
Co-Inhibition of Androgen Receptor and PARP as a Novel Treatment Paradigm in Prostate Cancer—Where Are We Now?

Abstract
:Simple Summary
Abstract
1. Introduction
2. Limitations of NAA Monotherapy in Advanced Prostate Cancer
Sequencing Considerations in Second and Subsequent Lines of Therapy
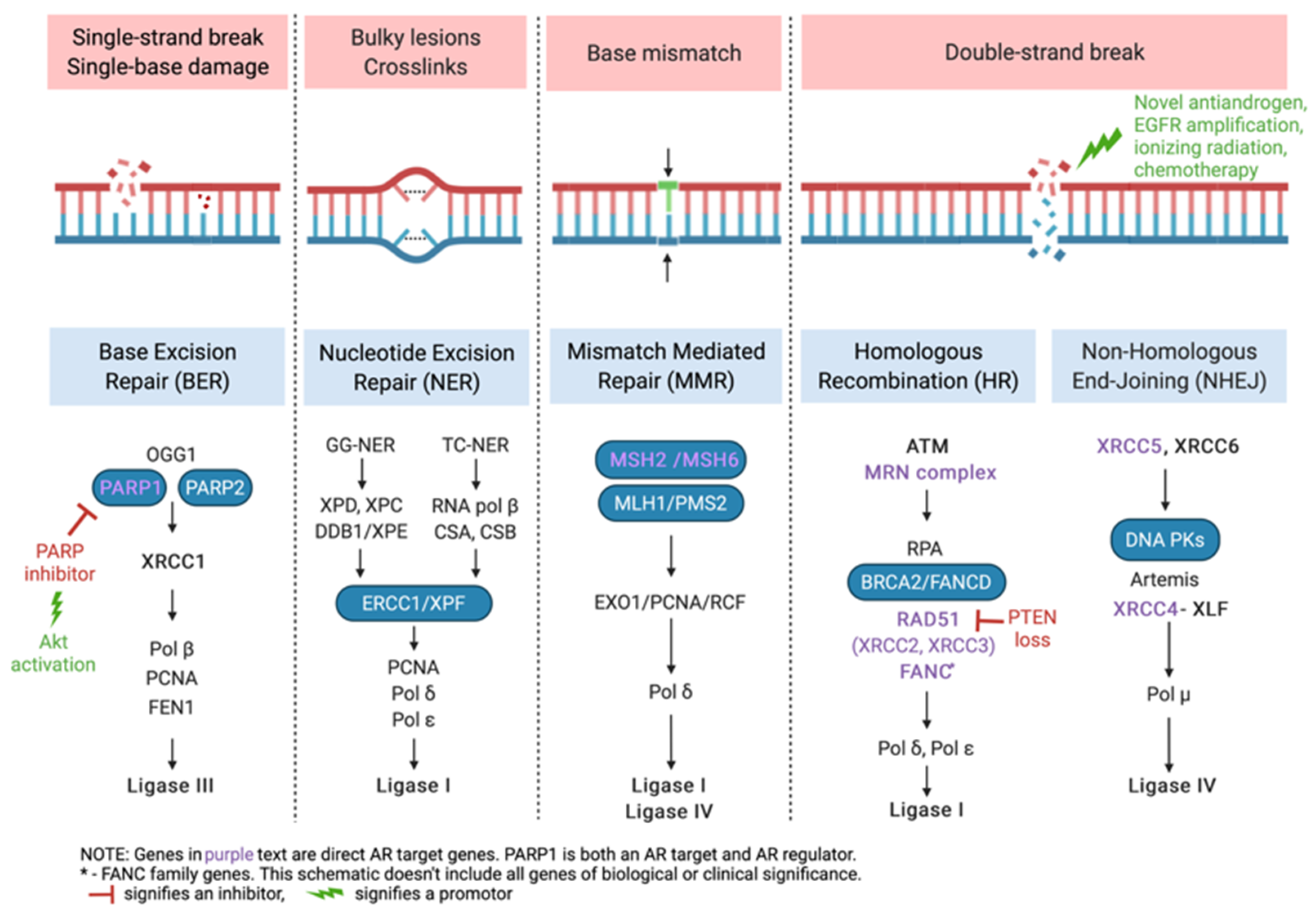
3. DNA Repair and HRR Gene Alterations in Prostate Cancer
4. PARP Inhibitor Monotherapy in Prostate Cancer
5. Synthetic Lethality by Co-Inhibition of AR and PARP
5.1. Summary of Clinical Trial Data on Synthetic Lethality with NAA and PARP Inhibitors
5.2. Ongoing Clinical Trials of AR and PARP Combination Therapies—Highlights and Key Considerations for the Near Future
6. Discussion
Moving it Upstream—Does NAA and PARP Co-Inhibition Have a Role in Earlier Disease Settings?
