
Systemic Influences of Mammary Cancer on Monocytes in Mice

 , , , and
, , , and 
Abstract
Simple Summary
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
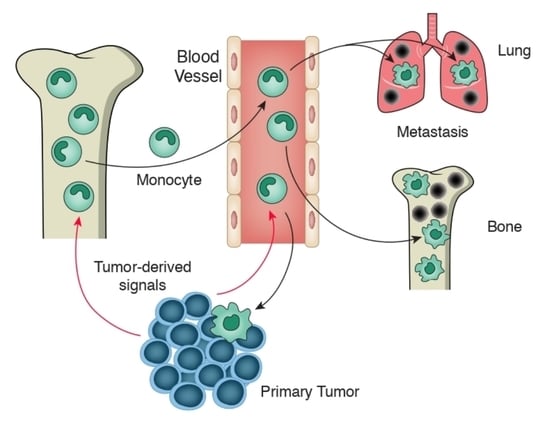
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Flow Cytometry and Cell Sorting
2.3. CFU Assays
2.4. Determination of Cells in DNA Synthesis
2.5. RNA-Seq of Monocytes
2.6. Motif Enrichment Analysis
2.7. Data Access
2.8. Statistical Analysis
3. Results
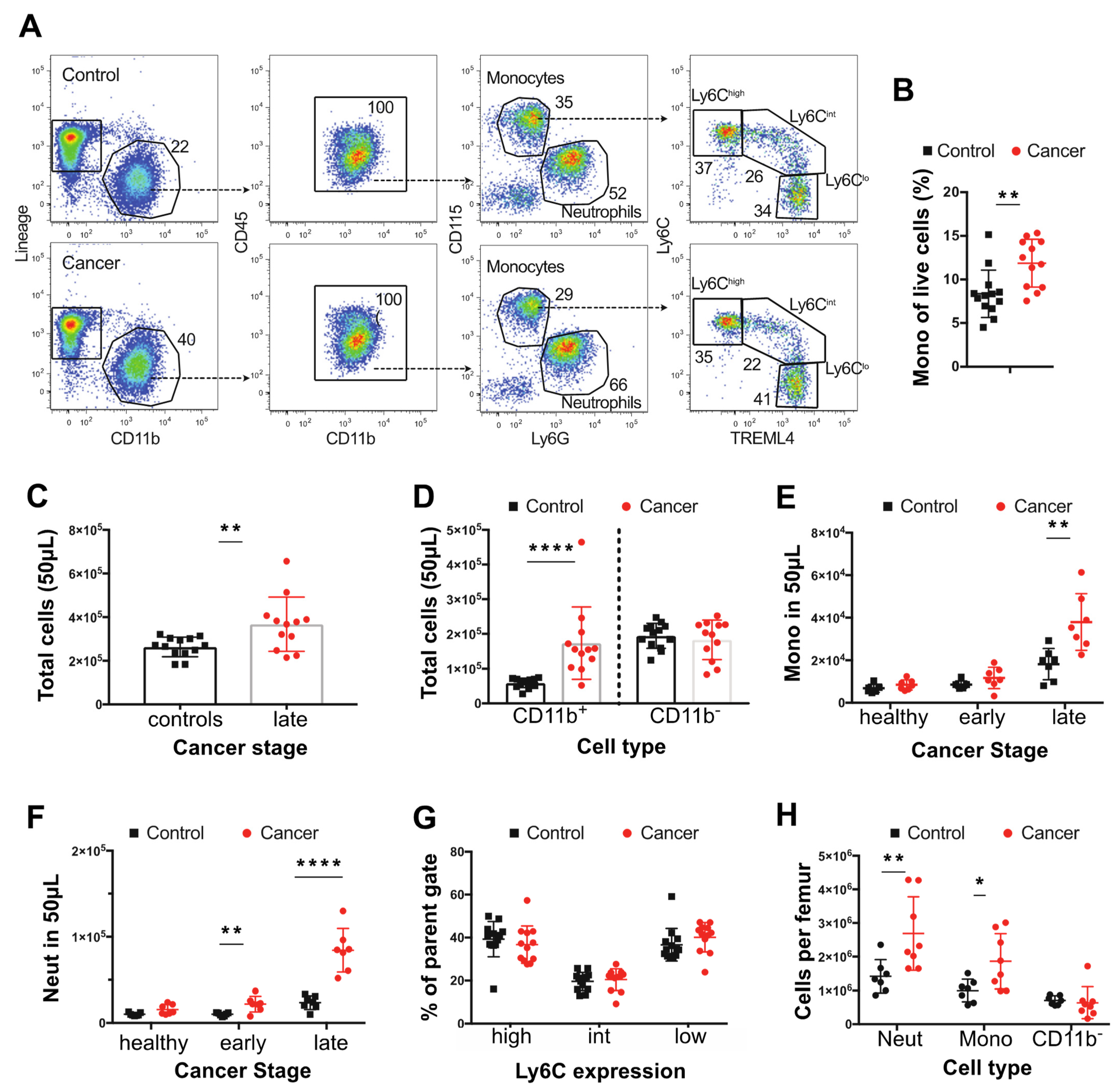
3.1. Tumor Bearing MMTV-PyMT Mice Show Increased Numbers of Monocytes
3.2. Tumor-Bearing Mice Exhibit Elevated Proliferation of BM Monocytes
3.3. Monocytes from the Blood of Mice with Tumors Are Transcriptionally Altered
3.4. Transcriptional Changes Occur within the Bone Marrow
3.5. Transcriptional Shifts in the Mouse PyMT Model Are Not Orthologous to Patients with Breast Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Geissmann, F.; Jung, S.; Littman, D.R. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef]
- Hettinger, J.; Richards, D.; Hansson, J.; Barra, M.M.; Joschko, A.-C.; Krijgsveld, J.; Feuerer, M. Origin of monocytes and macrophages in a committed progenitor. Nat. Immunol. 2013, 14, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Gu, Y.; Chakarov, S.; Bleriot, C.; Kwok, I.; Chen, X.; Shin, A.; Huang, W.; Dress, R.J.; Dutertre, C.-A.; et al. Fate mapping via Ms4a3-expression history traces monocyte-derived cells. Cell 2019, 178, 1509–1525.e19. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, A.; Coetzee, S.; Olsson, A.; Muench, D.; Berman, B.P.; Hazelett, D.J.; Salomonis, N.; Grimes, H.L.; Goodridge, H.S. Granulocyte-monocyte progenitors and monocyte-dendritic cell progenitors independently produce functionally distinct monocytes. Immunity 2017, 47, 890–902.e4. [Google Scholar] [CrossRef] [PubMed]
- Van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef]
- Ingersoll, M.A.; Spanbroek, R.; Lottaz, C.; Gautier, E.; Frankenberger, M.; Hoffmann, R.; Lang, R.; Haniffa, M.; Collin, M.; Tacke, F.; et al. Comparison of gene expression profiles between human and mouse monocyte subsets. Blood 2010, 115, e10–e19. [Google Scholar] [CrossRef]
- Zilionis, R.; Engblom, C.; Pfirschke, C.; Savova, V.; Zemmour, D.; Saatcioglu, H.D.; Krishnan, I.; Maroni, G.; Meyerovitz, C.V.; Kerwin, C.M.; et al. Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 2019, 50, 1317–1334.e10. [Google Scholar] [CrossRef] [PubMed]
- Zawada, A.M.; Rogacev, K.S.; Rotter, B.; Winter, P.; Marell, R.R.; Fliser, D.; Heine, G.H. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood 2011, 118, e50–e61. [Google Scholar] [CrossRef]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356, eaah4573. [Google Scholar] [CrossRef]
