
Uveal Melanoma Cell Line Proliferation Is Inhibited by Ricolinostat, a Histone Deacetylase Inhibitor

 , , , , ,
, , , , ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
Simple Summary
Abstract
1. Introduction
2. Results
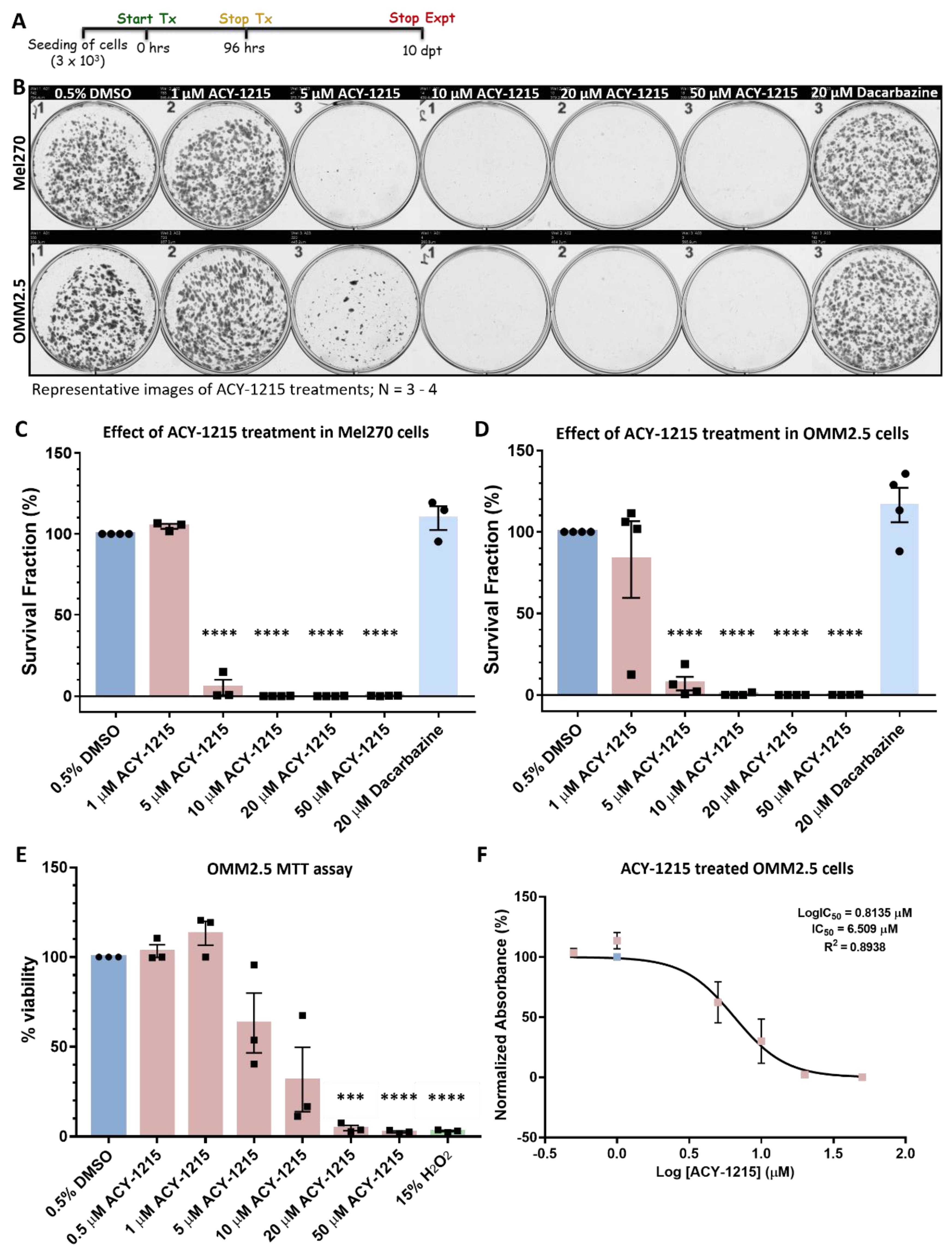
2.1. ACY-1215 Significantly Attenuates Long-Term Proliferation of Human Uveal Melanoma Cell Lines
2.2. Zebrafish OMM2.5 Xenografts Proved That ACY-1215 Is Efficacious In Vivo
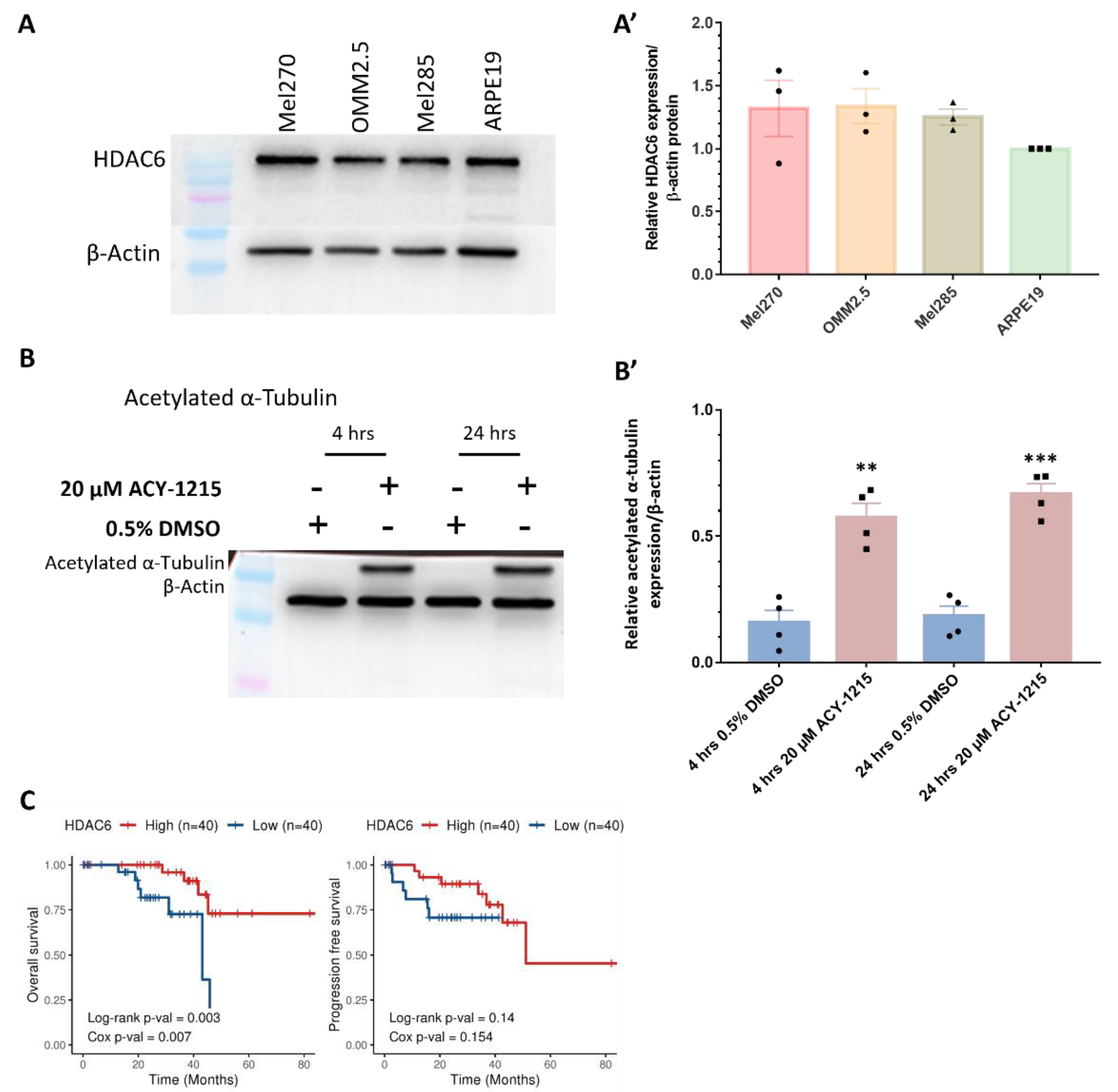
2.3. Analysis of ACY-1215 Targets in UM Patient Samples and UM Cells
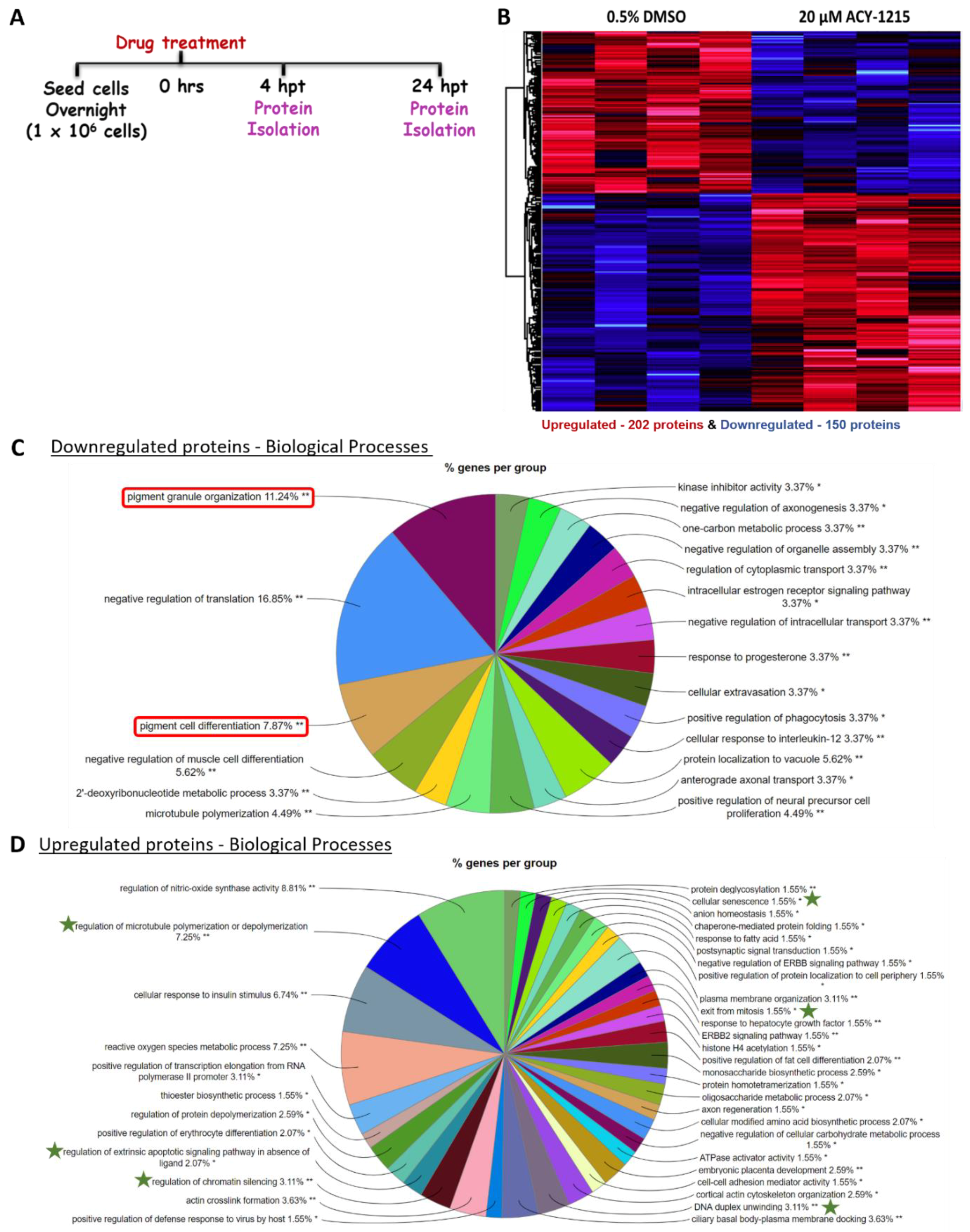
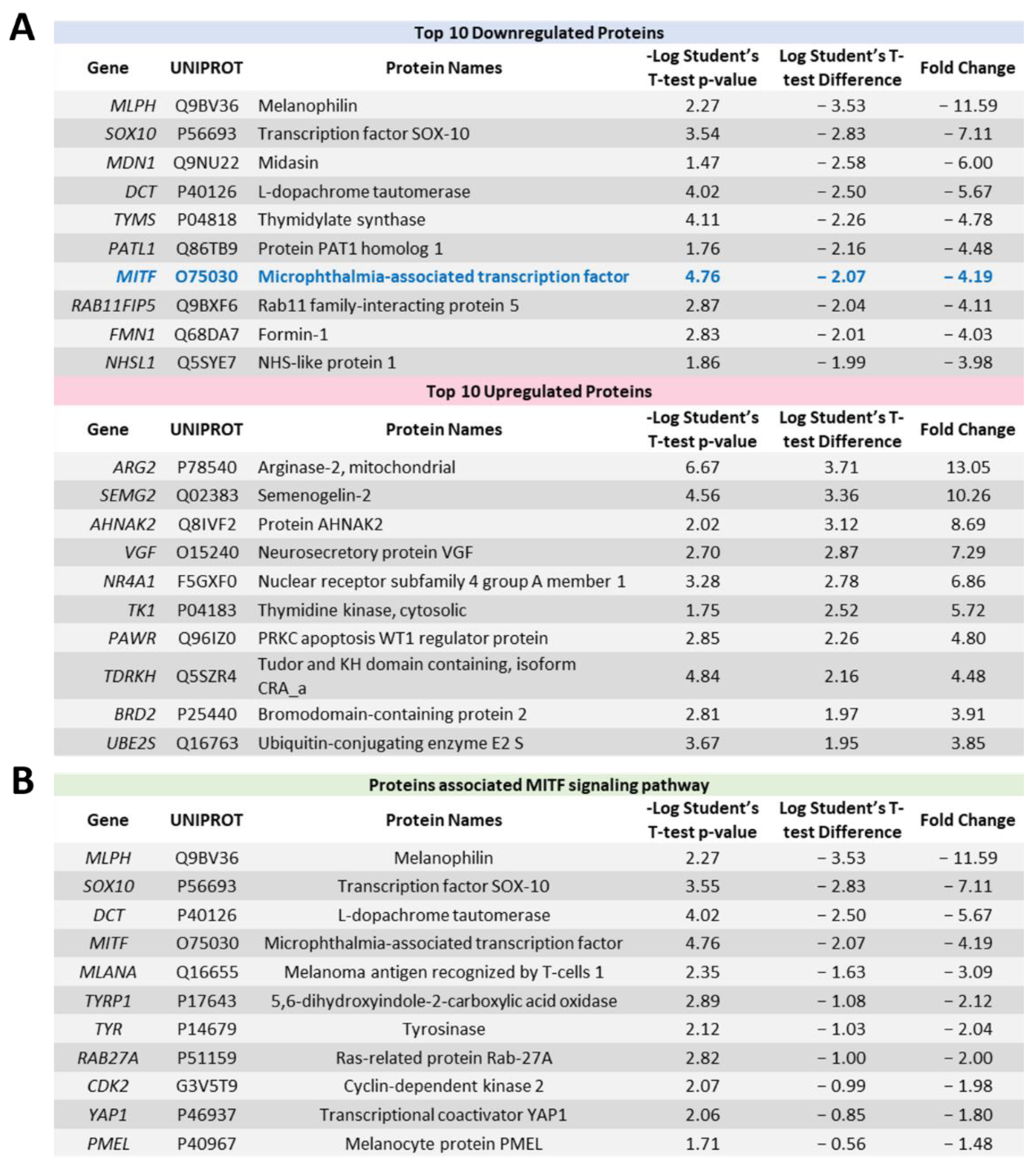
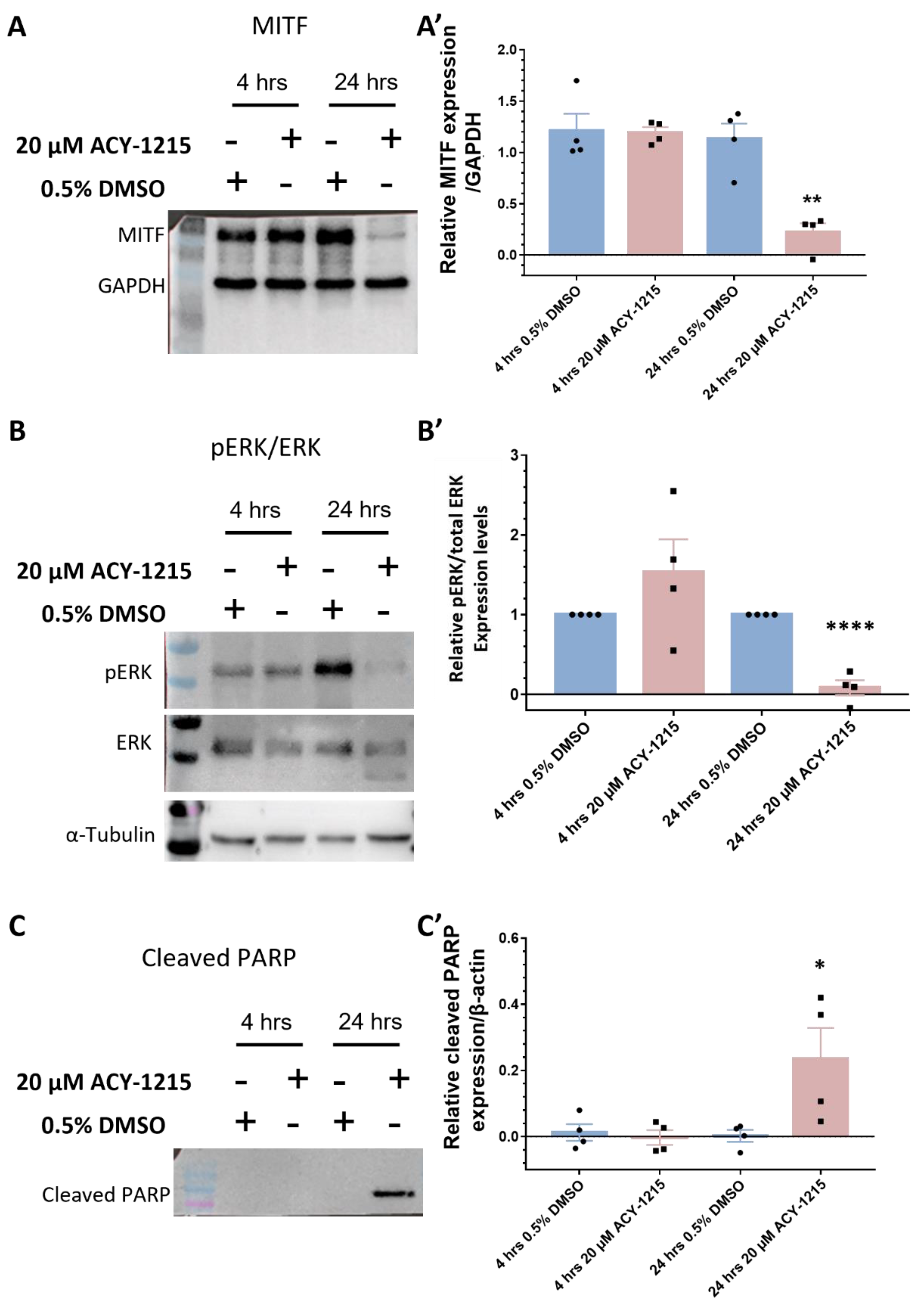
2.4. Proteome Profiling Uncovers Molecular Signals Altered in OMM2.5 Cells by ACY-1215
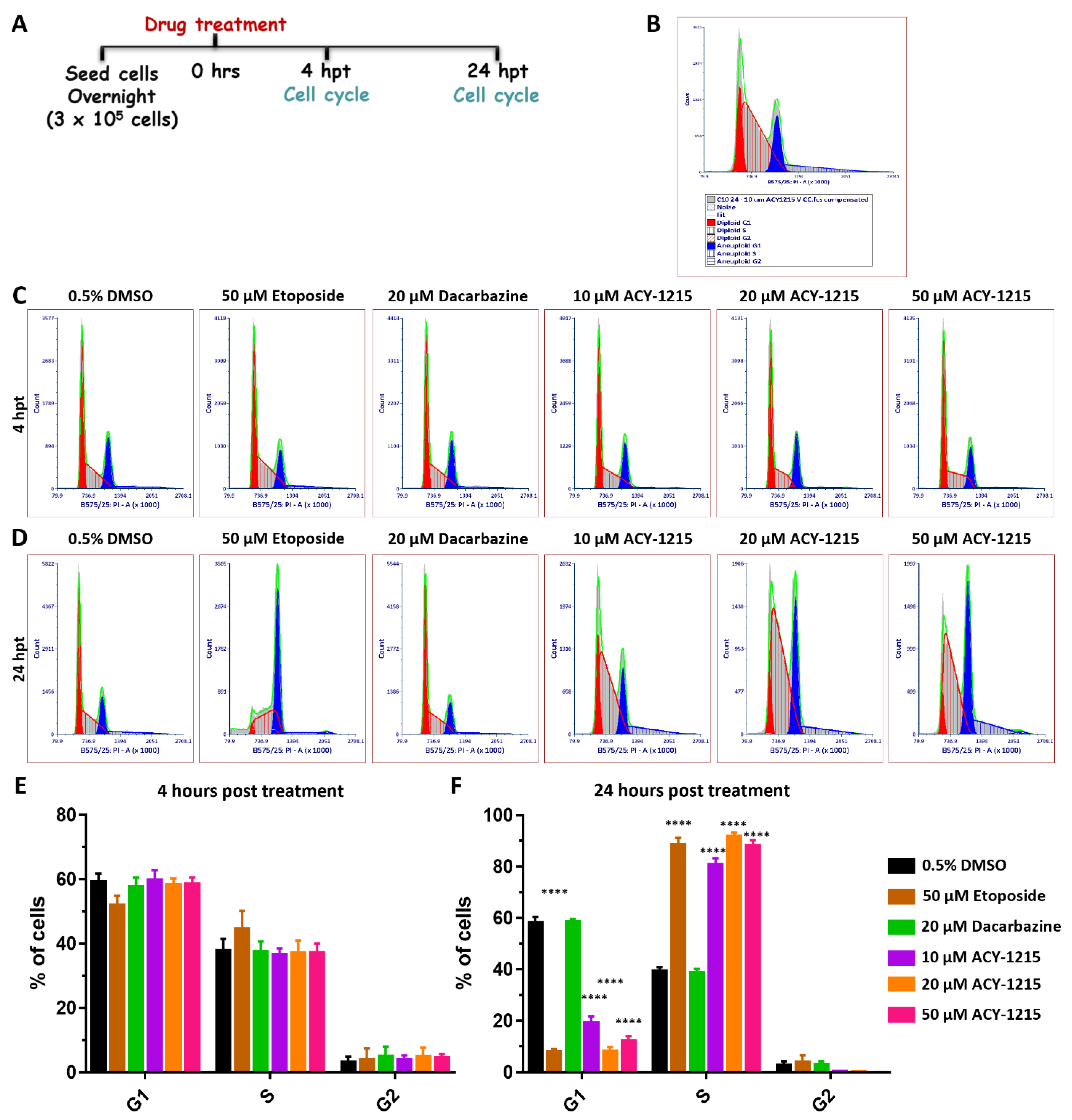
2.5. ACY-1215 Treatment Arrests OMM2.5 Cell Cycle Progression in S Phase
2.6. Elevated Apoptosis Results from ACY-1215 Treatment of OMM2.5 Cells
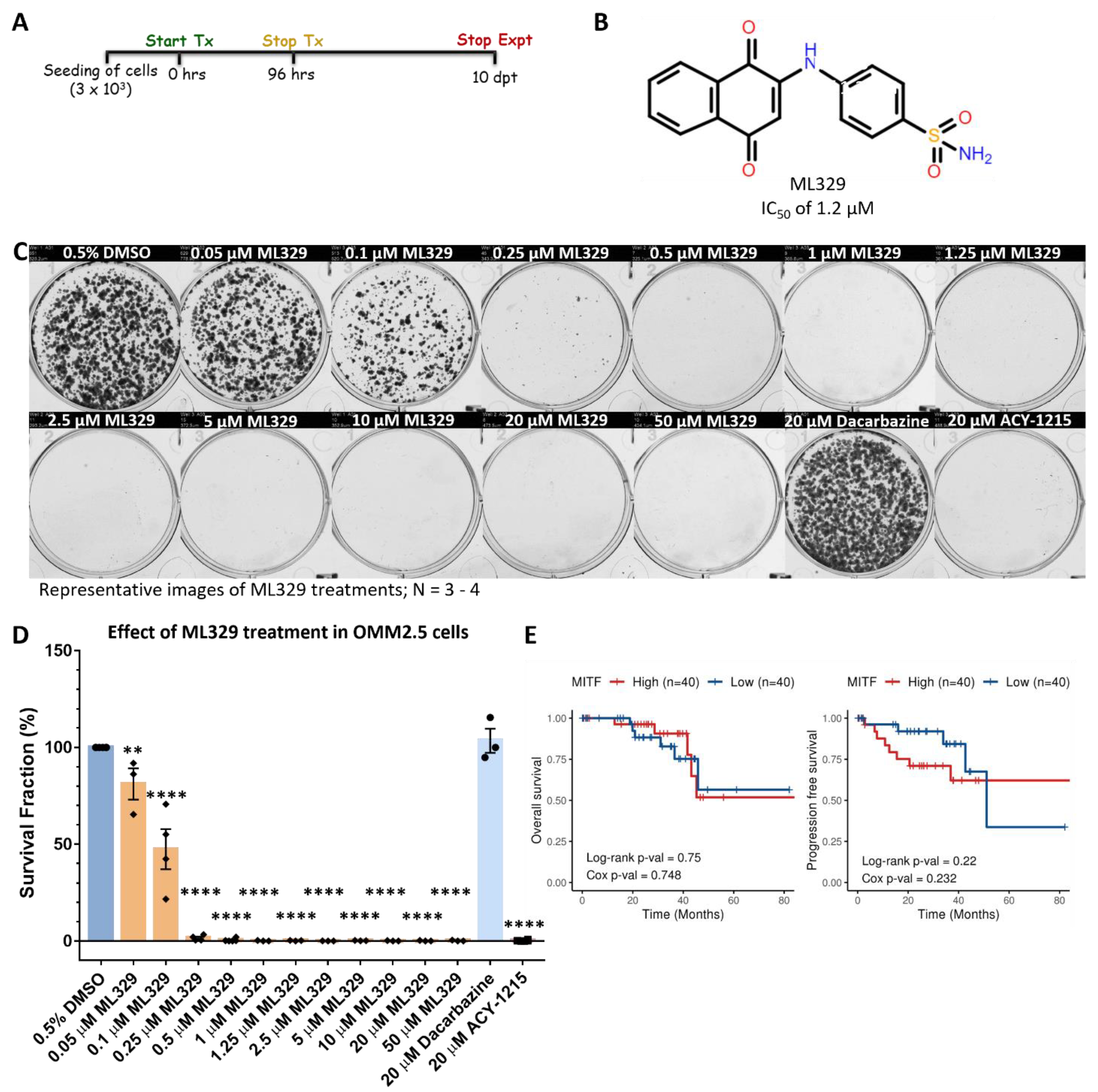
2.7. MITF Inhibitor Treatment Prevents OMM2.5 Cell Survival and Proliferation In Vitro
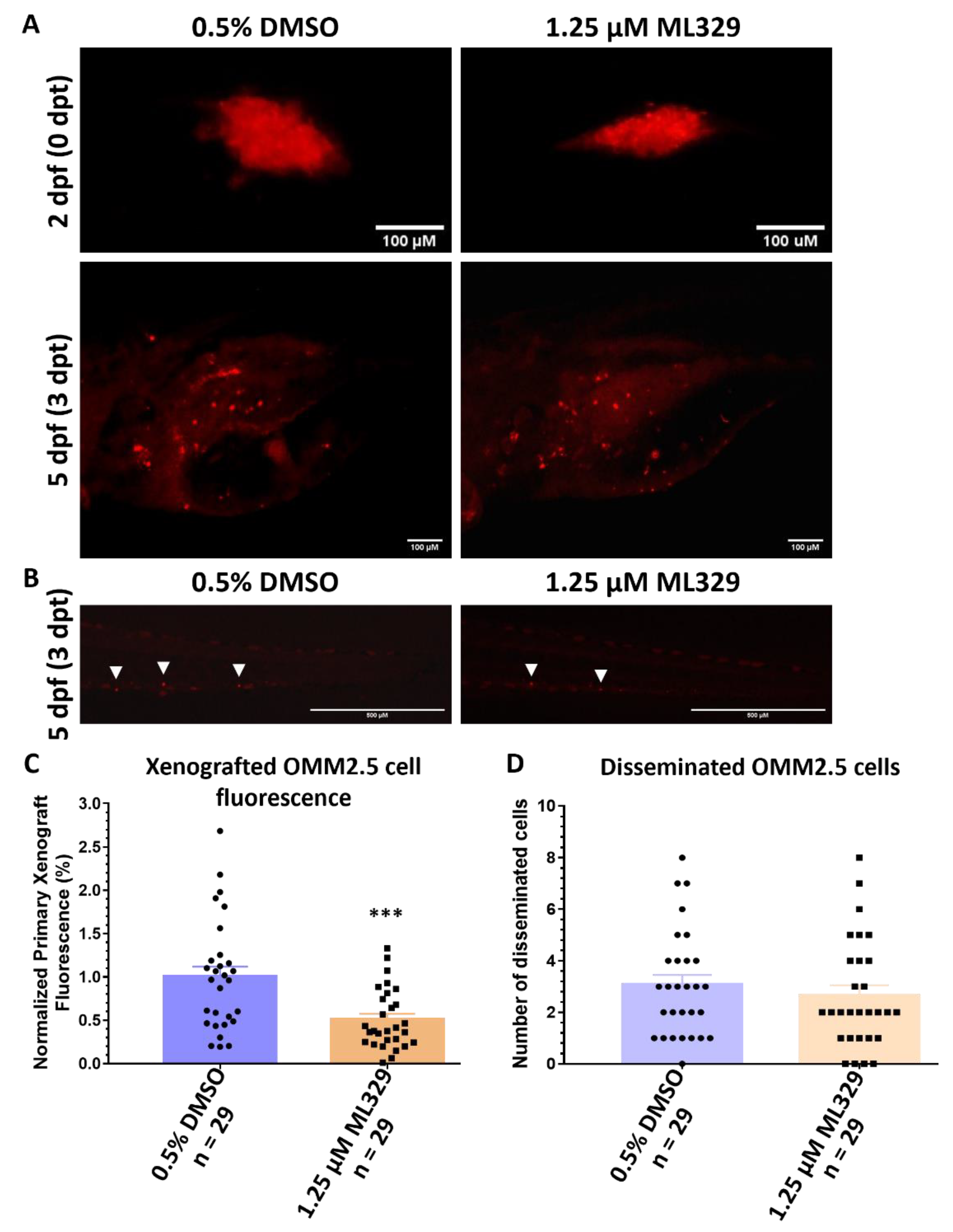
2.8. Inhibition of MITF Pathway Reduces OMM2.5 Cell Fluorescence In Vivo in Zebrafish Xenograft Models
3. Discussion
4. Methods
4.1. Cell Culture
4.2. Clonogenic Assay
4.3. MTT Assay
4.4. OMM2.5 Zebrafish Xenografts
4.5. Proteome Profiling and Mass Spectrometry Analysis
4.6. Western Blot Analysis
4.7. Flow Cytometry Analysis
4.8. TCGA Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grisanti, S.; Tura, A. Uveal Melanoma. In Noncutaneous Melanoma; Scott, J.F., Gerstenblith, M.R., Eds.; Codon Publications: Brisbane, Australia, 2018; Chapter 1. [Google Scholar]
- Jager, M.J.; Shields, C.L.; Cebulla, C.M.; Abdel-Rahman, M.H.; Grossniklaus, H.E.; Stern, M.H.; Carvajal, R.D.; Belfort, R.N.; Jia, R.; Shields, J.A.; et al. Uveal melanoma. Nat. Rev. Dis. Prim. 2020, 6, 24. [Google Scholar] [CrossRef] [PubMed]
- Baily, C.; O’Neill, V.; Dunne, M.; Cunningham, M.; Gullo, G.; Kennedy, S.; Walsh, P.M.; Deady, S.; Horgan, N. Uveal Melanoma in Ireland. Ocul. Oncol. Pathol. 2019, 5, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Krantz, B.A.; Dave, N.; Komatsubara, K.M.; Marr, B.P.; Carvajal, R.D. Uveal melanoma: Epidemiology, etiology, and treatment of primary disease. Clin. Ophthalmol. 2017, 11, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Kaliki, S.; Shields, C.L. Uveal melanoma: Relatively rare but deadly cancer. Eye 2017, 31, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Manson, D.K.; Marr, B.P.; Carvajal, R.D. Treatment of uveal melanoma: Where are we now? Ther. Adv. Med. Oncol. 2018, 10, 1758834018757175. [Google Scholar] [CrossRef]
- Slater, K.; Hoo, P.S.; Buckley, A.M.; Piulats, J.M.; Villanueva, A.; Portela, A.; Kennedy, B.N. Evaluation of oncogenic cysteinyl leukotriene receptor 2 as a therapeutic target for uveal melanoma. Cancer Metastasis. Rev. 2018, 37, 335–345. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Schwartz, G.K.; Tezel, T.; Marr, B.; Francis, J.H.; Nathan, P.D. Metastatic disease from uveal melanoma: Treatment options and future prospects. Br. J. Ophthalmol. 2017, 101, 38–44. [Google Scholar] [CrossRef]