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2018, 68, 394–424, Erratum in CA Cancer J. Clin. 2020, 70, 313. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.; Ma, J.; Zou, Z.; Jemal, A. Cancer Statistics, 2014. CA Cancer J. Clin. 2014, 64, 9–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Siegel, D.A.; King, J.B. Stage-Specific Incidence Rates and Trends of Prostate Cancer by Age, Race, and Ethnicity, United States, 2004–2014. Ann. Epidemiol. 2018, 28, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Sagaram, S.; Rao, A. Rapidly Evolving Treatment Paradigm and Considerations for Sequencing Therapies in Metastatic Prostate Cancer—a Narrative Review. Transl. Androl. Urol. 2021, 10, 3188198. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and Resistance to Enzalutamide and Abiraterone in Prostate Cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.H.; Gulati, R.; Azad, A.; Nadal, R.; Twardowski, P.; Vaishampayan, U.N.; Agarwal, N.; Heath, E.I.; Pal, S.K.; Rehman, H.; et al. Activity of Enzalutamide in Men with Metastatic Castration Resistant Prostate Cancer Is Affected by Prior Treatment with Abiraterone and/or Docetaxel. Prostate Cancer Prostatic Dis. 2015, 18, 122–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, K.N.; Annala, M.; Sunderland, K.; Khalaf, D.; Finch, D.; Oja, C.D.; Vergidis, J.; Zulfiqar, M.; Beja, K.; Vandekerkhove, G.; et al. A Randomized Phase II Cross-over Study of Abiraterone + Prednisone (ABI) vs Enzalutamide (ENZ) for Patients (Pts) with Metastatic, Castration-Resistant Prostate Cancer (MCRPC). J. Clin. Oncol. 2017, 35, 5002. [Google Scholar] [CrossRef]
- de Bono, J.S.; Chowdhury, S.; Feyerabend, S.; Elliott, T.; Grande, E.; Melhem-Bertrandt, A.; Baron, B.; Hirmand, M.; Werbrouck, P.; Fizazi, K. Antitumour Activity and Safety of Enzalutamide in Patients with Metastatic Castration-Resistant Prostate Cancer Previously Treated with Abiraterone Acetate Plus Prednisone for ≥ 24 Weeks in Europe. Eur. Urol. 2018, 74, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Becker, D.J.; Iyengar, A.D.; Punekar, S.R.; Ng, J.; Zaman, A.; Loeb, S.; Becker, K.D.; Makarov, D. Treatment of Metastatic Castration-Resistant Prostate Cancer With Abiraterone and Enzalutamide Despite PSA Progression. Anticancer Res. 2019, 39, 2467–2473. [Google Scholar] [CrossRef]
- Khalaf, D.J.; Annala, M.; Taavitsainen, S.; Finch, D.L.; Oja, C.; Vergidis, J.; Zulfiqar, M.; Sunderland, K.; Azad, A.A.; Kollmannsberger, C.K.; et al. Optimal Sequencing of Enzalutamide and Abiraterone Acetate plus Prednisone in Metastatic Castration-Resistant Prostate Cancer: A Multicentre, Randomised, Open-Label, Phase 2, Crossover Trial. Lancet Oncol. 2019, 20, 1730–1739. [Google Scholar] [CrossRef]
- Terada, N.; Maughan, B.L.; Akamatsu, S.; Kobayashi, T.; Yamasaki, T.; Inoue, T.; Kamba, T.; Ogawa, O.; Antonarakis, E.S. Exploring the Optimal Sequence of Abiraterone and Enzalutamide in Patients with Chemotherapy-Naïve Castration-Resistant Prostate Cancer: The Kyoto-Baltimore Collaboration. Int. J. Urol. 2017, 24, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, K.; Kimura, T.; Onuma, H.; Kimura, S.; Yamamoto, T.; Sasaki, H.; Miki, J.; Miki, K.; Egawa, S. Lactate Dehydrogenase Predicts Combined Progression-Free Survival after Sequential Therapy with Abiraterone and Enzalutamide for Patients with Castration-Resistant Prostate Cancer. Prostate 2017, 77, 1144–1150. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, D.R.; Wu, Y.-M.; Lonigro, R.J.; Vats, P.; Cobain, E.; Everett, J.; Cao, X.; Rabban, E.; Kumar-Sinha, C.; Raymond, V.; et al. Integrative Clinical Genomics of Metastatic Cancer. Nature 2017, 548, 297–303. [Google Scholar] [CrossRef]