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Zhang, Y.; Fullerton, J.N.; Boelen, L.; Rongvaux, A.; Maini, A.; Bigley, V.; Flavell, R.A.; Gilroy, D.; Asquith, B.; et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic targets. Cancer Cell 2019, 35, 588–602.e10. [Google Scholar] [CrossRef] [PubMed]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Cancer 2008, 9, 239–252. [Google Scholar] [CrossRef]
- Stansfield, B.K.; Ingram, D.A. Clinical significance of monocyte heterogeneity. Clin. Transl. Med. 2015, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Zhang, Z.; Song, L.; Leung, Y.T.; Petri, M.A.; E Sullivan, K. Monocyte enhancers are highly altered in systemic lupus erythematosus. Epigenomics 2015, 7, 921–935. [Google Scholar] [CrossRef]
- Bergenfelz, C.; Larsson, A.-M.; Von Stedingk, K.; Gruvberger-Saal, S.; Aaltonen, K.; Jansson, S.; Jernström, H.; Janols, H.; Wullt, M.; Bredberg, A.; et al. Systemic monocytic-MDSCs are generated from monocytes and correlate with disease progression in breast cancer patients. PLoS ONE 2015, 10, e0127028. [Google Scholar] [CrossRef]
- Güç, E.; Pollard, J.W. Redefining macrophage and neutrophil biology in the metastatic cascade. Immunity 2021, 54, 885–902. [Google Scholar] [CrossRef]
- Arwert, E.; Harney, A.S.; Entenberg, D.; Wang, Y.; Sahai, E.; Pollard, J.W.; Condeelis, J.S. A unidirectional transition from migratory to perivascular macrophage is required for tumor cell intravasation. Cell Rep. 2018, 23, 1239–1248. [Google Scholar] [CrossRef]
- Movahedi, K.; Laoui, D.; Gysemans, C.; Baeten, M.; Stangé, G.; Van den Bossche, J.; Mack, M.; Pipeleers, D.; Veld, P.I.; De Baetselier, P.; et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010, 70, 5728–5739. [Google Scholar] [CrossRef]
- Franklin, R.A.; Liao, W.; Sarkar, A.; Kim, M.V.; Bivona, M.R.; Liu, K.; Pamer, E.G.; Li, M.O. The cellular and molecular origin of tumor-associated macrophages. Science 2014, 344, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.; Deng, Y.; Im, J.H.; Muschel, R.J.; Zou, Y.; Li, J.; Lang, R.A.; Pollard, J.W. A distinct macrophage population mediates metastatic breast cancer cell extravasation, establishment and growth. PLoS ONE 2009, 4, e6562. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.-Z.; Pollard, J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015, 15, 73–86. [Google Scholar] [CrossRef]
- Zhu, Y.; Herndon, J.M.; Sojka, D.K.; Kim, K.W.; Knolhoff, B.L.; Zuo, C.; Cullinan, D.R.; Luo, J.; Bearden, A.R.; Lavine, K.J.; et al. Tissue resident macrophages in pancreatic ductal adenocarcinoma originate from embryonic hematopoiesis and promote tumor progression. Immunity 2017, 47, 323–338.e6. [Google Scholar] [CrossRef]
- Friebel, E.; Kapolou, K.; Unger, S.; Núñez, N.G.; Utz, S.; Rushing, E.J.; Regli, L.; Weller, M.; Greter, M.; Tugues, S.; et al. Single-cell mapping of human brain cancer reveals tumor-specific instruction of tissue-invading leukocytes. Cell 2020, 181, 1626–1642.e20. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; de Visser, K.E. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Watanabe, A.; Aburatani, H.; Maru, Y. Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nat. Cell Biol. 2006, 8, 1369–1375. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Cao, X. Characteristics and significance of the pre-metastatic niche. Cancer Cell 2016, 30, 668–681. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Doughty-Shenton, D.; Cassetta, L.; Fragkogianni, S.; Brownlie, D.; Kato, Y.; Carragher, N.; Pollard, J.W. Monocytes differentiate to immune suppressive precursors of metastasis-associated macrophages in mouse models of metastatic breast cancer. Front. Immunol. 2018, 8, 2004. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.-Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef]
- Gil-Bernabé, A.M.; Ferjančič, Š.; Tlalka, M.; Zhao, L.; Allen, P.D.; Im, J.H.; Watson, K.; Hill, S.A.; Amirkhosravi, A.; Francis, J.L.; et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 2012, 119, 3164–3175. [Google Scholar] [CrossRef]
- Cassetta, L.; Baekkevold, E.S.; Brandau, S.; Bujko, A.; Cassatella, M.A.; Dorhoi, A.; Krieg, C.; Lin, A.; Loré, K.; Marini, O.; et al. Deciphering myeloid-derived suppressor cells: Isolation and markers in humans, mice and non-human primates. Cancer Immunol. Immunother. 2019, 68, 687–697. [Google Scholar] [CrossRef]
- Feng, A.-L.; Zhu, J.-K.; Sun, J.-T.; Yang, M.-X.; Neckenig, M.R.; Wang, X.-W.; Shao, Q.-Q.; Song, B.-F.; Yang, Q.-F.; Kong, B.-H.; et al. CD16+ monocytes in breast cancer patients: Expanded by monocyte chemoattractant protein-1 and may be useful for early diagnosis. Clin. Exp. Immunol. 2011, 164, 57–65. [Google Scholar] [CrossRef]
- Chittezhath, M.; Dhillon, M.K.; Lim, J.Y.; Laoui, D.; Shalova, I.N.; Teo, Y.L.; Chen, J.; Kamaraj, R.; Raman, L.; Lum, J.; et al. Molecular profiling reveals a tumor-promoting phenotype of monocytes and macrophages in human cancer progression. Immunity 2014, 41, 815–829. [Google Scholar] [CrossRef]
- Hamm, A.; Prenen, H.; Van Delm, W.; Di Matteo, M.; Wenes, M.; Delamarre, E.; Schmidt, T.; Weitz, J.; Sarmiento, R.; Dezi, A.; et al. Tumour-educated circulating monocytes are powerful candidate biomarkers for diagnosis and disease follow-up of colorectal cancer. Gut 2016, 65, 990–1000. [Google Scholar] [CrossRef]
- Lin, E.Y.; Jones, J.G.; Li, P.; Zhu, L.; Whitney, K.D.; Muller, W.J.; Pollard, J.W. Progression to malignancy in the polyoma middle t oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am. J. Pathol. 2003, 163, 2113–2126. [Google Scholar] [CrossRef]
- Muller, W.J.; Sinn, E.; Pattengale, P.K.; Wallace, R.; Leder, P. Single-step induction of mammary adenocarcinoma in transgenic mice bearing the activated c-neu oncogene. Cell 1988, 54, 105–115. [Google Scholar] [CrossRef]
- Casbon, A.-J.; Reynaud, D.; Park, C.; Khuc, E.; Gan, D.D.; Schepers, K.; Passegue, E.; Werb, Z. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl. Acad. Sci. USA 2015, 112, E566–E575. [Google Scholar] [CrossRef]