- Rodriguez-Vidal, C.; Fernandez-Diaz, D.; Fernandez-Marta, B.; Lago-Baameiro, N.; Pardo, M.; Silva, P.; Paniagua, L.; Blanco-Teijeiro, M.J.; Pineiro, A.; Bande, M. Treatment of Metastatic Uveal Melanoma: Systematic Review. Cancers 2020, 12, 2557. [Google Scholar] [CrossRef]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in Combination with Dacarbazine in Patients with Metastatic Uveal Melanoma: A Phase III, Multicenter, Randomized Trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef]
- O’Neill, P.A.; Butt, M.; Eswar, C.V.; Gillis, P.; Marshall, E. A prospective single arm phase II study of dacarbazine and treosulfan as first-line therapy in metastatic uveal melanoma. Melanoma Res. 2006, 16, 245–248. [Google Scholar] [CrossRef]
- Schinzari, G.; Rossi, E.; Cassano, A.; Dadduzio, V.; Quirino, M.; Pagliara, M.; Blasi, M.A.; Barone, C. Cisplatin, dacarbazine and vinblastine as first line chemotherapy for liver metastatic uveal melanoma in the era of immunotherapy: A single institution phase II study. Melanoma Res. 2017, 27, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Woodman, S.E. Metastatic uveal melanoma: Biology and emerging treatments. Cancer J. 2012, 18, 148–152. [Google Scholar] [CrossRef] [PubMed]
- Chua, V.; Mattei, J.; Han, A.; Johnston, L.; LiPira, K.; Selig, S.M.; Carvajal, R.D.; Aplin, A.E.; Patel, S.P. The Latest on Uveal Melanoma Research and Clinical Trials: Updates from the Cure Ocular Melanoma (CURE OM) Science Meeting (2019). Clin. Cancer Res. 2021, 27, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Pandiani, C.; Beranger, G.E.; Leclerc, J.; Ballotti, R.; Bertolotto, C. Focus on cutaneous and uveal melanoma specificities. Genes Dev. 2017, 31, 724–743. [Google Scholar] [CrossRef] [PubMed]
- Nathan, P.; Hassel, J.C.; Rutkowski, P.; Baurain, J.F.; Butler, M.O.; Schlaak, M.; Sullivan, R.J.; Ochsenreither, S.; Dummer, R.; Kirkwood, J.M.; et al. Overall Survival Benefit with Tebentafusp in Metastatic Uveal Melanoma. N. Engl. J. Med. 2021, 385, 1196–1206. [Google Scholar] [CrossRef]
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 2011, 875824. [Google Scholar] [CrossRef]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- McClure, J.J.; Li, X.; Chou, C.J. Advances and Challenges of HDAC Inhibitors in Cancer Therapeutics. Adv. Cancer Res. 2018, 138, 183–211. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Jenke, R.; Ressing, N.; Hansen, F.K.; Aigner, A.; Buch, T. Anticancer Therapy with HDAC Inhibitors: Mechanism-Based Combination Strategies and Future Perspectives. Cancers 2021, 13, 634. [Google Scholar] [CrossRef]
- Lu, X.; Ning, Z.; Li, Z.; Cao, H.; Wang, X. Development of chidamide for peripheral T-cell lymphoma, the first orphan drug approved in China. Intractable Rare Dis. Res. 2016, 5, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Moschos, M.M.; Dettoraki, M.; Androudi, S.; Kalogeropoulos, D.; Lavaris, A.; Garmpis, N.; Damaskos, C.; Garmpi, A.; Tsatsos, M. The Role of Histone Deacetylase Inhibitors in Uveal Melanoma: Current Evidence. Anticancer Res. 2018, 38, 3817–3824. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, H.; Olofsson Bagge, R.; Ullenhag, G.; Carneiro, A.; Helgadottir, H.; Ljuslinder, I.; Levin, M.; All-Eriksson, C.; Andersson, B.; Stierner, U.; et al. Concomitant use of pembrolizumab and entinostat in adult patients with metastatic uveal melanoma (PEMDAC study): Protocol for a multicenter phase II open label study. BMC Cancer 2019, 19, 415. [Google Scholar] [CrossRef] [PubMed]
