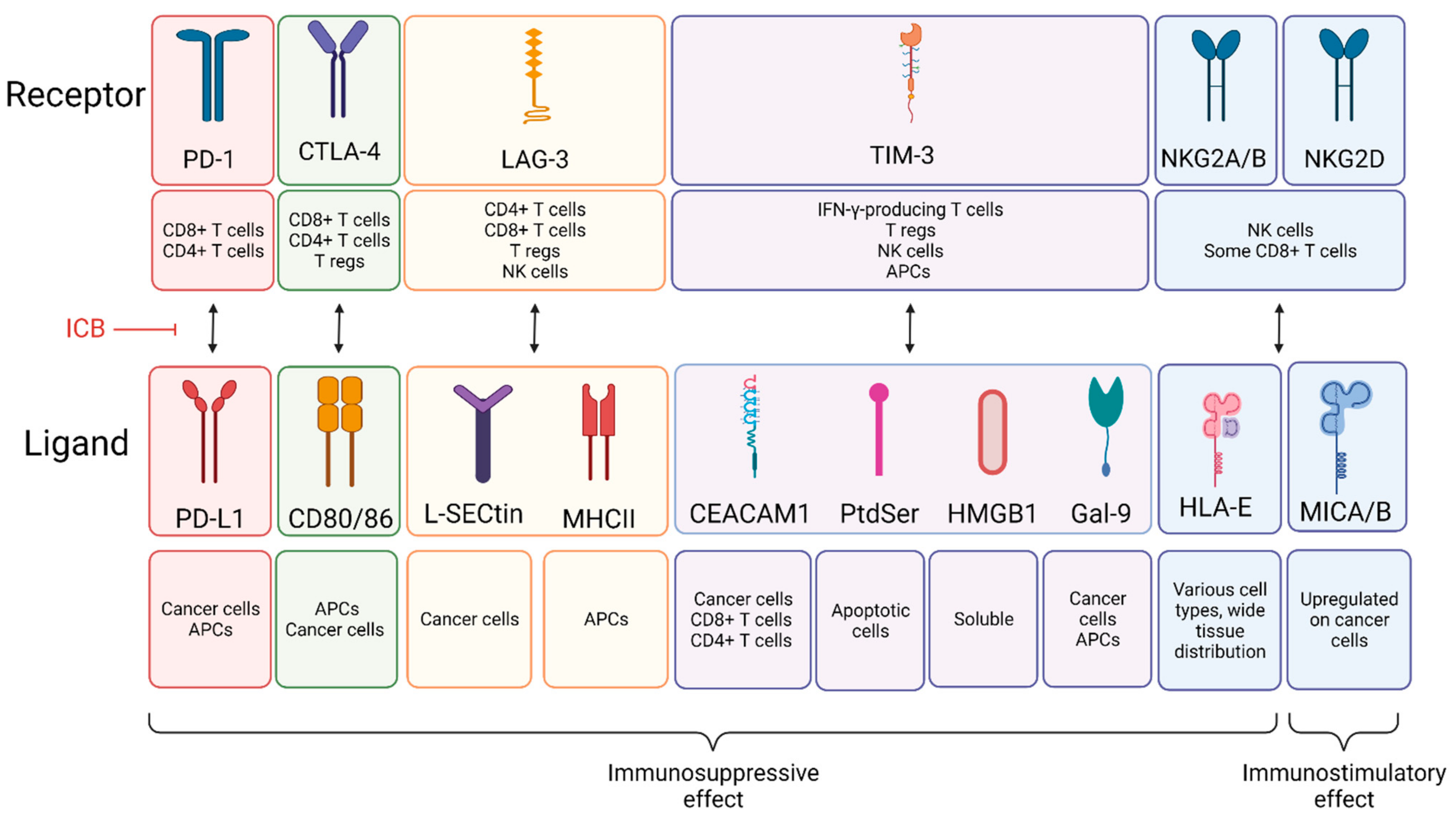
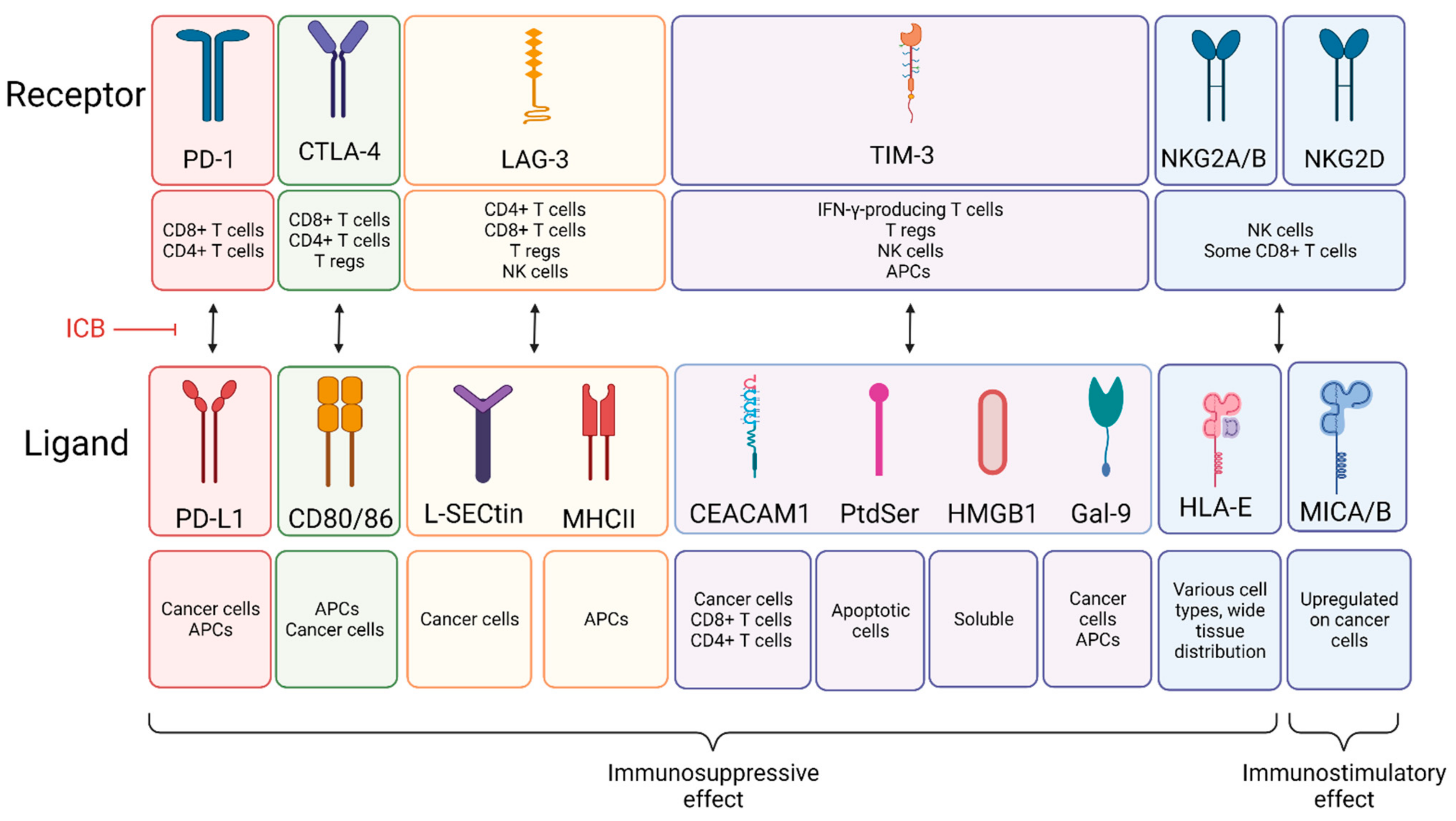
2.1. Immune Checkpoint Blockade
2.1.1. Anti-PD-1 and Anti-PD-L1
2.1.2. Anti-CTLA-4
2.1.3. Anti-LAG-3
2.1.4. Anti-TIM-3
2.1.5. Anti-NKG2
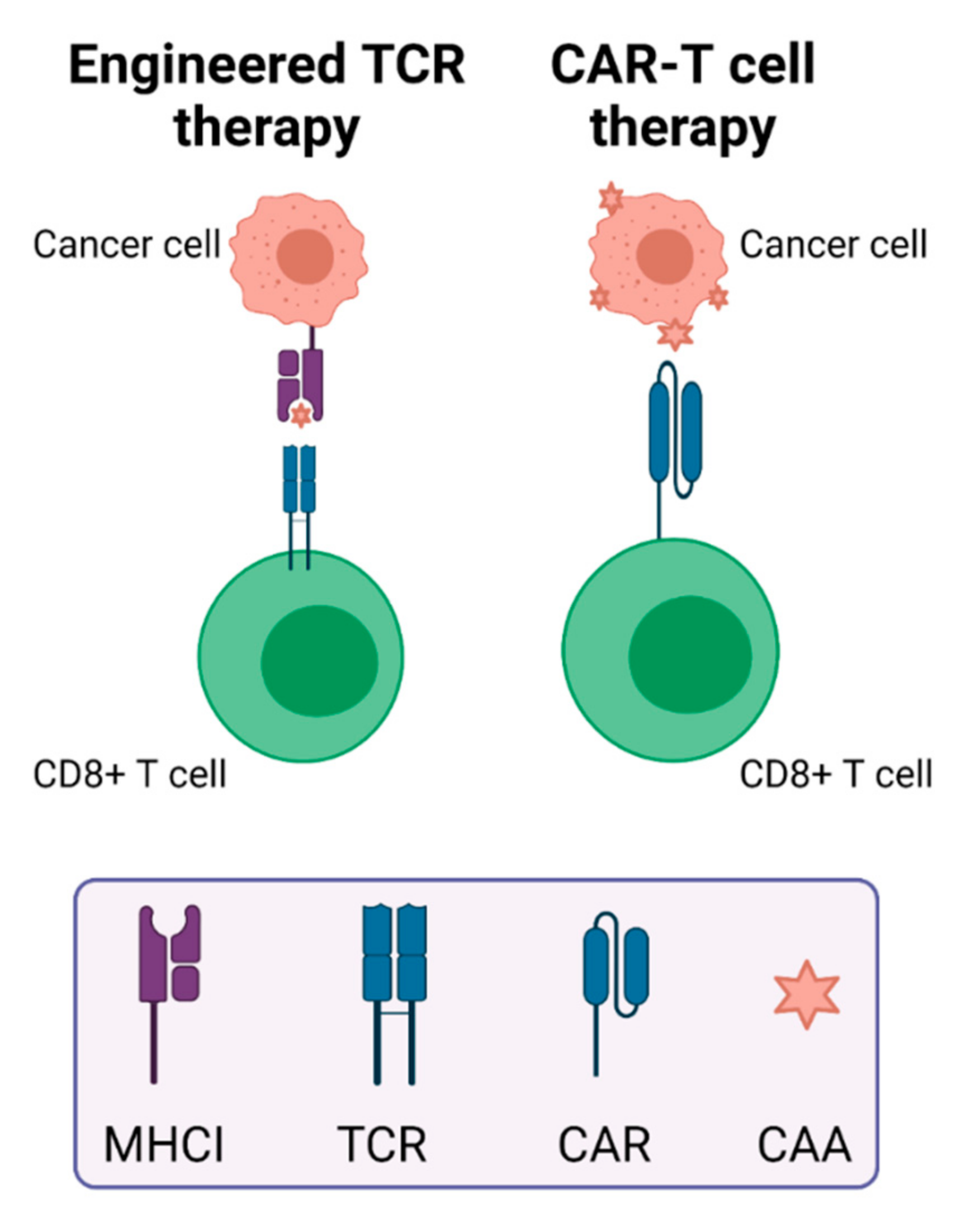
2.2. Adoptive Cell Therapies
2.2.1. Tumor-Infiltrating Lymphocyte Therapy
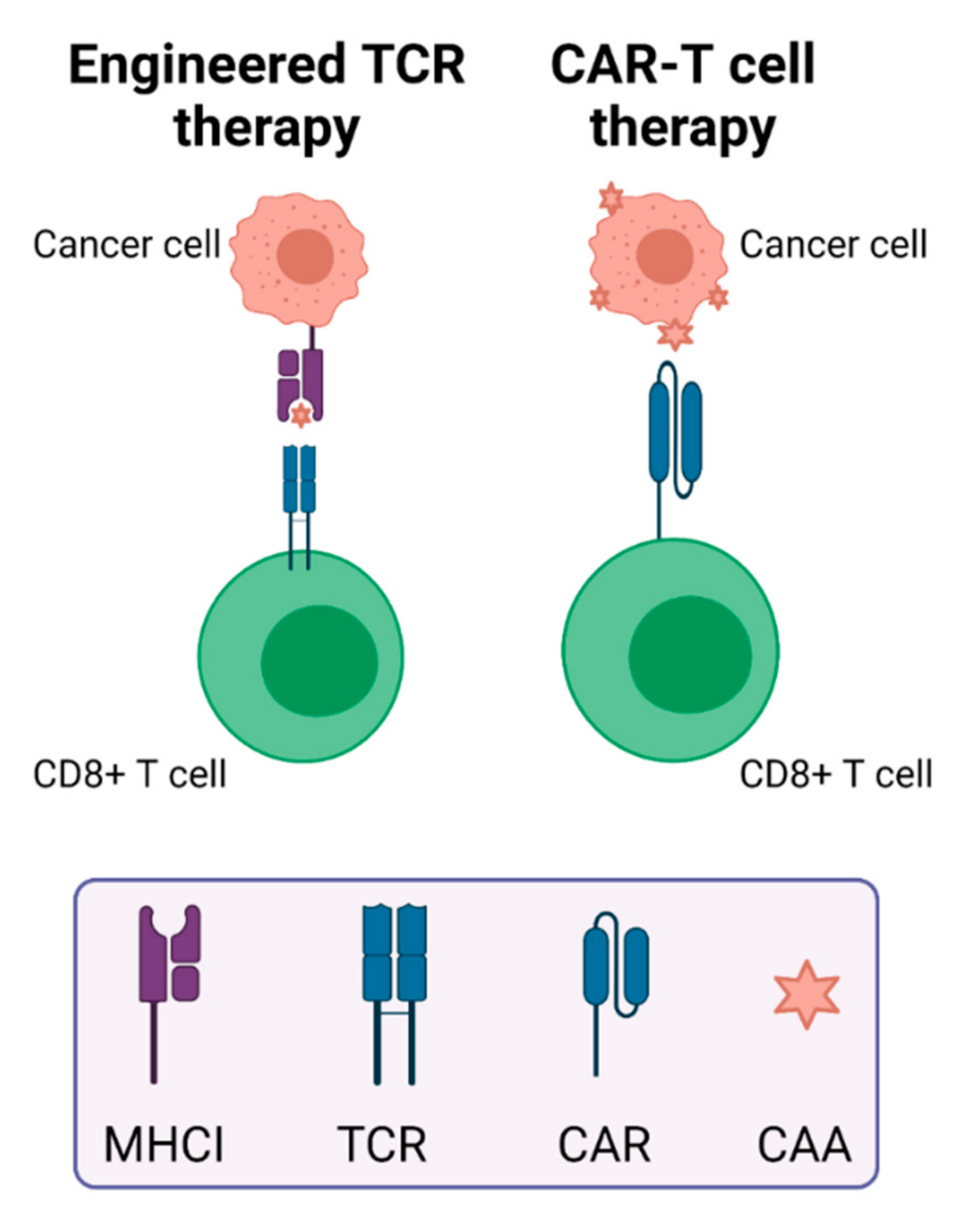
2.2.2. Engineered T Cell Therapy
2.2.3. Natural Killer Cell Therapy
2.3. Monoclonal Antibodies
2.3.1. Naked Monoclonal Antibodies
2.3.2. Conjugated Monoclonal Antibodies
2.3.3. Bispecific Antibodies
2.3.4. Nanobodies
2.4. Oncolytic Virus Therapy
2.5. Vaccines
2.5.1. Whole Tumor
2.5.2. Peptide
2.5.3. Viral Vector
2.5.4. Dendritic Cell Therapy
2.6. Immune System Modulators
2.6.1. Interleukins
2.6.2. Interferons
2.6.3. cGAS-STING
2.6.4. Immunomodulators
2.7. Targeting the Immunosuppressive TME
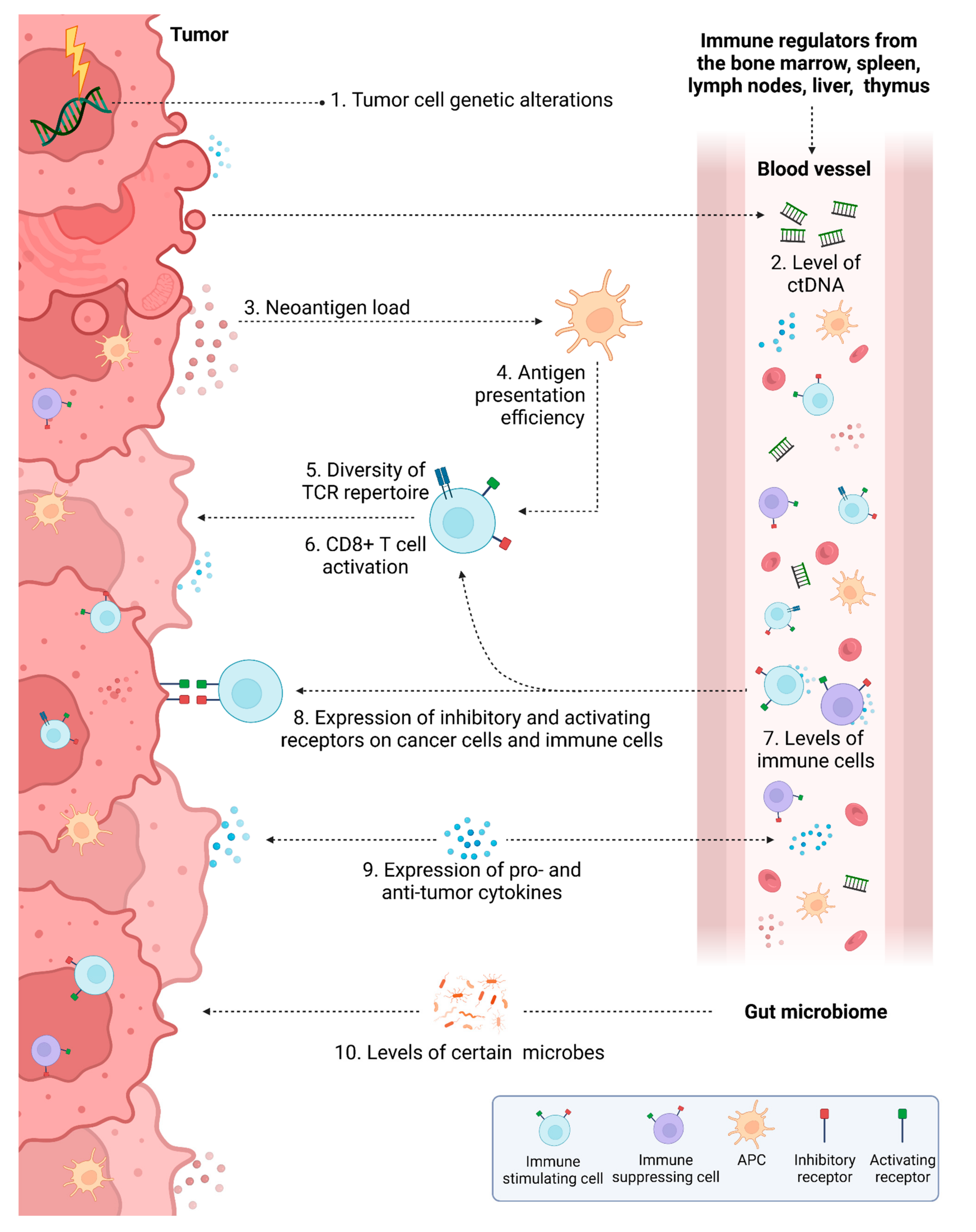
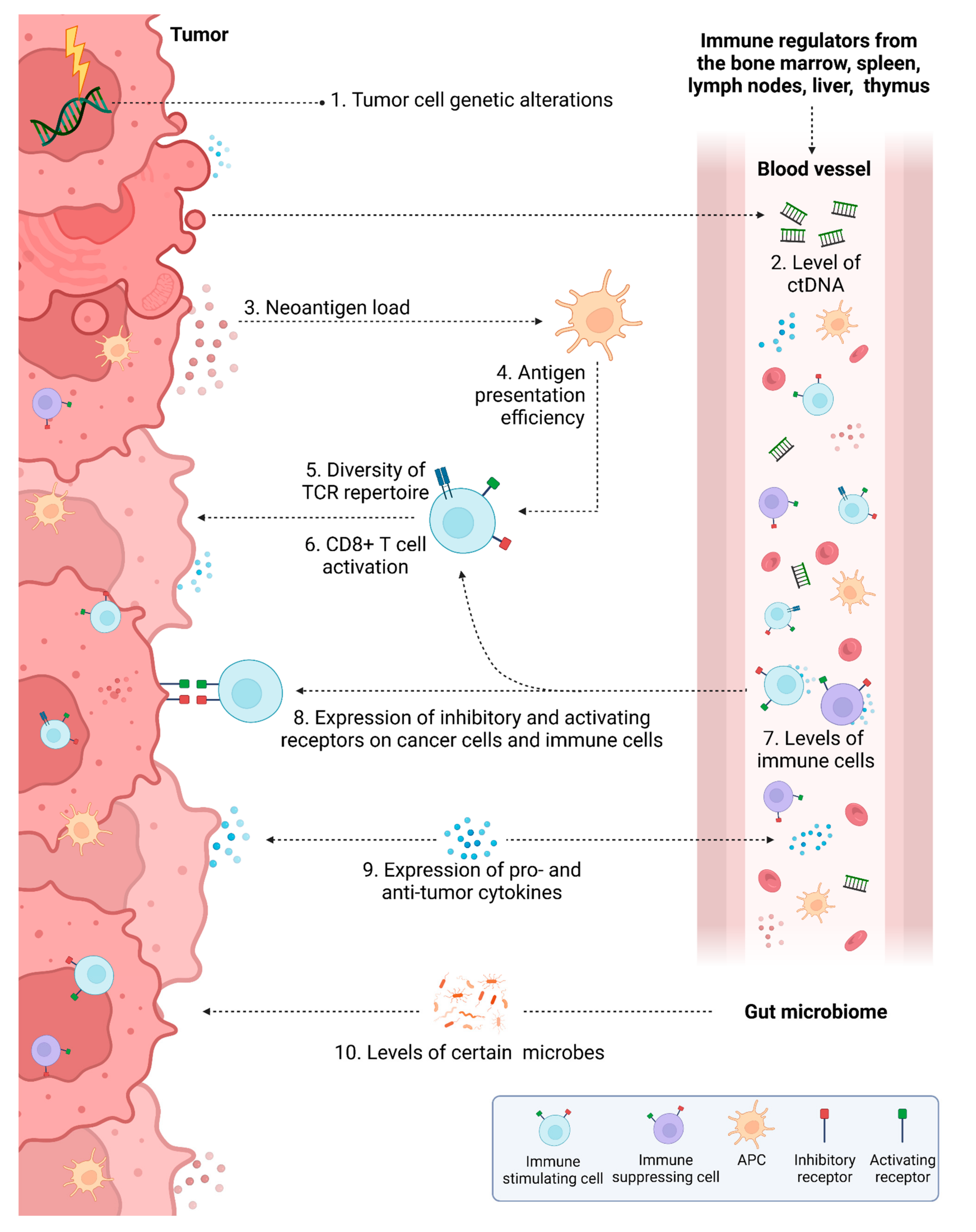
3. Predictive Biomarkers for Response to IT in Colorectal Cancer
3.1. Prediction Based on Genetic Alterations
3.1.1. Chromosomal Instability
3.1.2. Microsatellite Instability
3.1.3. CpG Island Methylation
3.1.4. Tumor Mutational Burden and Neoantigen Load
3.1.5. Other Specific Genetic Alterations
3.2. Prediction Based on the Tumor Microenvironment
3.2.1. PD-L1 Expression
3.2.2. Tumor-Infiltrating Lymphocytes
3.2.3. Immune Status of the Tumor Microenvironment
3.2.4. Diversity of T Cell Repertoires in TME
3.2.5. Tumor-Associated Macrophages
3.2.6. The Gut Microbiota
3.3. Liquid Biomarkers
3.3.1. Peripheral Blood Cells
3.3.2. Circulating Tumor DNA
3.3.3. Cytokines
3.3.4. Exosomes
3.4. Other Factors Influencing Response to IT
4. Sensitizing CRC to IT
5. Discussion and Open Questions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| Abbreviation | Meaning | Abbreviation | Meaning |
| 5-FU | 5-fluorouracil | mAbs | monoclonal antibodies |
| ACT | adoptive cell therapies | MAIT | mucosal-associated invariant T |
| ADCC | antibody-dependent cell-mediated cytotoxicity | MANAs | mutation-associated neoantigens |
| APCs | antigen-presenting cells | mCRC | metastatic CRC |
| bsAbs | bispecific antibodies | MDSCs | myeloid-derived suppressor cells |
| bsTCEs | T cell-engaging bsAbs | miRNA | microRNA |
| CAAs | cancer-associated antigens | MMR | mismatch repair |
| CAR-T cell | chimeric antigen receptor T cell | MSI | microsatellite instable |
| CEA | carcinoembryonic antigen | MSI-H | MSI-high |
| cGAS | cyclic GMP-AMP synthase | MSS | microsatellite stable |
| CIMP | CpG island methylator phenotype | Nbs | nanobodies |
| CIN | chromosomal instability | NDV | Newcastle disease virus |
| CRC | colorectal cancer | NK | natural killer |
| ctDNA | circulating tumor DNA | NKG2 | Natural Killer Group 2 |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 | NLR | neutrophil-to-lymphocyte |
| DCs | dendritic cells | NSCLC | non-small-cell lung cancer |
| dMMR | MMR deficiency | PD-1 | programmed cell death protein 1 |
| DR5 | death receptor 5 | PD-L1 | programmed cell death ligand 1 |
| dsDNA | double stranded DNA | PFS | progression-free survival |
| EpCAM/CD326 | epithelial cell adhesion molecule | pMMR | proficient MMR |
| EphA2 | ephrin type-A receptor 2 | PtdSer | phosphatidyl serine |
| FLT3L | Flt-3 Ligand | SNPs | single-nucleotide polymorphisms |
| Gal-9 | galectin-9 | STING | stimulator of interferon genes |
| GUCY2C | guanylyl cyclase C | STRs | short tandem repeats |
| HAVCR2 | hepatitis A virus cellular receptor 2 | T regs | regulatory T cells |
| HLA | human leukocyte antigen | TAAs | tumor-associated antigens |
| HMGB1 | high mobility group box 1 protein | TAG | tumor-associated glycoprotein |
| ICB | immune checkpoint blockade | TAMs | tumor-associated macrophages |
| IFNs | interferons | TCR | T cell receptor |
| ILs | interleukins | TGFβR2 | transforming growth factor β receptor type 2 |
| IMiDs | immunomodulatory drugs | TIL | tumor-infiltrating lymphocyte |
| IT | immunotherapy | TIM-3 | T cell immunoglobulin- and mucin-domain-containing-3 |
| LAG-3 | lymphocyte activation gene-3 | TMB-H | high tumor mutational burden |
| lncRNA | long non-coding RNA | TME | tumor microenvironment |
References
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.-H.; Chen, Y.-X.; Fang, J.-Y. Comprehensive review of targeted therapy for colorectal cancer. Signal Transduct. Target Ther. 2020, 5, 22. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.F.; Ibrahim, A.E.K.; Arends, M.J. Molecular pathological classification of colorectal cancer. Virchows Arch. 2016, 469, 125–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nojadeh, J.N.; Sharif, S.B.; Sakhinia, E. Microsatellite instability in colorectal cancer. EXCLI J. 2018, 17, 159–168. [Google Scholar] [PubMed]
- Tintelnot, J.; Stein, A. Immunotherapy in colorectal cancer: Available clinical evidence, challenges and novel approaches. World J. Gastroenterol. 2019, 25, 3920–3928. [Google Scholar] [CrossRef]