- Picelli, S.; Faridani, O.R.; Bjorklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Reimand, J.; Kull, M.; Peterson, H.; Hansen, J.; Vilo, J. g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 2007, 35, W193–W200. [Google Scholar] [CrossRef]
- Heinz, S.; Benner, C.; Spann, N.; Bertolino, E.; Lin, Y.C.; Laslo, P.; Cheng, J.X.; Murre, C.; Singh, H.; Glass, C.K. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 2010, 38, 576–589. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.-L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, F.; Rauch, P.; Ueno, T.; Gorbatov, R.; Marinelli, B.; Lee, W.W.; Dutta, P.; Wei, Y.; Robbins, C.; Iwamoto, Y.; et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 2012, 209, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Robbins, C.S.; Chudnovskiy, A.; Rauch, P.; Figueiredo, J.-L.; Iwamoto, Y.; Gorbatov, R.; Etzrodt, M.; Weber, G.F.; Ueno, T.; van Rooijen, N.; et al. Extramedullary hematopoiesis generates Ly-6C high monocytes that infiltrate atherosclerotic lesions. Circulation 2012, 125, 364–374. [Google Scholar] [CrossRef] [PubMed]
- Hunter, K.W.; Crawford, N.P.; Alsarraj, J. Mechanisms of metastasis. Breast Cancer Res. 2008, 10 (Suppl. 1), S2. [Google Scholar] [CrossRef] [PubMed]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Prim. 2018, 4, 17105. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Rafii, S.; Lyden, D. Preparing the “soil”: The premetastatic niche. Cancer Res. 2006, 66, 11089–11093. [Google Scholar] [CrossRef]
- Hiratsuka, S.; Watanabe, A.; Sakurai, Y.; Akashi-Takamura, S.; Ishibashi, S.; Miyake, K.; Shibuya, M.; Akira, S.; Aburatani, H.; Maru, Y. The S100A8–serum amyloid A3–TLR4 paracrine cascade establishes a pre-metastatic phase. Nat. Cell Biol. 2008, 10, 1349–1355. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Psaila, B.; Lyden, D. Bone marrow cells in the ‘pre-metastatic niche’: Within bone and beyond. Cancer Metastasis Rev. 2006, 25, 521–529. [Google Scholar] [CrossRef]
- Keklikoglou, I.; Cianciaruso, C.; Güç, E.; Squadrito, M.L.; Spring, L.M.; Tazzyman, S.; Lambein, L.; Poissonnier, A.; Ferraro, G.B.; Baer, C.; et al. Chemotherapy elicits pro-metastatic extracellular vesicles in breast cancer models. Nat. Cell Biol. 2019, 21, 190–202. [Google Scholar] [CrossRef]
- Mo, H.; Shi, Y.; Han, X.; Liu, P.; Yang, J.; Zhang, C.; Yang, S.; Qin, Y.; Gui, L.; Shi, Y.; et al. Absolute monocyte count is a prognostic indicator in a patient with diffuse large B-cell lymphoma after autologous peripheral blood stem cell transplant. Leuk. Lymphoma 2014, 56, 515–517. [Google Scholar] [CrossRef]
- Cassetta, L.; Bruderek, K.; Skrzeczynska-Moncznik, J.; Osiecka, O.; Hu, X.; Rundgren, I.M.; Lin, A.; Santegoets, K.; Horzum, U.; Godinho-Santos, A.; et al. Differential expansion of circulating human MDSC subsets in patients with cancer, infection and inflammation. J. Immunother. Cancer 2020, 8, e001223. [Google Scholar] [CrossRef] [PubMed]