- Ny, L.; Jespersen, H.; Karlsson, J.; Alsen, S.; Filges, S.; All-Eriksson, C.; Andersson, B.; Carneiro, A.; Helgadottir, H.; Levin, M.; et al. The PEMDAC phase 2 study of pembrolizumab and entinostat in patients with metastatic uveal melanoma. Nat. Commun. 2021, 12, 5155. [Google Scholar] [CrossRef]
- Nencetti, S.; Cuffaro, D.; Nuti, E.; Ciccone, L.; Rossello, A.; Fabbi, M.; Ballante, F.; Ortore, G.; Carbotti, G.; Campelli, F.; et al. Identification of histone deacetylase inhibitors with (arylidene)aminoxy scaffold active in uveal melanoma cell lines. J Enzyme Inhib. Med. Chem. 2021, 36, 34–47. [Google Scholar] [CrossRef]
- Amengual, J.E.; Lue, J.K.; Ma, H.; Lichtenstein, R.; Shah, B.; Cremers, S.; Jones, S.; Sawas, A. First-in-Class Selective HDAC6 Inhibitor (ACY-1215) Has a Highly Favorable Safety Profile in Patients with Relapsed and Refractory Lymphoma. Oncologist 2021, 26, e184–e366. [Google Scholar] [CrossRef]
- Lee, E.K.; Tan-Wasielewski, Z.; Matulonis, U.A.; Birrer, M.J.; Wright, A.A.; Horowitz, N.; Konstantinopoulos, P.A.; Curtis, J.; Liu, J.F. Results of an abbreviated Phase Ib study of the HDAC6 inhibitor ricolinostat and paclitaxel in recurrent ovarian, fallopian tube, or primary peritoneal cancer. Gynecol. Oncol. Rep. 2019, 29, 118–122. [Google Scholar] [CrossRef]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef]
- Li, T.; Zhang, C.; Hassan, S.; Liu, X.; Song, F.; Chen, K.; Zhang, W.; Yang, J. Histone deacetylase 6 in cancer. J. Hematol. Oncol. 2018, 11, 111. [Google Scholar] [CrossRef]
- Boyault, C.; Sadoul, K.; Pabion, M.; Khochbin, S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007, 26, 5468–5476. [Google Scholar] [CrossRef]
- Auzmendi-Iriarte, J.; Saenz-Antonanzas, A.; Mikelez-Alonso, I.; Carrasco-Garcia, E.; Tellaetxe-Abete, M.; Lawrie, C.H.; Sampron, N.; Cortajarena, A.L.; Matheu, A. Characterization of a new small-molecule inhibitor of HDAC6 in glioblastoma. Cell Death Dis. 2020, 11, 417. [Google Scholar] [CrossRef] [PubMed]
- Moufarrij, S.; Srivastava, A.; Gomez, S.; Hadley, M.; Palmer, E.; Austin, P.T.; Chisholm, S.; Roche, K.; Yu, A.; Li, J.; et al. Combining DNMT and HDAC6 inhibitors increases anti-tumor immune signaling and decreases tumor burden in ovarian cancer. Sci. Rep. 2020, 10, 3470. [Google Scholar] [CrossRef]
- Kuroki, H.; Anraku, T.; Kazama, A.; Shirono, Y.; Bilim, V.; Tomita, Y. Histone deacetylase 6 inhibition in urothelial cancer as a potential new strategy for cancer treatment. Oncol. Lett. 2021, 21, 64. [Google Scholar] [CrossRef]
- Jager, M.J.; Magner, J.A.; Ksander, B.R.; Dubovy, S.R. Uveal Melanoma Cell Lines: Where do they come from? (An American Ophthalmological Society Thesis). Trans. Am. Ophthalmol. Soc. 2016, 114, T5. [Google Scholar] [PubMed]
- Chen, P.W.; Murray, T.G.; Uno, T.; Salgaller, M.L.; Reddy, R.; Ksander, B.R. Expression of MAGE genes in ocular melanoma during progression from primary to metastatic disease. Clin. Exp. Metastasis 1997, 15, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Ksander, B.R.; Rubsamen, P.E.; Olsen, K.R.; Cousins, S.W.; Streilein, J.W. Studies of tumor-infiltrating lymphocytes from a human choroidal melanoma. Investig. Ophthalmol. Vis. Sci. 1991, 32, 3198–3208. [Google Scholar]