- Boyd, L.K.; Mao, X.; Lu, Y.-J. The Complexity of Prostate Cancer: Genomic Alterations and Heterogeneity. Nat. Rev. Urol. 2012, 9, 652–664. [Google Scholar] [CrossRef]
- Boerrigter, E.; Groen, L.N.; Van Erp, N.P.; Verhaegh, G.W.; Schalken, J.A. Clinical Utility of Emerging Biomarkers in Prostate Cancer Liquid Biopsies. Expert Rev. Mol. Diagn. 2020, 20, 219–230. [Google Scholar] [CrossRef] [Green Version]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus Prednisone in Metastatic, Castration-Sensitive Prostate Cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Evans, C.P.; Kim, C.-S.; Kimura, G.; et al. Enzalutamide in Men with Chemotherapy-Naïve Metastatic Castration-Resistant Prostate Cancer: Extended Analysis of the Phase 3 PREVAIL Study. Eur. Urol. 2017, 71, 151–154. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-Free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef]
- Fizazi, K.; Shore, N.; Tammela, T.L.; Ulys, A.; Vjaters, E.; Polyakov, S.; Jievaltas, M.; Luz, M.; Alekseev, B.; Kuss, I.; et al. Darolutamide in Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2019, 380, 1235–1246. [Google Scholar] [CrossRef]
- Hussain, M.; Fizazi, K.; Saad, F.; Rathenborg, P.; Shore, N.; Ferreira, U.; Ivashchenko, P.; Demirhan, E.; Modelska, K.; Phung, D.; et al. Enzalutamide in Men with Nonmetastatic, Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2018, 378, 2465–2474. [Google Scholar] [CrossRef]
- Ryan, C.J.; Smith, M.R.; Fizazi, K.; Saad, F.; Mulders, P.F.A.; Sternberg, C.N.; Miller, K.; Logothetis, C.J.; Shore, N.D.; Small, E.J.; et al. Abiraterone Acetate plus Prednisone versus Placebo plus Prednisone in Chemotherapy-Naive Men with Metastatic Castration-Resistant Prostate Cancer (COU-AA-302): Final Overall Survival Analysis of a Randomised, Double-Blind, Placebo-Controlled Phase 3 Study. Lancet Oncol. 2015, 16, 152–160. [Google Scholar] [CrossRef]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Allen, E.M.V.; et al. Genomic Correlates of Clinical Outcome in Advanced Prostate Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, M.J.; Heller, G.; Bryce, A.H.; Armstrong, A.J.; Beltran, H.; Hahn, O.M.; McGary, E.C.; Mehan, P.T.; Goldkorn, A.; Roth, B.J.; et al. Alliance A031201: A Phase III Trial of Enzalutamide (ENZ) versus Enzalutamide, Abiraterone, and Prednisone (ENZ/AAP) for Metastatic Castration Resistant Prostate Cancer (MCRPC). J. Clin. Oncol. 2019, 37, 5008. [Google Scholar] [CrossRef]
- Attard, G.; Borre, M.; Gurney, H.; Loriot, Y.; Andresen-Daniil, C.; Kalleda, R.; Pham, T.; Taplin, M.-E. Abiraterone Alone or in Combination With Enzalutamide in Metastatic Castration-Resistant Prostate Cancer With Rising Prostate-Specific Antigen During Enzalutamide Treatment. J. Clin. Oncol. 2018, 36, 2639–2646. [Google Scholar] [CrossRef]
- Wang, X.; Yang, H.; Hu, X.; Wang, W.; Yu, X.; Wang, S.; Zhang, X.; Liu, L. Comparing the Clinical Efficacy and Safety of Abiraterone and Enzalutamide in Metastatic Castration-Resistant Prostate Cancer: A Systematic Review and Meta-Analysis. J. Oncol. Pharm. Pract. 2021, 27, 614–622. [Google Scholar] [CrossRef]
- Demirci, A.; Bilir, C.; Gülbağcı, B.; Hacıbekiroğlu, İ.; Bayoğlu, İ.V.; Bilgetekin, İ.; Koca, S.; Çınkır, H.Y.; Akdeniz, N.; Gül, D.; et al. Comparison of Real-Life Data of Abiraterone Acetate and Enzalutamide in Metastatic Castration-Resistant Prostate Cancer. Sci. Rep. 2021, 11, 14131. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide and Overall Survival in Prostate Cancer. Eur. Urol. 2021, 79, 150–158. [Google Scholar] [CrossRef]
- Houtgraaf, J.H.; Versmissen, J.; van der Giessen, W.J. A Concise Review of DNA Damage Checkpoints and Repair in Mammalian Cells. Cardiovasc. Revascularization Med. 2006, 7, 165–172. [Google Scholar] [CrossRef]