- Boland, C.R.; Goel, A. Microsatellite instability in colorectal cancer. Gastroenterology 2010, 138, 2073–2087.e3. [Google Scholar] [CrossRef]
- Berger, M.D.; Lenz, H.J. The safety of monoclonal antibodies for treatment of colorectal cancer. Expert Opin. Drug Saf. 2016, 15, 799–808. [Google Scholar] [CrossRef]
- Golshani, G.; Zhang, Y. Advances in immunotherapy for colorectal cancer: A review. Therap. Adv. Gastroenterol. 2020, 13, 1–11. [Google Scholar] [CrossRef]
- Marcus, L.; Lemery, S.J.; Keegan, P.; Pazdur, R. FDA Approval Summary: Pembrolizumab for the Treatment of Microsatellite Instability-High Solid Tumors. Clin. Cancer Res. 2019, 25, 3753–3758. [Google Scholar] [CrossRef] [Green Version]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.-J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): An open-label, multicentre, phase 2 study. Lancet Oncol. 2017, 18, 1182–1191. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Kavousanaki, M.; Ioannou, M.; Boumpas, D.; Verginis, P. The negative costimulatory molecule PD-1 modulates the balance between immunity and tolerance via miR-21. Eur. J. Immunol. 2011, 41, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar] [PubMed]
- Van Coillie, S.; Wiernicki, B.; Xu, J. Molecular and Cellular Functions of CTLA-4. Adv. Exp. Med. Biol. 2020, 1248, 7–32. [Google Scholar] [PubMed]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrego, F.; Masilamani, M.; Marusina, A.I.; Tang, X.; Coligan, J.E. The CD94/NKG2 family of receptors: From molecules and cells to clinical relevance. Immunol. Res. 2006, 35, 263–278. [Google Scholar] [CrossRef]
- Lanier, L.L. Up on the tightrope: Natural killer cell activation and inhibition. Nat. Immunol. 2008, 9, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, H.; Ding, J.; Liu, H.; Li, H.; Lu, M.; Miao, Y.; Li, L.; Zheng, J. Combination Therapy with EpCAM-CAR-NK-92 Cells and Regorafenib against Human Colorectal Cancer Models. J. Immunol. Res. 2018, 2018, 4263520. [Google Scholar] [CrossRef] [Green Version]
- Goodin, S. Development of monoclonal antibodies for the treatment of colorectal cancer. Am. J. Health Syst. Pharm. 2008, 65 (Suppl. 4), S3–S7. [Google Scholar] [CrossRef]
- Ning, S.-T.; Lee, S.-Y.; Wei, M.-F.; Peng, C.-L.; Lin, S.Y.-F.; Tsai, M.-H.; Lee, P.-C.; Shih, Y.-H.; Lin, C.-Y.; Luo, T.-Y.; et al. Targeting Colorectal Cancer Stem-Like Cells with Anti-CD133 Antibody-Conjugated SN-38 Nanoparticles. ACS Appl. Mater. Interfaces 2016, 8, 17793–17804. [Google Scholar] [CrossRef]
- Koganemaru, S.; Kuboki, Y.; Koga, Y.; Kojima, T.; Yamauchi, M.; Maeda, N.; Kagari, T.; Hirotani, K.; Yasunaga, M.; Matsumura, Y.; et al. U3-1402, a Novel HER3-Targeting Antibody-Drug Conjugate, for the Treatment of Colorectal Cancer. Mol. Cancer Ther. 2019, 18, 2043–2050. [Google Scholar] [CrossRef] [Green Version]
- Lédel, F.; Stenstedt, K.; Hallström, M.; Ragnhammar, P.; Edler, D. HER3 expression in primary colorectal cancer including corresponding metastases in lymph node and liver. Acta Oncol. 2015, 54, 480–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takegawa, N.; Yonesaka, K. HER2 as an Emerging Oncotarget for Colorectal Cancer Treatment After Failure of Anti-Epidermal Growth Factor Receptor Therapy. Clin. Colorectal Cancer 2017, 16, 247–251. [Google Scholar] [CrossRef] [PubMed]
- Stenger, M. Trastuzumab Deruxtecan-nxki in HER2-Positive Metastatic Colorectal Cancer: DESTINY-CRC01. 2021. Available online: https://ascopost.com/news/may-2021/trastuzumab-deruxtecan-nxki-in-her2-positive-metastatic-colorectal-cancer-destiny-crc01/ (accessed on 10 July 2021).
- Labrijn, A.F.; Janmaat, M.L.; Reichert, J.M.; Parren, P.W.H.I. Bispecific antibodies: A mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019, 18, 585–608. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Guo, H.-F.; Xu, H.; Cheung, N.-K.V. Development of a Tetravalent Anti-GPA33/Anti-CD3 Bispecific Antibody for Colorectal Cancers. Mol. Cancer Ther. 2018, 17, 2164–2175. [Google Scholar] [CrossRef] [Green Version]
- Rageul, J.; Mottier, S.; Jarry, A.; Shah, Y.; Théoleyre, S.; Masson, D.; Gonzalez, F.J.; Laboisse, C.L.; Denis, M.G. KLF4-dependent, PPARgamma-induced expression of GPA33 in colon cancer cell lines. Int. J. Cancer 2009, 125, 2802–2809. [Google Scholar] [CrossRef]
- Mathur, D.; Root, A.R.; Bugaj-Gaweda, B.; Bisulco, S.; Tan, X.; Fang, W.; Kearney, J.C.; Lucas, J.; Guffroy, M.; Golas, J.; et al. A Novel GUCY2C-CD3 T-Cell Engaging Bispecific Construct (PF-07062119) for the Treatment of Gastrointestinal Cancers. Clin. Cancer Res. 2020, 26, 2188–2202. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, M.; Chang, C.-H.; Huang, Y.-C.; Chen, Y.-C.; Chi, M.-S.; Hao, H.-C.; Chang, Y.-C.; Takeda, S.; Chi, K.-H.; Wang, Y.-S. Pharmacologically upregulated carcinoembryonic antigen-expression enhances the cytolytic activity of genetically-modified chimeric antigen receptor NK-92MI against colorectal cancer cells. BMC Immunol. 2018, 19, 27. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Pe, L.; Xia, H.; Tang, Q.; Bi, F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol. Cancer 2021, 20, 143. [Google Scholar] [CrossRef]