- Slater, K.; Heeran, A.B.; Garcia-Mulero, S.; Kalirai, H.; Sanz-Pamplona, R.; Rahman, A.; Al-Attar, N.; Helmi, M.; O’Connell, F.; Bosch, R.; et al. High Cysteinyl Leukotriene Receptor 1 Expression Correlates with Poor Survival of Uveal Melanoma Patients and Cognate Antagonist Drugs Modulate the Growth, Cancer Secretome, and Metabolism of Uveal Melanoma Cells. Cancers 2020, 12, 2950. [Google Scholar] [CrossRef]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Depetter, Y.; Geurs, S.; De Vreese, R.; Goethals, S.; Vandoorn, E.; Laevens, A.; Steenbrugge, J.; Meyer, E.; de Tullio, P.; Bracke, M.; et al. Selective pharmacological inhibitors of HDAC6 reveal biochemical activity but functional tolerance in cancer models. Int. J. Cancer. 2019, 145, 735–747. [Google Scholar] [CrossRef]
- Cosenza, M.; Civallero, M.; Marcheselli, L.; Sacchi, S.; Pozzi, S. Ricolinostat, a selective HDAC6 inhibitor, shows anti-lymphoma cell activity alone and in combination with bendamustine. Apoptosis 2017, 22, 827–840. [Google Scholar] [CrossRef]
- Tan, Y.; Zhang, S.; Zhu, H.; Chu, Y.; Zhou, H.; Liu, D.; Huo, J. Histone deacetylase 6 selective inhibitor ACY1215 inhibits cell proliferation and enhances the chemotherapeutic effect of 5-fluorouracil in HCT116 cells. Ann. Transl. Med. 2019, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Czyz, M. Pro-survival role of MITF in melanoma. J. Investig. Dermatol. 2015, 135, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Kim, S.; Lee, M.W.; Jeon, H.J.; Ryu, H.; Kim, J.M.; Lee, H.J. MITF Promotes Cell Growth, Migration and Invasion in Clear Cell Renal Cell Carcinoma by Activating the RhoA/YAP Signal Pathway. Cancers 2021, 13, 2920. [Google Scholar] [CrossRef]
- Loercher, A.E.; Tank, E.M.; Delston, R.B.; Harbour, J.W. MITF links differentiation with cell cycle arrest in melanocytes by transcriptional activation of INK4A. J. Cell Biol. 2005, 168, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Mooy, C.; Vissers, K.; Luyten, G.; Mulder, A.; Stijnen, T.; de Jong, P.; Bosman, F. DNA flow cytometry in uveal melanoma: The effect of pre-enucleation irradiation. Br. J. Ophthalmol. 1995, 79, 174–177. [Google Scholar] [CrossRef][Green Version]
- Ehlers, J.P.; Worley, L.; Onken, M.D.; Harbour, J.W. Integrative genomic analysis of aneuploidy in uveal melanoma. Clin. Cancer Res. 2008, 14, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Faloon, P.W.; Bennion, M.; Weiner, W.S.; Smith, R.A.; Wurst, J.; Weiwer, M.; Hartland, C.; Mosher, C.M.; Johnston, S.; Porubsky, P.; et al. A Small Molecule Inhibitor of the MITF Molecular Pathway. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information: Bethesda, Maryland, 2010. [Google Scholar]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, N.E.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]
- Levinzon, L.; Madigan, M.; Nguyen, V.; Hasic, E.; Conway, M.; Cherepanoff, S. Tumour Expression of Histone Deacetylases in Uveal Melanoma. Ocul. Oncol. Pathol. 2019, 5, 153–161. [Google Scholar] [CrossRef]
- Putcha, P.; Yu, J.; Rodriguez-Barrueco, R.; Saucedo-Cuevas, L.; Villagrasa, P.; Murga-Penas, E.; Quayle, S.N.; Yang, M.; Castro, V.; Llobet-Navas, D.; et al. HDAC6 activity is a non-oncogene addiction hub for inflammatory breast cancers. Breast Cancer Res. 2015, 17, 149. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Yao, T.; Jia, R. Benefits of Zebrafish Xenograft Models in Cancer Research. Front. Cell Dev. Biol. 2021, 9, 616551. [Google Scholar] [CrossRef]