- Reed, E. DNA Damage and Repair in Translational Oncology: An Overview. Clin. Cancer Res. 2010, 16, 4511–4516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vítor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davar, D.; Beumer, J.H.; Hamieh, L.; Tawbi, H. Role of PARP Inhibitors in Cancer Biology and Therapy. Curr. Med. Chem. 2012, 19, 3907–3921. [Google Scholar] [CrossRef] [PubMed]
- Boehler, C.; Gauthier, L.R.; Mortusewicz, O.; Biard, D.S.; Saliou, J.-M.; Bresson, A.; Sanglier-Cianferani, S.; Smith, S.; Schreiber, V.; Boussin, F.; et al. Poly(ADP-Ribose) Polymerase 3 (PARP3), a Newcomer in Cellular Response to DNA Damage and Mitotic Progression. Proc. Natl. Acad. Sci. USA 2011, 108, 2783–2788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmukh, D.; Qiu, Y. Role of PARP-1 in Prostate Cancer. Am. J. Clin. Exp. Urol. 2015, 3, 1–12. [Google Scholar]
- Chaudhuri, A.R.; Nussenzweig, A. The Multifaceted Roles of PARP1 in DNA Repair and Chromatin Remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Haince, J.-F.; McDonald, D.; Rodrigue, A.; Déry, U.; Masson, J.-Y.; Hendzel, M.J.; Poirier, G.G. PARP1-Dependent Kinetics of Recruitment of MRE11 and NBS1 Proteins to Multiple DNA Damage Sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar-Quesada, R.; Muñoz-Gámez, J.A.; Martín-Oliva, D.; Peralta, A.; Valenzuela, M.T.; Matínez-Romero, R.; Quiles-Pérez, R.; Murcia, J.M.; de Murcia, G.; de Almodóvar, M.R.; et al. Interaction between ATM and PARP-1 in Response to DNA Damage and Sensitization of ATM Deficient Cells through PARP Inhibition. BMC Mol. Biol. 2007, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Haince, J.-F.; Kozlov, S.; Dawson, V.L.; Dawson, T.M.; Hendzel, M.J.; Lavin, M.F.; Poirier, G.G. Ataxia Telangiectasia Mutated (ATM) Signaling Network Is Modulated by a Novel Poly(ADP-Ribose)-Dependent Pathway in the Early Response to DNA-Damaging Agents. J. Biol. Chem. 2007, 282, 16441–16453. [Google Scholar] [CrossRef] [Green Version]
- Luijsterburg, M.S.; de Krijger, I.; Wiegant, W.W.; Shah, R.G.; Smeenk, G.; de Groot, A.J.L.; Pines, A.; Vertegaal, A.C.O.; Jacobs, J.J.L.; Shah, G.M.; et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-Homologous End-Joining. Mol. Cell 2016, 61, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Jubin, T.; Kadam, A.; Jariwala, M.; Bhatt, S.; Sutariya, S.; Gani, A.R.; Gautam, S.; Begum, R. The PARP Family: Insights into Functional Aspects of Poly (ADP-Ribose) Polymerase-1 in Cell Growth and Survival. Cell Prolif. 2016, 49, 421–437. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.J.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA Repair Defect in BRCA Mutant Cells as a Therapeutic Strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific Killing of BRCA2-Deficient Tumours with Inhibitors of Poly(ADP-Ribose) Polymerase. Nature 2005, 434. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.L.; Kaur, H.B.; Salles, D.C.; Murali, S.; Schaeffer, E.M.; Lanchbury, J.S.; Isaacs, W.B.; Brown, R.; Richardson, A.L.; Cussenot, O.; et al. Homologous Recombination Deficiency (HRD) Score in Germline BRCA2- versus ATM-Altered Prostate Cancer. Mod. Pathol. 2021, 34, 1185–1193. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Nicolosi, P.; Ledet, E.; Yang, S.; Michalski, S.; Freschi, B.; O’Leary, E.; Esplin, E.D.; Nussbaum, R.L.; Sartor, O. Prevalence of Germline Variants in Prostate Cancer and Implications for Current Genetic Testing Guidelines. JAMA Oncol. 2019, 5, 523–528. [Google Scholar] [CrossRef] [Green Version]
- U.S. Food and Drug Administration. Rucaparib. Available online: https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm533891.htm (accessed on 14 March 2018).