- Michel, M.; Kaps, L.; Maderer, A.; Galle, P.; Moehler, M. The Role of p53 Dysfunction in Colorectal Cancer and Its Implication for Therapy. Cancers 2021, 13, 2296. [Google Scholar] [CrossRef]
- Hidalgo, M.; Martinez-Garcia, M.; Le Tourneau, C.; Massard, C.; Garralda, E.; Boni, V.; Taus, A.; Albanell, J.; Sablin, M.-P.; Altet, M.; et al. First-in-Human Phase I Study of Single-agent Vanucizumab, A First-in-Class Bispecific Anti-Angiopoietin-2/Anti-VEGF-A Antibody, in Adult Patients with Advanced Solid Tumors. Clin. Cancer Res. 2018, 24, 1536–1545. [Google Scholar] [CrossRef] [Green Version]
- Patnaik, A.; Gordon, M.; Tsai, F.; Papadopoulous, K.; Rasco, D.; Beeram, S.M.; Fu, S.; Janku, F.; Hynes, S.M.; Gundala, S.R.; et al. A phase I study of LY3164530, a bispecific antibody targeting MET and EGFR, in patients with advanced or metastatic cancer. Cancer Chemother. Pharmacol. 2018, 82, 407–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Safaie Qamsari, E.; Safaie Ghaderi, S.; Zarei, B.; Dorostkar, R.; Bagheri, S.; Jadidi-Niaragh, F.; Somi, M.H.; Yousefi, M. The c-Met receptor: Implication for targeted therapies in colorectal cancer. Tumour Biol. 2017, 39, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.S.; Burns, T.F.; McDonald, E.R., 3rd; Jiang, W.; Meng, R.; Krantz, I.D.; Kao, G.; Gan, D.D.; Zhou, J.Y.; Muschel, R.; et al. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 1997, 17, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Xiao, T.; Xiao, Y.; Wang, W.; Tang, Y.Y.; Xiao, Z.; Su, M. Targeting EphA2 in cancer. J. Hematol. Oncol. 2020, 13, 114. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Jin, C.; Rajabi, H.; Pitroda, S.P.; Alam, M.; Ahmad, R.; Raina, D.; Hasegawa, M.; Suzuki, Y.; Tagde, A.; et al. MUC1-C activates the TAK1 inflammatory pathway in colon cancer. Oncogene 2015, 34, 5187–5197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mita, A.C.; Mita, M.M.; Nawrocki, S.T.; Giles, F.J. Survivin: Key regulator of mitosis and apoptosis and novel target for cancer therapeutics. Clin. Cancer Res. 2008, 14, 5000–5005. [Google Scholar] [CrossRef] [Green Version]
- Cai, Y.; Ma, W.; Huang, X.; Cao, L.; Li, H.; Jiang, Y.; Lu, N.; Yin, Y. Effect of survivin on tumor growth of colorectal cancer in vivo. Int. J. Clin. Exp. Pathol. 2015, 8, 13267–13672. [Google Scholar]
- Sherman, E.J.; Mitchell, D.C.; Garner, A.L. The RNA-binding protein SART3 promotes miR-34a biogenesis and G(1) cell cycle arrest in lung cancer cells. J Biol Chem. 2019, 294, 17188–17196. [Google Scholar] [CrossRef]
- Bartnik, A.; Nirmal, A.J.; Yang, S.Y. Peptide Vaccine Therapy in Colorectal Cancer. Vaccines 2012, 1, 1–16. [Google Scholar] [CrossRef]
- Briukhovetska, D.; Dörr, J.; Endres, S.; Libby, P.; Dinarello, C.A.; Kobold, S. Interleukins in cancer: From biology to therapy. Nat. Rev. Cancer 2021, 21, 481–499. [Google Scholar] [CrossRef]
- Reichert, T.E.; Watkins, S.; Stanson, J.; Johnson, J.T.; Whiteside, T.L. Endogenous IL-2 in cancer cells: A marker of cellular proliferation. J. Histochem. Cytochem. 1998, 46, 603–611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polin, R.A.; Abman, S.H.; Rowitch, D.; Benitz, W.E. Fetal and Neonatal Physiology, 5th ed.; Elsevier: Philadelphia, PA, USA, 2017. [Google Scholar]
- Rébé, C.; Ghiringhelli, F. Interleukin-1β and Cancer. Cancers 2020, 12, 1791. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.J.; Houston, A.; Brint, E. IL-1 Family Members in Cancer; Two Sides to Every Story. Front. Immunol. 2019, 10, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kienzl, M.; Hasenoehrl, C.; Valadez-Cosmes, P.; Maitz, K.; Sarsembayeva, A.; Sturm, E.; Heinemann, A.; Karg, I.J.; Schicho, R. IL-33 reduces tumor growth in models of colorectal cancer with the help of eosinophils. Oncoimmunology 2020, 9, 1776059. [Google Scholar] [CrossRef]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef]
- Castro, F.; Cardoso, A.P.; Gonçalves, R.M.; Serre, K.; Oliveira, M.J. Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol. 2018, 9, 847. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; An, X.; Zhang, X.; Qiao, Y.; Zheng, T.; Li, X. STING: A master regulator in the cancer-immunity cycle. Mol. Cancer 2019, 18, 152. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Chen, P.; Wang, L.; Li, W.; Chen, B.; Liu, Y.; Wang, H.; Zhao, S.; Ye, L.; He, Y.; et al. cGAS-STING, an important pathway in cancer immunotherapy. J. Hematol. Oncol. 2020, 13, 81. [Google Scholar] [CrossRef]
- CCR5-blockade in Metastatic Colorectal Cancer. Available online: https://ClinicalTrials.gov/show/NCT01736813 (accessed on 11 October 2021).