- Gamble, J.T.; Elson, D.J.; Greenwood, J.A.; Tanguay, R.L.; Kolluri, S.K. The Zebrafish Xenograft Models for Investigating Cancer and Cancer Therapeutics. Biology 2021, 10, 252. [Google Scholar] [CrossRef]
- Fazio, M.; Ablain, J.; Chuan, Y.; Langenau, D.M.; Zon, L.I. Zebrafish patient avatars in cancer biology and precision cancer therapy. Nat. Rev. Cancer 2020, 20, 263–273. [Google Scholar] [CrossRef]
- van der Ent, W.; Burrello, C.; Teunisse, A.F.; Ksander, B.R.; van der Velden, P.A.; Jager, M.J.; Jochemsen, A.G.; Snaar-Jagalska, B.E. Modeling of human uveal melanoma in zebrafish xenograft embryos. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6612–6622. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.; Giuliano, C.J.; Palladino, A.; John, K.M.; Abramowicz, C.; Yuan, M.L.; Sausville, E.L.; Lukow, D.A.; Liu, L.; Chait, A.R.; et al. Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials. Sci. Transl. Med. 2019, 11, eaaw8412. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Zhang, F.; Maguire, A.; Byrne, T.; Weiner-Gorzel, K.; Bridgett, S.; O’Toole, S.; O’Leary, J.; Beggan, C.; Fitzpatrick, P.; et al. HDAC6 Degradation Inhibits the Growth of High-Grade Serous Ovarian Cancer Cells. Cancers 2020, 12, 3734. [Google Scholar] [CrossRef] [PubMed]
- Wloga, D.; Joachimiak, E.; Fabczak, H. Tubulin Post-Translational Modifications and Microtubule Dynamics. Int. J. Mol. Sci. 2017, 18, 2207. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Feige, E.; Poling, L.L.; Levy, C.; Widlund, H.R.; Khaled, M.; Kung, A.L.; Fisher, D.E. Pharmacologic suppression of MITF expression via HDAC inhibitors in the melanocyte lineage. Pigment. Cell Melanoma Res. 2008, 21, 457–463. [Google Scholar] [CrossRef]
- Simmons, J.L.; Pierce, C.J.; Al-Ejeh, F.; Boyle, G.M. MITF and BRN2 contribute to metastatic growth after dissemination of melanoma. Sci. Rep. 2017, 7, 10909. [Google Scholar] [CrossRef]
- Vachtenheim, J. The Many Roles of MITF in Melanoma. Single-Cell Biol. 2017, 6, 162. [Google Scholar] [CrossRef]
- Yajima, I.; Kumasaka, M.Y.; Thang, N.D.; Goto, Y.; Takeda, K.; Iida, M.; Ohgami, N.; Tamura, H.; Yamanoshita, O.; Kawamoto, Y.; et al. Molecular Network Associated with MITF in Skin Melanoma Development and Progression. J. Skin Cancer 2011, 2011, 730170. [Google Scholar] [CrossRef]
- Kawakami, A.; Fisher, D.E. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef]
- Goding, C.R.; Arnheiter, H. MITF-the first 25 years. Genes Dev. 2019, 33, 983–1007. [Google Scholar] [CrossRef] [PubMed]
- Vachtenheim, J.; Borovansky, J. Transcription physiology of pigment formation in melanocytes: Central role of MITF. Exp. Dermatol. 2010, 19, 617–627. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, S.A.; Finlay, G.J.; Baguley, B.C.; Askarian-Amiri, M.E. Signaling Pathways in Melanogenesis. Int. J. Mol. Sci. 2016, 17, 1144. [Google Scholar] [CrossRef] [PubMed]
- Shibahara, S.; Takeda, K.; Yasumoto, K.; Udono, T.; Watanabe, K.; Saito, H.; Takahashi, K. Microphthalmia-associated transcription factor (MITF): Multiplicity in structure, function, and regulation. J. Investig. Dermatol. Symp. Proc. 2001, 6, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Levy, C.; Khaled, M.; Fisher, D.E. MITF: Master regulator of melanocyte development and melanoma oncogene. Trends Mol. Med. 2006, 12, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Garraway, L.A.; Widlund, H.R.; Rubin, M.A.; Getz, G.; Berger, A.J.; Ramaswamy, S.; Beroukhim, R.; Milner, D.A.; Granter, S.R.; Du, J.; et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature 2005, 436, 117–122. [Google Scholar] [CrossRef]