- Abida, W.; Campbell, D.; Patnaik, A.; Sautois, B.; Shapiro, J.; Vogelzang, N.J.; Bryce, A.H.; McDermott, R.; Ricci, F.; Rowe, J.; et al. Preliminary Results from the TRITON2 Study of Rucaparib in Patients (Pts) with DNA Damage Repair (DDR)-Deficient Metastatic Castration-Resistant Prostate Cancer (MCRPC): Updated Analyses. Ann. Oncol. 2019, 30, v327–v328. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Full Prescribing Information for Lynparza (Olaparib). Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/208558s013lbl.pdf (accessed on 14 June 2020).
- Smith, M.R.; Sandhu, S.K.; Kelly, W.K.; Scher, H.I.; Efstathiou, E.; Lara, P.N.; Yu, E.Y.; George, D.J.; Chi, K.N.; Saad, F.; et al. Pre-Specified Interim Analysis of GALAHAD: A Phase II Study of Niraparib in Patients (Pts) with Metastatic Castration-Resistant Prostate Cancer (MCRPC) and Biallelic DNA-Repair Gene Defects (DRD). Ann. Oncol. 2019, 30, v884–v885. [Google Scholar] [CrossRef]
- De Bono, J.S.; Mehra, N.; Higano, C.S.; Saad, F.; Buttigliero, C.; Mata, M.; Chen, H.-C.; Healy, C.G.; Paccagnella, M.L.; Czibere, A.; et al. TALAPRO-1: A Phase II Study of Talazoparib (TALA) in Men with DNA Damage Repair Mutations (DDRmut) and Metastatic Castration-Resistant Prostate Cancer (MCRPC)—First Interim Analysis (IA). J. Clin. Oncol. 2020, 38, 119. [Google Scholar] [CrossRef]
- Bolla, M.; Gonzalez, D.; Warde, P.; Dubois, J.B.; Mirimanoff, R.-O.; Storme, G.; Bernier, J.; Kuten, A.; Sternberg, C.; Gil, T.; et al. Improved Survival in Patients with Locally Advanced Prostate Cancer Treated with Radiotherapy and Goserelin. N. Engl. J. Med. 1997, 337, 295–300. [Google Scholar] [CrossRef] [Green Version]
- Zelefsky, M.J.; Reuter, V.E.; Fuks, Z.; Scardino, P.; Shippy, A. Influence of Local Tumor Control on Distant Metastases and Cancer Related Mortality after External Beam Radiotherapy for Prostate Cancer. J. Urol. 2008, 179, 1368–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.U.; Hunt, D.; McGowan, D.G.; Amin, M.B.; Chetner, M.P.; Bruner, D.W.; Leibenhaut, M.H.; Husain, S.M.; Rotman, M.; Souhami, L.; et al. Radiotherapy and Short-Term Androgen Deprivation for Localized Prostate Cancer. N. Engl. J. Med. 2011, 365, 107–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollack, A.; Salem, N.; Ashoori, F.; Hachem, P.; Sangha, M.; von Eschenbach, A.C.; Meistrich, M.L. Lack of Prostate Cancer Radiosensitization by Androgen Deprivation. Int. J. Radiat. Oncol. Biol. Phys. 2001, 51, 1002–1007. [Google Scholar] [CrossRef]
- Wo, J.Y.; Zietman, A.L. Why Does Androgen Deprivation Enhance the Results of Radiation Therapy? Urol. Oncol. 2008, 26, 522–529. [Google Scholar] [CrossRef]
- Al-Ubaidi, F.L.T.; Schultz, N.; Loseva, O.; Egevad, L.; Granfors, T.; Helleday, T. Castration Therapy Results in Decreased Ku70 Levels in Prostate Cancer. Clin. Cancer Res. 2013, 19, 1547–1556. [Google Scholar] [CrossRef] [Green Version]
- Debruyne, F.M.J.; Witjes, W.P.J.; Schulman, C.C.; van Cangh, P.J.; Oosterhof, G.O.N. A Multicentre Trial of Combined Neoadjuvant Androgen Blockade with Zoladex and Flutamide Prior to Radical Prostatectomy in Prostate Cancer. Eur. Urol. 1994, 26, 4. [Google Scholar] [CrossRef]
- Abbas, F.; Kaplan, M.; Soloway, M.S. Induction Androgen Deprivation Therapy before Radical Prostatectomy for Prostate Cancer--Initial Results. Br. J. Urol. 1996, 77, 423–428. [Google Scholar] [CrossRef]
- Goldenberg, S.L.; Klotz, L.H.; Srigley, J.; Jewett, M.A.; Mador, D.; Fradet, Y.; Barkin, J.; Chin, J.; Paquin, J.M.; Bullock, M.J.; et al. Randomized, Prospective, Controlled Study Comparing Radical Prostatectomy Alone and Neoadjuvant Androgen Withdrawal in the Treatment of Localized Prostate Cancer. Canadian Urologic Oncology Group. J. Urol. 1996, 156, 873–877. [Google Scholar] [CrossRef]