- Mukaida, N. CCR5 antagonist, an ally to fight against metastatic colorectal cancer. Transl. Cancer Res. 2016, 5, S309–S312. [Google Scholar] [CrossRef]
- Fan, J.; Shang, D.; Han, B.; Song, J.; Chen, H.; Yang, J.-M. Adoptive Cell Transfer: Is it a Promising Immunotherapy for Colorectal Cancer? Theranostics 2018, 8, 5784–5800. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Overman, M.J.; Ernstoff, M.S.; Morse, M.A. Where We Stand With Immunotherapy in Colorectal Cancer: Deficient Mismatch Repair, Proficient Mismatch Repair, and Toxicity Management. Am. Soc. Clin. Oncol. Educ. Book 2018, 38, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Johdi, N.A.; Sukor, N.K. Colorectal Cancer Immunotherapy: Options and Strategies. Front. Immunol. 2020, 11, 1624. [Google Scholar] [CrossRef] [PubMed]
- FDA Approves First-Line Immunotherapy for Patients with MSI-H/dMMR Metastatic Colorectal Cancer. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-line-immunotherapy-patients-msi-hdmmr-metastatic-colorectal-cancer (accessed on 11 October 2021).
- Administration USFaD. FDA Grants Nivolumab Accelerated Approval for MSI-H or dMMR Colorectal Cancer. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-nivolumab-accelerated-approval-msi-h-or-dmmr-colorectal-cancer (accessed on 11 October 2021).
- Shiu, K.-K.; Andre, T.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.J.A.; Smith, D.M.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. KEYNOTE-177: Phase III randomized study of pembrolizumab versus chemotherapy for microsatellite instability-high advanced colorectal cancer. J. Clin. Oncol. 2021, 39 (Suppl. 3), 6. [Google Scholar] [CrossRef]
- André, F.D.A.; Shiu, K.-K.; Kim, T.W.; Jensen, B.V.; Jensen, L.H.; Punt, C.; Smith, D.; Garcia-Carbonero, R.; Benavides, M.; Gibbs, P.; et al. Pembrolizumab in Microsatellite-Instability-High Advanced Colorectal Cancer. N. Engl. J. Med. 2020, 383, 2207–2218. [Google Scholar] [CrossRef]
- Hu, H.; Kang, L.; Zhang, J.; Wu, Z.; Wang, H.; Huang, M.; Lan, P.; Wu, X.; Wang, C.; Cao, W.; et al. Neoadjuvant PD-1 blockade with toripalimab, with or without celecoxib, in mismatch repair-deficient or microsatellite instability-high, locally advanced, colorectal cancer (PICC): A single-centre, parallel-group, non-comparative, randomised, phase 2 trial. Lancet Gastroenterol. Hepatol. 2021, 7, 38–48. [Google Scholar] [CrossRef]
- Assessing Adjuvant Immunotherapy for MSI-H Colorectal Cancer. 2018. Available online: https://dailynews.ascopubs.org/do/10.5555/ADN.18.190032/full/ (accessed on 11 October 2021).
- Hirano, H.; Takashima, A.; Hamaguchi, T.; Shida, D.; Kanemitsu, Y. Current status and perspectives of immune checkpoint inhibitors for colorectal cancer. Jpn. J. Clin. Oncol. 2020, 51, 10–19. [Google Scholar] [CrossRef]
- Fukuoka, S.; Hara, H.; Takahashi, N.; Kojima, T.; Kawazoe, A.; Asayama, M.; Yoshii, T.; Kotani, D.; Tamura, H.; Mikamoto, Y.; et al. Regorafenib Plus Nivolumab in Patients With Advanced Gastric or Colorectal Cancer: An Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). J. Clin. Oncol. 2020, 38, 2053–2061. [Google Scholar] [CrossRef]
- Wang, C.; Chevalier, D.; Saluja, J.; Sandhu, J.; Lau, C.; Fakih, M. Regorafenib and Nivolumab or Pembrolizumab Combination and Circulating Tumor DNA Response Assessment in Refractory Microsatellite Stable Colorectal Cancer. Oncologist 2020, 25, e1188–e1194. [Google Scholar] [CrossRef]
- Bocobo, A.G.; Wang, R.; Behr, S.; Carnevale, J.C.; Cinar, P.; Collisson, E.A.; Fong, L.; Kidder, W.A.; Ko, A.H.; Kolli, K.P.; et al. Phase II study of pembrolizumab plus capecitabine and bevacizumab in microsatellite stable (MSS) metastatic colorectal cancer (mCRC): Interim analysis. J. Clin. Oncol. 2021, 39 (Suppl. 3), 77. [Google Scholar] [CrossRef]
- Research, N.C.F.C. Beating Colorectal Cancer’s Immunotherapy Resistance. Available online: https://news.harvard.edu/gazette/story/2021/10/new-way-to-overcome-colorectal-cancers-resistance-to-immune-response/ (accessed on 11 October 2021).
- Hospital, M.G. Research Points to a Strategy for Overcoming Colorectal Cancers’ Immunotherapy Resistance. Available online: https://medicalxpress.com/news/2021-10-strategy-colorectal-cancers-immunotherapy-resistance.html (accessed on 11 October 2021).
- Sobhani, N.; Tardiel-Cyril, D.R.; Davtyan, A.; Generali, D.; Roudi, R.; Li, Y. CTLA-4 in Regulatory T Cells for Cancer Immunotherapy. Cancers 2021, 13, 1440. [Google Scholar] [CrossRef] [PubMed]
- Chalabi, M.; Fanchi, L.F.; Dijkstra, K.K.; Berg, J.G.V.D.; Aalbers, A.G.; Sikorska, K.; Lopez-Yurda, M.; Grootscholten, C.; Beets, G.L.; Snaebjornsson, P.; et al. Neoadjuvant immunotherapy leads to pathological responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat. Med. 2020, 26, 566–576. [Google Scholar] [CrossRef] [PubMed]
- An Investigational Immuno-therapy Study of Nivolumab, and Nivolumab in Combination With Other Anti-cancer Drugs, in Colon Cancer That Has Come Back or Has Spread. Available online: https://clinicaltrials.gov/ct2/show/NCT02060188 (accessed on 11 October 2021).
- Study of Nivolumab and Relatlimab in Patients With Microsatellite Stable (MSS) Advanced Colorectal Cancer. Available online: https://clinicaltrials.gov/ct2/show/NCT03642067 (accessed on 11 October 2021).
- Study of TSR-033 With an Anti-programmed Cell Death-1 Receptor (PD-1) in Participants With Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03250832 (accessed on 11 October 2021).
- Huang, Y.-H.; Zhu, C.; Kondo, Y.; Anderson, A.C.; Gandhi, A.; Russell, A.F.; Dougan, S.K.; Petersen, B.-S.; Melum, E.; Pertel, T.; et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 2015, 517, 386–390. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Li, A.; Krishnamurthy, N.; Lemmon, M.A. Phosphatidylserine binding directly regulates TIM-3 function. Biochem. J. 2021, 478, 3331–3349. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.; Anderson, A.C.; Kuchroo, V.C. TIM3 comes of age as an inhibitory receptor. Nat. Rev. Immunol. 2020, 20, 173–185. [Google Scholar] [CrossRef]
- Yang, R.; Sun, L.; Li, C.-F.; Wang, Y.-H.; Yao, J.; Li, H.; Yan, M.; Chang, W.-C.; Hsu, J.-M.; Cha, J.-H.; et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat. Commun. 2021, 12, 832. [Google Scholar] [CrossRef]
- A Study of TSR-022 in Participants With Advanced Solid Tumors (AMBER). Available online: https://clinicaltrials.gov/ct2/show/NCT02817633 (accessed on 11 October 2021).
- Sun, R.; Huang, H.; Wang, X.; Zhang, Y.; Zheng, X.; Wei, H. Up-regulation of NKG2F receptor, a functionally unknown killer receptor, of human natural killer cells by interleukin-2 and interleukin-15. Oncol. Rep. 2010, 24, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Cen, D.; Gan, H.; Sun, Y.; Huang, N.; Xiong, H.; Jin, Q.; Su, L.; Wang, K.; Yan, G.; et al. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol. Ther. 2019, 27, 1114–1125. [Google Scholar] [CrossRef] [Green Version]
- NKG2 | Colorectal Cancer: ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=Colorectal+Cancer+&term=NKG2&cntry=&state=&city=&dist= (accessed on 11 October 2021).
- alloSHRINK - Standard cHemotherapy Regimen and Immunotherapy With Allogeneic NKG2D-based CYAD-101 Chimeric Antigen Receptor T-cells. Available online: https://ClinicalTrials.gov/show/NCT03692429 (accessed on 11 October 2021).
- Dose Escalation and Dose Expansion Phase I Study to Assess the Safety and Clinical Activity of Multiple Doses of NKR-2 Administered Concurrently With FOLFOX in Colorectal Cancer With Potentially Resectable Liver Metastases. Available online: https://ClinicalTrials.gov/show/NCT03310008 (accessed on 11 October 2021).
- Haplo Allogeneic NKG2DL-targeting Chimeric Antigen Receptor-grafted γδ T Cells for Relapsed or Refractory Solid Tumour. Available online: https://ClinicalTrials.gov/show/NCT04107142 (accessed on 11 October 2021).
- Hepatic Transarterial Administrations of NKR-2 in Patients With Unresectable Liver Metastases From Colorectal Cancer. Available online: https://ClinicalTrials.gov/show/NCT03370198 (accessed on 11 October 2021).
- NKG2D CAR-T(KD-025) in the Treatment of Relapsed or Refractory NKG2DL+ Tumors. Available online: https://ClinicalTrials.gov/show/NCT04550663 (accessed on 11 October 2021).
- Phase 1b Study to Evaluate the Addition of a Pembrolizumab Treatment After Treatment With CYAD-101 With a FOLFOX Preconditioning in Metastatic Colorectal Cancer Patients. Available online: https://ClinicalTrials.gov/show/NCT04991948 (accessed on 11 October 2021).
- Adoptive Cell Transfer: National Cancer Institute. Available online: https://www.cancer.gov/publications/dictionaries/cancer-terms/def/adoptive-cell-transfer (accessed on 11 October 2021).
- T-cell Transfer Therapy National Cancer Institute. 2020. Available online: https://www.cancer.gov/about-cancer/treatment/types/immunotherapy/t-cell-transfer-therapy (accessed on 11 October 2021).
- Rosenberg, S.A.; Spiess, P.; Lafreniere, R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science 1986, 233, 1318–1321. [Google Scholar] [CrossRef]
- Tumor Infiltrating Lymphocyte | Colorectal Cancer: ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=Colorectal+Cancer&term=tumor+infiltrating+lymphocyte&cntry=&state=&city=&dist= (accessed on 11 October 2021).
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Yang, C.; Cheng, H.; Huang, S.; Zheng, Y. CAR-T cells for Colorectal Cancer: Target-selection and strategies for improved activity and safety. J. Cancer 2021, 12, 1804–1814. [Google Scholar] [CrossRef] [PubMed]
- Chmielowski, B.; Ejadi, S.; Funke, R.; Stallings-Schmitt, T.; Denker, M.; Walter Frolich, M.; Franzusoff, A.J.; Abedi, M.; Cristea, M.C. A phase Ia/Ib, open-label first-in-human study of the safety, tolerability, and feasibility of gene-edited autologous NeoTCR-T cells (NeoTCR-P1) administered to patients with locally advanced or metastatic solid tumors. J. Clin. Oncol. 2020, 38 (Suppl. 15), TPS3151. [Google Scholar] [CrossRef]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Hermunen, K.; Soveri, L.-M.; Boisen, M.K.; Mustonen, H.K.; Dehlendorff, C.; Haglund, C.H.; Johansen, J.S.; Osterlund, P. Postoperative serum CA19-9, YKL-40, CRP and IL-6 in combination with CEA as prognostic markers for recurrence and survival in colorectal cancer. Acta Oncol. 2020, 59, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Campos-Da-Paz, M.; Dórea, J.G.; Galdino, A.S.; Lacava, Z.G.M.; Santos, M.D.F.M.A. Carcinoembryonic Antigen (CEA) and Hepatic Metastasis in Colorectal Cancer: Update on Biomarker for Clinical and Biotechnological Approaches. Recent Pat. Biotechnol. 2018, 12, 269–279. [Google Scholar] [CrossRef] [PubMed]
- CAR-pNK Cell Immunotherapy in MUC1 Positive Relapsed or Refractory Solid Tumor. Available online: https://ClinicalTrials.gov/show/NCT02839954 (accessed on 11 October 2021).