- Vachtenheim, J.; Ondrusova, L. Microphthalmia-associated transcription factor expression levels in melanoma cells contribute to cell invasion and proliferation. Exp. Dermatol. 2015, 24, 481–484. [Google Scholar] [CrossRef]
- Aida, S.; Sonobe, Y.; Yuhki, M.; Sakata, K.; Fujii, T.; Sakamoto, H.; Mizuno, T. MITF suppression by CH5552074 inhibits cell growth in melanoma cells. Cancer Chemother. Pharm. 2017, 79, 1187–1193. [Google Scholar] [CrossRef]
- Wiedemann, G.M.; Aithal, C.; Kraechan, A.; Heise, C.; Cadilha, B.L.; Zhang, J.; Duewell, P.; Ballotti, R.; Endres, S.; Bertolotto, C.; et al. Microphthalmia-Associated Transcription Factor (MITF) Regulates Immune Cell Migration into Melanoma. Transl. Oncol. 2019, 12, 350–360. [Google Scholar] [CrossRef]
- Hsiao, Y.J.; Chang, W.H.; Chen, H.Y.; Hsu, Y.C.; Chiu, S.C.; Chiang, C.C.; Chang, G.C.; Chen, Y.J.; Wang, C.Y.; Chen, Y.M.; et al. MITF functions as a tumor suppressor in non-small cell lung cancer beyond the canonically oncogenic role. Aging 2020, 13, 646–674. [Google Scholar] [CrossRef]
- Guhan, S.M.; Artomov, M.; McCormick, S.; Njauw, C.; Stratigos, A.J.; Shannon, K.; Ellisen, L.W.; Tsao, H. Cancer risks associated with the germline MITF(E318K) variant. Sci. Rep. 2020, 10, 17051. [Google Scholar] [CrossRef] [PubMed]
- Nooron, N.; Ohba, K.; Takeda, K.; Shibahara, S.; Chiabchalard, A. Dysregulated Expression of MITF in Subsets of Hepatocellular Carcinoma and Cholangiocarcinoma. Tohoku J. Exp. Med. 2017, 242, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, J.; Shen, H.; Lu, J.; Li, C.; Hu, D.N.; Dong, X.D.; Yan, D.; Tu, L. Epigenetics, microRNAs, and carcinogenesis: Functional role of microRNA-137 in uveal melanoma. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1193–1199. [Google Scholar] [CrossRef]
- Lee, D.H.; Won, H.R.; Ryu, H.W.; Han, J.M.; Kwon, S.H. The HDAC6 inhibitor ACY1215 enhances the anticancer activity of oxaliplatin in colorectal cancer cells. Int. J. Oncol. 2018, 53, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.W.; Shin, D.H.; Lee, D.H.; Won, H.R.; Kwon, S.H. A potent hydroxamic acid-based, small-molecule inhibitor A452 preferentially inhibits HDAC6 activity and induces cytotoxicity toward cancer cells irrespective of p53 status. Carcinogenesis 2018, 39, 72–83. [Google Scholar] [CrossRef]
- Cao, J.; Lv, W.; Wang, L.; Xu, J.; Yuan, P.; Huang, S.; He, Z.; Hu, J. Ricolinostat (ACY-1215) suppresses proliferation and promotes apoptosis in esophageal squamous cell carcinoma via miR-30d/PI3K/AKT/mTOR and ERK pathways. Cell Death Dis. 2018, 9, 817. [Google Scholar] [CrossRef]
- Ruan, Y.; Wang, L.; Lu, Y. HDAC6 inhibitor, ACY1215 suppress the proliferation and induce apoptosis of gallbladder cancer cells and increased the chemotherapy effect of gemcitabine and oxaliplatin. Drug Dev. Res. 2021, 82, 598–604. [Google Scholar] [CrossRef] [PubMed]
- Boulares, A.H.; Yakovlev, A.G.; Ivanova, V.; Stoica, B.A.; Wang, G.; Iyer, S.; Smulson, M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J. Biol. Chem. 1999, 274, 22932–22940. [Google Scholar] [CrossRef]