- Aus, G.; Abrahamsson, P.A.; Ahlgren, G.; Hugosson, J.; Lundberg, S.; Schain, M.; Schelin, S.; Pedersen, K. Hormonal Treatment before Radical Prostatectomy: A 3-Year Followup. J. Urol. 1998, 159, 2013–2016. [Google Scholar] [CrossRef]
- Antonelli, A.; Palumbo, C.; Veccia, A.; Grisanti, S.; Triggiani, L.; Zamboni, S.; Furlan, M.; Simeone, C.; Magrini, S.; Berruti, A. Biological Effect of Neoadjuvant Androgen-Deprivation Therapy Assessed on Specimens from Radical Prostatectomy: A Systematic Review. Minerva Urol. Nefrol. 2018, 70, 370–379. [Google Scholar] [CrossRef] [Green Version]
- Cha, E.K.; Eastham, J.A. Chemotherapy and Novel Therapeutics before Radical Prostatectomy for High-Risk Clinically Localized Prostate Cancer. Urol. Oncol. 2015, 33, 217–225. [Google Scholar] [CrossRef]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.-F.; Cai, L.; Zheng, D.; et al. Androgen Receptor Signaling Regulates DNA Repair in Prostate Cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- BioRender. DNA Repair Mechanisms. Available online: Https://App.Biorender.Com/Biorender-Templates (accessed on 23 January 2022).
- Asim, M.; Tarish, F.; Zecchini, H.I.; Sanjiv, K.; Gelali, E.; Massie, C.E.; Baridi, A.; Warren, A.Y.; Zhao, W.; Ogris, C.; et al. Synthetic Lethality between Androgen Receptor Signalling and the PARP Pathway in Prostate Cancer. Nat. Commun. 2017, 8, 374. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen Receptor Inhibitor-Induced “BRCAness” and PARP Inhibition Are Synthetically Lethal for Castration-Resistant Prostate Cancer. Sci. Signal. 2017, 10, 480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, P.; Choudhary, G.S.; Alswillah, T.; Xiong, X.; Heston, W.D.; Magi-Galuzzi, C.; Zhang, J.; Klein, E.A.; Almasan, A. The TMPRSS2-ERG Gene Fusion Blocks XRCC4-Mediated Non-Homologous End-Joining Repair and Radiosensitizes Prostate Cancer Cells to PARP Inhibition. Mol. Cancer Ther. 2015, 14, 1896–1906. [Google Scholar] [CrossRef] [Green Version]
- Beltran, H.; Wyatt, A.W.; Chedgy, E.C.; Donoghue, A.; Annala, M.; Warner, E.W.; Beja, K.; Sigouros, M.; Mo, F.; Fazli, L.; et al. Impact of Therapy on Genomics and Transcriptomics in High-Risk Prostate Cancer Treated with Neoadjuvant Docetaxel and Androgen Deprivation Therapy. Clin. Cancer Res. 2017, 23, 6802–6811. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, J.F.; Schiewer, M.J.; Dean, J.L.; Schrecengost, R.S.; de Leeuw, R.; Han, S.; Ma, T.; Den, R.B.; Dicker, A.P.; Feng, F.Y.; et al. A Hormone–DNA Repair Circuit Governs the Response to Genotoxic Insult. Cancer Discov. 2013, 3, 1254–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ta, H.Q.; Gioeli, D. The Convergence of DNA Damage Checkpoint Pathways and Androgen Receptor Signaling in Prostate Cancer. Endocr. Relat. Cancer 2014, 21, R395–R407. [Google Scholar] [CrossRef] [Green Version]
- Clarke, N.; Wiechno, P.; Alekseev, B.; Sala, N.; Jones, R.; Kocak, I.; Chiuri, V.E.; Jassem, J.; Fléchon, A.; Redfern, C.; et al. Olaparib Combined with Abiraterone in Patients with Metastatic Castration-Resistant Prostate Cancer: A Randomised, Double-Blind, Placebo-Controlled, Phase 2 Trial. Lancet Oncol. 2018, 19, 975–986. [Google Scholar] [CrossRef]
- Rao, A.; Morris, D.; Assikis, V.J.; Jha, G.G.; Ryan, C.J.; Ablaza, A.-J.; Habeck, J.; Loehr, A.; Xiao, J.; Gangolli, E.A. Rucaparib plus Enzalutamide in Patients (Pts) with Metastatic Castration-Resistant Prostate Cancer (MCRPC): Pharmacokinetics (PK) and Safety Data from the Phase Ib RAMP Study. J. Clin. Oncol. 2021, 39, 79. [Google Scholar] [CrossRef]