- Radiofrquency Ablation Combined With Cytokine-induced Killer Cells for Colorectal Cancer Liver Metastases. Available online: https://ClinicalTrials.gov/show/NCT02419677 (accessed on 11 October 2021).
- A Phase Ib Study of Immunotherapy with Ex Vivo Pre-Activated and Expanded Cb-Nk Cells in Combination with Cetuximab, in Colorectal Cancer Patients with Minimal Residual Disease (Mrd). Available online: https://ClinicalTrials.gov/show/NCT05040568 (accessed on 11 October 2021).
- The Study of CIK Plus S-1 and Bevacizumab as Maintenance Treatment for Patients With Advanced Colorectal Cancer. Available online: https://ClinicalTrials.gov/show/NCT02487992 (accessed on 11 October 2021).
- Chemotherapy Combined With CIK Treating Colon Cancer. Available online: https://ClinicalTrials.gov/show/NCT03084809 (accessed on 11 October 2021).
- Touchefeu, Y.; Bailly, C.; Frampas, E.; Eugène, T.; Rousseau, C.; Bourgeois, M.; Bossard, C.; Faivre-Chauvet, A.; Rauscher, A.; Masson, D.; et al. Promising clinical performance of pretargeted immuno-PET with anti-CEA bispecific antibody and gallium-68-labelled IMP-288 peptide for imaging colorectal cancer metastases: A pilot study. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 874–882. [Google Scholar] [CrossRef] [PubMed]
- Baysal, H.; De Pauw, I.; Zaryouh, H.; Peeters, M.; Vermorken, J.B.; Lardon, F.; De Waele, J.; Wouters, A. The Right Partner in Crime: Unlocking the Potential of the Anti-EGFR Antibody Cetuximab via Combination With Natural Killer Cell Chartering Immunotherapeutic Strategies. Front. Immunol. 2021, 12, 737311. [Google Scholar] [CrossRef] [PubMed]
- Price, T.J.; Peeters, M.; Kim, T.W.; Li, J.; Cascinu, S.; Ruff, P.; Suresh, A.S.; Thomas, A.; Tjulandin, S.; Zhang, K.; et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): A randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014, 15, 569–579. [Google Scholar] [CrossRef] [Green Version]
- Kol, A.; Van Scheltinga, A.T.; Pool, M.; Gerdes, C.; De Vries, E.; De Jong, S. ADCC responses and blocking of EGFR-mediated signaling and cell growth by combining the anti-EGFR antibodies imgatuzumab and cetuximab in NSCLC cells. Oncotarget 2017, 8, 45432–45446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppenheim, D.E.; Spreafico, R.; Etuk, A.; Malone, D.; Amofah, E.; Peña-Murillo, C.; Murray, T.; McLaughlin, L.; Choi, B.S.; Allan, S.; et al. Glyco-engineered anti-EGFR mAb elicits ADCC by NK cells from colorectal cancer patients irrespective of chemotherapy. Br. J. Cancer 2014, 110, 1221–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fakih, M.; Vincent, M. Adverse events associated with anti-EGFR therapies for the treatment of metastatic colorectal cancer. Curr. Oncol. 2010, 17 (Suppl. 1), S18–S30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Jiang, C.; Yang, L.; Huang, J.; Peng, R.; Wang, X.; He, W.; Bai, L.; Zhou, Y.; Zhang, B.; et al. First-line cetuximab improves the efficacy of subsequent bevacizumab for RAS wild-type left-sided metastatic colorectal cancer: An observational retrospective study. Sci. Rep. 2020, 10, 12336. [Google Scholar] [CrossRef] [PubMed]
- Kamba, T.; McDonald, D.M. Mechanisms of adverse effects of anti-VEGF therapy for cancer. Br. J. Cancer 2007, 96, 1788–1795. [Google Scholar] [CrossRef] [PubMed]
- Riva, P.; Marangolo, M.; Tison, V.; Armaroli, L.; Moscatelli, G.; Franceschi, G.; Spinelli, A.; Vecchietti, G.; Morigi, P.; Tassini, R.; et al. Treatment of metastatic colorectal cancer by means of specific monoclonal antibodies conjugated with iodine-131: A phase II study. Int. J. Rad. Appl. Instrum. B 1991, 18, 109–119. [Google Scholar] [CrossRef]
- Tsai, M.-H.; Pan, C.-H.; Peng, C.-L.; Shieh, M.-J. Panitumumab-Conjugated Pt-Drug Nanomedicine for Enhanced Efficacy of Combination Targeted Chemotherapy against Colorectal Cancer. Adv. Healthc. Mater. 2017, 6, 1700111. [Google Scholar] [CrossRef]
- Keam, S.J. Trastuzumab Deruxtecan: First Approval. Drugs 2020, 80, 501–508. [Google Scholar] [CrossRef]
- DS-8201a in Human Epidermal Growth Factor Receptor2 (HER2)-Expressing Colorectal Cancer (DESTINY-CRC01). Available online: https://ClinicalTrials.gov/show/NCT03384940 (accessed on 11 October 2021).
- Bacac, M.; Fauti, T.; Sam, J.; Colombetti, S.; Weinzierl, T.; Ouaret, D.; Bodmer, W.; Lehmann, S.; Hofer, T.; Hosse, R.J.; et al. A Novel Carcinoembryonic Antigen T-Cell Bispecific Antibody (CEA TCB) for the Treatment of Solid Tumors. Clin Cancer Res. 2016, 22, 3286–3297. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Yang, Y.; Zhou, P.; Ma, H.; Zhao, X.; He, X.; Wang, T.; Zhang, J.; Liu, Y.; Zhang, T. Targeting CD133high Colorectal Cancer Cells In Vitro and In Vivo With an Asymmetric Bispecific Antibody. J. Immunother. 2015, 38, 217–228. [Google Scholar] [CrossRef]
- Douglass, J.; Hsiue, E.H.-C.; Mog, B.J.; Hwang, M.S.; Di Napoli, S.R.; Pearlman, A.H.; Miller, M.S.; Wright, K.M.; Azurmendi, P.A.; Wang, Q.; et al. Bispecific antibodies targeting mutant RAS neoantigens. Sci. Immunol. 2021, 6, eabd5515. [Google Scholar] [CrossRef]
- Hsiue, E.H.-C.; Wright, K.M.; Douglass, J.; Hwang, M.S.; Mog, B.J.; Pearlman, A.H.; Paul, S.; Di Napoli, S.R.; Konig, M.F.; Wang, Q.; et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021, 371, eabc8697. [Google Scholar] [CrossRef]
- Skora, A.D.; Douglass, J.; Hwang, M.S.; Tam, A.J.; Blosser, R.L.; Gabelli, S.B.; Cao, J.; Diaz, L.A.; Papadopoulos, N.; Kinzler, K.W.; et al. Generation of MANAbodies specific to HLA-restricted epitopes encoded by somatically mutated genes. Proc. Natl. Acad. Sci. USA 2015, 112, 9967–9972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bruin, R.C.G.; Veluchamy, J.P.; Lougheed, S.M.; Schneiders, F.L.; Lopez-Lastra, S.; Lameris, R.; Stam, A.G.; Sebestyen, Z.; Kuball, J.; Molthoff, C.F.M.; et al. A bispecific nanobody approach to leverage the potent and widely applicable tumor cytolytic capacity of Vγ9Vδ2-T cells. Oncoimmunolog 2017, 7, e1375641. [Google Scholar] [CrossRef] [PubMed]
- Mueller, T.; Freystein, J.; Lucas, H.; Schmoll, H.-J. Efficacy of a Bispecific Antibody Co-Targeting VEGFA and Ang-2 in Combination with Chemotherapy in a Chemoresistant Colorectal Carcinoma Xenograft Model. Molecules 2019, 24, 2865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Wen, Y.; Liu, Y.; Tan, X.; Chen, X.; Zhu, X.; Wei, C.; Chen, L.; Wang, Z.; Liu, J.; et al. Hollow mesoporous ruthenium nanoparticles conjugated bispecific antibody for targeted anti-colorectal cancer response of combination therapy. Nanoscale 2019, 11, 9661–9678. [Google Scholar] [CrossRef]
- A Study of XmAb®22841 Monotherapy & in Combination w/ Pembrolizumab in Subjects w/ Selected Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT03849469 (accessed on 11 October 2021).
- Chames, P.; Van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Moradi, A.; Pourseif, M.M.; Jafari, B.; Parvizpour, S.; Omidi, Y. Nanobody-based therapeutics against colorectal cancer: Precision therapies based on the personal mutanome profile and tumor neoantigens. Pharmacol. Res. 2020, 156, 104790. [Google Scholar] [CrossRef]
- First in Human Trial of TAS266 in Patients With Advanced Solid Tumors. Available online: https://ClinicalTrials.gov/show/NCT01529307 (accessed on 11 October 2021).
- αPD1-MSLN-CAR T Cells for the Treatment of MSLN-positive Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT04503980 (accessed on 11 October 2021).
- Wrobel, P.; Ahmed, S. Current status of immunotherapy in metastatic colorectal cancer. Int. J. Colorectal. Dis. 2019, 34, 13–25. [Google Scholar] [CrossRef]
- Geevarghese, S.K.; Geller, D.A.; De Haan, H.A.; Hörer, M.; Knoll, A.E.; Mescheder, A.; Nemunaitis, J.; Reid, T.R.; Sze, D.Y.; Tanabe, K.K.; et al. Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum. Gene Ther. 2010, 21, 1119–1128. [Google Scholar] [CrossRef] [Green Version]
- Pexastimogene Devacirepvec: National Cancer Institute. Available online: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/pexastimogene-devacirepvec (accessed on 11 October 2021).
- A Trial of JX-594 in Refractory Colorectal Carcinoma. Available online: https://clinicaltrials.gov/ct2/show/NCT01469611 (accessed on 11 October 2021).
- Park, S.H.; Breitbach, C.J.; Lee, J.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Moon, A.; Mun, J.-H.; Sommermann, E.M.; Avidal, L.M.; et al. Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Mol. Ther. 2015, 23, 1532–1540. [Google Scholar] [CrossRef]
- Monge, B.M.; Xie, C.; Steinberg, S.M.; Fioraventi, S.; Walker, M.; Mabry-Hrones, D.; Wood, B.J.; Kleiner, E.D.; Greten, T.F. A phase I/II study of Pexa-Vec oncolytic virus in combination with immune checkpoint inhibition in refractory colorectal cancer. J. Clin. Oncol. 2020, 38 (Suppl. 4), 117. [Google Scholar]
- Song, H.; Zhong, L.-P.; He, J.; Huang, Y.; Zhao, Y.-X. Application of Newcastle disease virus in the treatment of colorectal cancer. World J. Clin. Cases 2019, 7, 2143–2154. [Google Scholar] [CrossRef] [PubMed]
- Puccini, A.; Battaglin, F.; Iaia, M.L.; Lenz, H.-J.; Salem, E.M. Overcoming resistance to anti-PD1 and anti-PD-L1 treatment in gastrointestinal malignancies. J. Immunother. Cancer 2020, 8, e000404. [Google Scholar] [CrossRef] [PubMed]
- Fifis, T.; Lam, I.; Lin, D.; Malcontenti-Wilson, C.; Christophi, C.; Loveland, B. Vaccination with in vitro grown whole tumor cells induces strong immune responses and retards tumor growth in a murine model of colorectal liver metastases. Vaccine 2008, 26, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, W.; Steinlein, P.; Buschle, M.; Schweighoffer, T.; Herbst, E.; Mechtler, K.; Kirlappos, H.; Birnstie, M.L. Transloading of tumor cells with foreign major histocompatibility complex class I peptide ligand: A novel general strategy for the generation of potent cancer vaccines. Proc. Natl. Acad. Sci. USA 1996, 93, 9759–9763. [Google Scholar] [CrossRef] [Green Version]
- de Weger, V.A.; Turksma, A.W.; Voorham, Q.J.M.; Euler, Z.; Bril, H.; van den Eertwegh, A.J.; Bloemena, E.; Pinedo, H.M.; Vermorken, J.B.; van Tinteren, H.; et al. Clinical effects of adjuvant active specific immunotherapy differ between patients with microsatellite-stable and microsatellite-instable colon cancer. Clin. Cancer Res. 2012, 18, 882–889. [Google Scholar] [CrossRef] [Green Version]
- Harris, J.E.; Ryan, L.; Hoover, H.C., Jr.; Stuart, R.K.; Oken, M.M.; Benson, A.B.; Mansour, E.; Haller, D.G.; Manola, J.; Hanna, M.G., Jr. Adjuvant active specific immunotherapy for stage II and III colon cancer with an autologous tumor cell vaccine: Eastern Cooperative Oncology Group Study E5283. J. Clin. Oncol. 2000, 18, 148–157. [Google Scholar] [CrossRef]
- Tagliamonte, M.; Petrizzo, A.; Tornesello, M.L.; Buonaguro, F.M.; Buonaguro, L. Antigen-specific vaccines for cancer treatment. Hum. Vaccin. Immunother. 2014, 10, 3332–3346. [Google Scholar] [CrossRef]
- George, D.J.; Nabhan, C.; Devries, T.; Whitmore, J.B.; Gomella, L.G. Survival Outcomes of Sipuleucel-T Phase III Studies: Impact of Control-Arm Cross-Over to Salvage Immunotherapy. Cancer Immunol. Res. 2015, 3, 1063–1069. [Google Scholar] [CrossRef] [Green Version]
- Mirlekar, B.; Pylayeva-Gupta, Y. IL-12 Family Cytokines in Cancer and Immunotherapy. Cancers 2021, 13, 167. [Google Scholar] [CrossRef]
- Cytokines and Their Side Effects: American Cancer Society. Available online: https://www.cancer.org/treatment/treatments-and-side-effects/treatment-types/immunotherapy/cytokines.html (accessed on 11 October 2021).