- Kaufmann, S.H.; Desnoyers, S.; Ottaviano, Y.; Davidson, N.E.; Poirier, G.G. Specific proteolytic cleavage of poly (ADP-ribose) polymerase: An early marker of chemotherapy-induced apoptosis. Cancer Res. 1993, 53, 3976–3985. [Google Scholar]
- Wu, J.Y.; Moses, N.; Bai, W.; Zhang, X.M. Implications of the HDAC6-ERK1 feed-forward loop in immunotherapy. J. Immunol. Sci. 2018, 2, 59–68. [Google Scholar] [CrossRef][Green Version]
- Wu, J.Y.; Xiang, S.; Zhang, M.; Fang, B.; Huang, H.; Kwon, O.K.; Zhao, Y.; Yang, Z.; Bai, W.; Bepler, G.; et al. Histone deacetylase 6 (HDAC6) deacetylates extracellular signal-regulated kinase 1 (ERK1) and thereby stimulates ERK1 activity. J. Biol. Chem. 2018, 293, 1976–1993. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.A.; Zhang, M.; Xiang, S.; Hu, C.; Wu, J.Y.; Zhang, S.; Ryan, M.; Cox, A.D.; Der, C.J.; Fang, B.; et al. Extracellular signal-regulated kinase (ERK) phosphorylates histone deacetylase 6 (HDAC6) at serine 1035 to stimulate cell migration. J. Biol. Chem. 2013, 288, 33156–33170. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Z.H.; Zhou, B.; Chu, Y.; Huo, J.; Tan, Y.; Liu, D. Histone deacetylase 6 is overexpressed and promotes tumor growth of colon cancer through regulation of the MAPK/ERK signal pathway. OncoTargets Ther. 2019, 12, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Peng, U.; Wang, Z.; Pei, S.; Ou, Y.; Hu, P.; Liu, W.; Song, J. ACY-1215 accelerates vemurafenib induced cell death of BRAF-mutant melanoma cells via induction of ER stress and inhibition of ERK activation. Oncol. Rep. 2017, 37, 1270–1276. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chuang, M.J.; Wu, S.T.; Tang, S.H.; Lai, X.M.; Lai, H.C.; Hsu, K.H.; Sun, K.H.; Sun, G.H.; Chang, S.Y.; Yu, D.S.; et al. The HDAC inhibitor LBH589 induces ERK-dependent prometaphase arrest in prostate cancer via HDAC6 inactivation and down-regulation. PLoS ONE 2013, 8, e73401. [Google Scholar] [CrossRef]
- Lim, J.A.; Juhnn, Y.S. Isoproterenol increases histone deacetylase 6 expression and cell migration by inhibiting ERK signaling via PKA and Epac pathways in human lung cancer cells. Exp. Mol. Med. 2016, 48, e204. [Google Scholar] [CrossRef]
- Boru, G.; Cebulla, C.M.; Sample, K.M.; Massengill, J.B.; Davidorf, F.H.; Abdel-Rahman, M.H. Heterogeneity in Mitogen-Activated Protein Kinase (MAPK) Pathway Activation in Uveal Melanoma with Somatic GNAQ and GNA11 Mutations. Invest Ophthalmol. Vis. Sci. 2019, 60, 2474–2480. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.; Tan, L.; Porter, D.; Jager, M.J.; Emery, C.; Bastian, B.C. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene 2014, 33, 4724–4734. [Google Scholar] [CrossRef]
- Steeb, T.; Wessely, A.; Ruzicka, T.; Heppt, M.V.; Berking, C. How to MEK the best of uveal melanoma: A systematic review on the efficacy and safety of MEK inhibitors in metastatic or unresectable uveal melanoma. Eur. J. Cancer 2018, 103, 41–51. [Google Scholar] [CrossRef]
- Sagoo, M.S.; Harbour, J.W.; Stebbing, J.; Bowcock, A.M. Combined PKC and MEK inhibition for treating metastatic uveal melanoma. Oncogene 2014, 33, 4722–4723. [Google Scholar] [CrossRef] [PubMed]