- Rao, A.; Ryan, C.J.; Morris, D.; Assikis, V.; Jha, G.; Ablaza, A.-J.; Habeck, J.; Loehr, A.; Xiao, J.; Gangolli, E.A. Abstract 445: Genomic Characteristics and Response to Rucaparib and Enzalutamide in the Phase 1b RAMP Study of Metastatic Castration-Resistant Prostate Cancer (MCRPC) Patients. Cancer Res. 2021, 81, 445. [Google Scholar] [CrossRef]
- Rao, A.; Ryan, C.J.; VanderWeele, D.J.; Heller, G.; Lewis, L.D.; Watt, C.; Chen, R.C.; Grubb, R.; Hahn, O.M.; Beltran, H.; et al. CASPAR (Alliance A031902): A Randomized, Phase III Trial of Enzalutamide (ENZ) with Rucaparib (RUCA)/Placebo (PBO) as a Novel Therapy in First-Line Metastatic Castration-Resistant Prostate Cancer (MCRPC). J. Clin. Oncol. 2021, 39, TPS181. [Google Scholar] [CrossRef]
- ClinicalTrials.gov. A Clinical Study Evaluating The Benefit of Adding Rucaparib to Enzalutamide for Men With Metastatic Prostate Cancer That Has Become Resistant To Testosterone-Deprivation Therapy. Available online: https://clinicaltrials.gov/ct2/show/NCT04455750 (accessed on 17 September 2020).
- AstraZeneca. A Randomised, Double-Blind, Placebo-Controlled, Multicentre Phase III Study of Olaparib Plus Abiraterone Relative to Placebo Plus Abiraterone as First-Line Therapy in Men with Metastatic Castration-Resistant Prostate Cancer (PROpel Study) 2021. Available online: https://fdaaa.trialstracker.net/trial/NCT03732820/ (accessed on 23 January 2022).
- Janssen Research & Development, LLC. A Phase 3 Randomized, Placebo-Controlled, Double-Blind Study of Niraparib in Combination with Abiraterone Acetate and Prednisone Versus Abiraterone Acetate and Prednisone in Subjects With Metastatic Prostate Cancer 2021. Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2020.38.6_suppl.TPS257 (accessed on 23 January 2022).
- Agarwal, N.; Shore, N.D.; Dunshee, C.; Karsh, L.I.; Azad, A.; Fay, A.P.; Carles, J.; Sullivan, B.; Di Santo, N.; Elmeliegy, M.; et al. TALAPRO-2: A Placebo-Controlled Phase III Study of Talazoparib (TALA) plus Enzalutamide (ENZA) for Patients with First-Line Metastatic Castration-Resistant Prostate Cancer (MCRPC). J. Clin. Oncol. 2020, 38, TPS264. [Google Scholar] [CrossRef]
- Pfizer. A Phase 3, Randomized, Double-Blind, Placebo-Controlled Study of Talazoparib with Enzalutamide in Metastatic Castration-Resistant Prostate Cancer (NCT03395197) 2021. Available online: https://ascopubs.org/doi/abs/10.1200/JCO.2021.39.15_suppl.TPS5089 (accessed on 23 January 2022).
- Jackson, W.C.; Hartman, H.E.; Dess, R.T.; Birer, S.R.; Soni, P.D.; Hearn, J.W.D.; Reichert, Z.R.; Kishan, A.U.; Mahal, B.A.; Zumsteg, Z.S.; et al. Addition of Androgen-Deprivation Therapy or Brachytherapy Boost to External Beam Radiotherapy for Localized Prostate Cancer: A Network Meta-Analysis of Randomized Trials. J. Clin. Oncol. 2020, 38, 3024–3031. [Google Scholar] [CrossRef]
- Bolla, M.; Collette, L.; Blank, L.; Warde, P.; Dubois, J.B.; Mirimanoff, R.-O.; Storme, G.; Bernier, J.; Kuten, A.; Sternberg, C.; et al. Long-Term Results with Immediate Androgen Suppression and External Irradiation in Patients with Locally Advanced Prostate Cancer (an EORTC Study): A Phase III Randomised Trial. Lancet 2002, 360, 103–106. [Google Scholar] [CrossRef]
- Attard, G. LBA4_PR–Abiraterone Acetate plus Prednisolone (AAP) with or without Enzalutamide (ENZ) Added to Androgen Deprivation Therapy (ADT) Compared to ADT Alone for Men with High-Risk Non-Metastatic (M0) Prostate Cancer (PCa): Combined Analysis from Two Comparisons in the STAMPEDE Platform Protocol. Ann. Oncol. 2021, 32, S1283–S1346. [Google Scholar] [CrossRef]