- Chulpanova, D.S.; Kitaeva, K.V.; Green, A.R.; Rizvanov, A.A.; Solovyeva, V.V. Molecular Aspects and Future Perspectives of Cytokine-Based Anti-cancer Immunotherapy. Front. Cell Dev. Biol. 2020, 8, 402. [Google Scholar] [CrossRef]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferst, I.R.; et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β, and TNF-α: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, B.S.; Rautela, J.; Hertzog, P.J. Antitumour actions of interferons: Implications for cancer therapy. Nat. Rev. Cancer 2016, 16, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark. Res. 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Slattery, M.L.; Lundgreen, A.; Bondurant, K.L.; Wolff, R.K. Interferon-signaling pathway: Associations with colon and rectal cancer risk and subsequent survival. Carcinogenesis 2011, 32, 1660–1667. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, M.R.; Merlino, G. The two faces of interferon-γ in cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, M.R. The Interferon-Gamma Paradox in Cancer. J. Interferon. Cytokine Res. 2019, 39, 30–38. [Google Scholar] [CrossRef]
- Hanada, T.; Kobayashi, T.; Chinen, T.; Saeki, K.; Takaki, H.; Koga, K.; Minoda, Y.; Sanada, T.; Yoshioka, T.; Mimata, H.; et al. IFNgamma-dependent, spontaneous development of colorectal carcinomas in SOCS1-deficient mice. J. Exp. Med. 2006, 203, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Wiesenfeld, M.; O’Connell, M.J.; Wieand, H.S.; Gonchoroff, N.J.; Donohue, J.H.; Jr, R.J.F.; Krook, E.J.; Mailliard, A.J.; Gerstner, J.B.; Pazdur, R. Controlled clinical trial of interferon-gamma as postoperative surgical adjuvant therapy for colon cancer. J. Clin. Oncol. 1995, 13, 2324–2329. [Google Scholar] [CrossRef]
- Study of Gamma Interfereon in Metastatic Colorectal Carcinoma. Available online: https://ClinicalTrials.gov/show/NCT00786643 (accessed on 11 October 2021).
- Qiu, Y.; Su, M.; Liu, L.; Tang, Y.; Pan, Y.; Sun, J. Clinical Application of Cytokines in Cancer Immunotherapy. Drug Des. Dev. Ther. 2021, 15, 2269–2287. [Google Scholar] [CrossRef]
- Chon, H.J.; Kim, H.; Noh, J.H.; Yang, H.; Lee, W.S.; Kong, S.J.; Lee, S.J.; Lee, Y.S.; Kim, W.R.; Kim, J.H.; et al. STING signaling is a potential immunotherapeutic target in colorectal cancer. J. Cancer 2019, 10, 4932–4938. [Google Scholar] [CrossRef]
- Chen, S.Y.; Chen, S.; Feng, W.; Li, Z.; Luo, X.; Zhu, X. A STING-related prognostic score predicts high-risk patients of colorectal cancer and provides insights into immunotherapy. Ann. Transl. Med. 2021, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Endo, A.; Fang, F.G.; Huang, K.; Bao, X.; Choi, H.; Majumder, U.; Shen, Y.Y.; Mathieu, S.; Zhu, X.; et al. E7766, a Macrocycle-Bridged Stimulator of Interferon Genes (STING) Agonist with Potent Pan-Genotypic Activity. ChemMedChem 2021, 16, 1740–1743. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.K. Medicine. How thalidomide works against cancer. Science 2014, 343, 256–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.M.; Henry, J.Y.; Meyer, B.; Bartlett, J.B.; Dalgleish, A.G.; Galustian, C. Inhibition of metastatic potential in colorectal carcinoma in vivo and in vitro using immunomodulatory drugs (IMiDs). Br. J. Cancer 2009, 101, 803–812. [Google Scholar] [CrossRef]
- Goldstein, M.J.; Leighton, J.C.; Chapman, A.; Muskett, A.; Sharan, K.; Sansom, J.; Mitchell, E.P. Phase I trial of thalidomide and capecitabine for treatment of metastatic colorectal cancer. J. Clin. Oncol. 2006, 24 (Suppl. 18), 13552. [Google Scholar] [CrossRef]
- Kroschinsky, F.; Stölzel, F.; von Bonin, S.; Beutel, G.; Kochanek, M.; Kiehl, M.; Schellongowski, P. New drugs, new toxicities: Severe side effects of modern targeted and immunotherapy of cancer and their management. Crit. Care 2017, 21, 89. [Google Scholar] [CrossRef] [Green Version]
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res. GCR 2012, 5, 19–27. [Google Scholar]
- Wu, C.-E.; Yeh, D.-W.; Pan, Y.-R.; Huang, W.-K.; Chen, M.-H.; Chang, J.; Chen, J.-S.; Wang, Y.-C.; Yeh, C.-N. Chromosomal Instability May Not Be a Predictor for Immune Checkpoint Inhibitors from a Comprehensive Bioinformatics Analysis. Life 2020, 10, 276. [Google Scholar] [CrossRef]
- Walther, A.; Houlston, R.; Tomlinson, I. Association between chromosomal instability and prognosis in colorectal cancer: A meta-analysis. Gut 2008, 57, 941–950. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Liu, J.; Huang, X.; Liu, H.; Wei, C.; Ru, H.; Qin, H.; Lai, H.; Meng, Y.; Wu, G.; Xie, W.; et al. Immune landscape and prognostic immune-related genes in KRAS-mutant colorectal cancer patients. J. Transl. Med. 2021, 19, 27. [Google Scholar] [CrossRef]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunology. Immunotherapy 2017, 66, 1175–1187. [Google Scholar]
- Zhang, L.; Shay, J.W. Multiple Roles of APC and its Therapeutic Implications in Colorectal Cancer. JNCI J. Natl. Cancer Inst. 2017, 109, djw332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grasso, C.S.; Giannakis, M.; Wells, D.K.; Hamada, T.; Mu, X.J.; Quist, M.; Nowak, J.A.; Nishihara, R.; Qian, Z.R.; Inamura, K.; et al. Genetic Mechanisms of Immune Evasion in Colorectal Cancer. Cancer Discov. 2018, 8, 730–749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, K.; Wang, J.; Zhu, J.; Jiang, J.; Shou, J.; Chen, X. p53 induces TAP1 and enhances the transport of MHC class I peptides. Oncogene 1999, 18, 7740–7747. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Li, L.; Jiang, X.; Li, Q. Mismatch repair deficiency/microsatellite instability-high as a predictor for anti-PD-1/PD-L1 immunotherapy efficacy. J. Hematol. Oncol. 2019, 12, 54. [Google Scholar] [CrossRef]
- Hamada, T.; Soong, T.R.; Masugi, Y.; Kosumi, K.; Nowak, J.A.; Da Silva, A.; Mu, X.J.; Twombly, T.S.; Koh, H.; Yang, J.; et al. TIME (Tumor Immunity in the MicroEnvironment) classification based on tumor CD274 (PD-L1) expression status and tumor-infiltrating lymphocytes in colorectal carcinomas. Oncoimmunology 2018, 7, e1442999. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Park, H.E.; Cho, N.-Y.; Lee, H.S.; Kang, G.H. Characterisation of PD-L1-positive subsets of microsatellite-unstable colorectal cancers. Br. J. Cancer 2016, 115, 490–496. [Google Scholar] [CrossRef] [Green Version]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden-High Solid Tumors. Clin. Cancer Res. 2021, 27, 4685–4689. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef] [Green Version]
- Nusrat, M.; Roszik, J.; Katkhuda, R.; Menter, D.; Raghav, K.P.S.; Morris, V.K.; Sharma, P.; Allison, J.P.; Blando, J.M.; Maru, D.M.; et al. Association of PIK3CA mutations (mut) with immune engagement and clinical benefit from immunotherapy in microsatellite stable (MSS) colorectal cancer (CRC) patients (pts). J. Clin. Oncol. 2019, 37 (Suppl. 15), 3604. [Google Scholar] [CrossRef]
- Nassif, N.T.; Lobo, G.P.; Wu, X.; Henderson, A.C.J.; Morrison, C.D.; Eng, C.; Jalaludin, B.; Segelov, E. PTEN mutations are common in sporadic microsatellite stable colorectal cancer. Oncogene 2004, 23, 617–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidotto, T.; Melo, C.; Castelli, E.; Koti, M.; dos Reis, R.B.; Squire, J.A. Emerging role of PTEN loss in evasion of the immune response to tumours. Br. J. Cancer 2020, 122, 1732–1743. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Huang, L.; Li, S.L.; Gu, J.; Cui, X.; Zhou, Y. PTEN loss correlates with T cell exclusion across human cancers. BMC Cancer 2021, 21, 429. [Google Scholar] [CrossRef]
- Kim, B.-G.; Malek, E.; Choi, S.H.; Ignatz-Hoover, J.J.; Driscoll, J.J. Novel therapies emerging in oncology to target the TGF-β pathway. J. Hematol. Oncol. 2021, 14, 55. [Google Scholar] [CrossRef]
- Bai, X.; Yi, M.; Jiao, Y.; Chu, Q.; Wu, K. Blocking TGF-β Signaling To Enhance The Efficacy Of Immune Checkpoint Inhibitor. Onco. Targets Ther. 2019, 12, 9527–9538. [Google Scholar] [CrossRef] [Green Version]
- Grady, W.M.; Myeroff, L.L.; Swinler, S.E.; Rajput, A.; Thiagalingman, S.; Lutterbaugh, J.D.; Neumann, A.; Brattain, M.G.; Chang, J.; Kim, S.-J.; et al. Mutational Inactivation of Transforming Growth Factor β Receptor Type II in Microsatellite Stable Colon Cancers. Cancer Res. 1999, 59, 320–324. [Google Scholar]
- Li, T.; Wang, H.; Xu, J.; Li, C.; Zhang, Y.; Wang, G.; Liu, Y.; Cai, S.; Fang, W.; Li, J.; et al. TGFBR2 mutation predicts resistance to immune checkpoint inhibitors in patients with non-small cell lung cancer. Ther. Adv. Med. Oncol. 2021, 13, 1–14. [Google Scholar] [CrossRef]
- Hu, Y.; Chen, W.; Yan, Z.; Ma, J.; Zhu, F.; Hue, J. Prognostic value of PD-L1 expression in patients with pancreatic cancer: A PRISMA-compliant meta-analysis. Medicine 2019, 98, e14006. [Google Scholar] [CrossRef]
- Droeser, R.A.; Hirt, C.; Viehl, C.T.; Frey, D.M.; Nebiker, C.; Huber, X.; Zlobec, I.; Eppenberger-Castori, S.; Tzankov, A.; Rosso, R.; et al. Clinical impact of programmed cell death ligand 1 expression in colorectal cancer. Eur. J. Cancer 2013, 49, 2233–2242. [Google Scholar] [CrossRef]
- Klein, S.; Mauch, C.; Brinker, K.; Noh, K.-W.; Knez, S.; Büttner, R.; Quaas, A.; Helbig, D. Tumor infiltrating lymphocyte clusters are associated with response to immune checkpoint inhibition in BRAF V600E/K mutated malignant melanomas. Sci. Rep. 2021, 11, 1834. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, W.; Yang, X.; Wang, H.; Peng, Y.; Yin, J.; Lu, Y.; Liu, L.; Shang, J.; Zhao, Q. An immune landscape based prognostic signature predicts the immune status and immunotherapeutic responses of patients with colorectal cancer. Life Sci. 2020, 261, 118368. [Google Scholar] [CrossRef] [PubMed]
- Correale, P.; Rotundo, M.S.; Del Vecchio, M.T.; Remondo, C.; Migali, C.; Ginanneschi, C.; Tsang, K.Y.; Licchetta, A.; Mannucci, S.; Loiacono, L.; et al. Regulatory (FoxP3+) T-cell tumor infiltration is a favorable prognostic factor in advanced colon cancer patients undergoing chemo or chemoimmunotherapy. J. Immunother. 2010, 33, 435–441. [Google Scholar] [CrossRef]