- Zuidervaart, W.; van Nieuwpoort, F.; Stark, M.; Dijkman, R.; Packer, L.; Borgstein, A.M.; Pavey, S.; van der Velden, P.; Out, C.; Jager, M.J.; et al. Activation of the MAPK pathway is a common event in uveal melanomas although it rarely occurs through mutation of BRAF or RAS. Br. J. Cancer 2005, 92, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Abdel Karim, N.; Eldessouki, I.; Taftaf, A.; Ayham, D.; Gaber, O.; Makramalla, A.; Correa, Z.M. GNQ-209P Mutation in Metastatic Uveal Melanoma and Treatment Outcome. Case Rep. Oncol. Med. 2018, 2018, 4256365. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef] [PubMed]
- Mouriaux, F.; Vincent, S.; Kherrouche, Z.; Maurage, C.A.; Planque, N.; Monte, D.; Labalette, P.; Saule, S. Microphthalmia transcription factor analysis in posterior uveal melanomas. Exp. Eye Res. 2003, 76, 653–661. [Google Scholar] [CrossRef]
- Iwamoto, S.; Burrows, R.C.; Grossniklaus, H.E.; Orcutt, J.; Kalina, R.E.; Boehm, M.; Bothwell, M.A.; Schmidt, R. Immunophenotype of conjunctival melanomas: Comparisons with uveal and cutaneous melanomas. Arch. Ophthalmol. 2002, 120, 1625–1629. [Google Scholar] [CrossRef][Green Version]
- Perrino, C.M.; Wang, J.F.; Collins, B.T. Microphthalmia transcription factor immunohistochemistry for FNA biopsy of ocular malignant melanoma. Cancer Cytopathol. 2015, 123, 394–400. [Google Scholar] [CrossRef]
- Maurya, D.K. ColonyCountJ: A User-Friendly Image J Add-on Program for Quantification of Different Colony Parameters in Clonogenic Assay. J. Clin. Toxicol. 2017, 7, 2161. [Google Scholar] [CrossRef]
- Rouhi, P.; Jensen, L.D.; Cao, Z.; Hosaka, K.; Lanne, T.; Wahlberg, E.; Steffensen, J.F.; Cao, Y. Hypoxia-induced metastasis model in embryonic zebrafish. Nat. Protoc. 2010, 5, 1911–1918. [Google Scholar] [CrossRef]
- Sundaramurthi, H.; Roche, S.L.; Grice, G.L.; Moran, A.; Dillion, E.T.; Campiani, G.; Nathan, J.A.; Kennedy, B.N. Selective Histone Deacetylase 6 Inhibitors Restore Cone Photoreceptor Vision or Outer Segment Morphology in Zebrafish and Mouse Models of Retinal Blindness. Front. Cell Dev. Biol. 2020, 8, 689. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pages, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sundaramurthi, H.; García-Mulero, S.; Tonelotto, V.; Slater, K.; Marcone, S.; Piulats, J.M.; Watson, R.W.; Tobin, D.J.; Jensen, L.D.; Kennedy, B.N. Uveal Melanoma Cell Line Proliferation Is Inhibited by Ricolinostat, a Histone Deacetylase Inhibitor. Cancers 2022, 14, 782. https://doi.org/10.3390/cancers14030782
Sundaramurthi H, García-Mulero S, Tonelotto V, Slater K, Marcone S, Piulats JM, Watson RW, Tobin DJ, Jensen LD, Kennedy BN. Uveal Melanoma Cell Line Proliferation Is Inhibited by Ricolinostat, a Histone Deacetylase Inhibitor. Cancers. 2022; 14(3):782. https://doi.org/10.3390/cancers14030782
Chicago/Turabian StyleSundaramurthi, Husvinee, Sandra García-Mulero, Valentina Tonelotto, Kayleigh Slater, Simone Marcone, Josep M. Piulats, Ronald William Watson, Desmond J. Tobin, Lasse D. Jensen, and Breandán N. Kennedy. 2022. "Uveal Melanoma Cell Line Proliferation Is Inhibited by Ricolinostat, a Histone Deacetylase Inhibitor" Cancers 14, no. 3: 782. https://doi.org/10.3390/cancers14030782
APA StyleSundaramurthi, H., García-Mulero, S., Tonelotto, V., Slater, K., Marcone, S., Piulats, J. M., Watson, R. W., Tobin, D. J., Jensen, L. D., & Kennedy, B. N. (2022). Uveal Melanoma Cell Line Proliferation Is Inhibited by Ricolinostat, a Histone Deacetylase Inhibitor. Cancers, 14(3), 782. https://doi.org/10.3390/cancers14030782