- Satoh, T.; Nakagawa, K.; Sugihara, F.; Kuwahara, R.; Ashihara, M.; Yamane, F.; Minowa, Y.; Fukushima, K.; Ebina, I.; Yoshioka, Y.; et al. Identification of an atypical monocyte and committed progenitor involved in fibrosis. Nature 2016, 541, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Rauch, P.; Chudnovskiy, A.; Berger, C.; Ryan, R.; Iwamoto, Y.; Marinelli, B.; Gorbatov, R.; et al. Origins of tumor-associated macrophages and neutrophils. Proc. Natl. Acad. Sci. USA 2012, 109, 2491–2496. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Retamozo, V.; Engblom, C.; Pittet, M.J. Remote control of macrophage production by cancer. OncoImmunology 2013, 2, e24183. [Google Scholar] [CrossRef][Green Version]
- Smith, H.O.; Anderson, P.S.; Kuo, D.Y.; Goldberg, G.L.; DeVictoria, C.L.; A Boocock, C.; Jones, J.G.; Runowicz, C.D.; Stanley, E.R.; Pollard, J.W. The role of colony-stimulating factor 1 and its receptor in the etiopathogenesis of endometrial adenocarcinoma. Clin. Cancer Res. 1995, 1, 313–325. [Google Scholar]
- Tang, R.; Beuvon, F.; Ojeda, M.; Mosseri, V.; Pouillart, P.; Scholl, S. M-CSF (Monocyte colony stimulating factor) and M-CSF receptor expression by breast tumor cells: M-CSF mediated recruitment of tumour infiltrating monocytes? J. Cell. Biochem. 1992, 50, 350–356. [Google Scholar] [CrossRef]
- Wu, X.; Thi, V.L.D.; Huang, Y.; Billerbeck, E.; Saha, D.; Hoffmann, H.-H.; Wang, Y.; Silva, L.A.V.; Sarbanes, S.; Sun, T.; et al. Intrinsic immunity shapes viral resistance of stem cells. Cell 2018, 172, 423–438.e25. [Google Scholar] [CrossRef]
- Mendoza, A.; Hong, S.-H.; Osborne, T.; Khan, M.A.; Campbell, K.; Briggs, J.; Eleswarapu, A.; Buquo, L.; Ren, L.; Hewitt, S.M.; et al. Modeling metastasis biology and therapy in real time in the mouse lung. J. Clin. Investig. 2010, 120, 2979–2988. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robinson, A.; Burgess, M.; Webb, S.; Louwe, P.A.; Ouyang, Z.; Skola, D.; Han, C.Z.; Batada, N.N.; González-Huici, V.; Cassetta, L.; et al. Systemic Influences of Mammary Cancer on Monocytes in Mice. Cancers 2022, 14, 833. https://doi.org/10.3390/cancers14030833
Robinson A, Burgess M, Webb S, Louwe PA, Ouyang Z, Skola D, Han CZ, Batada NN, González-Huici V, Cassetta L, et al. Systemic Influences of Mammary Cancer on Monocytes in Mice. Cancers. 2022; 14(3):833. https://doi.org/10.3390/cancers14030833
Chicago/Turabian StyleRobinson, Amy, Matthew Burgess, Sheila Webb, Pieter A. Louwe, Zhengyu Ouyang, Dylan Skola, Claudia Z. Han, Nizar N. Batada, Víctor González-Huici, Luca Cassetta, and et al. 2022. "Systemic Influences of Mammary Cancer on Monocytes in Mice" Cancers 14, no. 3: 833. https://doi.org/10.3390/cancers14030833
APA StyleRobinson, A., Burgess, M., Webb, S., Louwe, P. A., Ouyang, Z., Skola, D., Han, C. Z., Batada, N. N., González-Huici, V., Cassetta, L., Glass, C. K., Jenkins, S. J., & Pollard, J. W. (2022). Systemic Influences of Mammary Cancer on Monocytes in Mice. Cancers, 14(3), 833. https://doi.org/10.3390/cancers14030833