- Tang, Y.-Q.; Chen, T.-F.; Zhang, Y.; Zhao, X.-C.; Zhang, Y.-Z.; Wang, G.-Q.; Huang, M.-L.; Cai, S.-L.; Zhao, J.; Wei, B.; et al. The tumor immune microenvironment transcriptomic subtypes of colorectal cancer for prognosis and development of precise immunotherapy. Gastroenterol. Rep. 2020, 8, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Hazama, S.; Yamaguchi, R.; Imoto, S.; Takenouchi, H.; Inoue, Y.; Kanekiyo, S.; Shindo, Y.; Miyano, S.; Nakamura, Y.; et al. Characterization of the T cell repertoire by deep T cell receptor sequencing in tissues and blood from patients with advanced colorectal cancer. Oncol. Lett. 2016, 11, 3643–3649. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Cui, Y.; Zhang, Y.; Liu, Z.; Zhang, Q.; Wu, W.; Zheng, Z.; Li, S.; Zhang, Z.; Li, Y. A comprehensive study of immunology repertoires in both preoperative stage and postoperative stage in patients with colorectal cancer. Mol. Genet. Genom. Med. 2019, 7, e504. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Zhang, S.-Y.; Song, X.-Y.; Li, Y.; Ye, L.-L.; Zhou, Q.; Yang, W.-B. Tumor-associated macrophages: A promising target for a cancer immunotherapeutic strategy. Pharmacol. Res. 2020, 161, 105111. [Google Scholar] [CrossRef]
- Pinto, M.L.; Rios, E.; Durães, C.; Ribeiro, R.; Machado, J.C.; Mantovani, A.; Barbosa, M.A.; Carneiro, F.; Oliveira, M.J. The Two Faces of Tumor-Associated Macrophages and Their Clinical Significance in Colorectal Cancer. Front. Immunol. 2019, 10, 1875. [Google Scholar] [CrossRef] [Green Version]
- Larionova, I.; Tuguzbaeva, G.; Ponomaryova, A.; Stakheyeva, M.; Cherdyntseva, N.; Pavlov, V.; Choinzonov, E.; Kzhyshkowska, J. Tumor-Associated Macrophages in Human Breast, Colorectal, Lung, Ovarian and Prostate Cancers. Front. Oncol. 2020, 10, 566511. [Google Scholar] [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: Rationale, challenges and potential. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 361–375. [Google Scholar] [CrossRef] [PubMed]
- Mager, L.F.; Burkhard, R.; Pett, N.; Cooke, N.C.A.; Brown, K.; Ramay, H.; Paik, S.; Stagg, J.; Groves, R.A.; Gallo, M.; et al. Microbiome-derived inosine modulates response to checkpoint inhibitor immunotherapy. Science 2020, 369, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Cheng, S.; Kou, Y.; Wang, Z.; Jin, R.; Hu, H.; Zhang, X.; Gong, F.-F.; Li, J.; Lu, M.; et al. The Gut Microbiome Is Associated with Clinical Response to Anti-PD-1/PD-L1 Immunotherapy in Gastrointestinal Cancer. Cancer Immunol. Res. 2020, 8, 1251–1261. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Maeng, H.G.; Lee, S.J.; Kim, Y.J.; Kim, D.W.; Na Lee, H.; Namgung, J.H.; Oh, H.-M.; Kim, T.J.; Jeong, J.E.; et al. Diagnostic value of peripheral blood immune profiling in colorectal cancer. Ann. Surg. Treat Res. 2018, 94, 312–321. [Google Scholar] [CrossRef]
- Tang, Y.-P.; Xie, M.-Z.; Li, K.-Z.; Li, J.-L.; Cai, Z.-M.; Hu, B.-L. Prognostic value of peripheral blood natural killer cells in colorectal cancer. BMC Gastroenterol. 2020, 20, 31. [Google Scholar] [CrossRef] [Green Version]
- Saied, A.; Licata, A.L.; Burga, R.A.; Thorn, M.; McCormack, A.E.; Stainken, B.F.; Assanah, E.O.; Khare, P.D.; Davies, R.G.; Espat, N.J.; et al. Neutrophil: Lymphocyte ratios and serum cytokine changes after hepatic artery chimeric antigen receptor-modified T-cell infusions for liver metastases. Cancer Gene Ther. 2014, 21, 457–462. [Google Scholar] [CrossRef]
- Reece, M.; Saluja, H.; Hollington, P.; Karapetis, C.; Vatandoust, S.; Young, G.; Symonds, E.L. The Use of Circulating Tumor DNA to Monitor and Predict Response to Treatment in Colorectal Cancer. Front. Genet. 2019, 10, 1118. [Google Scholar] [CrossRef] [Green Version]
- Calu, V.; Ionescu, A.; Stanca, L.; Geicu, O.I.; Iordache, F.; Pisoschi, A.M.; Serban, A.I.; Bilteanu, L. Key biomarkers within the colorectal cancer related inflammatory microenvironment. Sci. Rep. 2021, 11, 7940. [Google Scholar] [CrossRef]
- Wang, M.; Zhai, X.; Li, J.; Guan, J.; Xu, S.; Li, Y.; Zhu, H. The Role of Cytokines in Predicting the Response and Adverse Events Related to Immune Checkpoint Inhibitors. Front. Immunol. 2021, 12, 2894. [Google Scholar] [CrossRef]
- Liu, C.; Liu, R.; Wang, B.; Lian, J.; Yao, Y.; Sun, H.; Zhang, C.; Fang, L.; Guan, X.; Shi, J.; et al. Blocking IL-17A enhances tumor response to anti-PD-1 immunotherapy in microsatellite stable colorectal cancer. J. Immunother. Cancer 2021, 9, e001895. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Chan, M.-H.; Li, C.-H.; Fang, C.-Y.; Hsiao, M.; Chen, C.-L. Exosomal Components and Modulators in Colorectal Cancer: Novel Diagnosis and Prognosis Biomarkers. Biomedicines 2021, 9, 931. [Google Scholar] [CrossRef] [PubMed]
- Clerici, S.P.; Peppelenbosch, M.; Fuhler, G.; Consonni, S.R.; Ferreira-Halder, C.V. Colorectal Cancer Cell-Derived Small Extracellular Vesicles Educate Human Fibroblasts to Stimulate Migratory Capacity. Front. Cell Dev. Biol. 2021, 9, 696373. [Google Scholar] [CrossRef] [PubMed]
- Zhou, E.; Li, Y.; Wu, F.; Guo, M.; Xu, J.; Wang, S.; Tan, Q.; Ma, P.; Song, S.; Jin, Y. Circulating extracellular vesicles are effective biomarkers for predicting response to cancer therapy. EBioMedicine 2021, 67, 103365. [Google Scholar] [CrossRef] [PubMed]
- Foy, J.P.; Bertolus, C.; Michallet, M.-C.; Deneuve, S.; Incitti, R.; Bendriss-Vermare, N.; Albaret, M.-A.; Ortiz-Curan, S.; Thomas, E.; Colombe, A.; et al. The immune microenvironment of HPV-negative oral squamous cell carcinoma from never-smokers and never-drinkers patients suggests higher clinical benefit of IDO1 and PD1/PD-L1 blockade. Ann. Oncol. 2017, 28, 1934–1941. [Google Scholar] [CrossRef]
- Lin, Y.-M.; Sung, W.-W.; Hsieh, M.-J.; Tsai, S.-C.; Lai, H.-W.; Yang, S.-M.; Shen, K.-H.; Chen, M.-K.; Lee, H.; Yeh, K.-T.; et al. High PD-L1 Expression Correlates with Metastasis and Poor Prognosis in Oral Squamous Cell Carcinoma. PLoS ONE 2015, 10, e0142656. [Google Scholar] [CrossRef] [Green Version]
- Szabo, G.; Catalano, N.; White, B.; Mandrekar, P. Acute alcohol consumption inhibits accessory cell function of monocytes and dendritic cells. Alcohol Clin. Exp. Res. 2004, 28, 824–828. [Google Scholar] [CrossRef]
- Wang, Z.; Aguilar, E.G.; Luna, J.I.; Dunai, C.; Khuat, L.T.; Le, C.; Mirsoian, A.; Minnar, C.M.; Stoffel, K.M.; Sturgill, I.R.; et al. Paradoxical effects of obesity on T cell function during tumor progression and PD-1 checkpoint blockade. Nat. Med. 2019, 25, 141–151. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, T.; Xuan, Q.; Zhao, H.; Qin, L.; Zhang, Q. Lymphocyte-Activation Gene-3 Expression and Prognostic Value in Neoadjuvant-Treated Triple-Negative Breast Cancer. J. Breast Cancer 2018, 21, 124–133. [Google Scholar] [CrossRef]
- Heidelberger, V.; Goldwasser, F.; Kramkimel, N.; Jouinot, A.; Huillard, O.; Boudou-Rouquette, P.; Chanal, J.; Arrondeau, J.; Franck, N.; Alexandre, J.; et al. Sarcopenic overweight is associated with early acute limiting toxicity of anti-PD1 checkpoint inhibitors in melanoma patients. Investig. New Drugs 2017, 35, 436–441. [Google Scholar] [CrossRef]
- Naik, G.S.; Waikar, S.S.; Johnson, A.E.W.; Buchbinder, E.I.; Haq, R.; Hodi, F.S.; Schoenfeld, J.D.; Ott, P.A. Complex inter-relationship of body mass index, gender and serum creatinine on survival: Exploring the obesity paradox in melanoma patients treated with checkpoint inhibition. J. Immunother. Cancer 2019, 7, 89. [Google Scholar] [CrossRef] [Green Version]
- McQuade, J.L.; Daniel, C.R.; Hess, K.R.; Mak, C.; Wang, D.Y.; Rai, R.R.; Park, J.J.; Haydu, L.E.; Spencer, C.; Wongchenko, M.; et al. Association of body-mass index and outcomes in patients with metastatic melanoma treated with targeted therapy, immunotherapy, or chemotherapy: A retrospective, multicohort analysis. Lancet Oncol. 2018, 19, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Rahman, O. Smoking and EGFR status may predict outcomes of advanced NSCLC treated with PD-(L)1 inhibitors beyond first line: A meta-analysis. Clin. Respir. J. 2018, 12, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.-Y.; Yang, C.-Y.; Liao, B.-C.; Ho, C.-C.; Liao, W.-Y.; Chen, K.-Y.; Tsai, T.-H.; Hsu, C.-L.; Hsu, W.-H.; Su, K.-Y.; et al. Tumor PD-L1 Expression and Clinical Outcomes in Advanced-stage Non-Small Cell Lung Cancer Patients Treated with Nivolumab or Pembrolizumab: Real-World Data in Taiwan. J. Cancer 2018, 9, 1813–1820. [Google Scholar] [CrossRef] [PubMed]
- Nishino, M.; Dahlberg, S.E.; Adeni, A.E.; Lydon, C.A.; Hatabu, H.; Janne, P.A.; Hodi, F.S.; Awad, M.M. Tumor Response Dynamics of Advanced Non-small Cell Lung Cancer Patients Treated with PD-1 Inhibitors: Imaging Markers for Treatment Outcome. Clin. Cancer Res. 2017, 23, 5737–5744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reis, H.; Metzenmacher, M.; Goetz, M.; Savvidou, N.; Darwiche, K.; Aigner, C.; Herold, T.; Eberhardt, W.E.; Skiba, C.; Hense, J.; et al. MET Expression in Advanced Non-Small-Cell Lung Cancer: Effect on Clinical Outcomes of Chemotherapy, Targeted Therapy, and Immunotherapy. Clin. Lung Cancer 2018, 19, e441–e463. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015, 348, 124–128. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Polanczyk, M.J.; Hopke, C.; Vandenbark, A.A.; Offner, H. Treg suppressive activity involves estrogen-dependent expression of programmed death-1 (PD-1). Int. Immunol. 2007, 19, 337–343. [Google Scholar] [CrossRef] [Green Version]
- Deshpande, R.P.; Sharma, S.; Watabe, K. The Confounders of Cancer Immunotherapy: Roles of Lifestyle, Metabolic Disorders and Sociological Factors. Cancers 2020, 12, 2983. [Google Scholar] [CrossRef]
- Fujiyoshi, K.; Chen, Y.; Haruki, K.; Ugai, T.; Kishikawa, J.; Hamada, T.; Liu, L.; Arima, K.; Borowsky, J.; Väyrynen, J.P.; et al. Smoking Status at Diagnosis and Colorectal Cancer Prognosis According to Tumor Lymphocytic Reaction. JNCI Cancer Spectr. 2020, 4, pkaa040. [Google Scholar] [CrossRef]
- Crockett, S.D.; Long, M.D.; Dellon, E.S.; Martin, C.F.; Galanko, J.A.; Sandler, R.S. Inverse relationship between moderate alcohol intake and rectal cancer: Analysis of the North Carolina Colon Cancer Study. Dis. Colon Rectum 2011, 54, 887–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, A.; Zhang, J.; Luo, P. Crosstalk Between the MSI Status and Tumor Microenvironment in Colorectal Cancer. Front. Immunol. 2020, 11, 2039. [Google Scholar] [CrossRef] [PubMed]
- Berzins, S.P.; Wallace, M.E.; Kannourakis, G.; Kelly, J. A Role for MAIT Cells in Colorectal Cancer. Front. Immunol. 2020, 11, 949. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.D.; Patel, R.S.; Cook, D.R.; Choi, M.Y.; Patil, A.; Liang, A.C.; Li, M.Z.; Haigis, K.M.; Elledge, S.J. The adaptive immune system is a major driver of selection for tumor suppressor gene inactivation. Science 2021, 373, 1327–1335. [Google Scholar] [CrossRef]
- Cancer Cells’ Unexpected Genetic Tricks for Evading the Immune System: Howard Hughes Medical Institute. Available online: https://www.hhmi.org/news/cancer-cells-unexpected-genetic-tricks-evading-immune-system (accessed on 11 October 2021).