{kind=link}
| Study | Study Type | Sequence (n) | Median PFS | Median PSA-PFS ** | Median OS |
|---|---|---|---|---|---|
| Khalaf et al. [10] | Prospective, randomized | AAP → ENZ (101) | - | 19·3 months (95% CI 16·0–30·5) | 28.8 months (95% CI 25.4–NR) |
| ENZ → AAP (101) | - | 15.2 months (95% CI 11.9–19.8) (p = 0.036) | 24.7 months (95% CI 18.8–34.0) (p = 0.23) | ||
| Terada et al. [11] ## | Retrospective | AAP → ENZ (113) | - | 15.0 months (95% CI 12.6–16.3) | 30.2 months (95% CI 25.0–NR) |
| ENZ → AAP (85) | - | 9.7 months (95% CI 7.7–11.8) (p < 0.001) | 29.5 months (95% CI 24.4–NR) (p = 0.599) | ||
| Mori et al. [12] | Retrospective | AAP → ENZ (23) | NR | 9 months | Not statistically different (p = 0.62) |
| ENZ → AAP (46) | 11 months (p = 0.043) | 7 months (p = 0.049) | |||
| Maughan et al. [11] | Retrospective | AAP → ENZ (65) | 19.5 months (95% CI 15.5–22.3) | 17.5 months (95% CI 14.0–19.5) | 33.3 months (95% CI 2.4–NR) |
| ENZ → AAP (16) | 13.0 months (95% CI 10.3–21.2) | 12.3 months (95% CI 8.9–20.5) | 29.9 months (95% CI 18.8–NR) |
| Study | Study Design | Arms (n) | Patient Selection | Primary Endpoint |
|---|---|---|---|---|
| CASPAR/ A031902 [75,76] | Phase 3, randomized, placebo-controlled, double-blinded | Enzalutamide + Rucaparib (496) Enzalutamide + Placebo (496) | Prior NAA allowed for mHSPC and nmCRPC Prior docetaxel allowed for mHSPC HRR mutation not required | Radiographic PFS and overall survival |
| PROPEL [77] | Phase 3, randomized, placebo-controlled, double-blinded | Abiraterone + Olaparib (360) Abiraterone + Placebo (360) | Prior NAA NOT allowed Prior docetaxel allowed for mHSPC HRR mutation not required | Radiographic PFS |
| MAGNITUDE [78] | Phase 3, randomized, placebo-controlled, double-blinded | HRR-mutant cohort Enzalutamide + Rucaparib (200) Enzalutamide + Placebo (200) | Prior NAA NOT allowed for mHSPC and nmCRPC Prior docetaxel allowed for mHSPC HRR mutation not required | Radiographic PFS in HRR-mutant patients |
| HRR-wt cohort Enzalutamide + Rucaparib (300) Enzalutamide + Placebo (300) | Prior NAA NOT allowed for mHSPC and nmCRPC Prior docetaxel allowed for mHSPC HRR mutation not required | Radiographic PFS in HRR-wt patients | ||
| TALAPRO-2 [79] | Phase 3, randomized, placebo-controlled, double-blinded | Enzalutamide + Talazoparib (509) Enzalutamide + Placebo (509) | Prior abiraterone allowed (no novel AR inhibitors) for mHSPC and nmCRPC Prior docetaxel allowed for mHSPC HRR mutation not required | Radiographic PFS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rao, A.; Moka, N.; Hamstra, D.A.; Ryan, C.J. Co-Inhibition of Androgen Receptor and PARP as a Novel Treatment Paradigm in Prostate Cancer—Where Are We Now? Cancers 2022, 14, 801. https://doi.org/10.3390/cancers14030801
Rao A, Moka N, Hamstra DA, Ryan CJ. Co-Inhibition of Androgen Receptor and PARP as a Novel Treatment Paradigm in Prostate Cancer—Where Are We Now? Cancers. 2022; 14(3):801. https://doi.org/10.3390/cancers14030801
Chicago/Turabian StyleRao, Arpit, Nagaishwarya Moka, Daniel A. Hamstra, and Charles J. Ryan. 2022. "Co-Inhibition of Androgen Receptor and PARP as a Novel Treatment Paradigm in Prostate Cancer—Where Are We Now?" Cancers 14, no. 3: 801. https://doi.org/10.3390/cancers14030801
APA StyleRao, A., Moka, N., Hamstra, D. A., & Ryan, C. J. (2022). Co-Inhibition of Androgen Receptor and PARP as a Novel Treatment Paradigm in Prostate Cancer—Where Are We Now? Cancers, 14(3), 801. https://doi.org/10.3390/cancers14030801