- Pu, D.; Yin, L.; Huang, L.; Qin, C.; Zhou, Y.; Wu, Q.; Li, Y.; Zhou, Q.; Li, L. Cyclooxygenase-2 Inhibitor: A Potential Combination Strategy With Immunotherapy in Cancer. Front. Oncol. 2021, 11, 248. [Google Scholar] [CrossRef]
- Yan, G.; Zhao, H.; Zhang, Q.; Zhou, Y.; Wu, L.; Lei, J.; Wang, X.; Zhang, J.; Zhang, X.; Zheng, L.; et al. A RIPK3-PGE(2) Circuit Mediates Myeloid-Derived Suppressor Cell-Potentiated Colorectal Carcinogenesis. Cancer Res. 2018, 78, 5586–5599. [Google Scholar] [CrossRef] [Green Version]
- Nivolumab, Ipilimumab and COX2-inhibition in Early Stage Colon Cancer: An Unbiased Approach for Signals of Sensitivity. Available online: https://clinicaltrials.gov/ct2/show/NCT03026140 (accessed on 11 October 2021).
- Koulouridi, A.; Messaritakis, I.; Gouvas, N.; Tsiaoussis, J.; Souglakos, J. Immunotherapy in Solid Tumors and Gut Microbiota: The Correlation-A Special Reference to Colorectal Cancer. Cancers 2020, 13, 43. [Google Scholar] [CrossRef]
- Saha, T.; Dash, C.; Jayabalan, R.; Khiste, S.; Kulkarni, A.; Kurmi, K.; Mondal, J.; Majumder, P.K.; Bardia, A.; Jang, H.L.; et al. Intercellular nanotubes mediate mitochondrial trafficking between cancer and immune cells. Nat. Nanotechnol. 2021, 17, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Sandhu, J.; Ouyang, C.; Ye, J.; Lee, P.P.; Fakih, M. Clinical Response to Immunotherapy Targeting Programmed Cell Death Receptor 1/Programmed Cell Death Ligand 1 in Patients With Treatment-Resistant Microsatellite Stable Colorectal Cancer With and Without Liver Metastases. JAMA Netw. Open 2021, 4, e2118416. [Google Scholar] [CrossRef]
- Carlsen, L.; Schorl, C.; Huntington, K.; Hernandez-Borrero, L.; Jhaveri, A.; Zhang, S.; Zhou, L.; El-Deiry, W.S. Pan-drug and drug-specific mechanisms of 5-FU, irinotecan (CPT-11), oxaliplatin, and cisplatin identified by comparison of transcriptomic and cytokine responses of colorectal cancer cells. Oncotarget 2021, 12, 2006–2021. [Google Scholar] [CrossRef]
- Groysman, L.; Carlsen, L.; Huntington, K.E.; Shen, W.H.; Zhou, L.; El-Deiry, W.S. Chemotherapy-induced cytokines and prognostic gene signatures vary across breast and colorectal cancer. Am. J. Cancer Res. 2021, 11, 6086–6106. [Google Scholar] [PubMed]
- Huntington, K.E.; Louie, A.; Zhou, L.; El-Deiry, W.S. A high-throughput customized cytokinome screen of colon cancer cell responses to small-molecule oncology drugs. Oncotarget 2021, 12, 1980–1991. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. Cancer Immunotherapy, Part 3: Challenges and Future Trends. Pharm. Ther. 2017, 42, 514–521. [Google Scholar]
- Liu, D.; Jenkins, R.W.; Sullivan, R.J. Mechanisms of Resistance to Immune Checkpoint Blockade. Am. J. Clin. Dermatol. 2019, 20, 41–54. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Target | Expressed by | Molecular Action | Ref. |
|---|---|---|---|
| PD-1 | CD8+ T cells, CD4+ T cells | Suppresses CD8/4+ T cell activity | [11,12] |
| PD-L1 | Cancer cells, APCs | Suppresses CD8/4+ T cell activity | [11,12] |
| CTLA-4 | CD8+ T cells, CD4+ T cells, T regs | Suppresses CD8/4+ T cell activity Enhances T reg activity | [13] |
| LAG-3 | CD8+ T cells, CD4+ T cells, T regs, NK cells | Suppresses CD8/4+ T cell activity Enhances T reg activity | [14] |
| TIM-3 | IFN-γ-producing T cells, T regs, NK cells, APCs | Suppresses T cell, NK cell, and APC activity May enhance T reg activity | [14] |
| NKG2 | NK cells, some CD8+ T cells | NKG1A/B: Suppresses NK cell activity NKG2D: Enhances NK cell activity | [15,16] |
| EpCAM/CD326 | Cancer cells | Upregulates oncogene expression and cell proliferation | [17] |
| EGFR | Cancer cells | Triggers cell proliferation | [18] |
| VEGF | Cancer cells | Stimulates angiogenesis | [18] |
| CD133 | Cancer stem cells | Play a role in chemotherapy resistance | [19] |
| HER3 | Cancer cells | Promotes cell proliferation | [20,21] |
| HER2 | Cancer cells | Promotes cell proliferation | [22,23] |
| CD3 | CD8+ T cells, CD4+ T cells | Used in bsTCEs to engage T cells | [24] |
| GPA33 | Cancer cells | Function unclear; overexpressed in CRC | [25,26] |
| GUCY2C | Cancer cells | Maintains intestinal homeostasis | [27] |
| CEA | Cancer cells | May inhibit cell differentiation, apoptosis, and anoikis | [28] |
| Mutant KRAS | Cancer cells | Mutation causes overactive cell proliferation | [29] |
| Mutant TP53 | Cancer cells | Mutation causes loss of tumor suppressive ability and possible gain of oncogenic properties | [30] |
| Angiopoietin-2 | Cancer cells | Stimulates angiogenesis | [31] |
| MET | Cancer cells | Promotes cellular proliferation, motility, migration and invasion | [32,33] |
| DR5 | Cancer cells | Induces apoptosis | [34] |
| EphA2 | Cancer cells | Controls cell–cell repulsion or adhesion | [35] |
| MUC-1 | Cancer cells | Associated with invasion, metastases | [36] |
| Survivin | Cancer cells | Inhibits cell death | [37,38] |
| SART3 | Cancer cells | Spliceosome recycling factor; RNA-binding protein; overexpressed in CRC | [39,40] |
| IL-2 | Cancer cells, CD4+ T cells | Stimulates T cells and NK cells | [41,42] |
| IL-12 | APCs | Stimulates T cells and NK cells | [41] |
| IL-11 | Epithelial cells, endothelial cells, fibroblasts, myeloid cells | Tumor-promoting cytokine | [41,43] |
| IL-6 | Epithelial cells, myeloid cells | Tumor-promoting cytokine | [41] |
| IL-1α | Epithelial cells, myeloid cells | Multi-functional: promotes inflammatory carcinogenesis; promotes antitumour immunity | [41,44,45] |
| IL-1β | Epithelial cells, myeloid cells | Multi-functional: promotes inflammation-induced carcinogenesis; recruits antineoplastic cells, may block metastatic outgrowth | [41,44,45] |
| IL-33 | Epithelial cells, endothelial cells, adipocytes, fibroblasts, DCs | Recruits, activates, and degranulates eosinophils | [46,47] |
| IFN-γ | Cancer cells, CD4+ T cells, CD8+ T cells, γδ T cells, NK cells | Likely a tumor-inhibiting cytokine | [48] |
| STING | Widely expressed in immune and non-immune cells | Stimulates IFN genes and cellular senescence | [49,50] |
| CCR5 | Cancer cells, MDSCs, T regs, monocytes, macrophages, DCs, Th1 cells, activated T cells, NK cells | Enhances cancer cell motility; enhances MDSC and T reg infiltration | [51,52] |
| Immunotherapy | Advantages | Disadvantages |
|---|---|---|
| Immune checkpoint blockade | Sensitive, specific, additional T cell activation mechanisms possible, can be combined with each other | Most are mAbs and confer the same disadvantages, systemic toxicity is likely, response rates are low in CRC |
| Adoptive cell therapy | Personalized, permanent T cell modification confers immune memory | Expensive, difficult to manufacture, GvHD, CRS and B cell aplasia common |
| Monoclonal antibody | Relatively inexpensive, specific, effective across cancer types, can be conjugated easily | Target identification expensive, inadequate pharmacokinetics and tissue accessibility, resistance development common |
| Oncolytic virus therapy | Specific to cancer cells, may prime immune system to boost response to other ITs | Anti-viral immunity may reduce efficacy |
| Cancer vaccines | Specific, can be personalized | Rejection is possible due to delivery of foreign antigens |
| Immune system modulators | Many are FDA-approved, small size facilitates access to cancer cells, relatively inexpensive, can stimulate general anti-cancer immune response | Low specificity possible, risk of immediate onset CRS (cytokine storm] |
| Number of Clinical Trials | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ICB | ACT | mAb | Conj. Ab | bsAb | Virus | Vaccine | IFN | IL | IMiD | STING | |
| Completed | 31 | 8 | 67 | 3 | 3 | 3 | 46 | 11 | 7 | 2 | 0 |
| Active, not recruiting | 46 | 6 | 19 | - | - | 1 | 4 | 1 | 4 | - | 0 |
| Recruiting | 111 | 15 | 52 | 2 | 8 | 2 | 18 | 4 | 5 | - | 1 |
| Not yet recruiting | 16 | 7 | 9 | 1 | 3 | - | 2 | 1 | - | - | 0 |
| Terminated | 11 | 4 | 29 | 1 | 1 | 1 | 11 | 2 | 5 | 1 | 0 |
| Withdrawn | 10 | 2 | 12 | 2 | 2 | 1 | 5 | 1 | 2 | - | 0 |
| Suspended | - | 1 | - | - | - | - | 1 | - | - | - | 0 |
| Unknown | 9 | 14 | 15 | - | 2 | - | 9 | 5 | 2 | 3 | 0 |
| Total | 234 | 57 | 203 | 9 | 19 | 8 | 96 | 25 | 25 | 6 | 1 |
| Search terms | |||||||||||
| ICB | immune checkpoint blockade OR checkpoint blockade OR immune checkpoint inhibitor OR anti-PD-1 OR anti-pdl1 OR anti-ctla4 OR anti-lag3 OR anti-tim3 OR anti-nkg2 OR PD-1 OR pdl1 OR ctla4 OR lag3 OR tim3 OR nkg2 | ||||||||||
| ACT | adoptive cell therapy OR adoptive cell transfer OR cellular adoptive immunotherapy OR t cell transfer therapy OR tumor-infiltrating lymphocyte OR engineered t cell OR t cell receptor therapy OR car t cell OR NK cell | ||||||||||
| mAb | monoclonal antibody OR monoclonal antibodies | ||||||||||
| Conj. Ab | conjugated antibody OR conjugated antibodies | ||||||||||
| bsAb | bispecific antibody OR bispecific antibodies | ||||||||||
| Virus | oncolytic virus | ||||||||||
| Vaccine | vaccine | ||||||||||
| IFN | interferon | ||||||||||
| IL | interleukin | ||||||||||
| IMiD | thalidomide | ||||||||||
| STING | sting | ||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carlsen, L.; Huntington, K.E.; El-Deiry, W.S. Immunotherapy for Colorectal Cancer: Mechanisms and Predictive Biomarkers. Cancers 2022, 14, 1028. https://doi.org/10.3390/cancers14041028
Carlsen L, Huntington KE, El-Deiry WS. Immunotherapy for Colorectal Cancer: Mechanisms and Predictive Biomarkers. Cancers. 2022; 14(4):1028. https://doi.org/10.3390/cancers14041028
Chicago/Turabian StyleCarlsen, Lindsey, Kelsey E. Huntington, and Wafik S. El-Deiry. 2022. "Immunotherapy for Colorectal Cancer: Mechanisms and Predictive Biomarkers" Cancers 14, no. 4: 1028. https://doi.org/10.3390/cancers14041028
APA StyleCarlsen, L., Huntington, K. E., & El-Deiry, W. S. (2022). Immunotherapy for Colorectal Cancer: Mechanisms and Predictive Biomarkers. Cancers, 14(4), 1028. https://doi.org/10.3390/cancers14041028